
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and origins of selectivity in the enantioselective oxa-Pictet–Spengler reaction: a cooperative catalytic complex from a hydrogen bond donor and chiral phosphoric acid†
Mark A.
Maskeri‡
 a,
Alexander C.
Brueckner‡
b,
Taisiia
Feoktistova
b,
Matthew J.
O'Connor
a,
Daniel M.
Walden
b,
Paul Ha-Yeon
Cheong
*b and
Karl A.
Scheidt
*a
a,
Alexander C.
Brueckner‡
b,
Taisiia
Feoktistova
b,
Matthew J.
O'Connor
a,
Daniel M.
Walden
b,
Paul Ha-Yeon
Cheong
*b and
Karl A.
Scheidt
*a
aDepartment of Chemistry, Center for Molecular Innovation and Drug Discovery, Northwestern University, Evanston, IL 60208, USA. E-mail: scheidt@northwestern.edu
bDepartment of Chemistry, Oregon State University, Corvallis, OR 97331, USA. E-mail: cheongh@oregonstate.edu
First published on 27th July 2020
Abstract
Enantioselective additions to oxocarbenium ions are high-value synthetic transformations but have proven challenging to achieve. In particular, the oxa-Pictet–Spengler reaction has only recently been rendered enantioselective. We report experimental and computational studies on the mechanism of this unusual transformation. Herein we reveal that this reaction is hypothesized to proceed through a self-assembled ternary hydrogen bonding complex involving the substrate, chiral phosphate ion, and a urea hydrogen-bond donor. The computed transition state reveals C2-symmetric grooves in the chiral phosphate that are occupied by the urea and substrate. Occupation of one of these grooves by the urea co-catalyst tunes the available reactive volume and enhances the stereoselectivity of the chiral phosphate catalyst.
Introduction
Considerable recent advances in stereoselective synthesis allow great control over the processes that effect carbon–carbon bond formations from high-energy carbocation species. Unlike many other reactions proceeding through the oxocarbenium ion, the oxa-Pictet–Spengler reaction has been curiously resistant to enantioselective catalysis.1,2 This reaction, which has the potential for broad applicability to pharmaceutical and material sciences,3 has only recently been rendered enantioselective,4 with excellent contributions from Seidel,5,6 List,7 and our own laboratory8 (Fig. 1A). Reports from the Seidel group furnish chiral polysubstituted tetrahydropyranoindoles (THPIs) through cooperative amine (NH2)/hydrogen bond donor9–12 (HBD) and bifunctional Brønsted acid (BA)/HBD chiral catalysis. Similarly, the List group utilized sterically-congested imidodiphosphate (IDP) catalysts to produce substituted isochromanes with high stereochemical fidelity.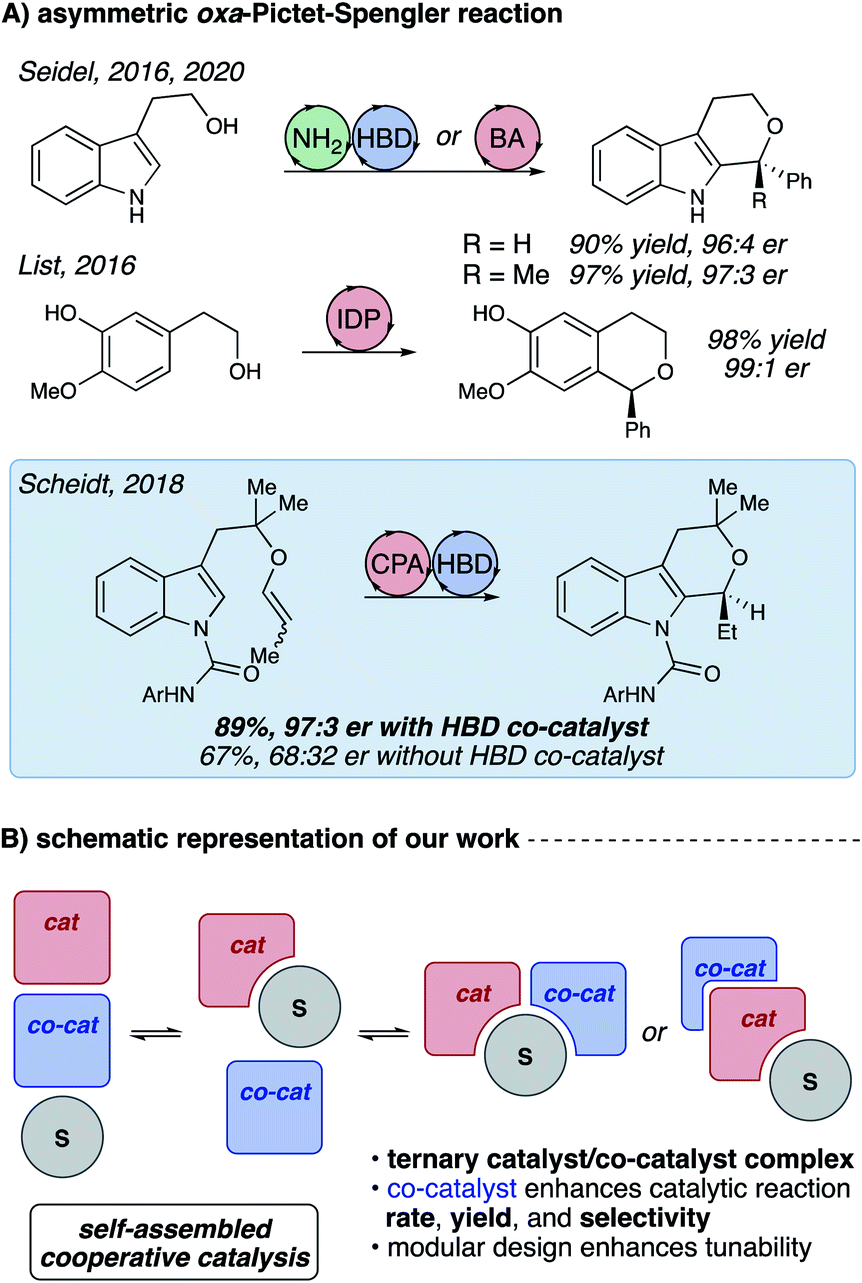 | ||
