
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–ligand bond strength determines the fate of organic ligands on the catalyst surface during the electrochemical CO2 reduction reaction†
James R.
Pankhurst
a,
Pranit
Iyengar
a,
Anna
Loiudice
a,
Mounir
Mensi
b and
Raffaella
Buonsanti
 *a
*a
aLaboratory of Nanochemistry for Energy (LNCE), Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne (EPFL), Rue de l'Industrie 17, 1950 Sion, Valais, Switzerland. E-mail: raffaella.buonsanti@epfl.ch
bInstitute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne (EPFL), Rue de l'Industrie 17, 1950 Sion, Valais, Switzerland
First published on 17th August 2020
Abstract
Colloidally synthesised nanocrystals (NCs) are increasingly utilised as catalysts to drive both thermal and electrocatalytic reactions. Their well-defined size and shape, controlled by organic ligands, are ideal to identify the parameters relevant to the activity, selectivity and stability in catalysis. However, the impact of the native surface ligands during catalysis still remains poorly understood, as does their fate. CuNCs are among the state-of-the-art catalysts for the electrochemical CO2 reduction reaction (CO2RR). In this work, we study CuNCs that are capped by different organic ligands to investigate their impact on the catalytic properties. We show that the latter desorb from the surface at a cathodic potential that depends on their binding strength with the metal surface, rather than their own electroreduction potentials. By monitoring the evolving surface chemistry in situ, we find that weakly bound ligands desorb very rapidly while strongly bound ligands impact the catalytic performance. This work provides a criterion to select labile ligands versus ligands that will persist on the surface, thus offering opportunity for interface design.
Introduction
Colloidally synthesised nanocrystals (NCs) are receiving increasing attention in both thermal and electrocatalysis because their well-defined size and shape are ideal to identify parameters relevant for activity, selectivity and stability in catalysis.1–6 Organic ligands are essential to their synthesis both as reactants, to tune the morphology, and as surface passivating agents, to assure solvent dispersibility.1–6 Surface and interface science normally considers organic species on catalytic surfaces a poison. However, numerous reports in thermal catalysis have been challenging this traditional view.7–11 Studies investigating surface chemistry in electrocatalysis are still comparatively scarce and mostly focus on the hydrogen and oxygen evolution reactions.12–15 In general, understanding the role of organic ligands still remains of utmost importance in order to correctly interpret the structure/property relations in colloidally synthesised catalysts and to potentially design interfaces that are beneficial for activity, selectivity and stability.In this work, we study the fate of different organic ligands during the electrochemical CO2 reduction reaction (CO2RR). This chemical transformation is gathering interest by researchers in various disciplines because of the promise to convert CO2 into value-added products while storing renewable energy, a process that would be a game changer in a sustainable society.16 Copper NCs (CuNCs) are at the forefront of current CO2RR research, primarily as copper is the only single metal that can produce multi-carbon products from the reaction.6,17–22 The tunability of size and shape offered by colloidal chemistry has allowed the identification of optimal geometries to direct selectivity towards C1 or C2 products.17,19,22 Furthermore, colloidal suspensions of these catalysts are appealing as they can be used to deposit CuNCs onto a variety of supports, including gas-diffusion electrodes that can operate at commercially-relevant current densities.23,24 In some of these studies, the organic ligands functionalising the surface of the NC catalysts were removed prior to catalytic testing using plasma, mild solvent-washing or hydrazine treatments.17,22,25 However, extreme care is needed to ensure that no undesirable modifications of the catalysts are caused by these ligand removal treatments, such as changes of exposed facets or of the copper oxidation state as well as deposition of poorly-understood carbonaceous material on the surface. In other studies, electrodesorption of the ligands during the initial stages of electrolysis was supposed or demonstrated.20,21,26 A similar scenario is found when looking at colloidal NCs other than copper in the CO2RR.27,28 Overall, a complete picture of the fate and role of the ligands in colloidal catalysts for CO2RR is clearly missing.
Here, we contribute to fill such a gap by studying the fate and elucidating the role of a variety of common ligands on CuNCs during the CO2RR. Specifically, we investigated oleylamine (OLAM), oleic acid (OLAC), dodecanethiol (DDT), trioctylphosphine (TOP), trioctylphosphine oxide (TOPO), and tetradecylphosphonic acid (TDPA). These ligands were chosen for three reasons. Firstly, they are commonly used in high-temperature colloidal synthesis of metal NCs.6 Secondly, they contain different functional groups that are expected to bind with different strengths to the copper surface, and could also undergo reduction at different potentials, thereby offering a platform to investigate the importance of these two parameters. Finally, these long-chain hydrocarbon ligands are expected to inhibit the catalyst activity whilst adsorbed, enabling us to study any correlations between ligand surface-coverage and catalyst poisoning.
By combining electrocatalytic and electroanalytical measurements with surface characterisation, we found that the ligands TOPO, OLAM, TDPA and OLAC rapidly desorb from the surface of the CuNCs and thus do not impact the catalytic behaviour in the CO2RR. On the contrary, DDT remains chemically adsorbed under the examined conditions. As corroborated by DFT calculations, the binding energy of the ligands on the copper surface is the key descriptor that explains the observed behaviour. By identifying the parameter that determines the ligand stability on the NC surface, this work provides a criterion with which labile versus persistent ligands can be selected, thereby offering opportunity for interface design in future CO2RR studies employing colloidally synthesised NCs.
