
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to substituted cyclobutenes by tandem [3,3]-sigmatropic rearrangement/[2 + 2] cycloaddition of dipropargylphosphonates under Ag/Co relay catalysis†
Qijian
Ni‡
 ab,
Xiaoxiao
Song‡
ab,
Chin Wen
Png
c,
Yongliang
Zhang
*c and
Yu
Zhao
*bd
ab,
Xiaoxiao
Song‡
ab,
Chin Wen
Png
c,
Yongliang
Zhang
*c and
Yu
Zhao
*bd
aKey Laboratory of Functionalized Molecular Solids, Ministry of Education, Anhui Key Laboratory of Molecule-Based Materials (State Key Laboratory Cultivation Base), College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241002, Anhui, China
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: zhaoyu@nus.edu.sg
cCentre for Life Sciences, National University of Singapore, #03-09, 28 Medical Drive, 117456, Singapore. E-mail: miczy@nus.edu.sg
dJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
First published on 16th October 2020
Abstract
We present herein an unconventional tandem [3,3]-sigmatropic rearrangement/[2 + 2] cycloaddition of simple dipropargylphosphonates to deliver a range of bicyclic polysubstituted cyclobutenes and cyclobutanes under Ag/Co relay catalysis. An interesting switch from allene–allene to allene–alkyne cycloaddition was observed based on the substitution of the substrates, which further diversified the range of compounds accessible from this practical method. Significantly, preliminary biological screening of these new compounds identified promising candidates as suppressors of cellular proliferation.
Introduction
Carbocyclic four-membered rings represent important structural motifs in a range of natural and synthetic bioactive molecules (Fig. 1). They also serve as valuable building blocks in organic synthesis.1 Accordingly, considerable efforts have been devoted to their preparation, with [2 + 2] cycloaddition representing the most straightforward approach.2,3 The cycloaddition of allenes, in particular, is capable of delivering four-membered rings with unsaturated substituents that can be further converted to various functionalities.4 Many elegant catalytic systems using Ru, Pd, Au, Co and organocatalysts were developed, which successfully promoted intra and intermolecular [2 + 2] cycloaddition of allenes with alkenes and even alkynes (Scheme 1a). However, all of these reported strategies required the use of pre-synthesized allene substrates that are sensitive compounds to handle. If reliable catalytic procedures can be developed to promote tandem allene formation from readily available substrates followed by cycloaddition in high efficiency, it will surely lead to more practical access to valuable four-membered carbocyclic compounds.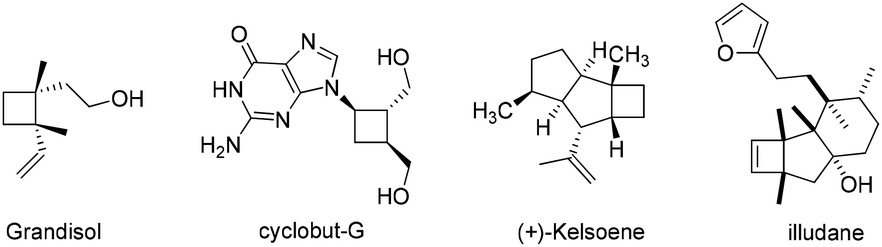 | ||
| Fig. 1 Examples of cyclobutanes/cyclobutenes in bioactive compounds. | ||
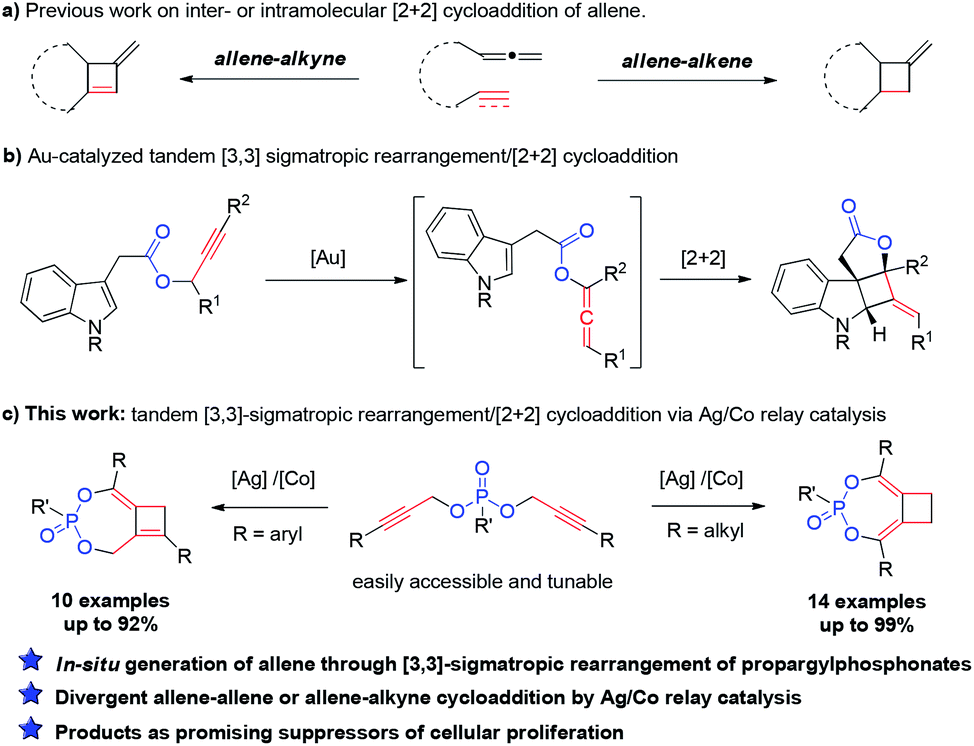 | ||
| Scheme 1 Tandem [3,3]-sigmatropic rearrangement/[2 + 2] cycloadditions. | ||
The [3,3]-sigmatropic rearrangement of propargylic carboxylates has been well-established under gold catalysis, which enabled a facile transformation of these simple substrates to allenylic esters.5 Building on this chemistry, the Zhang group developed an elegant tandem Au(I)-catalyzed [3,3]-rearrangement/[2 + 2] intramolecular cycloaddition of propargylic carboxylates with indoles (Scheme 1b).6 A conceptually related transformation was also disclosed by the Chan group.7 Despite these important advances, a general and modular strategy for effective rearrangement of new functionalities for allene generation in situ as well as cycloaddition with more diverse π-systems is still highly sought-after. To enable effective generation of different structural scaffolds, achieving divergent synthesis from the same family of substrates will be even more desirable. Herein, we present our discovery of an intriguing reactivity of easily accessible dipropargylphosphonates8 under silver/cobalt bimetallic relay catalysis. An unprecedented Ag-catalyzed [3,3]-sigmatropic rearrangement of propargylphosphonate was observed, which was followed by a cobalt-catalyzed intramolecular [2 + 2] cycloaddition to deliver bicyclo[5.2.0]phosphonates bearing poly-substituted cyclobutanes or cyclobutenes in high efficiency (Scheme 1c). Interestingly, either an allene–allene cycloaddition or allene–alkyne cycloaddition was observed based on the substitution of the substrates, which further diversified the range of compounds accessible from this practical method. More importantly, such structures were also demonstrated to be promising lead compounds as suppressors of cellular proliferation in cancer cells through biological screening.