| Fig. 1 (A) Existing strategies for catalytic enantioselective oxa-Pictet–Spengler reactions. (B) Two possible catalyst/substrate self-assembly motifs for the title reaction. | ||
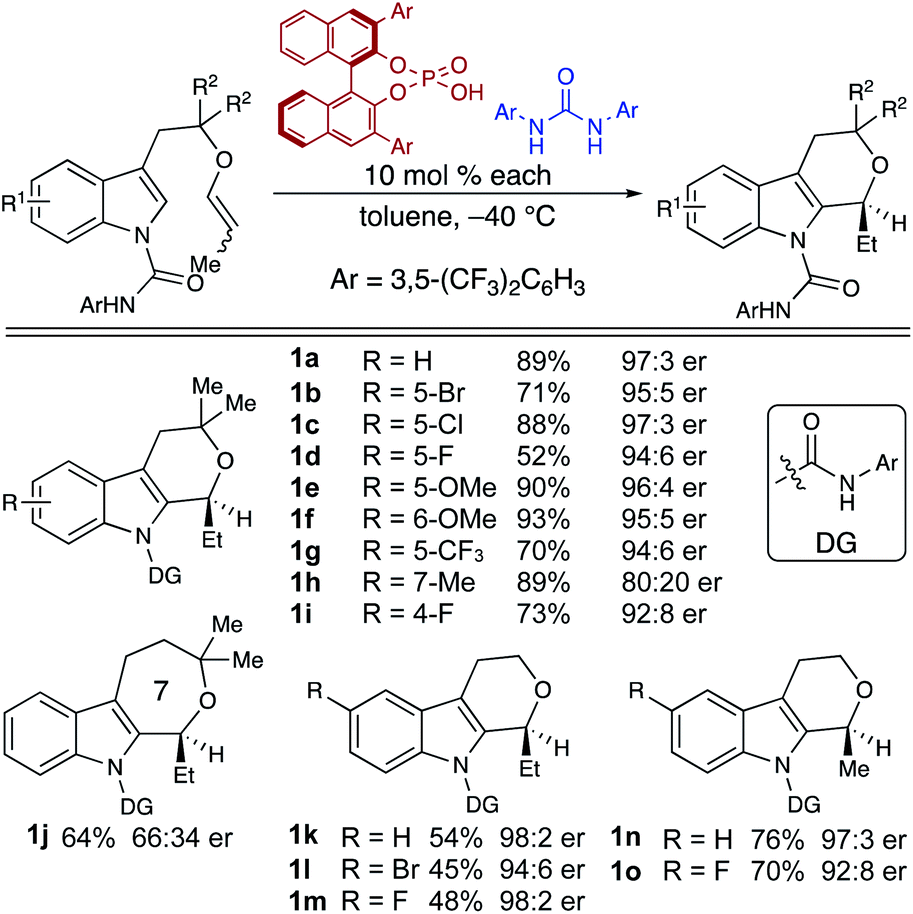
Our enantioselective oxa-Pictet–Spengler reaction (Fig. 1A) furnishes tetrahydropyranoindoles (THPIs) through cooperative chiral phosphoric acid (CPA)/hydrogen bond donor catalysis.8 This reaction demonstrates high levels of enantioselectivity and good-to-excellent yields for electronically-diverse substrates, with reaction rates substantially higher than previously published enantioselective oxa-Pictet–Spengler reactions (Table 1). Notably, the enhancements to reactivity and selectivity observed in this process were only observed in the presence of a hydrogen-bonding urea. These surprising observations compelled us to further examine the mechanism of this unusual transformation through experiments and computations.13
|
Catalyst control approaches to the synthesis of organic frameworks play a critical role in modern organic synthesis.14 In order to furnish sufficiently stable coordination between an enantioenriched catalyst and prochiral substrate, such catalytic systems rely on a variety of intermolecular interactions.15 These interactions vary wildly in strength depending on substrate and catalyst functional groups and play significant roles in the overall enantioenrichment of the product. Catalysts capable of supplementing primary interactions with hydrogen bonds, anion binding, or electrostatic interactions demonstrate particularly high levels of enantioinduction.16 This principle has been demonstrated to great effect in the context of nucleophilic additions to iminium ions (e.g., Jacobsen's asymmetric Strecker and Pictet–Spengler reactions) and for cyclic or stabilized oxocarbenium ions.17–26 Particularly prominent among organocatalysts are the CPAs.27 Since the seminal reports of Akiyama28 and Terada,29 CPAs have been successfully applied to a diverse range of reactions. In the context of oxocarbenium ions, CPAs have seen application in enantioselective trans/spiroacetalizations30,31 and Prins reactions,32,33 among others.
