
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully oxygen-tolerant atom transfer radical polymerization triggered by sodium pyruvate†
Grzegorz
Szczepaniak
 *ab,
Matylda
Łagodzińska
ac,
Sajjad
Dadashi-Silab
a,
Adam
Gorczyński
ad and
Krzysztof
Matyjaszewski
*a
*ab,
Matylda
Łagodzińska
ac,
Sajjad
Dadashi-Silab
a,
Adam
Gorczyński
ad and
Krzysztof
Matyjaszewski
*a
aDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, Pennsylvania 15213, USA. E-mail: km3b@andrew.cmu.edu
bFaculty of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
cDepartment of Chemistry, University of Oxford, South Parks Road, Oxford OX13QZ, UK
dFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
First published on 6th August 2020
Abstract
ATRP (atom transfer radical polymerization) is one of the most robust reversible deactivation radical polymerization (RDRP) systems. However, the limited oxygen tolerance of conventional ATRP impedes its practical use in an ambient atmosphere. In this work, we developed a fully oxygen-tolerant PICAR (photoinduced initiators for continuous activator regeneration) ATRP process occurring in both water and organic solvents in an open reaction vessel. Continuous regeneration of the oxidized form of the copper catalyst with sodium pyruvate through UV excitation allowed the chemical removal of oxygen from the reaction mixture while maintaining a well-controlled polymerization of N-isopropylacrylamide (NIPAM) or methyl acrylate (MA) monomers. The polymerizations of NIPAM were conducted with 250 ppm (with respect to the monomer) or lower concentrations of CuBr2 and a tris[2-(dimethylamino)ethyl]amine ligand. The polymers were synthesized to nearly quantitative monomer conversions (>99%), high molecular weights (Mn > 270![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), and low dispersities (1.16 < Đ < 1.44) in less than 30 min under biologically relevant conditions. The reported method provided a well-controlled ATRP (Đ = 1.16) of MA in dimethyl sulfoxide despite oxygen diffusion from the atmosphere into the reaction system.
000), and low dispersities (1.16 < Đ < 1.44) in less than 30 min under biologically relevant conditions. The reported method provided a well-controlled ATRP (Đ = 1.16) of MA in dimethyl sulfoxide despite oxygen diffusion from the atmosphere into the reaction system.
Introduction
According to the IUPAC report, reversible deactivation radical polymerization (RDRP) is one of the top ten emerging technologies in chemistry that could change the world.1 Atom transfer radical polymerization (ATRP) is one of the most widely used RDRP methods, providing access to well-defined, complex polymer architectures.2–7ATRP is catalyzed by transition metal complexes in their lower oxidation state. It is exceptionally tolerant to a wide variety of functional groups, solvents, and impurities. However, like any radical polymerization, ATRP is inhibited by oxygen. Recently, several avenues to design oxygen tolerant RDRP systems have been reviewed.8 The most active copper catalysts with highly negative redox potentials allow a well-controlled polymerization at a loading of only 10 ppm relative to the monomer.9–12 Even trace amounts of oxygen can inhibit polymerization by rapidly oxidizing the activator form of the catalyst CuI/L to the inactive CuII/L complex.13 Furthermore, oxygen molecules can react with the propagating carbon-based radicals, thus terminating the polymerization process.14 The sensitivity of ATRP to oxygen necessitates the use of specialized equipment or deoxygenation by inert gas sparging before the polymerization (Scheme 1). As a result, ATRP techniques can be cumbersome to non-experts. On top of that, inert gas sparging or freeze–pump–thaw degassing are often incompatible with the synthesis of hybrid biomacromolecules,15,16 as they may cause protein denaturation or a loss of enzymatic activity.17
 | ||
| Scheme 1 Approaches for oxygen scrubbing and achieving oxygen tolerance in ATRP. | ||
The CuI/L ATRP catalyst activates the dormant C(sp3)–X polymer chain end, resulting in the formation of the X–CuII/L complex and a carbon-centered radical. Both carbon-based radicals and CuI/L species react with molecular oxygen with diffusion control to form peroxy radicals or hydroperoxides and CuII/L complexes, respectively (Scheme 1). However, since CuI/L is at a concentration thousands to millions times higher than the concentration of propagating radicals, oxidation of the CuI/L activator to CuII/L is predominant. Thus, continuous regeneration of the oxidized form of the catalyst CuII/L with a reducing agent allows the chemical removal of oxygen from the reaction system (Scheme 1).