Results and discussion
Reaction discovery and optimization
In an effort to evaluate novel ligands prepared in our group, we chose dipropargylphosphonate 1a as the model substrate and tried to achieve an intramolecular hydroarylation-type reaction (Table 1). Initial studies focused on the use of a cationic rhodium catalyst that could be generated from [Cp*RhCl2]2 and AgBF4. As shown in entry 1, Table 1, instead of obtaining the proposed hydroarylation product, we were surprised to observe the formation of a completely unexpected bicyclic cyclobutane product 2a in moderate yield. The formation of 2a was believed to follow an interesting mechanism of tandem [3,3]-sigmatropic rearrangement/[2 + 2] cycloaddition. Based on our group's continuous interest in transition metal-catalyzed cycloadditions to access carbocycles and heterocycles of various ring sizes,9 as well as the synthetic utility of cyclobutanes, we decided to turn our attention to the optimization of 2a formation.|
|
||||
|---|---|---|---|---|
| Entry | [Ag] | Co-catalyst | Cat loading | Yieldb |
| a The reaction was performed by mixing the substrate and catalysts in the solvent under N2 on a 0.1 mmol scale. b Isolated yield. c 2.5 mol% [Cp*RhCl2]2. n.r. = no reaction. | ||||
| 1 | AgBF4 | [Cp*RhCl2]2c | 10 mol% | 47 |
| 2 | AgBF4 | — | 10 mol% | 62 |
| 3 | AgBF4 | Cu(OAc)2 | 10 mol% | 69 |
| 4 | AgBF4 | Ni(OAc)2·4H2O | 10 mol% | 65 |
| 5 | AgBF4 | Co(acac)2 | 10 mol% | 27 |
| 6 | AgBF4 | Co(acac)3 | 10 mol% | 69 |
| 7 | AgBF4 | Co(OAc)2 | 10 mol% | 90 |
| 8 | — | Co(OAc)2 | 10 mol% | n.r. |
| 9 | AgBF 4 | Co(OAc) 2 | 5 mol% | 99 |
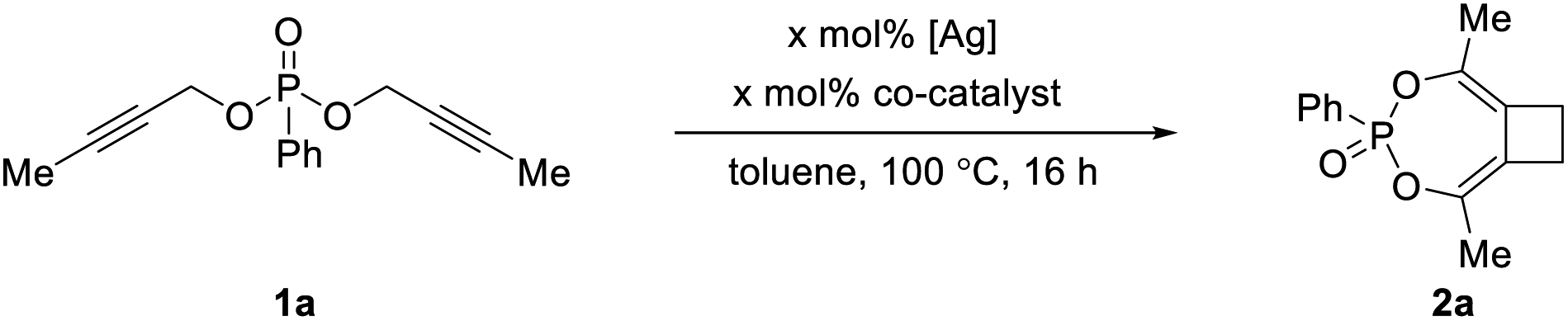
In a control experiment, the silver salt alone proved to be catalytically competent to produce 2a with an even higher yield of 62% (entry 2). Subsequent screening of different silver salts, however, led to no improvement, even though complete consumption of 1a was typically observed (see Table S1 in the ESI†). The [3,3]-sigmatropic rearrangement of 1a seemed to be facile under silver catalysis to generate the bis-allene intermediate, which might undergo decomposition resulting in reduced yield of 2a. Inspired by the reports on transition metal-catalyzed [2 + 2] cycloadditions, we examined the use of various metal salts as the co-catalyst to facilitate the [2 + 2] cycloaddition step (entries 3–7). In particular, the use of Co(OAc)2 was able to increase the yield of 2a to 90% (entry 7). As a control reaction, the use of Co(OAc)2 alone led to no formation of 2a at all (entry 8). Interestingly, lowering the loading of both catalysts to 5 mol% led to the formation of 2a in quantitative yield (entry 9), possibly by reducing the amount of the side product.