Results and discussion
Our initial hypothesis for the cooperative interaction between the three reaction components—chiral phosphoric acid, urea, and indole substrate—was that the substrate aryl carboxamide moiety served to recruit and self-assemble the catalysts through hydrogen bonding.34,35 Such an interaction would be expected to influence substrate–catalyst interactions. This proposal was constructed based on evidence of the importance of hydrogen bonding and the rate, yield, and selectivity enhancements observed in the HBD-co-catalyzed reactions (Fig. 2A). Subsequent DOSY studies using an unreactive substrate analogue indicate that the three reaction components diffuse together (see ESI†),36–38 suggesting the likelihood of a ternary complex. Additionally, diffusion constants extracted from the DOSY data using the spherical Stokes model suggest that all three components are packed into space approximately equal to the volume of a chiral phosphoric acid (∼5.5 Å in radius, see ESI†). Further evidence supporting interactions between the CPA and urea was garnered from a 31P NMR spectrographic analysis: the phosphorus resonance of the CPA is observed to shift in the presence of hydrogen bond donors, with larger shifts in the presence of unreactive substrate analogues and reaction products (see ESI†). As CPAs are known to engage in both Lewis acid and base interactions at the phosphate, the shifts support a model in which the three components are closely hydrogen-bonded in a ternary complex.§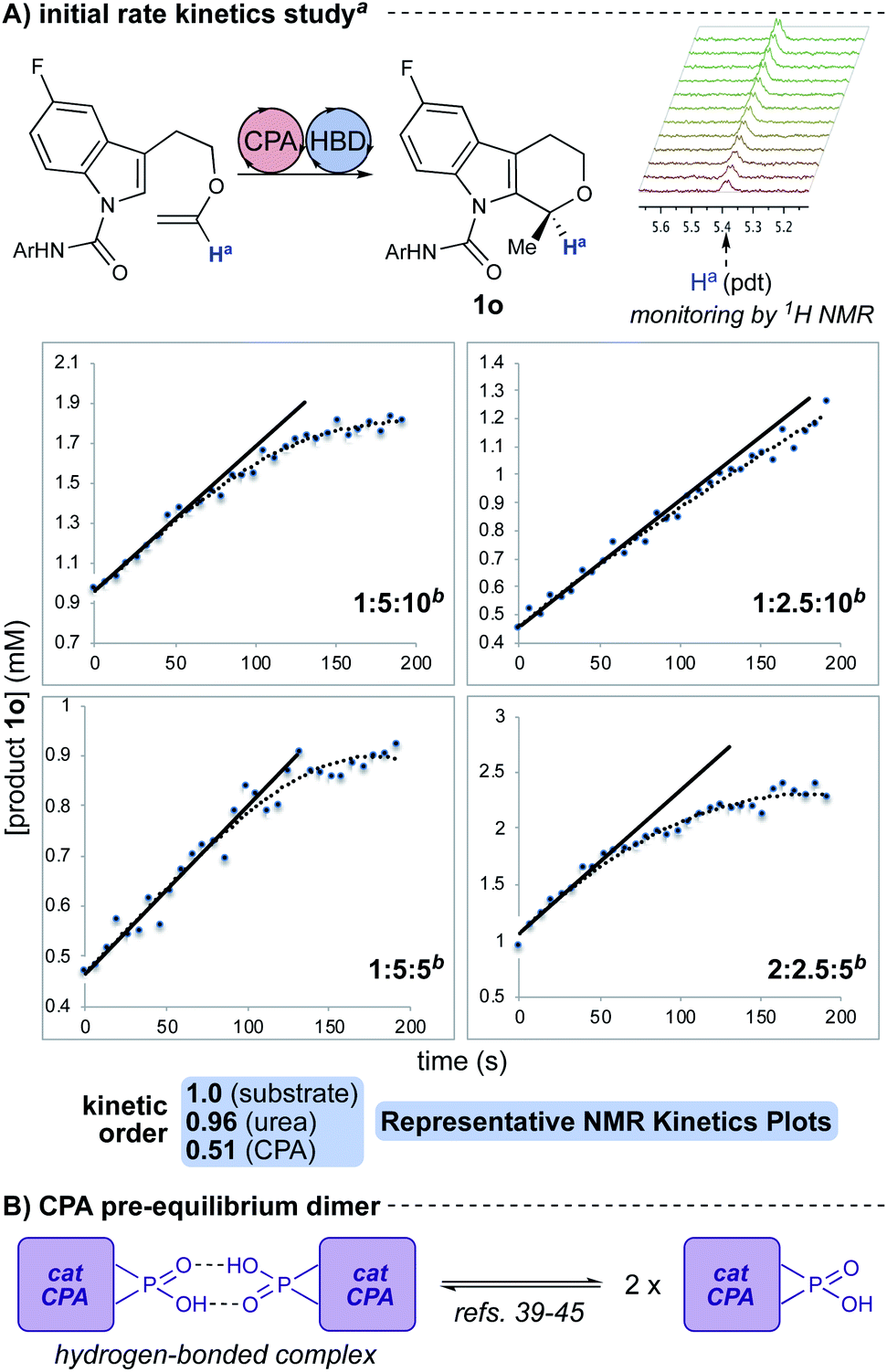 | ||
| Fig. 2 (A) Representative plots of 1H NMR reaction kinetics with initial rates analysis (see ESI†). Kinetic dependencies were found to be first order in substrate, first order in urea, and half order in CPA. aParent reaction conditions: 3,3′-(3,5-(CF3)2C6H3) CPA (10 mol%), 3,5-(CF3)2C6H3 urea (10 mol%), toluene-d8 (0.02 M), −40 °C. bRatios are: equiv. substrate: mol% CPA: mol% urea. (B) Cartoon representation of pre-equilibrium CPA dimer. | ||
An NMR reaction kinetics study of this reaction was performed with analysis by initial rates due to changes in reaction homogeneity8 (Fig. 2A; see ESI†). This analysis suggests that the reaction is first order in urea, first order in substrate, and half order in CPA. The fractional order in CPA is likely representative of a pre-equilibrium hydrogen-bonded CPA dimer (e.g., Fig. 2B).39–45 These kinetic dependencies lend further support to a hydrogen-bonded ternary complex as the catalytically-active species. With these data in hand, we engaged in computational analysis of the cooperative catalytic system.