In 1998, we demonstrated that a well-controlled ATRP could occur in the presence of a limited amount of oxygen using a zero-valent copper powder as a reducing agent.18 This concept was later extended to ATRP with copper wire19–21 or copper plate22–25 and other reducing agents, such as ascorbic acid,26–29 tin(II) 2-ethylhexanoate,30 tertiary amine,31 nitrogen-based ligands,32 phenols,33 alcohols,34 sodium dithionite,35 and zero-valent iron.36 Another area where significant progress has been made towards oxygen-tolerant ATRP is photoinduced polymerization.37–41 In photoinduced ATRP, catalyst regeneration occurs by excitation of the CuII/L complex, followed by a single electron donation from the amine-based ligand. Photoirradiation of a copper catalyst in the presence of an electron donor in excess enables removal of dissolved oxygen.42–54 Despite these great developments, the vast majority of reported methods are successful only when polymerization is performed in sealed vessels with a limited amount of oxygen in the reaction mixture. So far, only a few ATRP systems, mainly based on enzymatic degassing, can be carried out in a completely open reaction vessel, where oxygen continuously diffuses into the system from the atmosphere.55–58
In 2018, inspired by the works of Yagci59 and Stevens,60,61 we developed a “breathing ATRP” of oligo(ethylene oxide)methyl ether methacrylate that used glucose oxidase (GOx) as a highly efficient scavenger for oxygen.55 GOx catalyzes the oxidation of β-D-glucose to D-glucono-1,5-lactone and hydrogen peroxide. However, hydrogen peroxide reacts with CuI/L in a Fenton-type reaction to form a hydroxyl radical and the CuII/L complex. Hydroxyl radicals can initiate new polymer chains, decreasing average molecular weights (Mn) as compared to the theoretical values. To suppress this undesirable process, we developed a bio-inspired ATRP system in which GOx removed oxygen, while sodium pyruvate (SP) acted as a hydrogen peroxide scavenger and prevented the formation of new polymer chains. This study was later extended to “oxygen-fueled” ATRP by employing Horseradish peroxidase (HRP) as a catalyst for the generation of radicals from acetylacetone in the presence of hydrogen peroxide produced by GOx. This enzymatic cascade enabled a well-controlled ATRP in a reaction vessel open to the air.56 However, these high-performance biocatalytic systems created a new challenge: the synthesized polymers or polymer bioconjugates were contaminated with enzymes, which are particularly difficult to separate from biohybrids. Also, the methods were limited to aqueous media. Recently, Keitz et al. harnessed an even more complex biological system, microbial metabolism, to develop an aerobic ATRP in water.62 As with the enzymatic degassing, the use of cellular respiration machinery in bioconjugates synthesis can complicate the purification process. The development of efficient small molecule-based ATRP methods tolerant to oxygen that are compatible with water and organic solvents is therefore highly desirable.
Poly(N-isopropylacrylamide) (PNIPAM) is a temperature-responsive, biocompatible polymer that has a lower critical solution temperature in water of ∼32 °C.63 This feature is widely used in the design of controlled drug delivery systems,64 tissue engineering65 and biosensing.66,67 Low dispersity PNIPAM with varying molecular weights can be synthesized using a variety of ATRP techniques.68–72 However, the methods reported so far exhibit at least one critical flaw, such as the use of high loadings of copper catalysts, a relatively long reaction time, or oxygen intolerance. Recently, the disproportionation of CuI/Me6TREN in water was shown to enable the ATRP of N-isopropylacrylamide (NIPAM) in open-air conditions. However, the use of high copper concentration (2000–8000 ppm relative to NIPAM) was necessary to attain high monomer conversions and low dispersity values.73
Herein, we demonstrate the first fully oxygen tolerant, photoinduced ICAR ATRP of NIPAM with ppm level of Cu catalyst in water, enabling a quantitative conversion of the monomer in less than 30 min. This simplified, non-enzymatic ATRP system uses sodium pyruvate as both a hydrogen peroxide scavenger and a “fuel” for the continuous regeneration of the catalyst and can be easily transferred to organic solvents.
Results and discussion
Initial studies began by polymerizing NIPAM in water (targeting a degree of polymerization 200) under UV LED irradiation (λ = 394 nm, 2.6 mW cm−2), using 2-hydroxyethyl 2-bromoisobutyrate (HOBiB) as the initiator, CuBr2 as the precatalyst, and tris[2-(dimethylamino)ethyl]amine (Me6TREN) as the ligand (Table 1). The reactions were carried out in sealed vials with a septum (see Fig. S1 in the ESI†) at 6 °C and in the presence of limited amounts of oxygen (without degassing the reaction mixture). Table 1 shows the results of the polymerization of NIPAM and the effect of different components involved in the PICAR ATRP system.| No. | CuBr2b (equiv.) | Ligandb (equiv.) | SP (mM) | 1× PBS(Br)c | Time (h) | Conv.d (%) | M n,th | M n,GPC | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Reactions conditions: [NIPAM]/[HOBiB]/[CuBr2]/[ligand]: 200/1/0.05–0.10/0.15–0.60, [NIPAM] = 0.8 M, [HOBiB] = 4 mM, [NaBr] = 100 mM, in water at 6 °C, under UV LEDs (λ = 394 nm, 2.6 mW cm−2) in the presence of oxygen (non-degassed solution). b Relative to the initiator. c Modified PBS buffer containing bromide anions. ✓/− means only NaBr and KBr present in PBS(Br) were added to the reaction, but no Na2HPO4 nor KH2PO4. d Monomer conversion was determined by using 1H NMR spectroscopy. e See SEC traces in the ESI Fig. S2. f No NaBr. All measurements were analyzed using GPC (dimethylformamide as eluent) calibrated with linear poly(methyl methacrylate) standards. | |||||||||
| 1f | 0.05 | Me6TREN (0.30) | — | — | 12 | 16 | 3600 | 9500 | 1.86 |
| 2 | 0.05 | Me6TREN (0.30) | — | ✓ | 12 | 17 | 3900 | 2500 | 1.66 |