Substrate scope
With the optimal conditions in hand, we moved on to investigate the scope of this Ag/Co-catalyzed process (Scheme 2). Regarding the substituent on the alkyne, the reaction could tolerate a range of linear alkyl substituents to produce 2a–2f in high efficiency. Ether-containing substrates also worked well (2g–2i). When secondary or tertiary aliphatic substituents were introduced on the alkyne terminal position, the overall reaction efficiency dropped significantly, possibly due to steric hindrance upon [3,3]-sigmatropic rearrangement (e.g., a reduced 30% yield for the case of cyclopropyl-substituted 2j). To our delight, terminal dipropargylphosphonate underwent reaction smoothly to deliver 2k in good yield, the X-ray crystal structure of which confirmed the general structures of 2. In addition, the reaction was also operative with alkylphosphonates 1l–1m as well as phenyl phosphate 1n to produce 2l–2n in good yields.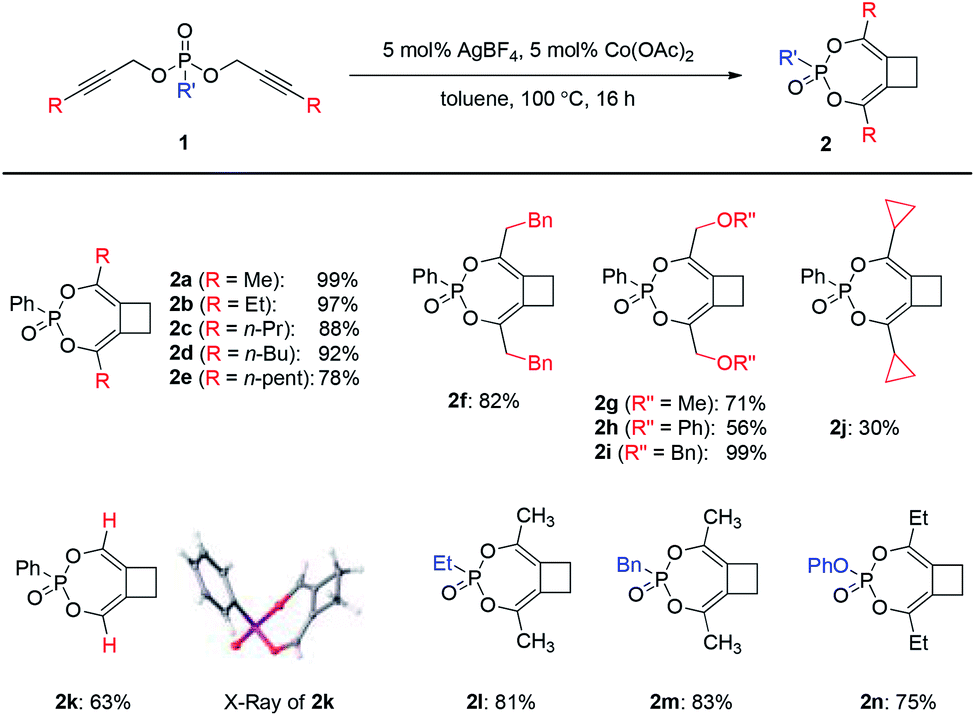 | ||
| Scheme 2 Tail-to-tail [2 + 2] cycloaddition of dipropargylphosphonates. | ||
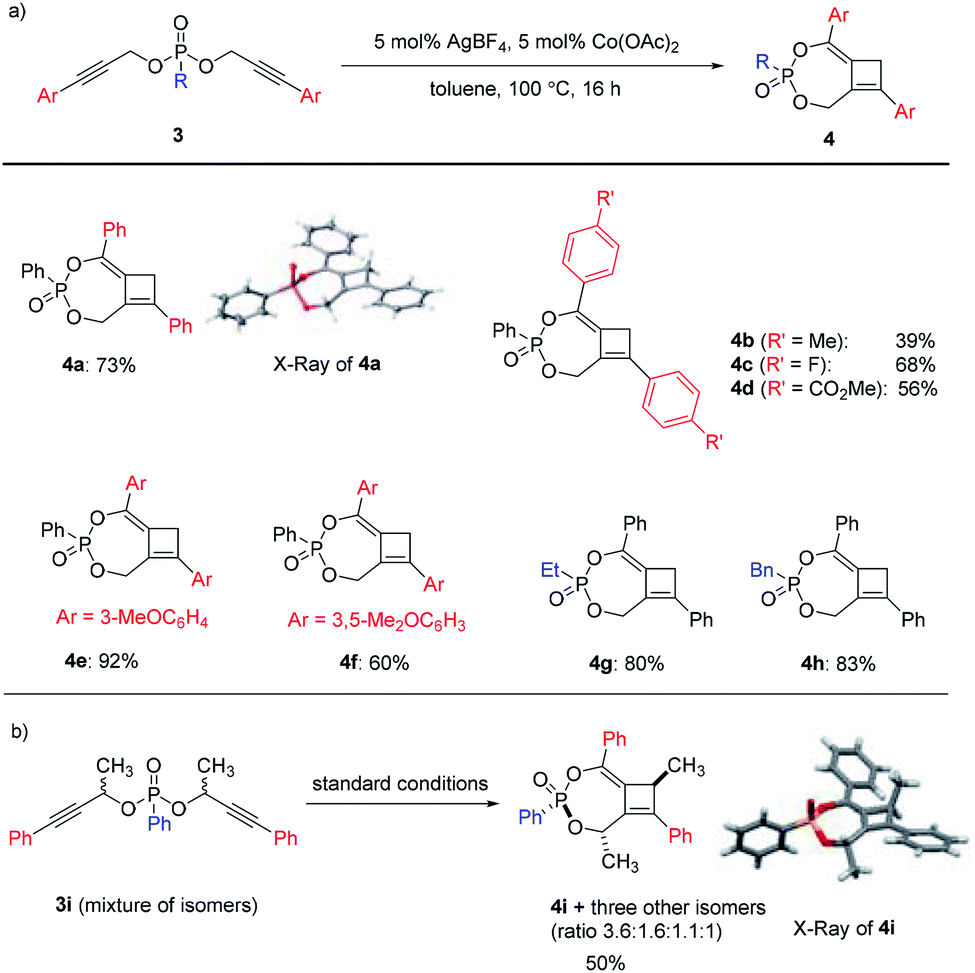
An interesting switch of reactivity was observed when we examined substrates bearing aryl-substituted alkynes 3 (Scheme 3a). Under the same reaction conditions, this class of substrates underwent a selective mono-[3,3]-sigmatropic rearrangement. A [2 + 2] cycloaddition of the allene–alkyne intermediate then produced bicyclo[5.2.0]cyclobutene 4 as the product, which was assigned by single crystal X-ray analysis of 4a. This class of polysubstituted cyclobutenes can serve as versatile and valuable building blocks in organic synthesis, and the current method represents an unprecedented catalytic tandem synthesis from readily available starting materials.
