
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA covalent organic cage compound acting as a supramolecular shadow mask for the regioselective functionalization of C60†‡
Viktoria
Leonhardt
ab,
Stefanie
Fimmel
b,
Ana-Maria
Krause
ab and
Florian
Beuerle
 *ab
*ab
aUniversität Würzburg, Institut für Organische Chemie, Am Hubland, 97074 Würzburg, Germany. E-mail: florian.beuerle@uni-wuerzburg.de
bUniversität Würzburg, Center for Nanosystems Chemistry (CNC), Bavarian Polymer Institute (BPI), Theodor-Boveri-Weg, 97074 Würzburg, Germany
First published on 7th July 2020
Abstract
A trigonal-bipyramidal covalent organic cage compound serves as an efficient host to form stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-complexes with C60 and C70. Fullerene encapsulation has been comprehensively studied by NMR and UV/Vis spectroscopy, mass spectrometry as well as single-crystal X-ray diffraction. Exohedral functionalization of encapsulated C60via threefold Prato reaction revealed high selectivity for the symmetry-matched all-trans-3 addition pattern.
:1-complexes with C60 and C70. Fullerene encapsulation has been comprehensively studied by NMR and UV/Vis spectroscopy, mass spectrometry as well as single-crystal X-ray diffraction. Exohedral functionalization of encapsulated C60via threefold Prato reaction revealed high selectivity for the symmetry-matched all-trans-3 addition pattern.
Introduction
Fullerenes act as spherically arranged electron-deficient polyolefins.1 Their unique electronic structures facilitate potential applications in medicine,2 photovoltaics3 and organic electronics.4 Synthetically, addition reactions, e.g., Diels–Alder,5 Bingel,6 and Prato7 reactions or other dipolar cycloadditions,8 exploit the inherent strain energy of the curved double bonds, thus leading to a multitude of exohedrally functionalized derivatives.9Ih symmetrical C60 as the most abundant fullerene gives only one monoadduct. However, the number of potential regioisomers quickly raises to 8 (ref. 9d) and 46 (ref. 9a and e) for bis and trisaddition, respectively. The precise spatial organization of the individual addends identifies such multiple adducts as highly promising building blocks for 3D molecular architectures, e.g., dendritic systems10 or extended metal–organic11 and supramolecular12 frameworks. However, regioselectivity is generally governed by a combination of statistical and kinetic factors.13 Thereby, the second addition step usually serves as a bottle neck for the selective formation of higher adducts and individual regioisomers have to be purified, if at all possible, by tedious HPLC chromatography. In some cases, specific addition patterns such as C60Cl6,14 C60Ph5H,15 or octahedral hexakisaddition9c,16 have been obtained in surprisingly high purity under thermodynamic control.Following a tether-directed remote functionalization approach that was introduced by the Diederich group,17 all seven sterically possible bisadducts for methanofullerenes have been obtained by means of a covalent fixation of the two reactive sites in suitable tether systems.9a For higher adducts however, only a few tethers for privileged addition patterns are so far available.18 Elaborate tether synthesis and limitations in postsynthetic modification further hamper this approach. In contrast, any supramolecular prealignment of the reactants or an in situ activation of specific double bonds is much more tempting. Seminal work by Guldi, Torres and coworkers on Prato reactions for a phthalocyanine aldehyde showed the highly selective formation of cis-1 bisadducts mediated by π–π interactions between the chromophores.19 More recently, von Delius and coworkers reported the preferred formation of trans-bisadducts for Bingel reactions on C60⊂[10]cyclo-paraphenylene.20 When applying more sophisticated cage structures with a spatially precise orientation of multiple pore windows as hosts, the regioselective formation of higher adducts appears feasible (Fig. 1).
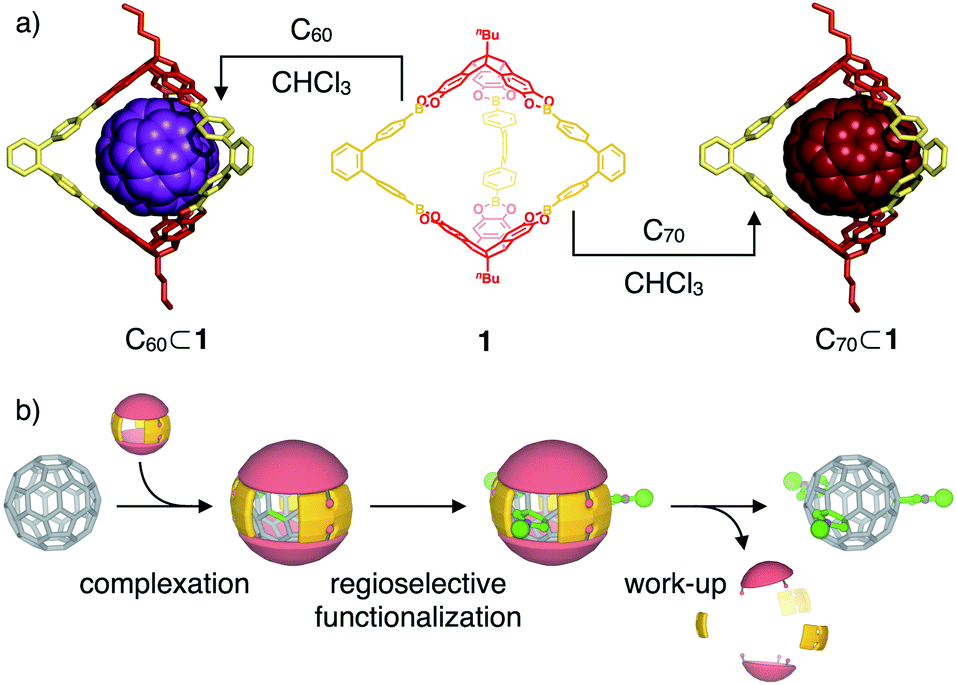 | ||
| Fig. 1 (a) Supramolecular complexation of fullerenes C60 and C70 within the cavity of covalent organic boronate ester cage 1 (molecular structures of inclusion complexes are derived from semiempirical PM6 modeling); (b) schematical representation for the use of C60-cage complexes as shadow masks for a regioselective exohedral C60 functionalization. | ||
In recent years, a large variety of organic and metallosupramolecular cage receptors for fullerenes have been reported.21 Selective binding was utilized for the separation of fullerene mixtures22 and electron transfer was studied for dye-attached complexes.23 Nitschke and coworkers reported on the selective encapsulation of Diels–Alder bisadduct mixtures within the cavity of an 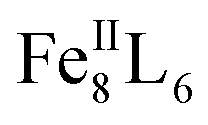 cage. However, the regioselectivity for the encapsulation has not been investigated.24 The Clever group used a bowl-shaped
cage. However, the regioselectivity for the encapsulation has not been investigated.24 The Clever group used a bowl-shaped 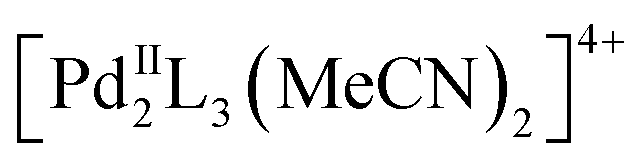 cage as a supramolecular protecting group for the selective monofunctionalization of encapsulated C60.25 The square-planar arrangement of the four cage windows for a metallosupramolecular fullerene sponge was recently utilized for the exclusive formation of all-e Bingel tetrakisadducts of C60, even under catalytic conditions.26 This masking strategy provides a significant improvement over the multistep orthogonal transposition approach27 as the so far only practicable synthetic route for this addition pattern. Despite these initial examples, no cage templates to control the inherently less selective Prato reaction have been reported so far.