| 3 | 0.05 | Me6TREN (0.30) | 100 | ✓/− | 0.5 | >99 | 22800 |
19500 |
1.42 |
| 4 | 0.05 | Me6TREN (0.30) | 100 | ✓ | 0.5 | >99 | 22800 |
31700 |
1.16 |
| 5 | 0.02 | Me6TREN (0.12) | 100 | ✓ | 0.5 | 99 | 22600 |
24800 |
1.21 |
| 6 | 0.01 | Me6TREN (0.06) | 100 | ✓ | 0.5 | 97 | 21700 |
27200 |
1.37 |
| 7 | 0.10 | Me6TREN (0.30) | 100 | ✓ | 0.5 | 99 | 22600 |
29700 |
1.17 |
| 8 | 0.05 | Me6TREN (0.15) | 100 | ✓ | 0.5 | 98 | 22400 |
29800 |
1.18 |
| 9 | 0.05 | Me6TREN (0.05) | 100 | ✓ | 0.5 | 99 | 22600 |
27000 |
1.49 |
| 10 | 0.05 | Me6TREN (0.30) | 200 | ✓ | 0.5 | >99 | 22800 |
31100 |
1.20 |
| 11 | 0.05 | Me6TREN (0.30) | 50 | ✓ | 0.5 | 98 | 22400 |
28000 |
1.16 |
| 12 | 0.05 | TPMA (0.30) | 100 | ✓ | 0.5 | 54 | 12300 |
17700 |
1.84 |
| 13 | 0.01 | TPMA*3 (0.30) | 100 | ✓ | 0.5 | 93 | 21200 |
25600 |
1.46 |
A set of control experiments was performed to evaluate the influence of SP on the ATRP process (Table 1, entries 1–3). The initial conditions used 250 ppm of CuBr2 (with respect to the monomer) with a six-fold excess of Me6TREN ligand to CuII and no SP. After 12 h of UV irradiation, the conversion of NIPAM measured by 1H NMR was only 16%. Furthermore, size exclusion chromatography (SEC) analysis showed that the polymer had a high dispersity (Đ) of 1.86 (Table 1, entry 1). The use of Cu-based ATRP catalysts in water typically results in a significant dissociation of the [X–CuII/L]+ deactivator to the “naked” [CuII/L]2+ dication and a free halide anion. The [CuII/L]2+ complex cannot act as a true deactivator, leading to poorly controlled polymerizations. To counteract this problem, aqueous ATRP is performed in the presence of halide anions to suppress the deactivator dissociation.74 The use of modified phosphate-buffered saline (PBS) solution containing bromide anions gave slightly better results (Table 1, entry 2; Đ = 1.66). To our delight, when both the Br-based PBS and SP were used (Table 1, entry 4), a quantitative conversion was achieved within 30 min, and the polymerization was well-controlled (Mn = 31700, Đ = 1.16). These experiments showed the critical role of SP. The reaction without the addition of buffer components (Na2HPO4 and KH2PO4 salts) reached quantitative monomer conversion with a similar rate of polymerization. However, the dispersity of the resulting polymer was 1.42 (Table 1, entry 3). For aqueous ATRP, the optimal pH is 7.5.75 UV irradiation induces the homolytic cleavage of SP (see further section on proposed mechanism), which leads to the protonation of the ligand, decreasing its ability to coordinate the metal center, resulting in a loss of control over the polymerization. Maintaining a constant pH ∼ 7.4 during the polymerization prevents this process.
Next, the performance of sodium pyruvate-based ATRP system was evaluated in the presence of varying amounts of CuBr2 (Table 1, entries 4–7). Despite decreasing the amount of CuBr2 to just 50 ppm relative to NIPAM, the reaction still proceeded to high monomer conversion (>97%) and yielding a polymer with a dispersity of 1.37 (Table 1, entry 6). Increasing the amount of CuBr2 to 1000 ppm did not improve the outcome, yielding similar control as with 250 ppm of CuBr2 (Table 1, entry 7).
In conventional photoinduced ATRP, a CuII complex in the excited state reacts with an amine-based ligand, which acts as an electron donor, resulting in the formation of the activator CuI/L and a radical cation from the donor.39,76 Since this process consumes the ligand, it must be present in excess. In our PICAR ATRP, SP is the dominant electron donor. However, in the presence of dissolved oxygen, the ligand oxidation may still occur during photoirradiation. This explains why the ratio [CuBr2]/[Me6TREN] = 1/1 was not sufficient to achieve well-controlled polymerization while maintaining high conversion (Table 1, entry 9). The use of a 1/3 or 1/6 ratio allowed much better control over polymerization of NIPAM (Table 1, entry 8 and 4).
Increasing the concentration of SP from 100 mM to 200 mM caused a slight increase in PNIPAM dispersity (Table 1, entry 10; Đ = 1.20). This could be attributed to a higher concentration of radicals resulting from the homolytic cleavage of SP under UV irradiation. The radicals thus formed could initiate new polymer chains or terminate polymerization by radical–radical coupling. Decreasing the SP concentration to 50 mM caused only a slight decrease in monomer conversion (98%), while maintaining the low Đ = 1.16 (Table 1, entry 11). However, further tests were carried out with a concentration of SP of 100 mM to make the polymerization more tolerant to oxygen.
Several hypotheses have been proposed to explain the difficulty of obtaining good control in the polymerization of acrylamides by ATRP.77–80 One possibility is the intramolecular cyclization reaction leading to the loss of C(sp3)–Br chain end. The ω–Br chain end functionality was shown to decrease as a function of reaction time and was dependent on the structure of the amide group.79 Low chain-end fidelity compromises the control over polymerization. The use of the pyridine-based ligands: the less active TPMA or the more active TPMA*3 ligands (TPMA = tris(2-pyridylmethyl)amine, TPMA*3 = tris([(4-methoxy-2,5-dimethyl)-2-pyridyl]methyl)amine)81 resulted in a significant decrease in control over the polymerization (Table 1, entry 12 and 13). TPMA is the most versatile ligand for aqueous ATRP of acrylates and methacrylates.75 However, for acrylamides, the [CuI/TPMA]+ catalyst does not provide a sufficiently high ATRP equilibrium constant (KATRP). Thus, the rate of the polymerization is slower, then the loss of C(sp3)–Br chain-ends via intramolecular cyclization. In turn, poor control provided by very active TPMA*3 ligand could be explained by the too high value of the ATRP equilibrium constant. Higher KATRP implies higher radical concentration at equilibrium, which favors bimolecular termination reactions, resulting in diminishing control over the polymerization.