Results and discussion
Colloidal CuNCs were synthesised according to a reported procedure, by heating copper(I) acetate in trioctylamine (TOA) in the presence of tetradecylphosphonic acid (TDPA).20 The resulting NCs are spherical, with an average diameter of 5–6 nm (Fig. 1A and S2†). These NCs were chosen instead of shaped NCs (i.e. cubes, octahedra), shown to be more selective towards certain products during CO2RR,19,22 because of their higher surface-to-volume ratio which facilitates surface chemistry studies. After synthesis, the surface is expected to be functionalised by a mixture of TOA and TDPA. In order to control the composition of the ligand shell, the native ligands were stripped using a mild chemical treatment with a Meerwein's salt.29Scheme 1 outlines this ligand exchange method and shows the molecular structures of the newly added ligands. Successful ligand stripping was confirmed by TEM imaging (Fig. 1B), which showed reduced interparticle distance, consistent with the removal of the long hydrocarbon ligands, while their colloidal stability is maintained in N,N-dimethylformamide.29 FT-IR spectroscopy also evidenced the disappearance of all organic signals arising from the ligands (Fig. 1D). Oleylamine (OLAM), oleic acid (OLAC), dodecanethiol (DDT), trioctylphosphine (TOP), trioctylphosphine oxide (TOPO), and TDPA were added as new organic ligands – yielding the CuNCs Cu-OLAM, Cu-OLAC, Cu-DDT, Cu-TOP, Cu-TOPO, and Cu-TDPA, respectively. As a control sample with a bare surface, Cu-TOPO was washed with acetone after deposition on the electrode to remove the TOPO ligands (details in the ESI†).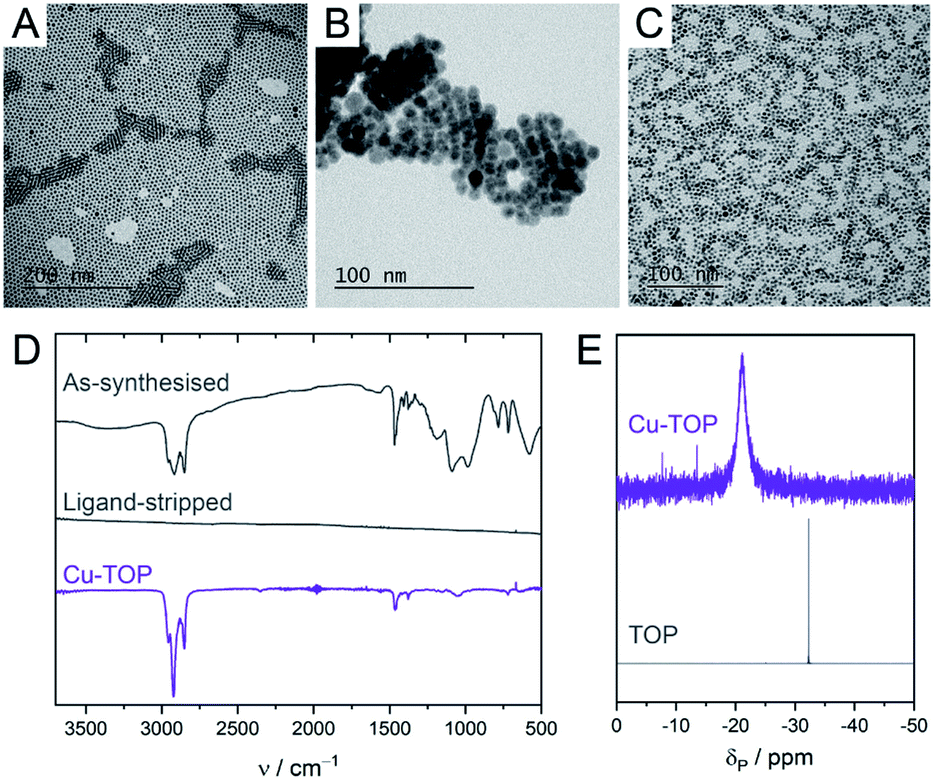 | ||
| Fig. 1 (A–C) TEM images of CuNCs as-synthesised (A), following ligand stripping with Meerwein's salt (B), and following re-ligation with TOP (C). (D) FT-IR spectra of as-synthesised CuNCs, ligand-stripped CuNCs and Cu-TOP NCs measured as a film. (E) Solution 31P{1H} NMR of free TOP and Cu-TOP NCs, measured in d6-benzene. | ||
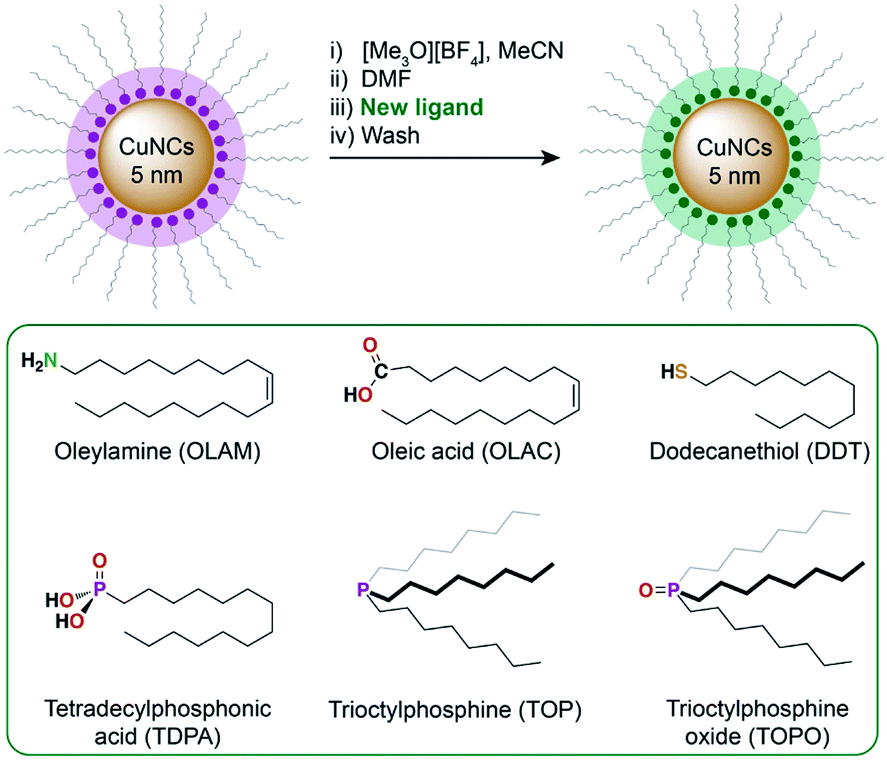 | ||
| Scheme 1 Overview of the ligand exchange procedure to prepare the CuNC catalysts. The ligands that were introduced using this method are shown in the green box. | ||
TEM imaging and UV-vis spectroscopy evidenced that the NCs had retained their size and shape, and no sintering was observed following the ligand exchange (Fig. 1C, S2 and S3†). XPS revealed that the ligand exchange did not alter the surface oxidation state of the CuNCs (Fig. S4†). Binding of the ligands to the NC surfaces was confirmed using a variety of spectroscopic techniques, including FT-IR and solution NMR (Fig. 1D, E and S5–S21†).
All of the CuNC catalysts were tested for CO2RR in a typical H-cell, using 0.1 M KHCO3 as the electrolyte (Fig. 2). For the initial screening, we selected a potential of −1.1 V vs. RHE and 1 hour electrolysis, as these conditions are in the range found to be optimal for activity and selectivity of CuNCs.17–22 Comparing the CuNCs capped with TOPO, OLAM, TDPA, OLAC and TOP, we observed that the product distributions were very similar, with formate being the major CO2RR product, followed by C2H4 and CO, and the rest being hydrogen (Fig. 2A). Near-identical intrinsic activities (JCO2RR) were also determined for all of these catalysts (Fig. 2B), after the currents were normalised by the electrochemically active surface area (ECSA) and contribution to the CO2RR (details in the ESI†). Whilst there were some important differences between the catalysts at the very beginning of the reaction (discussed below), the CuNCs capped with TOPO, OLAM, TDPA, OLAC and TOP ultimately displayed very similar selectivities and activities to the washed sample with no ligands when taken as an average after 10 minutes. Only Cu-DDT displayed clearly distinct catalytic behaviour, in that it produces much less methane, ethylene and formate in comparison to the other catalysts, and also has a much lower intrinsic activity.
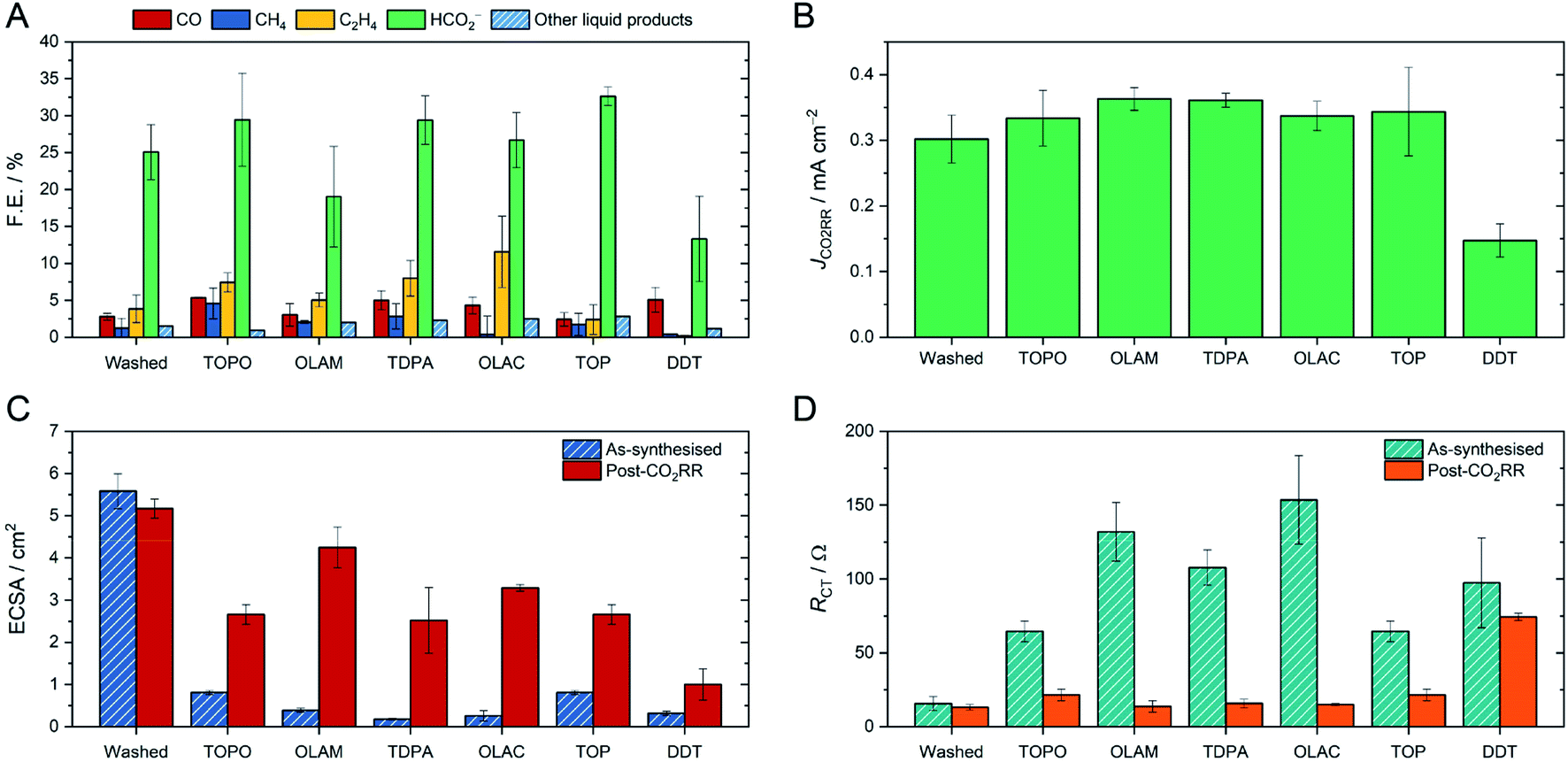 | ||
| Fig. 2 Overview of the CO2RR performance of CuNCs and their characterisation on the glassy carbon electrode before and after electrolysis at −1.1 V vs. RHE for 1 hour. (A) Faradaic efficiencies for CO2RR products; minor liquid products include acetate, ethylene glycol, ethanol and propanol, which were determined by HPLC. The rest of the faradaic efficiency is accounted for by hydrogen. (B) Intrinsic activities, where JCO2RR is the current normalised by the CO2RR contribution and the ECSA of catalyst. (C) ECSAs, determined by capacitance measurements. (D) Charge-transfer resistances (RCT), measured by EIS. | ||
We then proceeded to study changes occurring before and after the 1 hour CO2RR-electrolysis. First of all, we assessed the ECSAs of the catalysts (Fig. 2C). The ECSA is expected to change as a result of modified surface chemistry; specifically, to increase if long hydrophobic ligands are removed from the surface.13 Indeed, the washed sample had a high ECSA of 5.5 cm2, whereas those capped by organic ligands were much smaller (<1 cm2). Following CO2RR, the ECSA of the washed sample was unchanged, whilst the ECSAs of the ligand-capped catalysts became much higher and closer to the value of the washed sample. The exception was Cu-DDT, as the ECSA remained very small after CO2RR, suggesting that the DDT ligand is more persistent on the surface.
If an increase in ECSA corresponds to ligand detachment, then the charge transfer from the glassy carbon substrate to the CuNCs and from the CuNCs to the reactants should also be improved. We therefore turned to electrochemical impedance spectroscopy (EIS) to verify our hypothesis (Fig. 2D). Whilst other factors contribute to the total impedance in the cell, here we focus on the charge-transfer resistance at the working electrode (RCT/Ω) as this reflects the NC surface chemistry (Fig. S22†). Henckel et al. have previously shown how chemical removal of ligands from a surface lowers the RCT.30 For the washed CuNC sample, RCT was indeed very low both before and after CO2RR. In contrast, each ligand-capped CuNC sample displayed high RCT prior to electrocatalysis, consistent with high surface coverage by the ligands. Following CO2RR, the resistance decreased dramatically, except for Cu-DDT, in agreement with the observed changes in ECSA.
At this point, the data suggest that all of the ligands undergo detachment at some point during the CO2RR, with DDT being an exception. However, to be able to correctly and reliably interpret the electrocatalytic data and relate these to the surface chemistry, it becomes important to know when the detachment happens.
While one can rely on a variety of techniques to study in situ compositional and structural changes of the inorganic component during electrocatalysis (e.g. TEM, X-ray absorption and scattering techniques), changes in the organic shell are more challenging to monitor, due to the low surface concentration. Here, we thought to use EIS, run intermittently between periods of CO2RR electrolysis, as a quasi in situ technique to gain a kinetic picture of the changing surface chemistry (Fig. 3 and S23–S31†). From such experiments we found that for TOPO, OLAM and OLAC ligands, RCT drops immediately after applying a potential of −1.1 V vs. RHE, implying a very rapid loss of ligands from the surface (Fig. 3A). Cu-TDPA underwent a slower decrease in RCT, although the electrode finally reached similarly low resistance values. In contrast, Cu-TOP and Cu-DDT appeared to lose a fraction of ligands very rapidly at the beginning of the reaction, but thereafter RCT decreased only minutely. Overall, Cu-DDT once again experienced the smallest change during the CO2RR. A comparison of the evolving RCT values with the CO2RR chronoamperograms evidences a consistent correlation between the resistance decrease and the onset of product evolution from the electrode surface (Fig. S32†). The effect of the applied potential on the ligand stripping kinetics was also investigated and Cu-TOP is reported as a representative example (Fig. 3B). As the potential was lowered to more positive values, both the initial rate and the extent of the ligand stripping was drastically reduced. This demonstrates that the applied potential plays a crucial role in the destabilisation of the ligand binding on the surface.
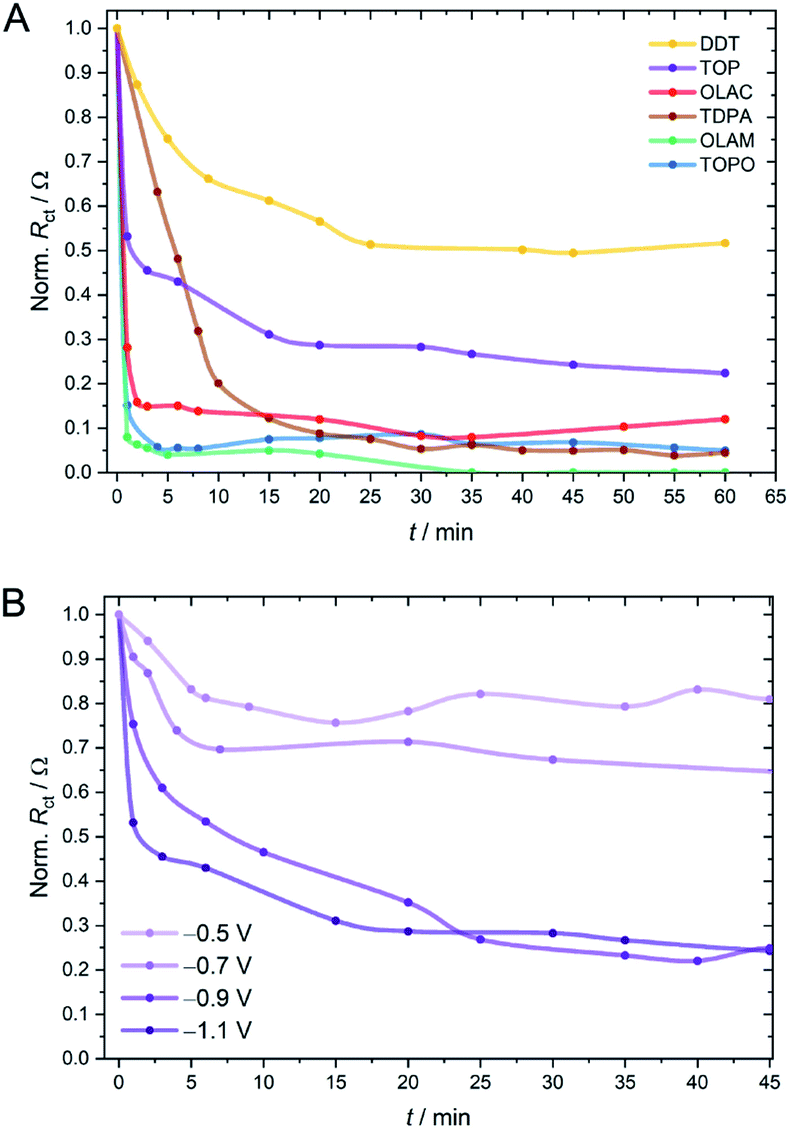 | ||
| Fig. 3 Changes in the charge-transfer resistance (RCT) over time on glassy carbon electrodes for (A) CuNCs capped by different ligands, where the potential in the CO2RR was −1.1 V vs. RHE and for (B) the Cu-TOP catalyst measured at different applied potentials under CO2RR conditions. In these experiments, electrolysis was stopped whilst impedance spectra were measured; t refers to the length of time the catalyst was held under CO2RR conditions. | ||
X-ray Photoelectron Spectroscopy (XPS) was then used to characterise the electrode surface before and after electrolysis (Fig. 4). For the as-synthesised CuNC samples, signature XPS peaks were observed for each ligand (i.e. P2s and P2p peaks for Cu-TOP, Cu-TOPO and Cu-TDPA, S2p for Cu-DDT and N1s for Cu-OLAM). Following CO2RR, only some trace residues of TOPO, OLAM and TDPA remained on the CuNC surface. These residues may be the reason why the ECSAs of the ligand-stripped catalysts do not reach the same size as the washed CuNCs. In contrast, Cu-TOP and Cu-DDT displayed more prominent peaks following CO2RR, indicating a higher retention of these ligands; these results were also confirmed by energy-dispersive X-ray spectroscopy (EDX), as discussed below. Thanks to the high signal-to-noise ratio of the DDT S2p peak, a reliable deconvolution was possible (Fig. 4 and S33†). In the as-synthesised sample, the broad peak can be deconvoluted into two major components, corresponding to the protonated form (thiol; S2p1/2 at 164.30 eV) and deprotonated form (thiolate; S2p1/2 at 162.85 eV) of DDT, respectively.31 One can assume that the thiolate is the one binding to the surface, while the thiol is simply physisorbed. Following CO2RR, the contribution from the thiol component is much lower. This suggests that the physisorbed DDT on the surface is readily desorbed at the beginning of the CO2RR while the more strongly bound, chemisorbed thiolate remains and poisons the catalyst. This detail accounts for the EIS behaviour of Cu-DDT in Fig. 3A, where the desorbing thiol leads to a reduction of RCT at the beginning of the reaction.
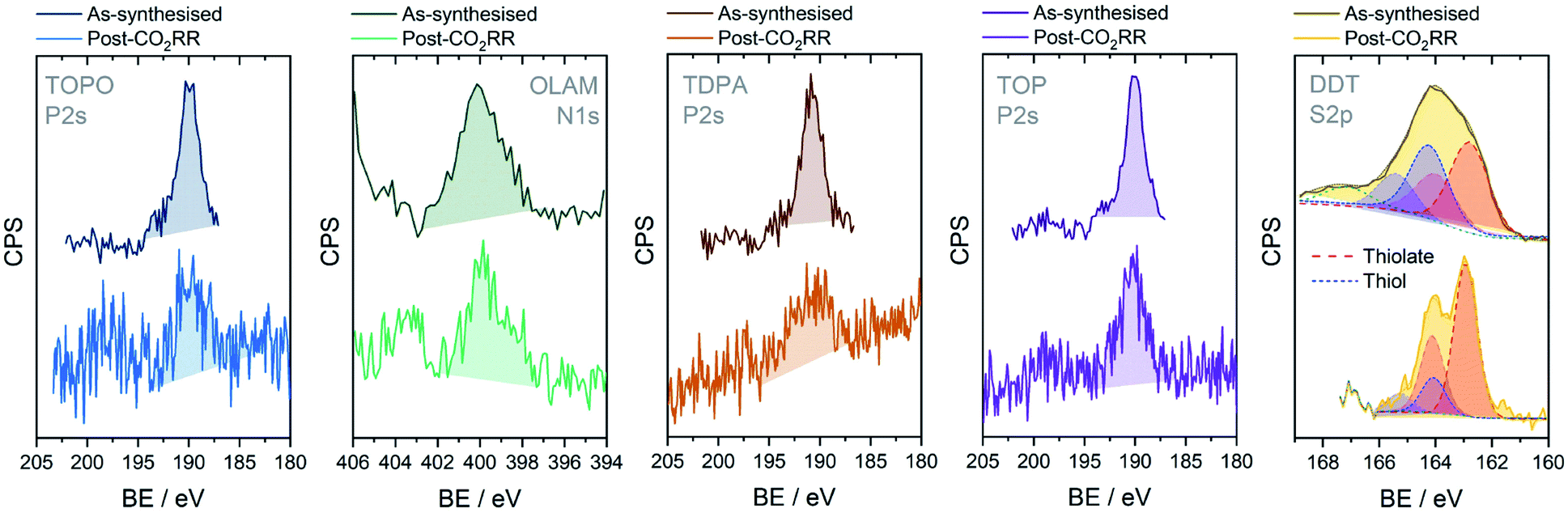 | ||
| Fig. 4 Characterisation of CuNC catalyst films by XPS before and after CO2RR electrolysis at −1.1 V vs. RHE. Signature XPS spectra are shown for each ligand: P2s for TOPO, TDPA and TOP; N1s for OLAM; and S2p for DDT. More detailed fitting for Cu-DDT can be found in Fig. S33.† For the as-synthesised Cu-DDT sample, there is also a small contribution from a sulfite species around 167 eV (unshaded peak). | ||
Cu-TOPO, Cu-TOP and Cu-DDT were also analysed by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) as representative examples of ligands that desorb rapidly, slowly and seemingly not at all, respectively (Fig. S34–S38†). From quantification of the ligands by EDX analysis, no P signal from TOPO could be detected post-CO2RR (Fig. S36†). For Cu-TOP, 36% of the initial TOP ligands are retained (Fig. S37†), corresponding nicely with the RCT data in Fig. 3A, where the resistance was measured around 35% of the initial value at the same time point (10 minutes). In comparison, the concentration of sulfur in the Cu-DDT sample was unchanged following CO2RR (Fig. S38†), confirming the high stability of the ligand on the surface.
Having established that organic ligands can have a different impact on the catalytic behaviour of the CuNCs depending on their fate during the reaction, it becomes important to elucidate the underlying phenomena driving their surface detachment. In particular, the next question becomes whether the ligands detach under cathodic potential due to electroreduction or electrodesorption. The reduction potential of the ligands and the binding energy to the Cu surface would be the determining factors of the two phenomena, respectively. Answering this question will facilitate a more predictive approach to the choice of ligands used in colloidal synthesis and, eventually, guide interface design for colloidally synthesised nanocatalysts. For example, it could steer the selection of ligands to be employed during the NC synthesis, towards those that rapidly desorb from the catalyst surface under the reaction conditions and do not interfere with the catalysis, thereby avoiding post-synthesis modification. Conversely, when the aim is to make use of organic ligands as co-catalysts at designed interfaces, binding groups that will ensure their persistence on the surface at the desired potentials can be selected.32–34
We conducted voltammetry studies to gain further insight into the redox behaviour of the ligands. For these studies, it was necessary to use 0.1 M [nBu4N][PF6] in acetonitrile as the supporting electrolyte, due to the wider potential window that is offered in comparison with aqueous electrolytes. In the latter, hydrogen evolution occurred before any signature from the ligands could be observed. Details on the potential conversion from Fc+/Fc to SHE are reported in the ESI.†
In conventional cyclic voltammetry (CV) experiments for the free ligands in solution, no redox activity was observed between −0.15 and −2.40 V vs. SHE (Fig. S39†). This result shows that a formal electroreduction or decomposition of the ligands does not take place at the potentials used in CO2RR conditions.
In order to verify that it is indeed electrodesorption that drives the ligand stripping, we performed square-wave voltammetry on the CuNCs (SWV, Fig. 5). CV was uninformative in this case due to its lower sensitivity, combined with the low ligand concentration in the CuNC samples.35 In these experiments, we expected to observe cathodic waves in correspondence to ligand desorption. Indeed, such waves were observed for all CuNC samples on sweeping the potential to increasingly negative values between −0.2 and −1.6 V vs. SHE, and the trend in the stripping potential is the following: TOPO < OLAM < TDPA ≲ OLAC < TOP < DDT (Fig. 5A). Interrogation of the binding strengths between the ligands and the Cu surface was conducted by DFT calculations (Fig. 5B, S40 and Table S3†). As the surface chemistry of CuNCs still remains poorly investigated, the binding interactions of ligands on NC surfaces are non-trivial and their elucidation requires dedicated computational modelling complemented by state-of-the-art spectroscopic studies.36 We therefore limited our calculations to very simplistic models by representing the NC surface with a single Cu atom. In this way we could investigate the Cu–ligand interactions whilst avoiding any incorrect assumptions about the surface. Metal–ligand bond enthalpies were calculated by fragmentation analysis. The data in Fig. 5 indicate a clear trend between the Cu–L binding strengths and the stripping potentials. Specifically, stronger bound ligands require larger negative potentials to induce electrodesorption. Fig. S41† shows the linear correlation between the binding strengths and the stripping potential.
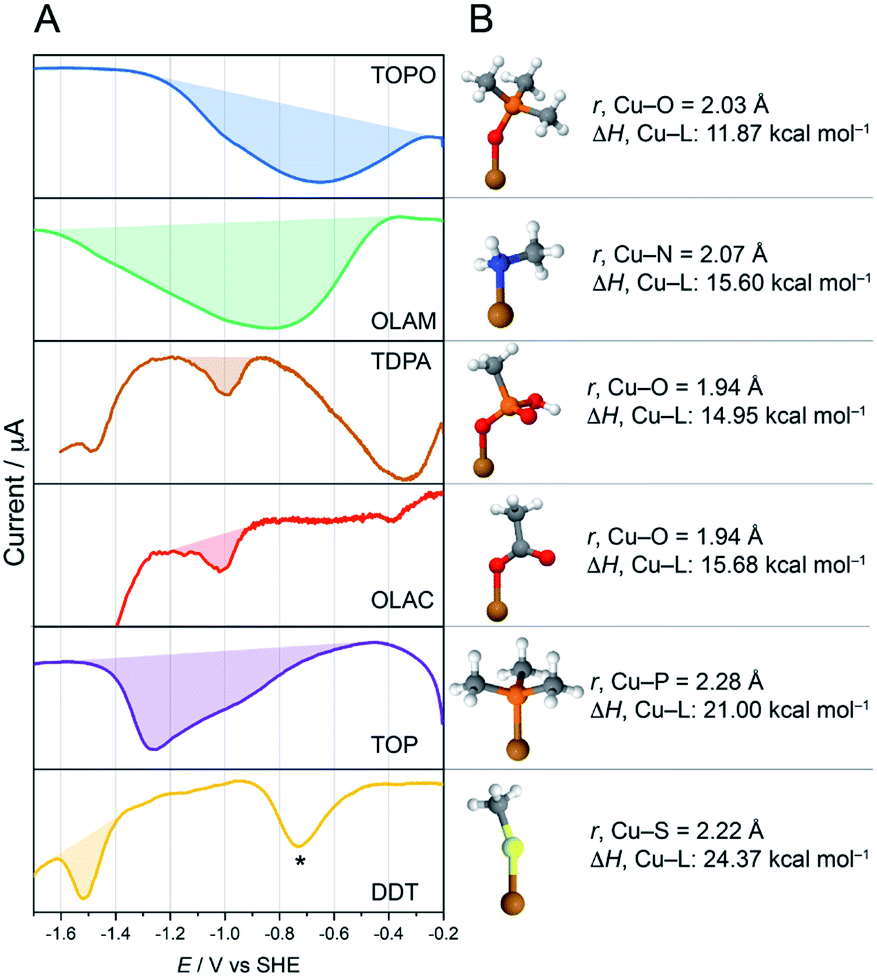 | ||
| Fig. 5 Investigating the stabilities of the metal–ligand bonds. (A) Square-wave voltammograms (SWVs) of CuNCs. The ligand desorption waves are shaded, and the peak potential is proportional to the metal–ligand binding strength. The symbol “*” indicates a trace impurity in the electrolyte, which is most likely oxygen as it can removed by sparging with N2.37 (B) Cu-ligand DFT models of the ligand binding, along with calculated bond lengths and bond strengths. In Cu-TDPA and Cu-OLAC, the bond energies are slightly lower than expected. However, in these simple models, the possibility of TDPA or OLAC to bind to the surface in a multidentate or bridging mode is not considered, and so the bond strengths in these cases are likely to be underestimated. | ||
With this information in hand, the rapid desorption observed for the weakly bound TOPO, OLAM, TDPA and OLAC ligands (Fig. 3A) can be explained by considering that the applied potential of −1.1 V vs. RHE is more negative than the ligand stripping potentials. The high stability of DDT on the CuNC surface is also explained from the perspective of its strongly negative ligand-stripping potential, which is more negative than the potential required for the CO2RR. Under the conditions that we have studied here, TOP can be considered a borderline case, considering that the ligand stripping potential is quite close to the CO2RR potential. Indeed, the SWV stripping peak for Cu-TOP actually spans a potential window that includes the CO2RR potential. Differences in the double layer between the organic and aqueous electrolytes along with local pH and concentration gradients during CO2RR will definitively impact ligand desorption from the CuNC surface. Nevertheless, the correlations between the electrocatalytic results, the surface characterisation, the SWVs and the DFT calculations indicate that solid guidelines can be derived from the obtained results.