Computational exploration of the CPA-mediated process in the absence of the urea co-catalyst is shown in Fig. 3. We discovered a low-energy “phosphate-acetal” complex ((R)-phosphate-acetal), analogous to other CPA-type acetals observed in the literature.7 In addition, this investigation found that the annulation proceeds through a stepwise process where a spirocyclization at the indole C3 is followed by a subsequent 1,2-shift and rearomatization. Such a sequence operates in gratifying agreement with prior computational studies on the Pictet–Spengler reaction mechanism.46,47
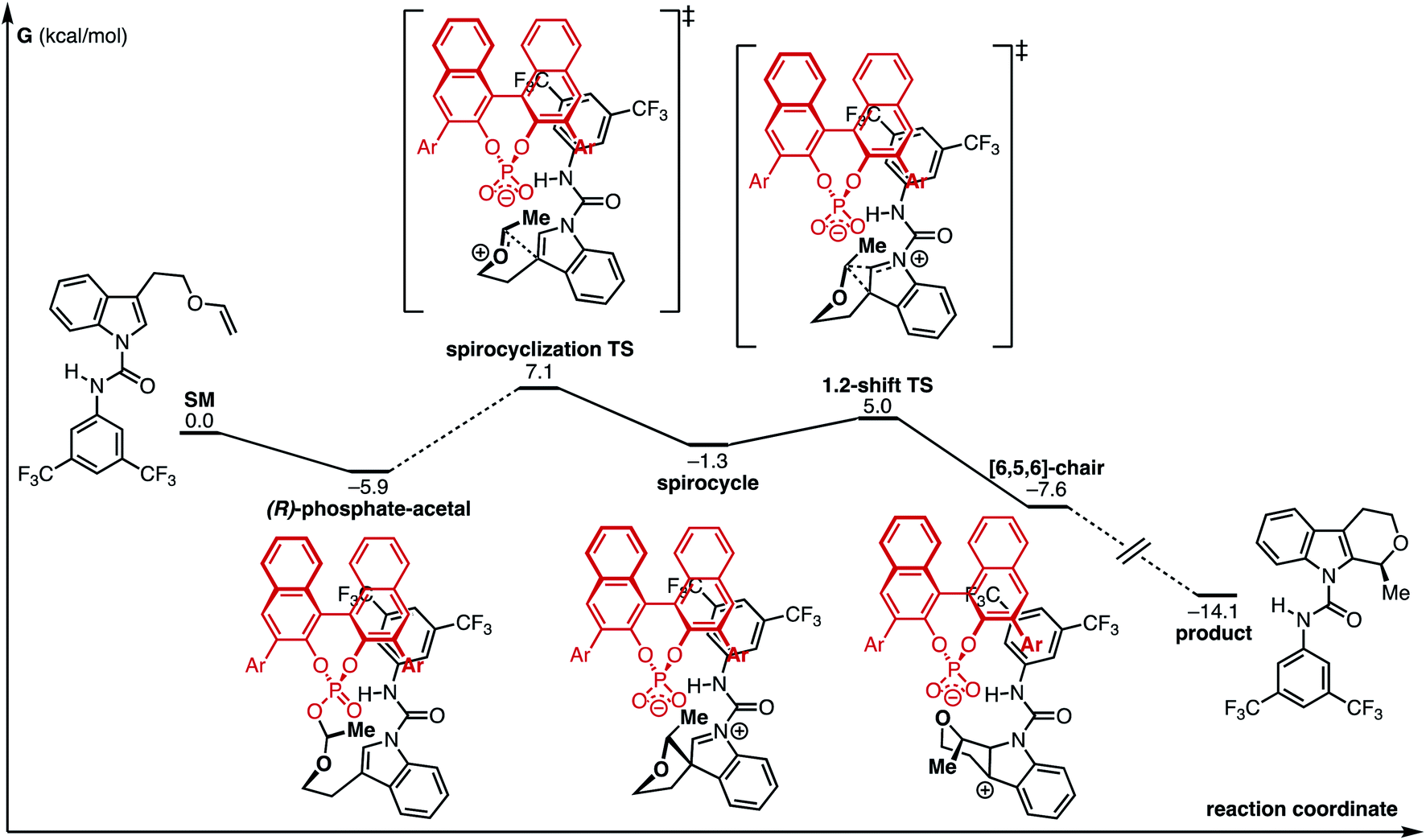 | ||
| Fig. 3 Computed reaction coordinate diagram of the CPA-mediated process. Ar = 3,5-(CF3)2C6H3. Energies are in kcal mol−1 and are relative to infinitely separated compounds. | ||
With the aforementioned experimental observations and preliminary computational investigation established, we sought to computationally investigate the key C–C bond forming transition state. Due to the high energy/short lifetime of the oxocarbenium ion preceding the transition state, we hypothesized that the spirocyclization transition state conformation may be closely related to that of the phosphate-acetal. We therefore performed a manual conformational search of both the (R)- and (S)-phosphate-acetal intermediates (see ESI†) and computed the key C–C bond forming transition structures that would lead directly from them.
In order to deduce the binding motif of the urea to the CPA-mediated spirocyclization transition state, we computed several model systems to identify critical interactions for the assembly of the catalyst/co-catalyst and catalyst/substrate complexes (see ESI†). Key to this analysis was a study of the coordination of the urea to two possible hydrogen bond acceptors—the CPA and the substrate carboxamide. The computations suggest that the urea–substrate interaction is weak compared to the urea–CPA interaction (ΔG = 24.9 and −2.1 kcal mol−1, respectively). In addition to the hydrogen bonding, π–π interactions between the CPA, urea, and substrate produced interactions consistently favored by 3–4 kcal mol−1 over non-π–π interaction models.
These observations from the model systems were confirmed in the full substrate/CPA/urea system (Fig. 4). Coordination of the urea to the CPA phosphate oxygen (Urea⋯CPA⋯Major-TS) in the spirocyclization transition state was the most energetically favorable. Alternative coordination of urea to the substrate carboxamide carbonyl was higher in energy by 5.6 kcal mol−1.¶
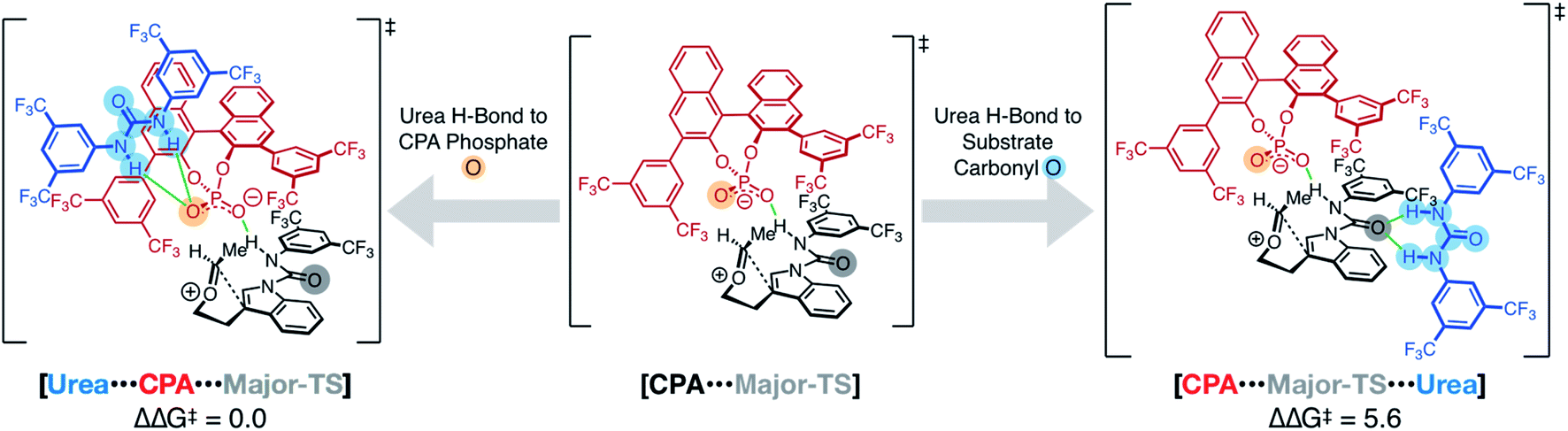 | ||
| Fig. 4 Computed transition structures of the CPA-mediated TS (middle) and urea + CPA mediated TS (left and right). Hydrogen bonding of the urea to the CPA is favored over urea coordination to the substrate carboxamide carbonyl by 5.6 kcal mol−1. | ||
Fusing these analyses into a cohesive whole, we located a pair of major and minor transition structures (Major-TS and Minor-TS, Fig. 5) that account for the critical stabilizing interactions observed in these model systems. The computed enantioselectivity of 1.8 kcal mol−1 agrees well with experimental selectivity of 1.6 kcal mol−1. All of the hydrogen bonding arrangements between the CPA, urea, and the substrate are consistent in both structures. The critical difference between the major and minor transition states and the origins of stereoselectivity arise from two factors.