Next, the kinetics of the polymerization of NIPAM was investigated in an open reaction vessel (Fig. 1A). The reaction was performed in a Br-based PBS buffer with [NIPAM]/[HOBiB]/[CuBr2]/[Me6TREN] molar ratios of 200/1/0.05/0.30, in the presence of SP (100 mM). A short inhibition period of 15 min was observed, followed by a well-controlled (Đ = 1.15), rapid polymerization with linear semi-logarithmic kinetics, that reached 97% monomer conversion in 15 minutes. We unexpectedly observed that polymerization in an open vessel (Fig. 1B) led to smaller deviation from the theoretical molecular weight value (Mn,th = 22400, Mn,GPC = 25400) than polymerization in a sealed vessel (Mn,th = 22800, Mn,GPC = 31700). Fast activation of initiators, leading to termination of initiating radicals could explain this deviation.3
 | ||
| Fig. 1 PICAR ATRP of NIPAM in an aqueous medium demonstrating well-controlled polymerizations achieved in an open reaction vessel. (A) Kinetics and (B) SEC results of the polymerizations. Reactions conditions: [NIPAM]/[HOBiB]/[CuBr2]/[Ligand]: 200/1/0.05/0.30, [NIPAM] = 0.8 M, [HOBiB] = 4 mM, [SP] = 100 mM, [NaBr] = 100 mM, in PBS(Br) at 6 °C, under UV LEDs (λ = 394 nm, 2.6 mW cm−2) in an open reaction vessel (purple line) or in a sealed vessel (black line). | ||
The performance of this system was further evaluated in a series of reactions in a closed vessel, with varying target degrees of polymerization (DP) of NIPAM (Table 2). The concentration of CuBr2 was maintained at 250 ppm relative to the monomer. The results showed a high degree of control for targeted DP = 100, 200, 400, 1000, and 2000 (Table 2, entries 1–5). In all cases, nearly quantitative monomer conversions were reached with Đ in the range 1.16–1.44. However, for higher DP = 4000 and 10000, a significant deviation from the theoretical molecular weights and higher Đ values were observed (Table 2, entries 6 and 7). Moreover, SEC traces of the polymers showed a significant tailing (ESI Fig. S5F and G†). The appearance of this tailing could be attributed to the continuous formation of new chains, plausibly generated by radicals formed from the photochemical homolytic cleavage of SP.
| No. | [NIPAM]/[HOBiB]/[CuBr2]/[Me6TREN] | Time (min) | Conv.b (%) | M n,th × 10−3 | M n,GPC × 10−3 | Đ |
|---|---|---|---|---|---|---|
| a Reaction conditions: [NIPAM] = 0.8 M, [HOBiB] = 0.08–8 mM, [CuBr2] = 0.2 mM, [Me6TREN] = 1.2 mM, [SP] = 100 mM, [NaBr] = 100 mM, in 1× PBS(Br) at 6 °C, under UV LEDs (λ = 394 nm, 2.6 mW cm−2) in the presence of oxygen (sealed vessel). b Monomer conversion was determined by using 1H NMR spectroscopy. c See SEC traces in the ESI Fig. S3. All measurements were analyzed using GPC (dimethylformamide as eluent) calibrated to poly(methyl methacrylate) standards. | ||||||
| 1 | 100/1/0.025/0.15 | 30 | 99 | 11.4 | 15.7 | 1.20 |
| 2 | 200/1/0.05/0.30 | 30 | 99 | 22.8 | 31.7 | 1.16 |
| 3 | 400/1/0.10/0.60 | 30 | 99 | 45.0 | 58.4 | 1.20 |
| 4 | 1000/1/0.25/1.50 | 30 | 99 | 112.1 | 135.8 | 1.32 |
| 5 | 2000/1/0.50/3.00 | 30 | 99 | 263.5 | 272.1 | 1.44 |
| 6 | 4000/1/1.00/6.00 | 20 | 97 | 438.7 | 278.1 | 1.58 |
| 7 | 10000/1/2.50/15.00 |
20 | 96 | 1085.0 | 361.4 | 1.67 |
Then, polymerizations of NIPAM were conducted in the open reaction vessel, targeting DP of 100–2000 (Table 3). A high level of control over polymers was achieved under PICAR ATRP conditions when oxygen continuously diffused into the reaction system from the atmosphere, reaching 65–97% monomer conversions and providing polymers with monomodal, narrow molecular weight distributions (1.15 < Đ < 1.32). In open-air conditions, the CuI/L activator is constantly oxidized to inactive CuII/L, which results in lower monomer conversions. In turn, the increased X–CuII/L deactivator concentration provides better control over polymerization.
| No. | [NIPAM]/[HOBiB]/[CuBr2]/[Me6TREN] | Time (min) | Conv.b (%) | M n,th × 10−3 | M n,GPC × 10−3 | Đ |
|---|---|---|---|---|---|---|
| a Reactions conditions: [NIPAM] = 0.8 M, [HOBiB] = 0.08–8 mM, [CuBr2] = 0.2 mM, [Me6TREN] = 1.2 mM, [SP] = 100 mM, [NaBr] = 100 mM, in 1× PBS(Br) at 6 °C, under UV LEDs (λ = 394 nm, 2.6 mW cm−2) in an open reaction vessel. b Monomer conversion was determined by using 1H NMR spectroscopy. c See SEC traces in the ESI Fig. S5. All measurements were analyzed using GPC (dimethylformamide as eluent) calibrated to poly(methyl methacrylate) standards. | ||||||
| 1 | 100/1/0.025/0.15 | 40 | 93 | 10.7 | 16.0 | 1.16 |
| 2 | 200/1/0.05/0.30 | 30 | 97 | 22.4 | 25.4 | 1.15 |
| 3 | 400/1/0.10/0.60 | 30 | 93 | 42.2 | 56.0 | 1.16 |
| 4 | 1000/1/0.25/1.50 | 30 | 65 | 73.7 | 94.7 | 1.28 |
| 5 | 2000/1/0.50/3.00 | 30 | 66 | 149.4 | 176.4 | 1.32 |
The promising results of PICAR ATRP in an aqueous medium prompted us to utilize this fully oxygen-tolerant system in an organic solvent, which would significantly extend the scope of this method toward hydrophobic monomers. Since the SP-triggered ATRP catalytic system is based on small molecules, it can be transferred to organic solvents much more easily compared to ATRP techniques based on enzymatic degassing. However, sodium pyruvate has limited solubility in organic solvents due to its ionic structure. We used a stoichiometric amount of tetrabutylammonium bromide (TBAB) to increase the solubility of SP in dimethyl sulfoxide (DMSO). This quaternary ammonium salt is commonly used as a phase transfer catalyst in many synthetic transformations.82
The investigation was started by preparing a reaction mixture that contained all components needed for the polymerization (Fig. 2). Methyl acrylate (MA), ethyl α-bromoisobutyrate (EBiB), CuBr2, and Me6TREN ligand were dissolved in DMSO with [MA]/[EBiB]/[CuIIBr2]/[Me6TREN] molar ratios of 200/1/0.05/0.3. SP and TBAB were added in the molar ratio of 1/1. Salt metathesis induced partial precipitation of NaBr and the formation of tetrabutylammonium pyruvate, which is well soluble in DMSO. The solution was filtered through a syringe filter to remove the precipitate, then transferred into an open reaction vessel. PICAR ATRP of MA under UV LED irradiation (λ = 365 nm, 3 × 50 mW cm−2) was performed at room temperature. After 3 h, the conversion of MA measured by 1H NMR was 84%. SEC analysis showed that the polymer had a low dispersity (Đ = 1.16), and a molecular weight close to the theoretical value (Mn,th = 14500, Mn,GPC = 16700), indicating a well-controlled polymerization (Fig. 2). This experiment shows that even without a time-consuming, careful optimization, a highly efficient, fully open-air ATRP system could be quickly developed. Further optimization of this method and its applicability to other non-polar monomers will be the subject of a forthcoming publication.
 | ||
| Fig. 2 Fully oxygen-tolerant PICAR ATRP in organic solvent. (A) Reaction mixture preparation and (B) SEC results of the polymerizations. Reactions conditions: [MA]/[HOBiB]/[CuBr2]/[Me6TREN]: 200/1/0.05/0.30, [MA] = 7.4 M, [HOBiB] = 37 mM, [SP] = 100 mM, [TBAB] = 100 mM in DMSO at rt, under UV LEDs (λ = 365 nm, 3 × 50 mW cm−2) in an open reaction vessel. | ||
To gain insights into the polymerization mechanism, we investigated the reactivity of SP toward Cu complexes. Recently we reported the sono-ATRP of MA in DMSO in the presence of sodium carbonate.83 In this system, ultrasonication triggered the homolytic cleavage of the in situ formed (CO3)–CuII/TPMA complex, generating CuI species and a radical carbonate anion. Haddleton et al. observed a similar phenomenon for the photoreduction of (HCO2)–CuII/Me6TREN complex.84,85 In addition, Vaida et al. showed that the UV excitation of pyruvic acid in an aqueous medium causes photodecarboxylation, which forms radicals as intermediates.86 Furthermore, α-keto acids can undergo decarboxylative acyl radical formation in transition metal-catalyzed radical cross-couplings.87,88
Based on the above results, we propose that the SP reacts with CuII species to yield a (CH3C(O)CO2)–CuII/L complex by a simple anion dissociation/association process (Scheme 2). Subsequent UV excitation causes the homolytic cleavage of the carbon–carbon bond in the pyruvate moiety. This photolysis induces decarboxylation, which leads to the reduction of CuII/L to CuI/L and the formation of the acyl radical. This radical can regenerate the activator or initiate a new polymer chain by addition to the monomer. The role of a buffer medium is to control the pH and, thus, the concentration of the formed acyl radicals.86 Furthermore, the reaction between the acyl radical and X–CuII/L deactivator leads to the formation of an acyl halide, which undergoes rapid hydrolysis in a buffer. This prevents the initiation of new polymer chains and the protonation of the ligand. A control experiment without the HOBiB initiator showed that SP could initiate ATRP on its own, but the polymer had a broad molecular weight distribution and a high Đ = 1.92. In order to confirm the role of SP in the catalytic system, UV-vis spectroscopy measurements were performed. Fig. S6A† shows a decrease in the absorbance of CuII/TPMA complex under UV irradiation in the presence of SP. Fig. S6B† shows that the absorbance of CuII/TPMA decreased much slower when SP was absent, which indicates that SP is necessary for the efficient reduction of CuII species. This in turn is critical for ATRP in an ambient atmosphere. The proposed mechanism shown in Scheme 2 can be considered equivalent to PICAR (photoinduced initiators for continuous activator regeneration) ATRP.89,90
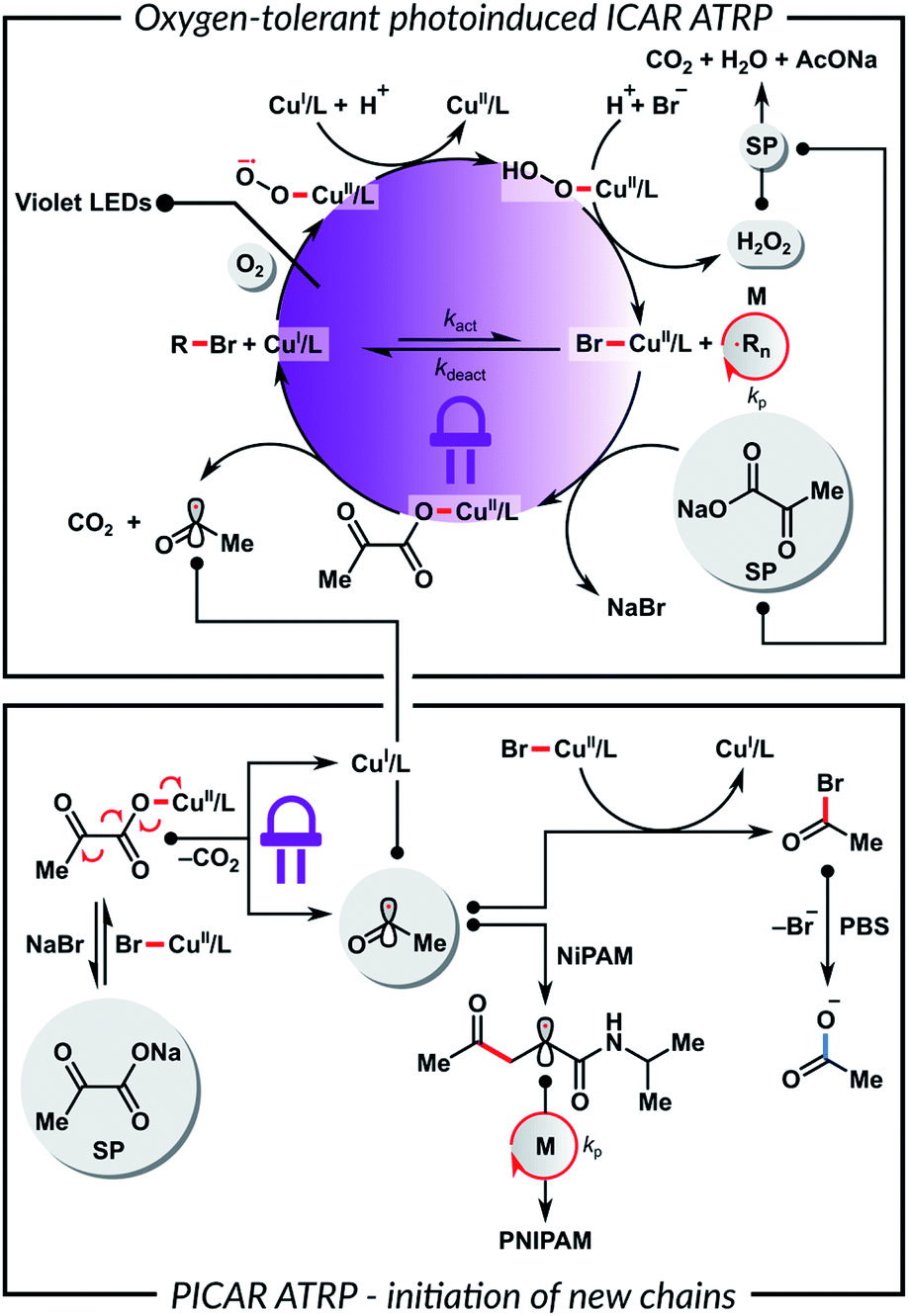 | ||
| Scheme 2 Proposed mechanism of PICAR ATRP. | ||
Conclusions
We have developed the first example of a photoinduced ATRP system that operates in an open reaction vessel and yields well-controlled polymerizations in both aqueous and organic solvents. Sodium pyruvate is the essential component in this novel method, acting as a hydrogen peroxide scavenger and enabling the continuous regeneration of the copper catalyst through UV excitation. This methodology allowed the synthesis of poly(N-isopropylacrylamide) in water with high monomer conversion (97%) and dispersity of 1.15 with 250 ppm of a catalyst in 30 min under an ambient atmosphere. Furthermore, the use of sodium pyruvate with tetrabutylammonium bromide enabled the polymerization of methyl acrylate in DMSO in a fully open vessel without compromising the control over the molecular weight distribution (Đ = 1.16). Non-experts can easily apply this straightforward and robust protocol for the synthesis of well-defined polymers. Expanding the scope of this methodology to more complex polymer architectures and polymer-based biohybrids is currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. S. gratefully acknowledges the Polish Ministry of Science and Higher Education (“Mobilnosc Plus” grant no. 1646/MOB/V/2017/0) for financial support. M. Ł. financial support from Christ Church, Oxford (“Summer Bursary” program) is gratefully acknowledged. A. G. support in the form of the post-doctoral fellowship (Polish National Agency for Academic Exchange – Bekker Programme no. PPN/BEK/2018/1/00261; Polish-U.S. Fulbright Commission – Senior Award no. PL/2019/SR/41) and Foundation for Polish Science (FNP) is gratefully acknowledged. Financial support from the NSF (CHE 2000391) is acknowledged.References
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- F. Lorandi and K. Matyjaszewski, Isr. J. Chem., 2020, 60, 108–123 CrossRef CAS.