 | ||
| Scheme 3 Allene-ynes [2 + 2] cycloaddition of dipropargylphosphonates. | ||
The scope of this transformation was then probed. A range of aryl-substituents containing electron-donating or electron-withdrawing groups on the alkyne terminal position were well tolerated, giving rise to 4b–4f in reasonable to excellent yields. Alkyl substituents on the phosphonates were also compatible with this catalytic system to produce 4g–4h in high yields. To further expand the scope of this methodology, substrate 3i derived from a secondary propargylic alcohol was also examined as a mixture of diastereomers (Scheme 3b). It is noteworthy that the product would possess three stereogenic centers and a complex mixture of isomers could be obtained. Interestingly, we were able to isolate the major diastereomer 4i after the reaction, the relative configuration of which was also unambiguously assigned by single crystal X-ray analysis.
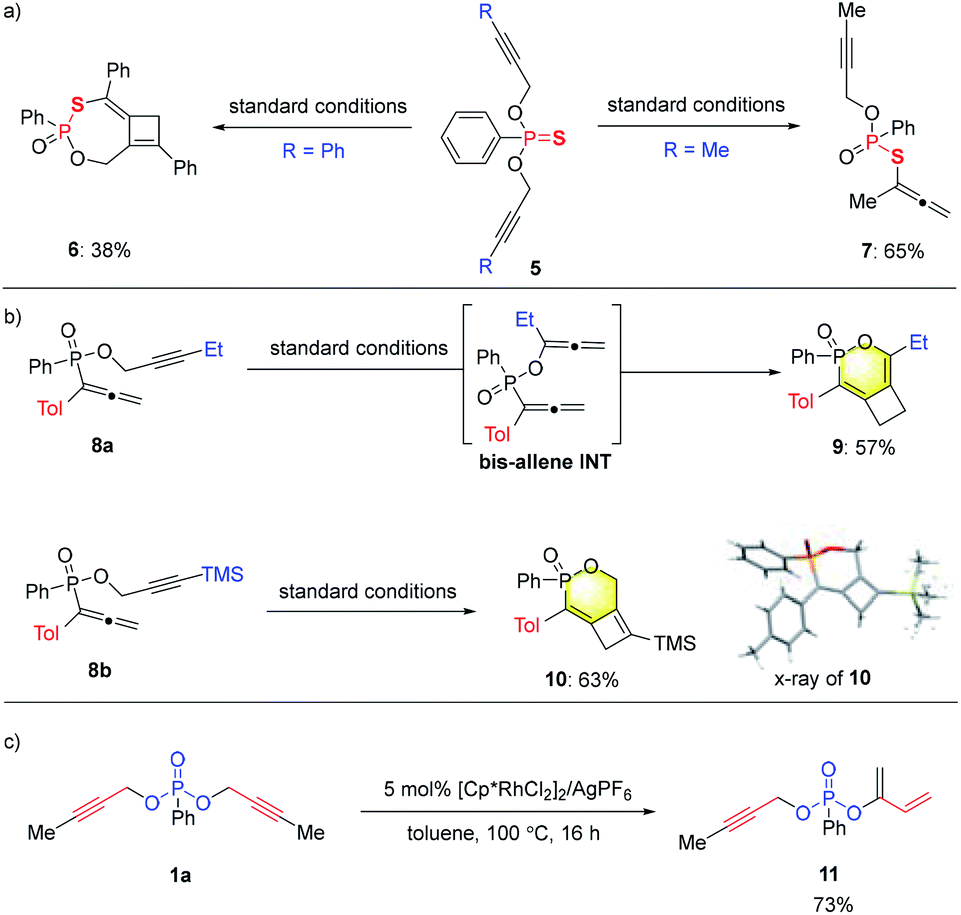
In addition to phosphonates, the analogous dipropargyl-phosphonothioates 5 were also prepared and tested for the tandem reaction (Scheme 4a). For a phenyl-substituted substrate, the tandem reaction proceeded smoothly under standard conditions to deliver cyclobutene 6, albeit with a lower yield of 38%. Surprisingly, the methyl-substituted substrate underwent the [3,3]-sigmatropic rearrangement step only once to produce 7 in 65% yield. While the exact reason for this change in reactivity is not clear, the incorporation of the sulfur substituent seemed to prohibit the sigmatropic rearrangement of the remaining propargylic ester unit.
 | ||
| Scheme 4 More product diversity by substrate/catalyst variation. | ||
In an attempt to prepare phosphonate substrates bearing mixed aryl/alkyl-substituted propargyl units, we prepared, to our surprise, propargyl phosphinates 8 (Tol = p-tolyl) with an allenyl substituent at the phosphorus center instead (Scheme 4b; see the ESI† for details). Interestingly, depending on the substituent on alkyne (ethyl or TMS in 8a or 8b), either [3,3] rearrangement followed by allene–allene cycloaddition or direct alkyne–allene cycloaddition resulted in efficient synthesis of 9 or 10, respectively. Cyclobutene 10 was unambiguously assigned by single crystal X-ray analysis. These studies further expanded the range of products (6,4 fused bicycles) from this catalytic system.