cage as a supramolecular protecting group for the selective monofunctionalization of encapsulated C60.25 The square-planar arrangement of the four cage windows for a metallosupramolecular fullerene sponge was recently utilized for the exclusive formation of all-e Bingel tetrakisadducts of C60, even under catalytic conditions.26 This masking strategy provides a significant improvement over the multistep orthogonal transposition approach27 as the so far only practicable synthetic route for this addition pattern. Despite these initial examples, no cage templates to control the inherently less selective Prato reaction have been reported so far.
Results and discussion
Recently, we utilized the unique orthogonal arrangement of the three aromatic wings in C3v symmetrical hexahydroxy tribenzotriquinacenes (TBTQs)28 for the design and synthesis of covalent organic cage compounds21f,29via dynamic covalent boronate ester formation. By varying the bite angle of the diboronic acid counterpart, a series of cages30 with trigonal-bipyramidal, tetrahedral or cubic shape and sizes ranging from 1.9 to 3.5 nm (solvodynamic diameters derived from DOSY NMR) were obtained. As fullerene binding was already shown for some π-extended TBTQ derivatives,31 we tested the host properties of these cages. After saturation with either C60 or C70 by standing over pristine fullerenes for 24 hours at room temperature, fullerene uptake was analysed by UV/Vis absorption spectroscopy. No additional absorption in the range of 350 to 600 nm, which would indicate fullerene complexation, was observed for CHCl3 solutions of a methoxy-protected TBTQ precursor and a large cubic cage (Fig. S5 and S6†). In case of trigonal-bipyramidal [2 + 3] cage 1 however, a linear correlation between the host concentration and additional absorption features characteristic for C60 and C70, respectively, clearly indicated uptake of both fullerenes (Fig. S7†).Further evidence for encapsulation was obtained by MALDI-TOF MS. The isotope patterns for the major signals at m/z = 2323.57 and 2442.67 are in excellent agreement with the respective 1:1 host–guest complexes C60⊂1 and C70⊂1 (Fig. 1). Remarkably, no signals for the empty cages besides small peaks for pristine C60 or C70 are detected for 1:1 mixtures (Fig. 2a and b). Semiempirical molecular modeling on the PM6 level revealed that the two TBTQ units in 1 are indeed perfectly preorganized for efficient encapsulation of one fullerene molecule. The distance of the two TBTQ closo-atoms in 1, C60⊂1 and C70⊂1 only slightly changes from 1.55 to 1.66 and 1.69 nm (Fig. S1†). In the case of the larger cubic assemblies, only empty cages and no complexes were detected in MALDI measurements (Fig. S10†). Here, the increased distance between two TBTQs does not allow any favourable interaction of one fullerene with two TBTQs, thus preventing complexation.
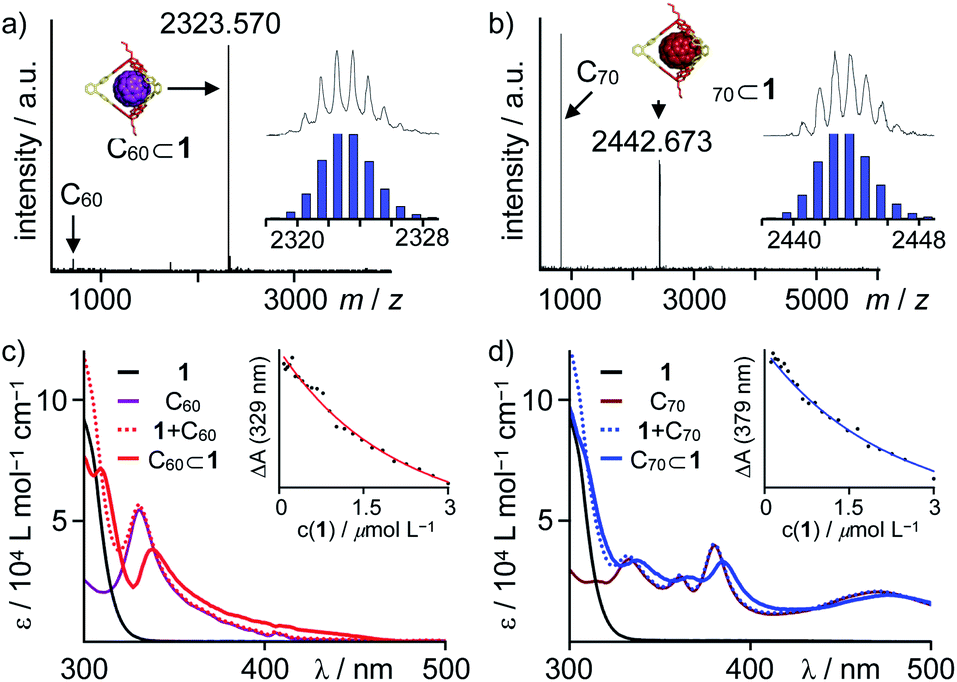 | ||
| Fig. 2 MALDI-TOF MS (TCNQ, CHCl3) for 1:1 mixtures of 1 and (a) C60 and (b) C70 (insets show comparison of measured and simulated isotope patterns); UV/Vis titration experiments for complex formation between 1 and (c) C60 (black 1, purple C60, dotted red 1 + C60, red C60⊂1 derived from global fit to 1:1 model, inset shows fit to 1:1 model at λ = 329 nm) and (d) C70 (black 1, brown C70, dotted blue 1 + C70, blue C70⊂1 derived from global fit to 1:1 model, inset shows fit to 1:1 model at λ = 379 nm). | ||
For a quantitative analysis, CHCl3 solutions of fullerenes C60 and C70 were titrated with a stock solution of 1 and absorption changes were plotted against cage concentration (see ESI† for details). Global fitting to a 1:1 binding model revealed complexation constants of 6.3 ± 0.4 × 105 M−1 and 5.3 ± 0.4 × 105 M−1 for C60 and C70, respectively (Fig. 2c and d). Apparently, there is no significant preference for either C60 or C70, which is presumably attributed to opposing effects of better size and shape matching for C60 and larger dispersion interactions for C70.