- T. G. Ribelli, F. Lorandi, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800616 CrossRef PubMed.
- T. G. Ribelli, M. Fantin, J. C. Daran, K. F. Augustine, R. Poli and K. Matyjaszewski, J. Am. Chem. Soc., 2018, 140, 1525–1534 CrossRef CAS PubMed.
- A. E. Enciso, F. Lorandi, A. Mehmood, M. Fantin, G. Szczepaniak, B. G. Janesko and K. Matyjaszewski, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202004724.
- M. Langerman and D. G. H. Hetterscheid, Angew. Chem., Int. Ed., 2019, 58, 12974–12978 CrossRef CAS PubMed.
- V. A. Bhanu and K. Kishore, Chem. Rev., 1991, 91, 99–117 CrossRef CAS.
- S. L. Baker, B. Kaupbayeva, S. Lathwal, S. R. Das, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2019, 20, 4272–4298 CrossRef CAS PubMed.
- M. S. Messina, K. M. M. Messina, A. Bhattacharya, H. R. Montgomery and H. D. Maynard, Prog. Polym. Sci., 2020, 100, 101186 CrossRef CAS.
- L. Zhang, Y. Zhang, J. Cheng, L. Wang, X. Wang, M. Zhang, Y. Gao, J. Hu, X. Zhang, J. Lü, G. Li, R. Tai and H. Fang, Sci. Rep., 2017, 7, 10176 CrossRef PubMed.
- K. Matyjaszewski, S. Coca, S. G. Gaynor, M. Wei and B. E. Woodworth, Macromolecules, 1998, 31, 5967–5969 CrossRef CAS.
- D. Konkolewicz, Y. Wang, P. Krys, M. Zhong, A. A. Isse, A. Gennaro and K. Matyjaszewski, Polym. Chem., 2014, 5, 4396–4417 RSC.
- D. Konkolewicz, Y. Wang, M. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 CrossRef CAS.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS PubMed.
- T. Zhang, E. M. Benetti and R. Jordan, ACS Macro Lett., 2019, 8, 145–153 CrossRef CAS.
- T. Zhang, Y. Du, J. Kalbacova, R. Schubel, R. D. Rodriguez, T. Chen, D. R. T. Zahn and R. Jordan, Polym. Chem., 2015, 6, 8176–8183 RSC.
- W. Yan, M. Fantin, N. D. Spencer, K. Matyjaszewski and E. M. Benetti, ACS Macro Lett., 2019, 8, 865–870 CrossRef CAS.
- Y. Che, T. Zhang, Y. Du, I. Amin, C. Marschelke and R. Jordan, Angew. Chem., Int. Ed., 2018, 57, 16380–16384 CrossRef CAS PubMed.
- K. Min, W. Jakubowski and K. Matyjaszewski, Macromol. Rapid Commun., 2006, 27, 594–598 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS PubMed.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- H. Kang, W. Jeong and D. Hong, Langmuir, 2019, 35, 7744–7750 CrossRef CAS PubMed.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- H. Dong and K. Matyjaszewski, Macromolecules, 2008, 41, 6868–6870 CrossRef CAS.
- Y. Kwak and K. Matyjaszewski, Polym. Int., 2009, 58, 242–247 CrossRef CAS.
- Y. Gnanou and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 351–359 CAS.
- Y. Wang, X. Li, F. Du, H. Yu, B. Jin and R. Bai, Chem. Commun., 2012, 48, 2800–2802 CAS.
- A. S. R. Oliveira, P. V Mendonça, A. C. Serra and J. F. J. Coelho, J. Polym. Sci., 2020, 58, 145–153 CrossRef CAS.
- A. Layadi, B. Kessel, W. Yan, M. Romio, N. D. Spencer, M. Zenobi-Wong, K. Matyjaszewski and E. M. Benetti, J. Am. Chem. Soc., 2020, 142, 3158–3164 CrossRef CAS PubMed.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- E. H. Discekici, A. Anastasaki, J. Read de Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS.
- R. A. Olson, A. B. Korpusik and B. S. Sumerlin, Chem. Sci., 2020, 11, 5142–5156 RSC.
- J. Mosnáček, A. Eckstein-Andicsová and K. Borská, Polym. Chem., 2015, 6, 2523–2530 RSC.
- Q. Yang, J. Lalevée and J. Poly, Macromolecules, 2016, 49, 7653–7666 CrossRef CAS.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. G. Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 11, 1486 CrossRef CAS PubMed.
- W. Yan, S. Dadashi-Silab, K. Matyjaszewski, N. D. Spencer and E. M. Benetti, Macromolecules, 2020, 53, 2801–2810 CrossRef CAS.
- M. Rolland, N. P. Truong, R. Whitfield and A. Anastasaki, ACS Macro Lett., 2020, 9, 459–463 CrossRef CAS.
- X. Pan, S. Lathwal, S. Mack, J. Yan, S. R. Das and K. Matyjaszewski, Angew. Chem., Int. Ed., 2017, 56, 2740–2743 CAS.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Macromolecules, 2017, 50, 7967–7977 CrossRef CAS.