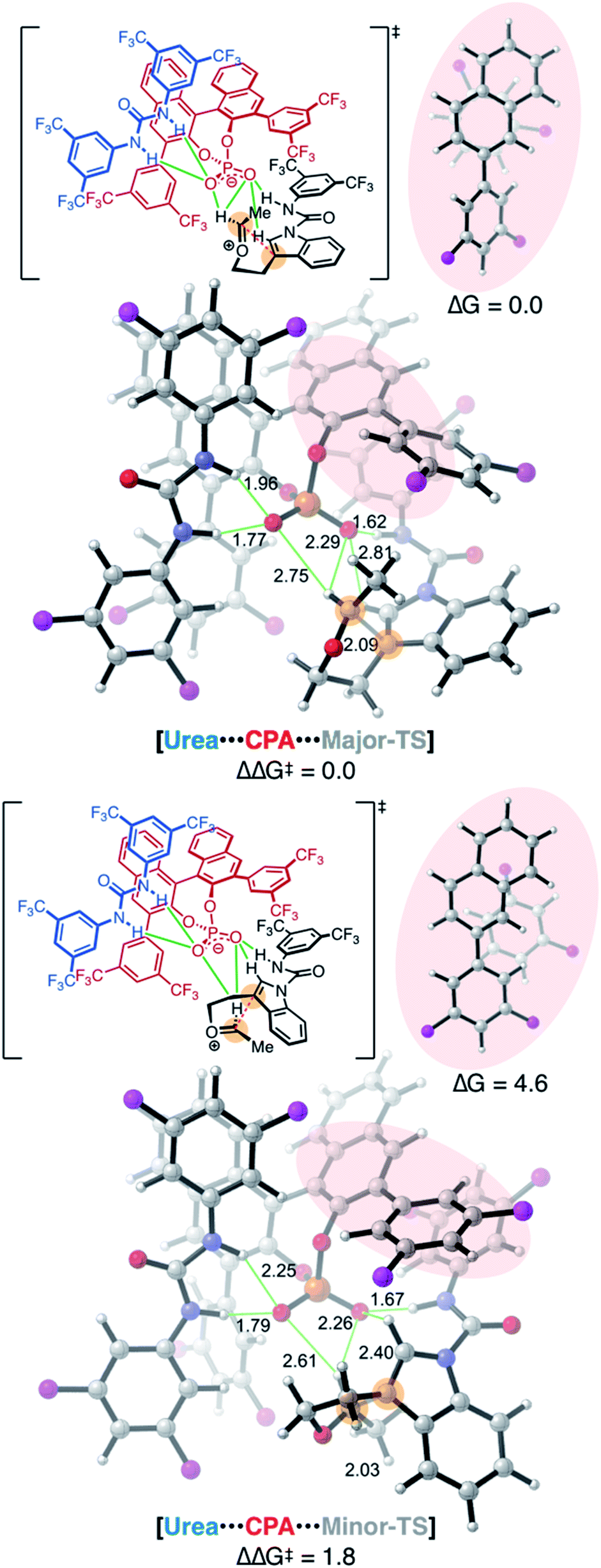 | ||
| Fig. 5 Computed major and minor transition structures of the CPA + urea mediated process. Trifluoromethyl (CF3) groups are represented as pink spheres. Pink insets show two model complexes of substrate aryl group and the CPA naphthyl and aryl groups as found in the Major-TS and Minor-TS. Capping hydrogen atoms were quantum mechanically optimized. Energies are in kcal mol−1 and distances in Å. | ||
Factor 1 substrate–CPA interactions
One key feature that differentiates the Major-TS from the Minor-TS is encapsulated in π–π stacking interactions between the substrate and the CPA (pink inset, Fig. 5). In the Major-TS, the substrate aryl carboxamide group is capable of adopting a stabilizing π-stacking arrangement with the CPA naphthyl backbone, a feature absent in the Minor-TS. The stabilizing capacity of this interaction was confirmed with a computed truncated model system of these aromatic groups, with which we found the arrangement in the Major-TS to be favored by 4.6 kcal mol−1, compared to that of the Minor-TS.Experimental observations from our original report demonstrate that the absence of the substrate aryl carboxamide group leads to no enantioselectivity.8 Selectivity is restored when the substrate contains groups that can engage in the requisite π–π arrangements. These observations underscore the importance of this interaction. Additionally, the Major-TS and Minor-TS reveal why the reaction synthetic scope is accommodating of substitution patterns on the indole and the intramolecular tether, as these groups do not sterically or electronically interact with the CPA but rather protrude into solvent-occupied space.