Ultimately, single crystals suitable for X-ray diffraction were obtained during reactivity studies (see below) for the C60 complex. C60⊂1 crystallizes in the monoclinic space group C2/c.†Fig. 3b shows an ORTEP representation of the 1:1 inclusion complex. Whereas the host could be nicely refined, much higher disorder was observed for the guest indicating a very low barrier for internal C60 rotation.
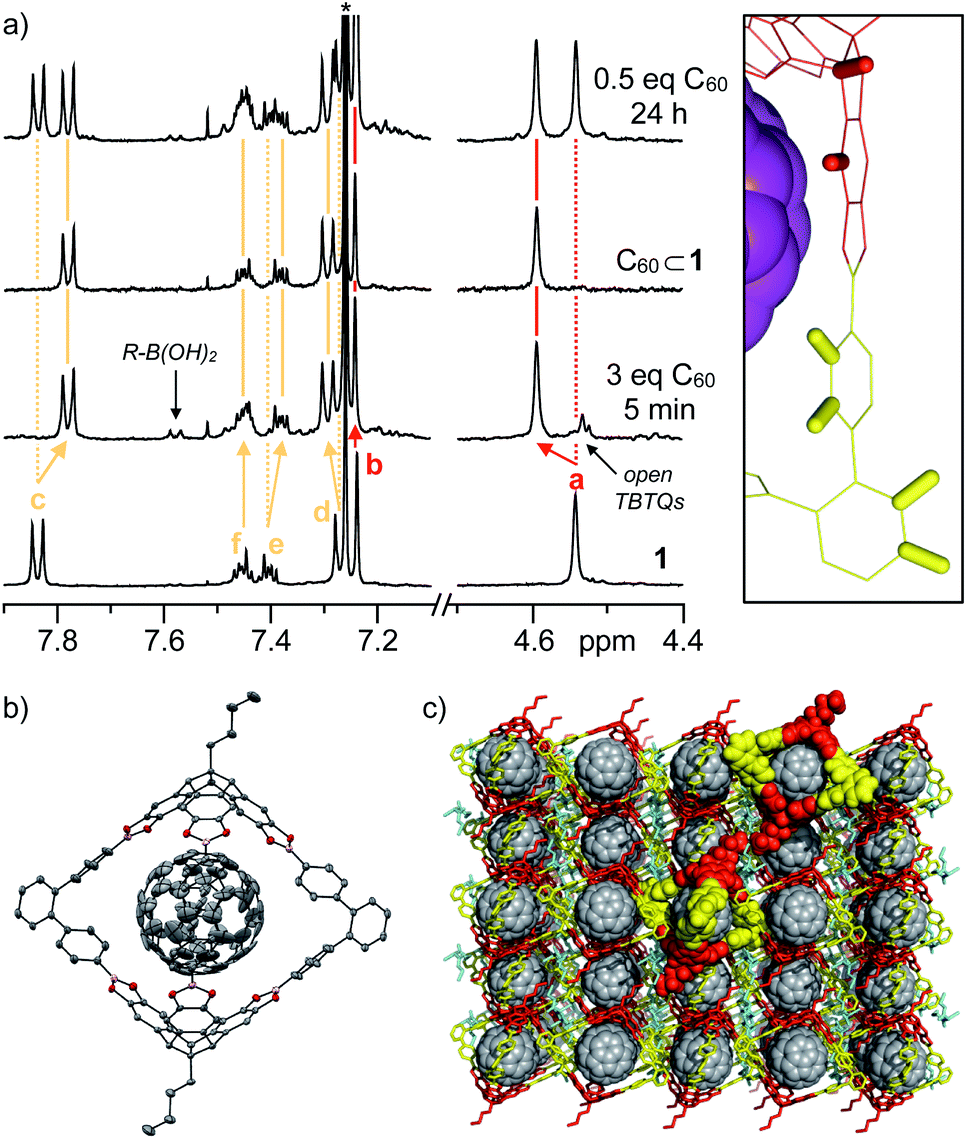 | ||
| Fig. 3 (a) 1H-NMR spectra (400 MHz, CDCl3, rt) for complexation of 1 with C60, (b) ORTEP representation of single-crystal X-ray structure of C60⊂1 (thermal ellipsoids were set at 50% probability, C = grey, O = red, B = purple, H atoms were omitted for clarity) and (c) packing of C60⊂1 in the solid state indicating the accessible C60 surface in the three cage windows. | ||
Specific shifting of cage protons in 1H NMR spectra of 1 in CDCl3 in the presence of 3 equivalents of solid C60 revealed instantaneous complex formation (Fig. 3a). The rather small shifts might be explained by the assumption that spherical C60 is freely rotating within the cavities and the fact, that the para and diamagnetic ring currents within the five- and six-membered rings of C60 are almost cancelling each other out. Directly after mixing, residual signals that can be attributed to free boronic acid and catechol moieties (Fig. 3a) suggest that encapsulation is achieved via partially opened cages and not via direct slipping. After one hour, cages 1 are again fully closed and quantitative complex formation is observed in case of equimolar mixtures or excess C60. For substoichiometric fullerene concentrations, the appearance of two separate sets of signals for both 1 and C60⊂1 suggests a rather high kinetic stability for the assemblies, at least on the NMR time scale. For C70⊂1, chemical shifts are more prominent, with the significant low-field shift for the aromatic TBTQ protons being particularly apparent (Fig. S2†). Therefore, we speculate that C70 is complexed in a more rigid fashion with the benzenoid hexagons of the equatorial belt being in close proximity to the axial TBTQ units and the fivefold rotational axes located along one of the cage windows. This model is also in accordance with semiempirical PM6 calculations (Fig. S1†). However, the exchange of the encapsulated C70 between the three windows should be fast, at least on the NMR time scale, as we did not observe any splitting of the signals for 1 in 1H-NMR. In the 13C-NMR spectrum of C60⊂1 (Fig. S3d†), a slight upfield shift from 143.24 to 141.65 ppm was observed for C60 upon complexation (Fig. S3e†). Due to the lower solubility of C70, no 13C-NMR spectrum could be measured in CDCl3. For C70⊂1 however, five signals at 129.01, 143.72, 146.18, 146.63 and 149.63 ppm appeared, which are in very good agreement with values reported for C70 in C6D6 (130.28, 144.77, 146.82, 147.52, 150.07).32
Based on these analytical data, we concluded that the 1:1 complex C60⊂1 forms with very high thermodynamic and kinetic stability. A space filling model from the X-ray structure (Fig. 3c) further reveals that the aromatic scaffold of 1 shields a significant part of the C60 surface apart from the three cage windows arranged in a trigonal planar fashion. Therefore, we speculated on the potential of C60⊂1 acting as a supramolecular template for exohedral C60 functionalization. Assuming that each window can only accommodate one addend due to steric constraints, a limitation to trisaddition was anticipated. Furthermore, trans-3 and neighbouring addition patterns, which approach the 120° angle between two individual cage windows, should be favoured (Fig. 6 and S16†).