- L. Fu, Z. Wang, S. Lathwal, A. E. Enciso, A. Simakova, S. R. Das, A. J. Russell and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 1248–1253 CrossRef CAS PubMed.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963–971 RSC.
- A. Marathianos, E. Liarou, A. Anastasaki, R. Whitfield, M. Laurel, A. M. Wemyss and D. M. Haddleton, Polym. Chem., 2019, 10, 4402–4406 RSC.
- S. Dadashi-Silab, G. Szczepaniak, S. Lathwal and K. Matyjaszewski, Polym. Chem., 2020, 11, 843–848 RSC.
- R. Whitfield, K. Parkatzidis, M. Rolland, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2019, 58, 13323–13328 CrossRef CAS PubMed.
- M. Rolland, R. Whitfield, D. Messmer, K. Parkatzidis, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2019, 8, 1546–1551 CrossRef CAS.
- A. E. Enciso, L. Fu, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 933–936 CrossRef CAS PubMed.
- A. E. Enciso, L. Fu, S. Lathwal, M. Olszewski, Z. Wang, S. R. Das, A. J. Russell and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 16157–16161 CrossRef CAS PubMed.
- L. A. Navarro, A. E. Enciso, K. Matyjaszewski and S. Zauscher, J. Am. Chem. Soc., 2019, 141, 3100–3109 CrossRef CAS PubMed.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 603–609 CrossRef CAS.
- F. Oytun, M. U. Kahveci and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1685–1689 CrossRef CAS.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 CrossRef CAS.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Angew. Chem., Int. Ed., 2016, 55, 4500–4503 CrossRef CAS PubMed.
- G. Fan, A. J. Graham, J. Kolli, N. A. Lynd and B. K. Keitz, Nat. Chem., 2020, 12, 638–646 CrossRef CAS PubMed.
- G.-F. Luo, W.-H. Chen and X.-Z. Zhang, ACS Macro Lett., 2020, 9, 872–881 CrossRef CAS.
- S. Qiao and H. Wang, Nano Res., 2018, 11, 5400–5423 CrossRef CAS.
- S. Lanzalaco and E. Armelin, Gels, 2017, 3, 36 CrossRef PubMed.
- O. Rifaie-Graham, J. Pollard, S. Raccio, S. Balog, S. Rusch, M. A. Hernández-Castañeda, P.-Y. Mantel, H.-P. Beck and N. Bruns, Nat. Commun., 2019, 10, 1369 CrossRef PubMed.
- S. Kim and H. D. Sikes, Polym. Chem., 2020, 11, 1424–1444 RSC.
- G. Masci, L. Giacomelli and V. Crescenzi, Macromol. Rapid Commun., 2004, 25, 559–564 CrossRef CAS.
- J. Ye and R. Narain, J. Phys. Chem. B, 2009, 113, 676–681 CrossRef CAS PubMed.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- P. Chmielarz, S. Park, A. Simakova and K. Matyjaszewski, Polymer, 2015, 60, 302–307 CrossRef CAS.
- P. Chmielarz, P. Krys, S. Park and K. Matyjaszewski, Polymer, 2015, 71, 143–147 CrossRef CAS.
- E. Liarou, Y. Han, A. M. Sanchez, M. Walker and D. M. Haddleton, Chem. Sci., 2020, 11, 5257–5266 RSC.
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 6371–6379 CrossRef CAS.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromolecules, 1999, 32, 4826–4831 CrossRef CAS.
- J. T. Rademacher, M. Baum, M. E. Pallack, W. J. Brittain and W. J. Simonsick, Macromolecules, 2000, 33, 284–288 CrossRef CAS.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC.
- F. De Bon, S. Marenzi, A. A. Isse, C. Durante and A. Gennaro, ChemElectroChem, 2020, 7, 1378–1388 CrossRef CAS.
- K. Schröder, R. T. Mathers, J. Buback, D. Konkolewicz, A. J. D. Magenau and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1037–1040 CrossRef.
- M. Makosza, Pure Appl. Chem., 2000, 72, 1399–1403 CAS.
- Z. Wang, F. Lorandi, M. Fantin, Z. Wang, J. Yan, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 161–165 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, F. Brandford-Adams, G. Nurumbetov, Q. Zhang, G. J. Clarkson, D. J. Fox, P. Wilson, K. Kempe and D. M. Haddleton, Chem. Commun., 2015, 51, 5626–5629 RSC.
- V. Nikolaou, A. Anastasaki, F. Brandford-Adams, R. Whitfield, G. R. Jones, G. Nurumbetov and D. M. Haddleton, Polym. Chem., 2016, 7, 191–197 RSC.
- E. C. Griffith, B. K. Carpenter, R. K. Shoemaker and V. Vaida, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11714–11719 CrossRef CAS PubMed.
- L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380–7391 RSC.
- F. Penteado, E. F. Lopes, D. Alves, G. Perin, R. G. Jacob and E. J. Lenardão, Chem. Rev., 2019, 119, 7113–7278 CrossRef CAS PubMed.
- T. G. Ribelli, D. Konkolewicz, X. Pan and K. Matyjaszewski, Macromolecules, 2014, 47, 6316–6321 CrossRef CAS.
- M. Ciftci, M. A. Tasdelen, W. Li, K. Matyjaszewski and Y. Yagci, Macromolecules, 2013, 46, 9537–9543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03179h |
| This journal is © The Royal Society of Chemistry 2020 |