Divergent reactivity with the methyl-substituted substrate 1a was also achieved under rhodium catalysis that is very similar to that in entry 1 of Table 1 (Scheme 4c). By the use of [Cp*RhCl2]2 in combination with AgPF6, a diene product 11 was produced as the major product of the reaction, which is likely formed from a sequence of mono [3,3]-sigmatropic rearrangement followed by a [1,3]-H shift.10 It's interesting to note that the formation of products 4, 7 or 11 all generate a P-stereogenic center through desymmetrization of the prochiral substrates via the key [3,3]-sigmatropic rearrangement step. Considerable efforts were spent on achieving enantioselectivity in these transformations. Some preliminary results were obtained, e.g., a 45% ee was obtained for the preparation of 11 (see the ESI† for details). Further investigation along these lines is underway.
Diverse synthetic application
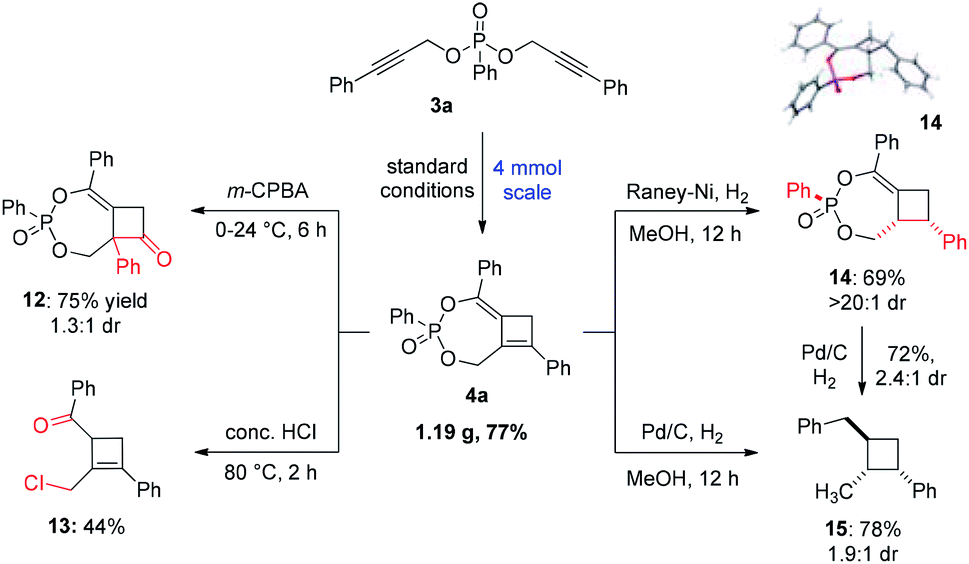
To demonstrate the robustness of this methodology, a gram-scale reaction of 3a was conducted that afforded 4a in a slightly improved 77% yield (Scheme 5). The synthetic utility of this polysubstituted cyclobutene was showcased in several derivatization studies to deliver highly valuable four-membered ring building blocks. Firstly, treatment of 4a with m-CPBA led to an efficient oxidative rearrangement to produce cyclobutanone 12 bearing an α-quaternary center in 75% yield. The new quaternary stereogenic center was introduced as a mixture of diastereomer relative to the P-stereocenter. Secondly, compound 4a could also undergo hydrolysis in the presence of conc. HCl to yield a highly functionalized trisubstituted cyclobutene 13 in 44% yield. Interestingly an alkyl chloride was introduced in addition to the ketone moiety, making this a highly versatile building block in synthesis. Lastly, hydrogenation using different catalysts could produce diverse reduction products. The reaction using RANEY®-Ni selectively reduced 4a to the corresponding cyclobutane 14 as a single diastereomer in good yield. Alternatively, reduction using Pd/C led to a global reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, benzyl ether and methylene ether to produce trisubstituted 15 in a good yield of 78%, albeit with a low 1.9
C bond, benzyl ether and methylene ether to produce trisubstituted 15 in a good yield of 78%, albeit with a low 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. The relative configuration of the major diastereomer of 15 was determined to be trans as shown based in the NOESY experiment. The reduction of 14 under similar conditions successfully yielded product 15 in 72% yield with a slightly improved dr of 2.4:1. Overall, compound 4a can serve as a versatile common precursor to access a wide range of valuable polysubstituted four-membered carbocycles.
:1 dr. The relative configuration of the major diastereomer of 15 was determined to be trans as shown based in the NOESY experiment. The reduction of 14 under similar conditions successfully yielded product 15 in 72% yield with a slightly improved dr of 2.4:1. Overall, compound 4a can serve as a versatile common precursor to access a wide range of valuable polysubstituted four-membered carbocycles.