The Bingel6 and Prato7 reactions are the two most common methods for exohedral functionalization of fullerenes. For the twofold Bingel reaction, there is an intrinsic preference for trans-3 and e-addition,13 whereas cis-addition is significantly disfavoured due to the steric demand of the ester substituents at the cyclopropane rings. For higher adducts, any pre-existing addends in e-position increasingly favour further e-addition, ultimately leading to the highly preferred Th symmetrical hexakisaddition pattern.33 Building upon this inherent selectivity, Ribas and coworkers reported on the exclusive formation of the all-equatorial tetrakisadduct for Bingel reactions on a complex of C60 within a tetragonal prismatic metallosupramolecular cage.26 For the Prato reaction however, a much broader distribution of all eight possible bisadducts is usually observed (Fig. S12b†).34 Only trans-1 and cis-1 isomers are formed in lower yields due to statistical (t1) and steric (c1) factors. Substantial cis-additions for smaller Prato addends generally result in very complex mixtures of higher adducts. In contrast to Bingel reactions, the isolation of specific regioisomers has only been reported in rare cases.34a
Initial investigations for Bingel reactions on C60⊂1 gave promising hints for the selective formation of specific bis and trisadducts at the early stages. At higher conversion however, an increasing proportion of the undesired background reaction on free C60 was observed due to partial decomposition of the cages in the basic medium. Under Prato conditions though, cage 1 is sufficiently stable to restrict functionalization to the encapsulated C60. As a model reaction, we chose the synthesis of N-methylfulleropyrrolidine multiple adducts C60(CH2NCH3CH2)n from sarcosine (Sar, N-methylglycine) and paraformaldehyde (Fig. 4a).
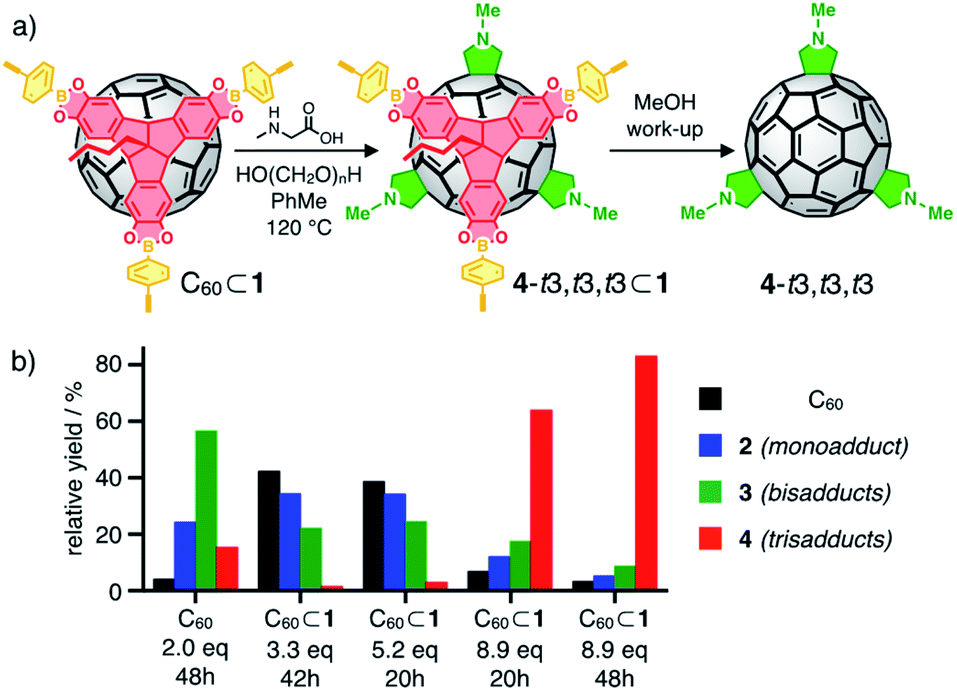 | ||
| Fig. 4 (a) Cage-templated synthesis of N-methylfulleropyrrolidine trisadducts; (b) relative yields (based on HPLC integration) for unreacted C60 (black) and Prato adducts 2 (mono, blue), 3 (bis, green) and 4 (tris, red) for control experiment and cage-templated reactions. | ||
The use of formaldehyde as carbonyl component prevents the occurrence of complex mixtures of stereoisomers at the pyrrolidine rings. The implementation of Me as a very small N-substituent also ensures that any selectivity effect is primarily based on the inherent properties of the template and not on the steric demand of the addends. Ultimately, comprehensive data on HPLC elution profiles and spectroscopic signatures are available34a,35 as reference for monoadduct 2 (n = 1), all regioisomeric bis (3, n = 2) and relevant trisadducts (4, n = 3).
To test the templating effect of cage 1, varying equivalents of Sar and excess formaldehyde were added to solutions of C60⊂1 in 1:1 dry CHCl3/toluene (or C60 in dry toluene as control) followed by heating under reflux (Fig. 4a, see ESI† for details) and reaction progress was monitored by MALDI-TOF MS. After specific time intervals, methanolic workup resulted in disassembly of the cages and the obtained fulleropyrrolidine mixtures were analyzed by analytical HPLC. As expected, multiple 1,3-dipolar cycloadditions on free C60 proceed rather unselective. After reaction with two equivalents Sar for eight hours, all eight regioisomeric bisadducts 3 and small amounts of trisadducts 4 were observed (Fig. 4b) in similar relative yields as previously reported (Fig. 5a and S12†).34a By contrast, reactions on C60⊂1 (Fig. 4b) proceed more slowly but with significantly higher selectivity. Reaction with up to 5 equivalents of Sar over 20 hours indicated low conversion with most of the C60 being unreacted (39% based on HPLC integration) or only converted to monoadduct 2 (34%). For the bisadducts 3 (24%), cis-addition is completely prevented and the symmetry-matched 3-t3 is formed as the main regioisomer (Fig. 5b).