Factor 2 substrate–urea interactions
Analysis of the packing of the urea and substrate around the CPA unveils a pair of binding “grooves”, analogous to enzyme binding clefts (Fig. 6A and B). In both the Major- and Minor-TS, the urea and the substrate each occupy one groove (Fig. 6C and D, respectively), producing a pseudo-C2-symmetric complex where the urea and substrate are linked via hydrogen bonding contacts to the CPA. In the Minor-TS, the substrate fully occupies one groove and extends into the second groove occupied by the urea, resulting in steric repulsion. This is in contrast to the Major-TS, where the substrate does not extend into the second groove and therefore minimizes contact with the urea. This occupancy of the urea co-catalyst in the second groove effectively tunes the available reactive volume and is critical for the observed enhanced selectivity induced by the urea co-catalyst. This “occupancy-induced” enhancement of selectivity model appears to be a new aspect of cooperative catalysis, since based on the available data, the urea co-catalyst alters the available reaction volume to induce selectivity. In this work, we hypothesize that a co-catalyst is also important in inducing selectivity. This aspect is similar to allosteric modulation of protein/enzyme substrate binding and catalysis. Moreover, this approach is potentially tunable and modular, as the electronic and steric parameters of the co-catalyst and the catalyst can be combined without the need for the resource-intensive syntheses of a new, singular, catalysts.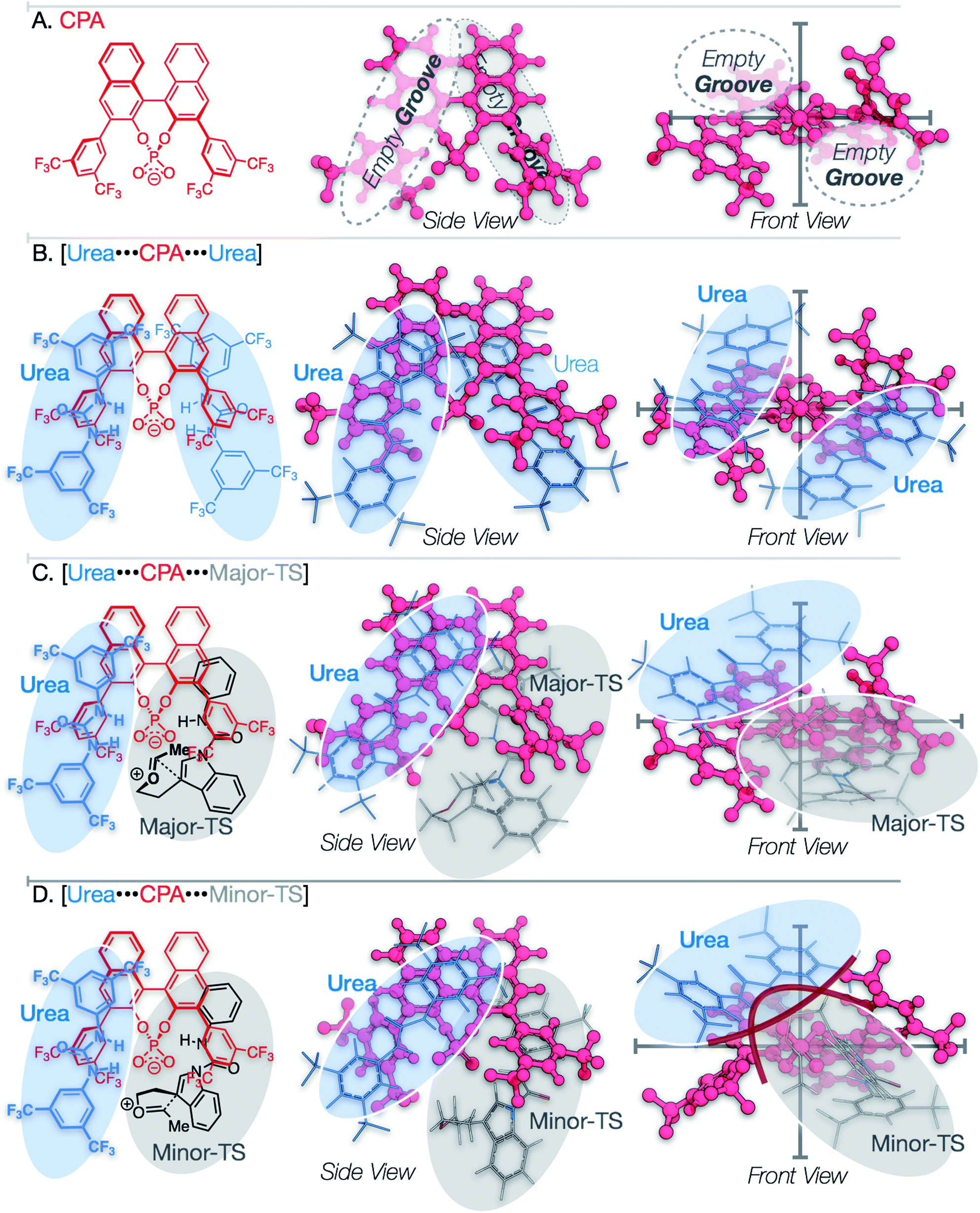 | ||
| Fig. 6 Visualization of CPA groove model. Trifluoromethyl groups omitted from substrate aryl ring to increase clarity in chemical diagrams. (A) CPA binding “grooves” along naphthyl backbone. (B) CPA with two ureas bound to the “grooves” along naphthyl backbone. (C) “Groove model” representation of Major-TS. (D) “Groove model” representation of Minor-TS. | ||
As a result of our computational studies and experimental observations, we propose the following as a revision to our initially proposed catalytic cycle (Fig. 7). The substrate recruits the urea and CPA by hydrogen bonding to form the proto-assembled complex. Subsequently, the CPA protonates the substrate enol ether, producing an oxocarbenium ion that is rapidly trapped by the chiral phosphate to form the transient phosphate-ketal I.7,48,49 This reversible phosphate-ketal formation is followed by reionization to form the key reactive oxocarbenium ion. Subsequent C–C bond-formation (Major-TS) rapidly funnels into spirocyclic intermediate II, which then undergoes a cationic 1,2-shift to produce the final connectivity at the indole C2 position. Deprotonation of the resulting tricycle restores aromaticity to the indole and furnishes the complete tetrahydropyranoindole product.