 | ||
| Scheme 5 Transformation of 4a into versatile functionalized cyclobutanes. | ||
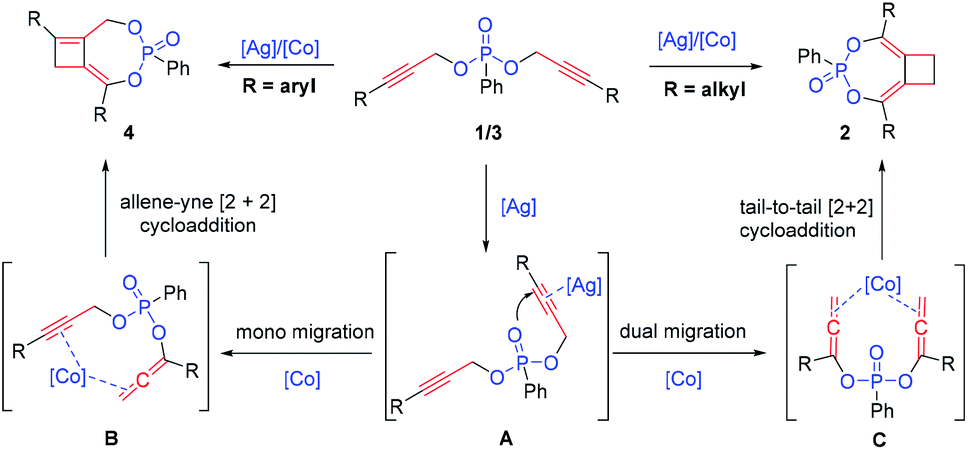
In an effort to better understand the reaction pathway, a series of 1H and 31P NMR measurements were carried out by heating substrate 1a with different catalysts in toluene-d8 (see the ESI† for details). By heating 1a with 5 mol% AgBF4 for 1 hour, we observed the formation of a new peak at 17.26 in 31P NMR and 5.16 and 1.98 ppm in 1H NMR. In contrast, by heating 1a with 5 mol% Co(OAc)2, no change in 31P NMR was observed at all, suggesting that the cobalt catalyst was not active for the initiation of the tandem reaction. For the reaction with AgBF4, continuous heating led to a gradual decrease of the originally observed intermediate with the detection of product 2a after 2 h. Based on these observations, we propose that this reaction is initiated by silver activation of the alkyne moiety (as in the postulated A, Scheme 6). The allene–alkyne B or bis-allene C, once formed, is likely activated by cobalt for the following facile [2 + 2] cycloaddition leading to higher yield of the desired products.4i,11
 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Biological studies
Considering that cyclobutane-containing compounds display significant biological activities (Fig. 1) and our products represent novel structures, efforts were then spent to examine the possible anti-proliferative effect of the newly developed compounds and in turn as potential anti-cancer agents. Thus, adenocarcinoma cells, human derived colorectal cancer cell line, DLD1, was subjected to treatment with 2h, 4a, or 6, for 48 hours in culture. At a dosage of 1.25 g ml−1, all three compounds suppressed DLD1 growth by more than 30% compared to the DMSO control group (Fig. 2). This prompted further confirmation and comparisons of the level of the anti-proliferation effect between these compounds. In order to establish controlled comparison, IC50 of each compound was determined, which was subsequently employed for other assays. As shown in Fig. 2B, the relative IC50 values of 2h, 4a and 6 were 0.008 mg ml−1, 0.01 mg ml−1 and 0.074 mg ml−1 respectively. Of note, treatment with 6 at the maximum test dosage of 0.15 mg ml−1 resulted in ∼89% suppression of proliferation compared to ∼72% and ∼87% suppression by 2h and 4a respectively. It is well understood that the loss of overall level of cellular proliferation could in part be caused by increased cell death after compound treatment. Of the three compounds, 6 showed significant induction of apoptosis in DLD1 cells (Fig. 2C). After treatment with 6 at the IC50 concentration, total DLD1 cell count was significantly reduced by 48% (Fig. 2C, left most bar chart). There were 2.1-fold more total apoptotic cells after treatment with 6 compared to the DMSO control (Fig. 2C, right most bar chart). Interestingly, the three compounds showed different kinetics for proliferation suppression (Fig. 2D). Based on results from the xCELLigence real-time cell analysis (RTCA) assay, cell proliferation was marginally inhibited by 2h and 4a at early time points compared to the DMSO control. On the other hand, upon treatment with 6, there was a significant and rapid loss of cell density based on impedance measurements in the assay. The low level of cell density was maintained throughout, to the end of the experiment, at 48 hours and did not recover. All these demonstrated that the three bicyclic compounds, particularly 6, are biologically functional and could induce the suppression or killing of colorectal cancer cells in vitro.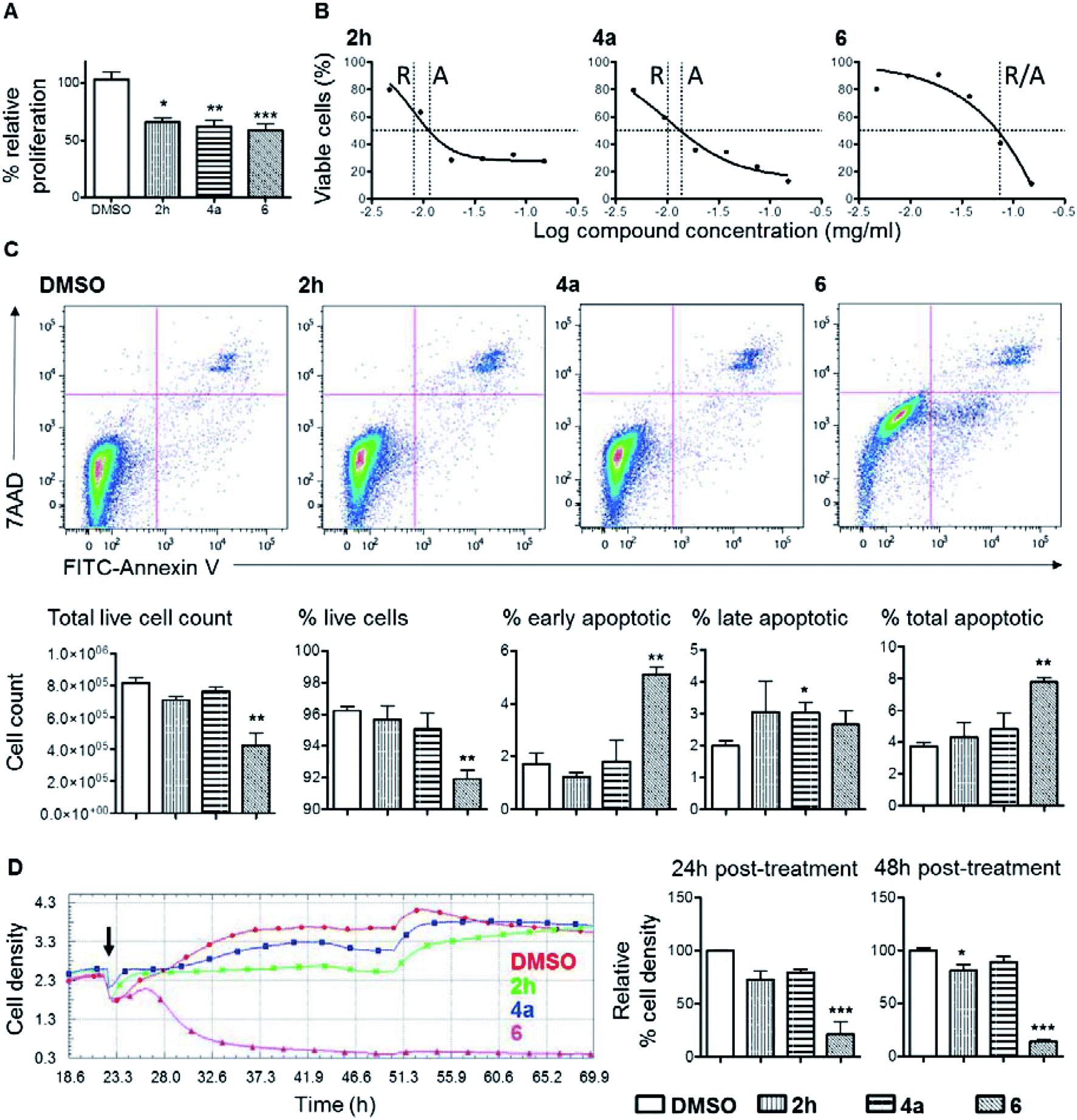 | ||
| Fig. 2 Evaluation of selected products for anti-cancer activities. | ||