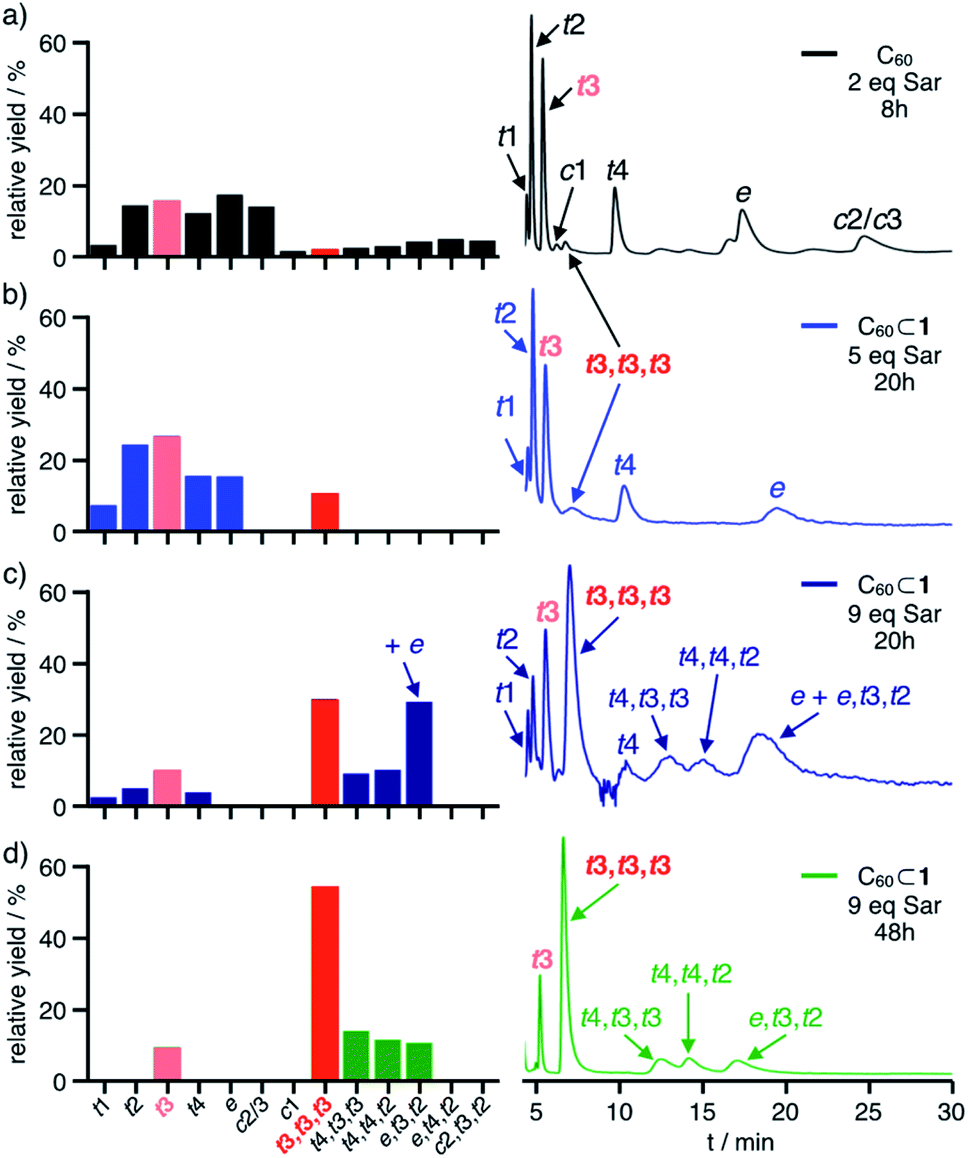 | ||
| Fig. 5 Relative yields (left, calculated from HPLC profiles) for bisadducts 3 and trisadducts 4 and HPLC chromatograms (right) for (a) control experiment with C60 (2.0 eq. Sar, 8 h) and cage-templated reactions with C60⊂1 and (b) 5 eq. Sar (20 h), (c) 9 eq. Sar (20 h) and (d) 9 eq. Sar (48 h, preparative scale); yields for 3-t3 and 4-t3,t3,t3 are indicated in light red and red, respectively. | ||
Intriguingly, even at low conversion significant amounts of D3-symmetrical 4-t3,t3,t3 (3%) are formed as the only detectable trisadduct. For this addition pattern, all three addends can be perfectly centred within the cage windows, thus minimizing any steric interactions between the addends and the cage walls.
By contrast, a control reaction on C60 with five equivalents of Sar for 20 hours resulted in a complex and inseparable mixture of many isomers with up to five pyrrolidine addends (Fig. S14a and d†). Reaction on C60⊂1 with 9 equivalents of Sar for 20 hours resulted in higher conversion and the predominant formation of trisadducts 4 (64%). According to MALDI-TOF MS for the reaction solution (Fig. S15a†), all fullerenes are still encapsulated and almost no higher adducts are formed. Remarkably, only four out of the possible 46 regioisomers for 4 were detected (Fig. 5c). Alongside the symmetry-matched 4-t3,t3,t3 (25%) as the main product, only 4-t4,t4,t2 (7%), 4-t4,t3,t3 (8%) and 4-e,t3,t2 (24% together with 3-e) were observed as side products. This selectivity can be nicely explained by molecular models for the trans- and e-bisadduct complexes 3⊂1 (see Fig. S17†), since any addition to double bonds centred in the third window leads exclusively to these four isomers (Fig. 6). Cage 1 acts as a supramolecular shadow mask that prevents over-functionalization and templates the addition in trans-3 and related positions. Indeed, no cis-adducts and only those four trisadducts, which best match the trigonal planar arrangement of the cage windows, were observed throughout the reaction time (Fig. 5).