Conclusions
Herein, we have studied a series of CuNC catalysts to determine the fate of the organic ligands during the CO2RR and to identify a predictive descriptor for it. Electrocatalytic measurements combined with surface characterisation techniques have evidenced that TOPO, OLAM, TDPA and OLAC behave as labile ligands that desorb at the reaction potential and thus impact neither the selectivity nor the activity of the catalysts. In contrast, DDT persists on the surface and TOP behaves as a borderline case under the examined conditions. Electroanalytical methods and DFT calculations have led us to conclude that TOPO, OLAM, TDPA and OLAC are electrodesorbed from the metal surface, rather than being formally reduced, and that their stripping potentials correlate with their binding energies to the copper surface.In summary, we propose that the metal–ligand bond strength is a good predictor of the fate of the ligand during the CO2RR and can direct the selection of the ligands depending on the targeted goal. Weakly bound ligands are desirable to use during the NC synthesis if one wishes them not to interfere with the catalytic behaviour. Instead, strongly bound ligands, such as those possessing thiolate or phosphine anchoring groups to the CuNCs, will provide opportunity for interface design wherein functional ligands could act as co-catalysts.32–34
Overall, this work is only the first step in understanding how ligands behave under CO2RR conditions, yet it could guide or enable future work looking at more in-depth modelling of how the ligands impact the double layer, and how that, in turn, influences ligand desorption energies and kinetics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council Starting Grant ERC-HYCAT under agreement number 715634. GAZNAT S. A. is acknowledged for the financial support for Pranit Iyengar. The authors thank Seyedeh Behnaz Varandili for ICP-OES measurements and Yannick T. Guntern for additional HPLC measurements.References
- Y. Kang, P. Yang, N. M. Markovic and V. R. Stamenkovic, Nano Today, 2016, 11, 587–600 CrossRef CAS.
- J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- Z. Niu, N. Becknell, Y. Yu, D. Kim, C. Chen, N. Kornienko, G. A. Somorjai and P. Yang, Nat. Mater., 2016, 15, 1188–1194 CrossRef CAS PubMed.
- P. Strasser, M. Gliech, S. Kuehl and T. Moeller, Chem. Soc. Rev., 2018, 47, 715–735 RSC.
- M. Cargnello, Chem. Mater., 2019, 31, 576–596 CrossRef CAS.
- J. Huang and R. Buonsanti, Chem. Mater., 2019, 31, 13–25 CrossRef CAS.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- J. L. Fiorio, N. López and L. M. Rossi, ACS Catal., 2017, 7, 2973–2980 CrossRef CAS.
- S. T. Marshall, M. O'Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwartz and J. W. Medlin, Nat. Mater., 2010, 9, 853–858 CrossRef CAS PubMed.
- K. R. Kahsar, D. K. Schwartz and J. W. Medlin, ACS Catal., 2013, 3, 2041–2044 CrossRef CAS.
- M. Makosch, W.-I. Lin, V. Bumbálek, J. Sá, J. W. Medlin, K. Hungerbühler and J. A. van Bokhoven, ACS Catal., 2012, 2, 2079–2081 CrossRef CAS.
- Y.-H. Chung, D. Y. Chung, N. Jung and Y.-E. Sung, J. Phys. Chem. Lett., 2013, 4, 1304–1309 CrossRef CAS PubMed.
- D. Ung and B. M. Cossairt, ACS Appl. Energy Mater., 2019, 2, 1642–1645 CrossRef CAS.
- Y.-H. Chung, S. J. Kim, D. Y. Chung, H. Y. Park, Y.-E. Sung, S. J. Yoo and J. H. Jang, Chem. Commun., 2015, 51, 2968–2971 RSC.
- E. E. Benson, H. Zhang, S. A. Schuman, S. U. Nanayakkara, N. D. Bronstein, S. Ferrere, J. L. Blackburn and E. M. Miller, J. Am. Chem. Soc., 2018, 140, 441–450 CrossRef CAS PubMed.
- R. F. Service, Science, 2019, 365, 1236–1239 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 10560–10565 CrossRef CAS PubMed.
- W. T. Osowiecki, J. J. Nussbaum, G. A. Kamat, G. Katsoukis, M. Ledendecker, H. Frei, A. T. Bell and A. P. Alivisatos, ACS Appl. Energy Mater., 2019, 2, 7744–7749 CrossRef CAS.
- P. Iyengar, J. Huang, G. L. De Gregorio, C. Gadiyar and R. Buonsanti, Chem. Commun., 2019, 55, 8796–8799 RSC.
- Y. Wang, H. Shen, K. J. T. Livi, D. Raciti, H. Zong, J. Gregg, M. Onadeko, Y. Wan, A. Watson and C. Wang, Nano Lett., 2019, 19, 8461–8468 CrossRef CAS PubMed.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- Z. Zhang, M. Chi, G. M. Veith, P. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Zhu, ACS Catal., 2016, 6, 6255–6264 CrossRef CAS.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- K. Manthiram, Y. Surendranath and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 7237–7240 CrossRef CAS PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
- E. L. Rosen, R. Buonsanti, A. Llordes, A. M. Sawvel, D. J. Milliron and B. A. Helms, Angew. Chem., Int. Ed., 2012, 51, 684–689 CrossRef CAS PubMed.
- D. A. Henckel, O. Lenz and B. M. Cossairt, ACS Catal., 2017, 7, 2815–2820 CrossRef CAS.
- S. Bandyopadhyay, S. Chattopadhyay and A. Dey, Phys. Chem. Chem. Phys., 2015, 17, 24866–24873 RSC.
- J. R. Pankhurst, Y. T. Guntern, M. Mensi and R. Buonsanti, Chem. Sci., 2019, 10, 10356–10365 RSC.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- M. Weber, S. Westendorf, B. Märker, K. Braun and M. Scheele, Phys. Chem. Chem. Phys., 2019, 21, 8992–9001 RSC.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. N. Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J.-M. Basset, Nat. Chem., 2017, 9, 890–895 CrossRef CAS PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details, TEM and SEM images, IR, UV-vis, NMR, XPS and EDX spectra, as well as additional electrochemistry data. See DOI: 10.1039/d0sc03061a |
| This journal is © The Royal Society of Chemistry 2020 |