Our data showed that 2h and 4a are weak suppressors of cellular proliferation compared to 6 which caused significant reduction in DLD1 proliferation and increased cell death (Fig. 2). Induction of cellular apoptosis is an important mechanism for any compound to function as an anti-tumour drug particularly for long term tumour eradication. Based on the TCRPs of the three compounds, it is clear that 6 worked rapidly and irreversibly to suppress DLD1 cell growth compared to the 2h and 4a where cell growth was retarded only at the early phase of the treatment (Fig. 2D). The TCRP of 6 showed a similar trend to other cytotoxic compounds that inhibited DNA synthesis and caused cycle arrest that eventually resulted in cell death.12 On the other hand, 2h and 4a displayed TCRP similar to that of compounds that act as antagonists of various G protein-coupled receptor signalling pathways. Further detail characterization of these compounds in other tumour cell types and in vivo models will be required to understand the mechanism of actions and the extent of their applications in cancer therapy.
Conclusions
In summary, we have developed an efficient preparation of bicyclo[5.2.0]-phosphonates via Ag/Co-catalyzed tandem [3,3]-sigmatropic rearrangement/[2 + 2] cycloaddition of dipropargyl-phosphonates. These products can serve as a versatile springboard for the construction of various polysubstituted cyclobutenes and cyclobutanes. Selected products are shown to be promising leads as suppressors of cellular proliferation. The development of a better understanding of the catalytic mechanism as well as other tandem cycloaddition reactions is currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. Z. is grateful for the generous financial support from the Ministry of Education of Singapore (R-143-000-A94-112 and R-144-000-420-112) and National University of Singapore (R-143-000-A57-114). Q. N. acknowledges the generous financial support of the Anhui Natural Science Foundation (1908085QB55). Y. Z. thanks the National Research Foundation, Prime Minister's Office, Singapore under its Campus of Research Excellence and Technological Enterprise (CREATE) Programme (R571-010-012-592) for financial support.Notes and references
- For selected reviews, see: (a) J. D. Winkler, C. M. Bowen and F. Liotta, Chem. Rev., 1995, 95, 2003–2020 CAS; (b) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1484 CAS; (c) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1538 CrossRef CAS; (d) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS; (e) A. Misale, S. Niyomchon and N. Maulide, Acc. Chem. Res., 2016, 49, 2444–2458 CrossRef CAS.
- For selected recent reviews, see: (a) S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef; (b) M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572–8574 CrossRef CAS; (c) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS; (d) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS; (e) T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CASFor selected examples on [2+2] cycloadditions, see: (f) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS; (g) A. Nishimura, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2012, 134, 15692–15695 CrossRef CAS; (h) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 9009–9013 CrossRef CAS; (i) J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, J. Am. Chem. Soc., 2017, 139, 9807–9810 CrossRef CAS; (j) D. Kossler, F. G. Perrin, A. A. Suleymanov, G. Kiefer, R. Scopelliti, K. Severin and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 11490–11493 CrossRef CAS.
- For other approaches to cyclobutanes/cyclobutenes, see: (a) H. Xu, W. Zhang, D. Shu, J. B. Werness and W. Tang, Angew. Chem., Int. Ed., 2008, 47, 8933–8936 CrossRef CAS; (b) M. Chaumontet, R. Piccardi, N. Audic, J. Hitce, J.-L. Peglion, E. Clot and O. Baudoin, J. Am. Chem. Soc., 2008, 130, 15157–15166 CrossRef CAS; (c) F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS; (d) F. Frébault, M. Luparia, M. T. Oliveira, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 5672–5676 CrossRef; (e) R. Liu, M. Zhang, T. P. Wyche, G. N. Winston-McPherson, T. S. Bugni and W. Tang, Angew. Chem., Int. Ed., 2012, 51, 7503–7506 CrossRef CAS; (f) Y. Deng, L. A. Massey, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7479–7483 CAS; (g) Y.-B. Bai, Z. Luo, Y. Wang, J.-M. Gao and L. Zhang, J. Am. Chem. Soc., 2018, 140, 5860–5865 CrossRef CAS.