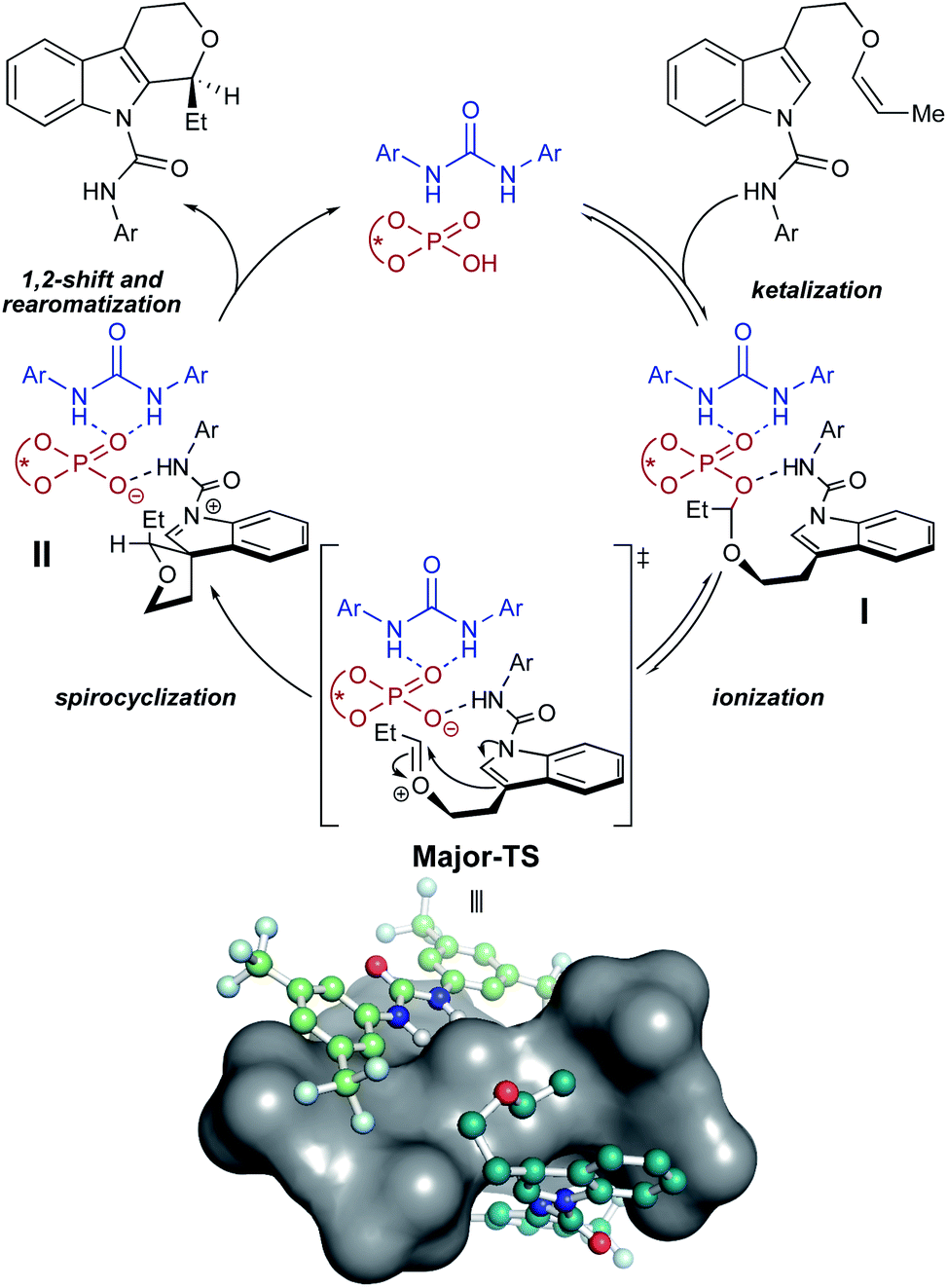 | ||
| Fig. 7 Proposed catalytic cycle. Major-TS has been visualized with a surface covering the CPA to illustrate the grooves identified in Fig. 6; hydrogen atoms omitted for clarity. | ||
Our computational models and proposed mechanism also demonstrate possible origins for the other two enhancements—yield and reaction rate—engendered by the urea co-catalyst. The rate enhancement can be attributed to hydrogen bonding between the urea and the CPA phosphate oxygen. Our computations, kinetics experiments, and literature precedence7 suggests that the spirocyclization step is rate-determining. Hydrogen bonding between the urea and phosphate oxygen likely lowers the barrier to ionization and spirocyclization, thereby increasing reaction rate. The positioning of the urea in the groove surrounding the intramolecular tether arm is also posed to shield the transient oxocarbenium from exogenous water, preventing decomposition of the key intermediate and boosting yield.
Conclusions
This new CPA–urea co-catalyst system here demonstrates that high levels of control can be achieved with high energy oxocarbenium ions. Despite the existing difficulty in controlling selectivity on such reactive functional groups, we demonstrate the applicability of this cooperative catalysis hydrogen bonding mode to induce selectivity on non-stabilized oxocarbenium ions. By accessing a self-assembled multi-catalyst/substrate complex, the overarching structure of the system is created in situ without extensive catalyst synthesis/engineering. The resulting interactions necessary for the reaction to function—and the inherent occupancy of the reactive volume of the catalyst—are nuanced and provide a window into the transient world of dynamic hydrogen bonding apart from canonical empirical evidence. This “occupancy-induced” selectivity model could be enabling for cooperative catalysis and holds potential for new and selective oxocarbenium ion-driven transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. A. S. thanks Northwestern and the National Institute of General Medical Sciences (GM R01 GM131431, GM R35 GM136440) for support of this work. P. H.-Y. C. gratefully acknowledges financial support from the Vicki & Patrick F. Stone family, the Bert & Emelyn Christensen family, and the National Science Foundation (NSF, CHE-1352663).Notes and references
- E. L. Larghi and T. S. Kaufman, Synthesis, 2006, 2, 187–220 Search PubMed.
- E. L. Larghi and T. S. Kaufman, Eur. J. Org. Chem., 2011, 2011, 5195–5231 CrossRef CAS.
- N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323–3335 RSC.
- Z. Zhu, A. Adili, C. Zhao and D. Seidel, SynOpen, 2019, 03, 77–90 CrossRef.
- Z. Zhu, M. Odagi, C. Zhao, K. A. Abboud, H. U. Kirm, J. Saame, M. Lokov, I. Leito and D. Seidel, Angew. Chem., Int. Ed., 2020, 59, 2028–2032 CrossRef CAS PubMed.
- C. Zhao, S. B. Chen and D. Seidel, J. Am. Chem. Soc., 2016, 138, 9053–9056 CrossRef CAS PubMed.
- S. Das, L. Liu, Y. Zheng, M. W. Alachraf, W. Thiel, C. K. De and B. List, J. Am. Chem. Soc., 2016, 138, 9429–9432 CrossRef CAS PubMed.
- M. A. Maskeri, M. J. O'Connor, A. A. Jaworski, A. V. Davies and K. A. Scheidt, Angew. Chem., Int. Ed., 2018, 57, 17225–17229 CrossRef CAS PubMed.
- A. Borovika, P.-I. Tang, S. Klapman and P. Nagorny, Angew. Chem., Int. Ed., 2013, 52, 13424–13428 CrossRef CAS PubMed.
- W.-T. Fan, N.-K. Li, L. Xu, C. Qiao and X.-W. Wang, Org. Lett., 2017, 19, 6626–6629 CrossRef CAS PubMed.
- W. H. Pace, D.-L. Mo, T. W. Reidl, D. J. Wink and L. L. Anderson, Angew. Chem., Int. Ed., 2016, 55, 9183–9186 CrossRef CAS PubMed.
- C. Palo-Nieto, A. Sau, R. Williams and M. C. Galan, J. Org. Chem., 2017, 82, 407–414 CrossRef CAS PubMed.
- R. Peters, Cooperative Catalysis, Wiley-VCH, Weinheim, 2015 Search PubMed.
- M. S. Taylor and E. N. Jacobsen, PNAS, 2004, 101, 5368–5373 CrossRef CAS PubMed.
- R. R. Knowles and E. N. Jacobsen, PNAS, 2010, 107, 20678–20685 CrossRef CAS PubMed.
- M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed.
- S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198–7199 CrossRef CAS PubMed.
- K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
- N. Z. Burns, M. R. Witten and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS PubMed.
- C. S. Yeung, R. E. Ziegler, J. A. Porco Jr and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 13614–13617 CrossRef CAS PubMed.
- C. R. Kennedy, D. Lehnherr, N. S. Rajapaksa, D. D. Ford, Y. Park and E. N. Jacobsen, J. Am. Chem. Soc., 2016, 138, 13525–13528 CrossRef CAS PubMed.
- M. D. Visco, J. Attard, Y. Guan and A. E. Mattson, Tetrahedron Lett., 2017, 58, 2623–2628 CrossRef CAS.
- M. S. Sigman, P. Vachal and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 1279–1281 CrossRef CAS PubMed.
- M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901–4902 CrossRef CAS.
- R. S. Klausen, C. R. Kennedy, A. M. Hyde and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 12299–12309 CrossRef CAS PubMed.
- M. S. Taylor and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 10558–10559 CrossRef CAS PubMed.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- I. Čorić and B. List, Nature, 2012, 483, 315–319 CrossRef PubMed.
- I. Čorić, S. Vellalath and B. List, J. Am. Chem. Soc., 2010, 132, 8536–8537 CrossRef PubMed.
- C. Lalli and P. van de Weghe, Chem. Commun., 2014, 50, 7495–7498 RSC.
- L. Liu, P. S. Kaib, A. Tap and B. List, J. Am. Chem. Soc., 2016, 138, 10822–10825 CrossRef CAS PubMed.
- S. Bhadra and H. Yamamoto, Chem. Rev., 2018, 118, 3391–3446 CrossRef CAS PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1993, 115, 4291–4299 CrossRef CAS.
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1992, 114, 3139–3141 CrossRef CAS.
- M. P. Crockett, H. Zhang, C. M. Thomas and J. A. Byers, Chem. Commun., 2019, 55, 14426–14429 RSC.
- D. Jansen, J. Gramüller, F. Niemeyer, T. Schaller, M. C. Letzel, S. Grimme, H. Zhu, R. M. Gschwind and J. Niemeyer, Chem. Sci., 2020, 11, 4381–4390 RSC.
- R. Mitra, H. Zhu, S. Grimme and J. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 11456–11459 CrossRef CAS PubMed.
- C. Detering, P. M. Tolstoy, N. S. Golubev, G. S. Denisov and H. H. Limbach, Dokl. Phys. Chem., 2001, 379, 191–193 CrossRef.
- J. DeFord, F. Chu and E. V. Anslyn, Tetrahedron Lett., 1996, 37, 1925–1928 CrossRef CAS.
- S. Rossi and S. E. Denmark, Lewis Base Catalysis in Organic Synthesis, Wiley, 2016 Search PubMed.
- D. B. Collum, A. J. McNeil and A. Ramirez, Angew. Chem., Int. Ed., 2007, 46, 3002–3017 CrossRef CAS PubMed.
- X. Sun, S. L. Kenkre, J. F. Remenar, J. H. Gilchrist and D. B. Collum, J. Am. Chem. Soc., 1997, 119, 4765–4766 CrossRef CAS.
- C. Zheng, Z.-L. Xia and S.-L. You, Chem, 2018, 4, 1952–1966 CAS.
- P. D. Bailey, J. Chem. Res., Synop., 1987, 202–203 CAS.
- N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. D. Toste, Nature, 2011, 470, 245–249 CrossRef CAS PubMed.
- Y. Y. Khomutnyk, A. J. Arguelles, G. A. Winschel, Z. Sun, P. M. Zimmerman and P. Nagorny, J. Am. Chem. Soc., 2016, 138, 444–456 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009, see ESI for full citation.†.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, PCCP Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, PCCP Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, spectra, computational procedures and coordinates. See DOI: 10.1039/d0sc03250f |
| ‡ These authors contributed equally to this work. |
| § Notes on computation. Manual, exhaustive conformation searches were performed to locate all relevant structures. The structures and thermal corrections for all conformers were optimized with Gaussian0922 (ref. 50) using PBE51-D3BJ52/6-31G(d)53 with the SMD implicit solvation model54 for toluene. Single point energies were obtained at the PBE/def2-TZVPP55,56 level of theory with ORCA 4.0.1.57 Final solvation corrections were computed using PBE-D3BJ/6-311++G(2df,p)58,59 in toluene using SMD. Computational model graphics were generated with CYLview.60 |
| ¶ The Bridge-model transition state previously conjectured in our original report of this transformation could not be located computationally. For additional computed model systems and analysis, please see the ESI. |
| This journal is © The Royal Society of Chemistry 2020 |