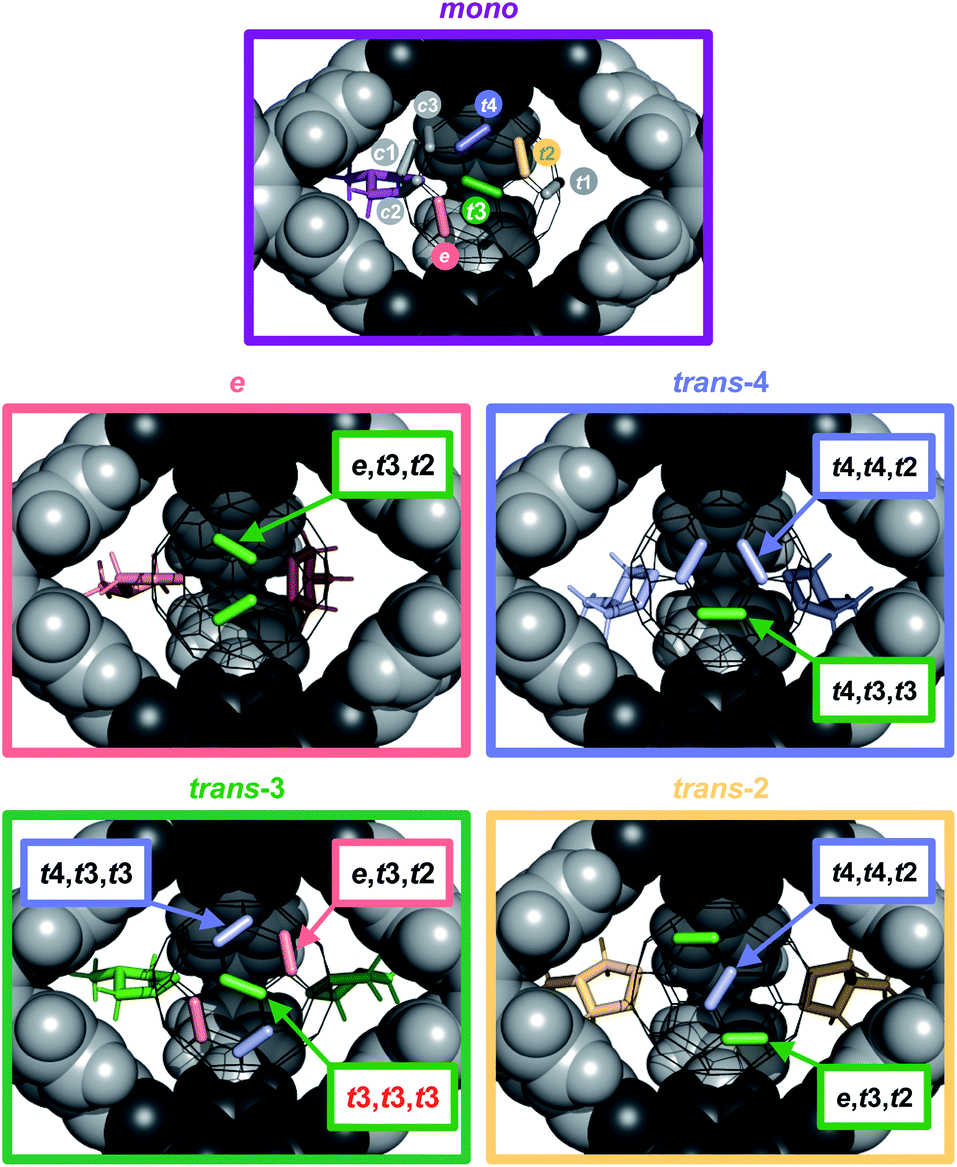 | ||
| Fig. 6 Accessibility of specific addition patterns in the cage windows for a second addition on 2⊂1 (top, first addend purple) and a third addition on 3-e⊂1 (middle left, e-addends in red), 3-t4⊂1 (middle right, t4-addends in blue), 3-t3⊂1 (bottom left, t3-addends in green) and 3-t2⊂1 (bottom right, t2-addends in yellow). | ||
For the non-templated reaction,34a nine different trisadducts 4 have been isolated in a combined yield of 4.4% (Fig. S24†) only after tedious chromatographic separation that involved several preparative TLC and HPLC steps. Thereby, 4-t3,t3,t3 was obtained as the minor isomer in only 0.2% yield. When running the cage-templated reaction (Fig. 4a) on a preparative scale, it proved advantageous to add up to 9 equivalents of Sar in three batches and to extend reaction times to 48 hours (see ESI† for details). For these prolonged reactions, the emerging decomposition of cage 1 initiated some overreaction outside of the cages (see Fig. S19a† for MALDI-TOF MS). Apparently, all mismatched isomers react faster, thus further simplifying the remaining mixture and increasing the relative yield for the matching 3-t3 and 4-t3,t3,t3 (Fig. 5d).
After methanolic work-up, all five remaining bis and trisadducts could be purified by one single automated flash chromatography run without the need for tedious HPLC separation. Finally, the four different trisadducts were isolated as pure regioisomers in a one-pot-reaction starting from pristine C60 with a combined yield of 16%. All products were analyzed by MALDI-TOF MS (Fig. S22†) and comparison of UV/Vis (Fig. S23†) and 1H-NMR spectra (Fig. S20 and S21†) to literature data.34a When compared to the rather nonselective control reaction, the selectivity for the cage-templated reaction is completely reversed and the isolated yield for 4-t3,t3,t3 as the main product is increased by a factor of 30 (Fig. S24†). The small pore windows of 1 enforce the formation of the unfavoured t3,t3,t3-addition pattern that would otherwise only be obtained in trace amounts. On the other hand, the cage walls prevent overreaction to higher adducts. In future work, we plan to optimize reaction conditions for a further increase in isolated amounts of pure trisadducts, modify the cage windows to enhance the selectivity and develop more stable cages to perform more sensitive reactions.
Conclusions
In summary, we have demonstrated the efficient fullerene encapsulation within the pores of trigonal bipyramidal covalent organic cage 1. Due to suitable preorganization of its two TBTQ moieties, strong 1:1 complexes C60⊂1 and C70⊂1 are formed in organic solvents and binding constants of 6.3 ± 0.4 × 105 and 5.3 ± 0.4 × 105 M−1, respectively, have been determined by UV/Vis titrations. As evidenced by an X-ray structure for C60⊂1, only three distinct parts of the C60 surface are accessible for exohedral functionalization. In prototypical Prato reactions on C60⊂1, remarkable selectivity for the symmetry-matched trisadduct 4-t3,t3,t3, which is formed only in trace amounts in cage-free reactions, was observed. Cage 1 acts as an efficient supramolecular shadow mask, whose small pore windows force the formation of an otherwise unfavoured trisaddition pattern. These exciting findings are the first example for a supramolecular control of the regioselectivity for the intrinsically nonselective Prato reaction. They will pave the way for novel applications of covalent organic cage compounds as effective templates for spatially precise reactions on large spherical π-systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Fonds der Chemischen Industrie (Liebig fellowship for FB and doctoral fellowship for SF), the DFG (BE4808/2-1) and the Collaborative Research Network “Solar Technologies Go Hybrid” of the Bavarian Ministry of Science, Research and the Arts.Notes and references
- R. Taylor and D. R. M. Walton, Nature, 1993, 363, 685–693 CrossRef CAS.
- (a) E. Castro, A. H. Garcia, G. Zavala and L. Echegoyen, J. Mater. Chem. B, 2017, 5, 6523–6535 RSC; (b) F. Beuerle, R. Lebovitz and A. Hirsch, in Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes, 2008, vol. 1, pp. 51–78 Search PubMed.
- (a) C.-Z. Li, H.-L. Yip and A. K.-Y. Jen, J. Mater. Chem., 2012, 22, 4161–4177 RSC; (b) G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolski and N. Martin, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC.
- M. Tsuda, T. Ishida, T. Nogami, S. Kurono and M. Ohashi, Chem. Commun., 1993, 1296–1298 RSC.
- C. Bingel, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- W. Zhang and T. M. Swager, J. Am. Chem. Soc., 2007, 129, 7714–7715 CrossRef CAS PubMed.
- (a) A. Hirsch and M. Brettreich, Fullerenes — Chemistry and Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) C. Thilgen and F. Diederich, Chem. Rev., 2006, 106, 5049–5135 CrossRef CAS PubMed; (c) W. Yan, S. M. Seifermann, P. Pierrat and S. Bräse, Org. Biomol. Chem., 2015, 13, 25–54 RSC; (d) A. Hirsch, I. Lamparth and H. R. Karfunkel, Angew. Chem., Int. Ed., 1994, 33, 437–438 CrossRef; (e) F. Djojo, A. Hirsch and S. Grimme, Eur. J. Org. Chem., 1999, 3027–3039 CrossRef CAS.
- (a) F. Hörmann and A. Hirsch, Chem.–Eur. J., 2013, 19, 3188–3197 CrossRef PubMed; (b) S. K. Dey, F. Beuerle, M. A. Olson and J. F. Stoddart, Chem. Commun., 2011, 47, 1425–1427 RSC; (c) A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J.-S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J.-F. Nierengarten and N. Martín, Nat. Chem., 2016, 8, 50–57 CrossRef PubMed; (d) S. Guerra, J. Iehl, M. Holler, M. Peterca, D. A. Wilson, B. E. Partridge, S. Zhang, R. Deschenaux, J.-F. Nierengarten and V. Percec, Chem. Sci., 2015, 6, 3393–3401 RSC; (e) B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS PubMed; (f) M. Riala, K. L. Maxouti, C. P. Ioannou and N. Chronakis, Org. Lett., 2016, 18, 1132–1135 CrossRef CAS PubMed; (g) F. Beuerle and A. Hirsch, Chem.–Eur. J., 2009, 15, 7447–7455 CrossRef CAS PubMed.
- (a) A. Kraft and F. Beuerle, Tetrahedron Lett., 2016, 57, 4651–4663 CrossRef CAS; (b) A. M. Rice, E. A. Dolgopolova and N. B. Shustova, Chem. Mater., 2017, 29, 7054–7061 CrossRef CAS; (c) A. L. Balch and K. Winkler, Chem. Rev., 2016, 116, 3812–3882 CrossRef CAS PubMed; (d) P. Peng, F.-F. Li, V. S. P. K. Neti, A. J. Metta-Magana and L. Echegoyen, Angew. Chem., Int. Ed., 2014, 53, 160–163 CrossRef CAS PubMed; (e) A. Kraft, P. Roth, D. Schmidt, J. Stangl, K. Müller-Buschbaum and F. Beuerle, Chem.–Eur. J., 2016, 22, 5982–5987 CrossRef CAS PubMed; (f) A. Kraft, C. Roger, D. Schmidt, J. Stangl, K. Müller-Buschbaum and F. Beuerle, Chem.–Eur. J., 2017, 23, 15864–15868 CrossRef CAS PubMed.
- (a) A. Kraft, M. Gsänger and F. Beuerle, Eur. J. Org. Chem., 2014, 523–528 CrossRef CAS; (b) A. Kraft, J. Stangl, A.-M. Krause, K. Müller-Buschbaum and F. Beuerle, Beilstein J. Org. Chem., 2017, 13, 1–9 CrossRef CAS PubMed; (c) E. Fernandez-Bartolome, J. Santos, A. Gamonal, S. Khodabakhshi, L. J. McCormick, S. J. Teat, E. C. Sañudo, J. S. Costa and N. Martín, Angew. Chem., Int. Ed., 2019, 58, 2310–2315 CrossRef CAS PubMed.
- F. Djojo, A. Herzog, I. Lamparth, F. Hampel and A. Hirsch, Chem.–Eur. J., 1996, 2, 1537–1547 CrossRef CAS.
- P. R. Birkett, A. G. Avent, A. D. Darwish, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Chem. Commun., 1993, 1230–1232 RSC.
- M. Sawamura, H. Iikura and E. Nakamura, J. Am. Chem. Soc., 1996, 118, 12850–12851 CrossRef CAS.
- (a) I. Lamparth, C. Maichle-Mössmer and A. Hirsch, Angew. Chem., Int. Ed., 1995, 34, 1607–1609 CrossRef CAS; (b) A. Hirsch and O. Vostrowsky, Eur. J. Org. Chem., 2001, 2001, 829–848 CrossRef.
- L. Isaacs, R. F. Haldimann and F. Diederich, Angew. Chem., Int. Ed., 1994, 33, 2339–2342 CrossRef.
- (a) U. Reuther, T. Brandmüller, W. Donaubauer, F. Hampel and A. Hirsch, Chem.–Eur. J., 2002, 8, 2261–2273 CrossRef CAS PubMed; (b) F. Beuerle, N. Chronakis and A. Hirsch, Chem. Commun., 2005, 3676–3678 RSC; (c) F. Beuerle and A. Hirsch, Chem.–Eur. J., 2009, 15, 7434–7446 CrossRef CAS PubMed.
- G. Bottari, O. Trukhina, A. Kahnt, M. Frunzi, Y. Murata, A. Rodríguez-Fortea, J. M. Poblet, D. M. Guldi and T. Torres, Angew. Chem., Int. Ed., 2016, 55, 11020–11025 CrossRef CAS PubMed.
- Y. Xu, R. Kaur, B. Wang, M. B. Minameyer, S. Gsanger, B. Meyer, T. Drewello, D. M. Guldi and M. von Delius, J. Am. Chem. Soc., 2018, 140, 13413–13420 CrossRef CAS PubMed.
- (a) T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250–5273 CrossRef CAS PubMed; (b) K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC; (c) C. García-Simón, M. Costas and X. Ribas, Chem. Soc. Rev., 2016, 45, 40–62 RSC; (d) C. Yu, Y. Jin and W. Zhang, Chem. Rec., 2015, 15, 97–106 CrossRef CAS PubMed; (e) D. Canevet, E. M. Pérez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 9248–9259 CrossRef CAS PubMed; (f) F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed; (g) C. García-Simón, A. Monferrer, M. Garcia-Borràs, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem. Commun., 2019, 55, 798–801 RSC; (h) V. Martínez-Agramunt, T. Eder, H. Darmandeh, G. Guisado-Barrios and E. Peris, Angew. Chem., Int. Ed., 2019, 58, 5682–5686 CrossRef PubMed.
- (a) C. Zhang, Q. Wang, H. Long and W. Zhang, J. Am. Chem. Soc., 2011, 133, 20995–21001 CrossRef CAS PubMed; (b) C. Fuertes-Espinosa, A. Gómez-Torres, R. Morales-Martínez, A. Rodríguez-Fortea, C. García-Simón, F. Gándara, I. Imaz, J. Juanhuix, D. Maspoch, J. M. Poblet, L. Echegoyen and X. Ribas, Angew. Chem., Int. Ed., 2018, 57, 11294–11299 CrossRef CAS PubMed; (c) M.-J. Li, C.-H. Huang, C.-C. Lai and S.-H. Chiu, Org. Lett., 2012, 14, 6146–6149 CrossRef CAS PubMed; (d) W. Sun, Y. Wang, L. Ma, L. Zheng, W. Fang, X. Chen and H. Jiang, J. Org. Chem., 2018, 83, 14667–14675 CrossRef CAS PubMed.
- (a) Y. Xu, B. Wang, R. Kaur, M. B. Minameyer, M. Bothe, T. Drewello, D. M. Guldi and M. von Delius, Angew. Chem., Int. Ed., 2018, 57, 11549–11553 CrossRef CAS PubMed; (b) M. Ortiz, S. Cho, J. Niklas, S. Kim, O. G. Poluektov, W. Zhang, G. Rumbles and J. Park, J. Am. Chem. Soc., 2017, 139, 4286–4289 CrossRef CAS PubMed; (c) S. Bähring, K. R. Larsen, M. Supur, K. A. Nielsen, T. Poulsen, K. Ohkubo, C. W. Marlatt, E. Miyazaki, K. Takimiya, A. H. Flood, S. Fukuzumi and J. O. Jeppesen, Chem. Commun., 2017, 53, 9898–9901 RSC; (d) J. Rio, S. Beeck, G. Rotas, S. Ahles, D. Jacquemin, N. Tagmatarchis, C. Ewels and H. A. Wegner, Angew. Chem., Int. Ed., 2018, 57, 6930–6934 CrossRef CAS PubMed.
- W. Brenner, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 75–78 CrossRef CAS PubMed.
- B. Chen, J. J. Holstein, S. Horiuchi, W. G. Hiller and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 8907–8913 CrossRef CAS PubMed.
- C. Fuertes-Espinosa, C. García-Simón, M. Pujals, M. Garcia-Borràs, L. Gómez, T. Parella, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem, 2020, 6, 169–186 CAS.
- R. Schwenninger, T. Müller and B. Kräutler, J. Am. Chem. Soc., 1997, 119, 9317–9318 CrossRef CAS.
- (a) D. Kuck, Angew. Chem., Int. Ed., 1984, 23, 508–509 CrossRef; (b) D. Kuck, Chem. Rev., 2006, 106, 4885–4925 CrossRef CAS PubMed; (c) A. Dhara and F. Beuerle, Synthesis, 2018, 50, 2867–2877 CrossRef CAS.
- (a) G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC; (b) T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS; (c) M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS PubMed.
- (a) S. Klotzbach, T. Scherpf and F. Beuerle, Chem. Commun., 2014, 50, 12454–12457 RSC; (b) S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed.
- (a) B. Bredenkötter, S. Henne and D. Volkmer, Chem.–Eur. J., 2007, 13, 9931–9938 CrossRef PubMed; (b) S. Henne, B. Bredenkötter, A. A. Dehghan Baghi, R. Schmid and D. Volkmer, Dalton Trans., 2012, 41, 5995–6002 RSC; (c) B. Bredenkötter, M. Grzywa, M. Alaghemandi, R. Schmid, W. Herrebout, P. Bultinck and D. Volkmer, Chem.–Eur. J., 2014, 20, 9100–9110 Search PubMed; (d) S. Henne, B. Bredenkötter, M. Alaghemandi, S. Bureekaew, R. Schmid and D. Volkmer, ChemPhysChem, 2014, 15, 3855–3863 CrossRef CAS PubMed; (e) T. Wang, Z.-Y. Li, A.-L. Xie, X.-J. Yao, X.-P. Cao and D. Kuck, J. Org. Chem., 2011, 76, 3231–3238 CrossRef CAS PubMed; (f) P. E. Georghiou, L. N. Dawe, H.-A. Tran, J. Strübe, B. Neumann, H.-G. Stammler and D. Kuck, J. Org. Chem., 2008, 73, 9040–9047 CrossRef CAS PubMed.
- R. Taylor, J. P. Hare, A. K. Abdul-Sada and H. W. Kroto, Chem. Commun., 1990, 1423–1425 RSC.
- A. Hirsch, I. Lamparth, T. Grösser and H. R. Karfunkel, J. Am. Chem. Soc., 1994, 116, 9385–9386 CrossRef CAS.
- (a) S. Marchesan, T. Da Ros and M. Prato, J. Org. Chem., 2005, 70, 4706–4713 CrossRef CAS PubMed; (b) Q. Lu, D. I. Schuster and S. R. Wilson, J. Org. Chem., 1996, 61, 4764–4768 CrossRef CAS PubMed.
- T. Nishimura, K. Tsuchiya, S. Ohsawa, K. Maeda, E. Yashima, Y. Nakamura and J. Nishimura, J. Am. Chem. Soc., 2004, 126, 11711–11717 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1913637. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03131c |
| ‡ C60⊂1: C60⊂C106H72B6O4·(C7H11BrO4)2, M = 2801.23 g mol−1, monoclinic, C2/c, a = 23.3258(10), b = 22.5169(10), c = 31.3889(10) Å, α = 90, β = 91.177(2), γ = 90°, V = 16482.7(12) Å3, Z = 4, ρcalc. = 1.129 g cm−3, μ(CuKα) = 1.129 mm−1, T = 100(2) K; 168505 independent measured reflections. F2 refinement, R1 = 0.1264, wR2 = 0.4437 (observed), 16220 independent observed reflections (Rint = 0.0337) [|F0| > 4σ(|F0|), 2Θ ≤ 144.72°], 1100 parameters, 490 restraints. |
| This journal is © The Royal Society of Chemistry 2020 |