- For a selected recent review, see: (a) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783–816 RSCFor selected examples on [2+2] cycloaddition of allenes, see: (b) S. Saito, K. Hirayama, C. Kabuto and Y. Yamamoto, J. Am. Chem. Soc., 2000, 122, 10776–10780 CrossRef CAS; (c) X. Jiang, X. Cheng and S. Ma, Angew. Chem., Int. Ed., 2006, 45, 8009–8013 CrossRef CAS; (d) M. R. Luzung, P. Mauleón and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 12402–12403 CrossRef CAS; (e) M. R. Siebert, J. M. Osbourn, K. M. Brummond and D. J. Tantillo, J. Am. Chem. Soc., 2010, 132, 11952–11966 CrossRef CAS; (f) M. L. Conner, Y. Xu and M. K. Brown, J. Am. Chem. Soc., 2015, 137, 3482–3485 CrossRef CAS; (g) Y. Qiu, B. Yang, C. Zhu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2016, 55, 6520–6524 CrossRef CAS; (h) J. M. Wiest, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2018, 57, 4647–4651 CrossRef CAS; (i) W. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2019, 58, 2500–2504 CrossRef CAS.
- For [3,3]-sigmatropic rearrangement with carboxylic esters, see: (a) B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 15978–15979 CrossRef CAS; (b) N. Marion and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2750–2752 CrossRef CAS; (c) P. Mauleón, J. L. Krinsky and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 4513–4520 CrossRef; (d) J. W. Cran and M. E. Krafft, Angew. Chem., Int. Ed., 2012, 51, 9398–9402 CrossRef CAS; (e) A. C. Jones, J. A. May, R. Sarpong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 2556–2591 CrossRef CAS; (f) V. Pirovano, E. Arpini, M. Dell'Acqua, R. Vicente, G. Abbiati and E. Rossi, Adv. Synth. Catal., 2016, 358, 403–409 CrossRef CAS.
- (a) L. Zhang, J. Am. Chem. Soc., 2005, 127, 16804–16805 CrossRef CAS; (b) Z. L. Niemeyer, S. Pindi, D. A. Khrakovsky, C. N. Kuzniewski, C. M. Hong, L. A. Joyce, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 12943–12946 CrossRef CAS.
- W. Rao, D. Susanti and P. W. H. Chan, J. Am. Chem. Soc., 2011, 133, 15248–15251 CrossRef CAS.
- The [3,3]-sigmatropic rearrangement of phosphorus compounds has been reported, see: (a) A. W. Sromek, A. V. Kel'in and V. Gevorgyan, Angew. Chem., Int. Ed., 2004, 43, 2280–2282 CrossRef CAS; (b) B. Chen and A. K. Mapp, J. Am. Chem. Soc., 2005, 127, 6712–6718 CrossRef CAS; (c) E. E. Lee and R. A. Batey, J. Am. Chem. Soc., 2005, 127, 14887–14893 CrossRef CAS; (d) M. L. Ferguson, T. D. Senecal, T. M. Groendyke and A. K. Mapp, J. Am. Chem. Soc., 2006, 128, 4576–4577 CrossRef CAS; (e) T. Schwier, A. W. Sromek, D. M. L. Yap, D. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 9868–9878 CrossRef CAS.
- (a) M. Wang, Z.-Q. Rong and Y. Zhao, Chem. Commun., 2014, 50, 15309–15312 RSC; (b) C. Ma, Y. Huang and Y. Zhao, ACS Catal., 2016, 6, 6408–6412 CrossRef CAS; (c) L.-C. Yang, Z.-Q. Rong, Y.-N. Wang, Z. Y. Tan, M. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 2927–2931 CrossRef CAS; (d) Y. Li-Cheng, T. Z. Yin, R. Zi-Qiang, L. Ruoyang, W. Ya-Nong and Z. Yu, Angew. Chem., Int. Ed., 2018, 57, 7860–7864 CrossRef; (e) Y.-N. Wang, L.-C. Yang, Z.-Q. Rong, T.-L. Liu, R. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1596–1600 CrossRef CAS.
- (a) M. J. Wanner, R. N. S. van der Haas, K. R. de Cuba, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2007, 46, 7485–7487 CrossRef CAS; (b) R. Hayashi, Z.-X. Ma and R. P. Hsung, Org. Lett., 2012, 14, 252–255 CrossRef CAS.
- For cobalt-catalyzed [2+2] cycloadditions, see: (a) K. C. Chao, D. K. Rayabarapu, C.-C. Wang and C.-H. Cheng, J. Org. Chem., 2001, 66, 8804–8810 CrossRef CAS; (b) G. Hilt, A. Paul and J. Treutwein, Org. Lett., 2010, 12, 1536–1539 CrossRef CAS; (c) J. Treutwein and G. Hilt, Angew. Chem., Int. Ed., 2008, 47, 6811–6813 CrossRef CAS; (d) A. Nishimura, E. Tamai, M. Ohashi and S. Ogoshi, Chem. – Eur. J., 2014, 20, 6613–6617 CrossRef CAS; (e) V. A. Schmidt, J. M. Hoyt, G. W. Margulieux and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 7903–7914 CrossRef CAS; (f) V. V. Pagar and T. V. RajanBabu, Science, 2018, 361, 68–72 CrossRef CAS.
- Y. A. Abassi, B. Xi, W. Zhang, P. Ye, S. L. Kirstein, M. R. Gaylord, S. C. Feinstein, X. Wang and X. Xu, Chem. Biol., 2009, 16, 712–723 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2005765–2005768 and 2036801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02972f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |