
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated electrocatalytic nESI-MS platform for quantification of fatty acid isomers directly from untreated biofluids†
Kavyasree
Chintalapudi
 and
Abraham K.
Badu-Tawiah
*
and
Abraham K.
Badu-Tawiah
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210, USA. E-mail: badu-tawiah.1@osu.edu
First published on 25th August 2020
Abstract
Positional isomers of alkenes are frequently transparent to the mass spectrometer and it is difficult to provide convincing data to support their presence. This work focuses on the development of a new reactive nano-electrospray ionization (nESI) platform that utilizes non-inert metal electrodes (e.g., Ir and Ru) for rapid detection of fatty acids by mass spectrometry (MS), with concomitant localization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to differentiate fatty acid isomers. During the electrospray process, the electrical energy (direct current voltage) is harnessed for in situ oxide formation on the electrode surface via electro-oxidation. The as-formed surface oxides are found to facilitate in situ epoxide formation at the CC bond position and the products are analyzed by MS in real-time. This phenomenon has been applied to analyze isomers of unsaturated fatty acids from complex serum samples, without pre-treatment.
C bond to differentiate fatty acid isomers. During the electrospray process, the electrical energy (direct current voltage) is harnessed for in situ oxide formation on the electrode surface via electro-oxidation. The as-formed surface oxides are found to facilitate in situ epoxide formation at the CC bond position and the products are analyzed by MS in real-time. This phenomenon has been applied to analyze isomers of unsaturated fatty acids from complex serum samples, without pre-treatment.
Introduction
Fatty acids (FAs), either as free or constituents of complex lipids, are involved in diverse aspects of eukaryotic cellular function such as energy storage, membrane structure and dynamics, and signal transduction.1–3 The length of the alkyl chain, degree of its saturation, and position of the double bond confer the specific biological activities unique to various FAs. FAs are implicated in chronic diseases (e.g., cardiovascular and heart disease, cancer, inflammation and autoimmune diseases),4–6 for which the normal/reference levels are yet to be defined.7 An efficient and accurate method of FA analysis is clinically useful for evaluating the nutritional status and detecting essential fatty acid deficiency, which is common in premature and low birth weight infants. In addition, FA structural identification and quantification are important in molecular biology and biomarker discovery.5,8,9Mass Spectrometry (MS) is a powerful tool for rapid identification of a spectrum of free (non-esterified) fatty acids (FFAs), not only for mass measurement but also for structural information and quantitation capabilities in complex samples. FA analysis in lipidomics is challenging because lipids are much more complex compared to other classes of biomolecules that can be considered as permutations of a fixed number of monomers (e.g., proteins and oligonucleotides).10 Lipid complexity increases when considering isomeric forms that differ only in double bond positions.11,12 Tandem MS (MS/MS) experiments, especially those involving low-energy collision-induced dissociation (CID), have not been effective in locating CC bond positions in the FAs, due to the high bond dissociation energies associated with cleaving a CC bond. Without characteristic fragment ions (also known as diagnostic ions), the CC locations cannot be identified using MS/MS.13 To tackle this problem, CC specific chemical derivatization reactions have been utilized prior to MS/MS. These include the reaction of ozone with the double bonds present in lipids resulting in the formation of ozonides that could be directly analyzed by MS.14–16 Upon application of CID on fatty acid ozonides, diagnostic ions are released to characterize the FAs in a mixture using MS/MS. Although the challenges of ozonolysis17 are well known (e.g., safety concerns and the need for external generators, independent of the mass spectrometer), this approach is still used because there are only a few other methods that are effective in terms of selectivity, yield and cleanness. Furthermore, a photochemical derivatization18 procedure has been proposed that utilizes acetone as the Paternò–Büchi (PB) reagent. While this method is effective in localizing CC bonds in FAs, challenges like competing side reactions and retro PB reactions can complicate data interpretation. Other recent developments in this area include photodissociation,19,20 plasma-based,21,22 and in-solution (bulk) epoxidation reactions.23,24 These methods either required special equipment or complex and time-consuming offline extraction procedures. This has motivated the need to develop simpler and greener analytical methods. In this work, we report a simple analytical procedure that integrates in-capillary liquid/liquid extraction with online electrocatalytic oxidation of the CC bond in a conventional nano-electrospray ionization (nESI) source.
There has been substantial literature on the formation of oxides on non-inert metals such as Ir and Ru upon application of voltage;25–28 this is in contrast to coinage metals (e.g., Ag) which tend to release only electrons (and sometimes the corresponding metal cation) when biased with DC potential.29 Therefore, we expected the use of non-inert electrodes in nESI to result in the formation of nascent oxides at the electrode surface due to the high electrical energy typically used in the electrospray process. The presence of the oxide should in turn open new reaction pathways besides those available solely by electron-transfer-driven mechanisms on inert electrodes (e.g., Pt or carbon).30–32 These expectations have been met, allowing electrocatalytic CC oxidation reactions to be integrated on a traditional nESI-MS platform for the first time. The use of non-inert metal electrodes (Ir and Ru) in nESI-MS permits the differentiation of positional isomers via the localization of CC bonds in FAs without sample preparation and with no instrument modifications. We were able to achieve the epoxidation reaction instantaneously using our nESI-MS platform fitted with non-inert Ir and Ru metal electrodes in MeOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution, without the onset of a corona (spray voltage was kept at 1.5 kV) and in the absence of additives such as acetone or epoxidation reagents. FA isomers, oleic acid and cis-vaccenic acid were successfully differentiated and quantified directly from serum at concentration levels as low as 1.2 μM.
:1, v/v) solution, without the onset of a corona (spray voltage was kept at 1.5 kV) and in the absence of additives such as acetone or epoxidation reagents. FA isomers, oleic acid and cis-vaccenic acid were successfully differentiated and quantified directly from serum at concentration levels as low as 1.2 μM.
Results and discussion
Strategy for in situ oxide generation and real-time reaction monitoring
The integrated electrocatalytic nESI-MS platform developed in this work is shown in Fig. 1a. After loading the glass capillary with ∼10 μL of the sample, it was fitted in front of the MS inlet and electric contact was made via a metal/alloy electrode. The applied electrical energy (1.5 kV) enabled in situ modification of the electrode surface to generate electrochemical oxides, the specific identity of which was effectively controlled by varying the target electrode (Pure Ir, Pt/Ir (90/10%), Pt/Ru (95/5%), etc.). Charged microdroplets containing reactants/products are generated concomitantly when the DC potential is applied enabling MS analysis in real-time. Control experiments were performed using a pure inert Pt electrode. It should be noted that the low flowrates30 (∼100 nL min−1; see the ESI† for details) in this nESI-MS platform provide long reagent residence time on the electrode allowing the electrocatalytic reactions to occur before the reactants/products are released into the charged microdroplets for MS detection. The MS detection method afforded both qualitative (from MS/MS fragmentation) and quantitative studies (using relative ion intensity measurements in the presence of an internal standard) on the reaction systems.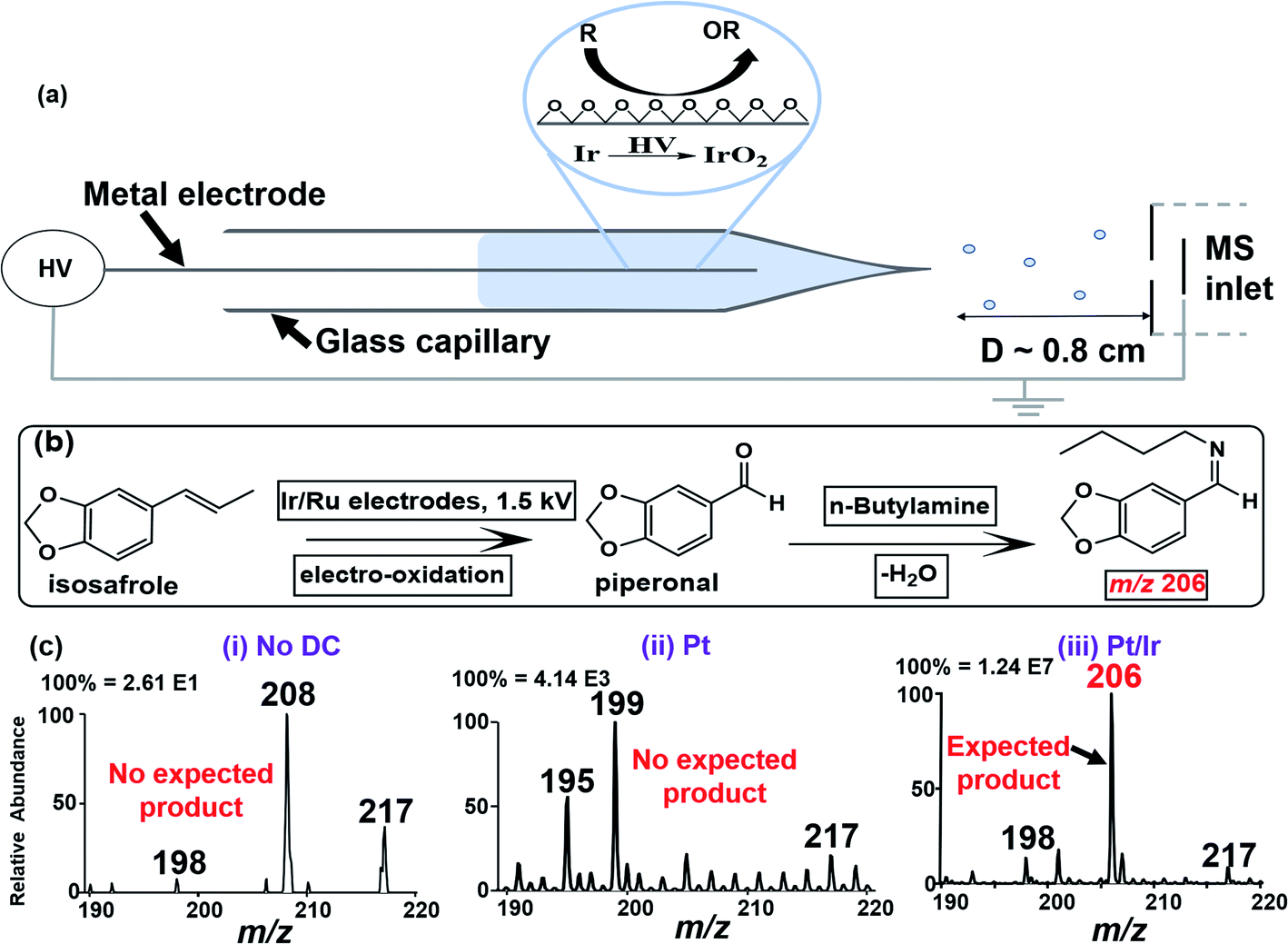 | ||
| Fig. 1 (a) Schematic illustration of the integrated electrocatalytic nESI-MS platform capable of in situ CC bond oxidation and ionization in a single step. (b) Reaction scheme for online nESI-MS-based electro-oxidation of isosafrole and subsequent reaction with n-butylamine yielding the corresponding Schiff base for MS detection. (c) nESI-MS analysis of isosafrole using (i) non-electrical gas-driven spray ionization and (ii) Pt and (iii) Ir/Pt (10/90%) anodes. The ions at m/z 195, 198, 199, 208, and 217 in (c) (i–iii) are unrelated background species. A Thermo Velos Pro linear ion trap mass spectrometer was used in positive ion mode and the DC voltage was 1.5 kV. | ||
To optimize our electrocatalytic nESI-MS platform, we used the oxidative cleavage of isosafrole (MW 162 g mol−1) to piperonal (MW 150 g mol−1). The use of isosafrole as the olefinic substrate was inspired by the work of Zanta et al. in which catalytic conversion of isosafrole to piperonal was achieved using Ti/RuO2 and Ti/IrO2 electrodes.33 To enable effective detection of piperonal by nESI-MS, we added n-butylamine (MW 73 g mol−1) to the starting solution containing isosafrole. n-Butylamine reacts in situ with piperonal derived from electrooxidation of isosafrole to produce the corresponding Schiff base (MW 205 g mol; Fig. 1b), which is more easily detected via protonation. Fig. 1c(iii) shows the result of this experiment where a peak at m/z 206 was detected instantaneously upon the application of DC potential to the Pt/Ir electrode inserted in the isosafrole/n-butylamine reaction mixture. The appearance of this ion (m/z 206; see Fig. S1 and Scheme S1† for MS/MS characterization) indicates that the electro-oxidation of isosafrole occurred during the nESI-MS analysis, and that the piperonal product reacted with the n-butylamine in real-time. We performed two control experiments to validate this interpretation: (1) nESI-MS analysis of isosafrole using non-electrical gas-driven spray ionization. That is, although the Pt/Ir electrode was in contact with isosafrole solution (3 min), no DC potential was applied. Instead, we used gas (N2) driven spray apparatus to push the solution out from the glass capillary to form ions. The results (Fig. 1c(i)) showed no product formation indicating there is no active (permanent) oxide layer at the surface of the Pt/Ir electrode. This suggests that active nascent oxides are formed upon the application of DC potential in the presence of a solvent (see Scheme S2† for details). (2) The second control experiment involved the use of a pure Pt electrode (instead of Pt/Ir alloy) on the nESI-MS platform. The result shown in Fig. 1c(ii) represents data recorded with the Pt electrode at its first use, where no expected product was observed at m/z 206 (see Fig. S2† for details). The absence of the electrocatalytic product when using the inert Pt electrode indicated limited oxide formation in the presence of electrical energy.
Encouraged by the successful oxidation of isosafrole on the Pt/Ir electrode, we further optimized the process by evaluating the effect of the electrode composition, solvent and voltage where the reaction progress was monitored for four minutes of continuous spraying (Fig. S3–S6†). We used the isosafrole/butylamine (200 μM) system in MeOH/H2O (1:1, v/v) on five different electrodes (Pt, Ir, Ag, Pt/Ir (90/10%) and Pt/Ru (95/5%)). Aprotic acetonitrile solutions were also tested but isosafrole oxidation was limited due to its inability to donate oxygen. Although product yield increased with applied voltage (Fig. S3†), we settled on 1.5 kV to avoid corona discharge, which can damage the tip and cause signal instabilities in nESI-MS. Using positive-ion mode analysis (at 1.5 kV and in MeOH/H2O (1:1, v/v)), product yield was monitored as a function of electrode type. The observed activity Ir > Pt/Ir > Pt/Ru > Pt > Ag (Fig. S5†) is consistent with the known oxophilicity among the transition metals.25 Hence, we chose to use the pure Ir electrode for all further studies, unless stated otherwise. This result (i.e., dependency of yield on electrode type) proves that the reaction is catalytic and supports our hypothesis regarding the formation of in situ oxides on surfaces of non-inert metals/alloys during nESI MS. Further evidence for in situ nascent oxide formation was derived from extensive X-ray photoelectron spectroscopic experiments (Fig. S7, S8, and Table S1†).
Epoxidation of unsaturated fatty acids and subsequent localization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bonds
C bonds
We applied the optimized electrocatalytic nESI-MS platform to analyze fatty acids with the main objective of localizing CC bonds and differentiating isomeric species. Surprisingly, while isosafrole underwent complete CC bond oxidative cleavage we observed epoxidation reactions for all fatty acids tested (Scheme S3†). For example, we observed instantaneous epoxidation of oleic acid (MW 282 g mol−1) when using the Ir electrode biased with −1.5 kV, without the use of any additives. The analysis was conducted in the negative-ion mode, and the epoxide product ions [M + O–H]− (m/z 297) were detected at a 10% intensity relative to the abundance of deprotonated [M − H]− ions at m/z 281 (Fig. S9†). The efficiency of the epoxidation reaction was improved >10× by increasing the ionic strength of the spray solvent via the addition of 1% NH4OH (Fig. 2a). Additional experiments utilizing other MS additives such as ammonium acetate [NH4CO2CH3] and ammonium carbonate [(NH4)2CO3] further proved ammonium hydroxide [NH4OH] to be superior (Fig. S10†). We also performed control studies for this oleic acid sample under traditional nESI-MS conditions using Ag and Pt electrodes for understanding the effectiveness of the reaction induced using the Ir electrode attached in the ESI (see Fig. S11†). A slight increase (<2×) in the oxidative effect was also observed for inert electrodes (e.g., Pt) in the presence of 1% NH4OH, but the oxidative power of the Pt/1% NH4OH pales when compared with that of the Ir/1% NH4OH reaction system. Upon application of CID on the fatty acid epoxides, diagnostic fragment ions were detected in MS/MS experiments that characterized the exact position of the CC bond in the fatty acid. For the oleic acid epoxide, characteristic CID product ions included m/z 177 and 155 (Fig. 2b and c), which were generated in the gas-phase via two distinct ring opening fragmentation pathways (see Scheme S4,† Fig. S12†). Compared to isosafrole, the formation of epoxide (as opposed to complete CC cleavage) might be attributed to two factors, that is differences in (1) the electron density and chemical environment of the alkene functional group,13,22 including rotational flexibility in the fatty acid, which can limit contact of the CC bond with the electrode surface and (2) the reactivity for anodic versus cathodic oxidation. Isosafrole was analyzed using positive potential (anodic oxidation), which is a more efficient means to generate oxide films on a metallic substrate. Due to their high propensity to form deprotonated [M − H]− ions, we had to use negative potential for fatty acid analysis, which led to a less efficient cathodic oxidation. Preliminary experiments have indicated minimal involvement/production of reactive oxygen species such as H2O2 or O2−˙ after the application of negative potential35 (see Fig. S13† for details).
 | ||
| Fig. 2 (a) Negative-ion mode nESI MS spectra of 200 μM oleic acid in the presence of 1% NH4OH in MeOH/H2O (1:1), showing the oleic acid and its epoxide at m/z 281 and 297, respectively. (b) Schematic illustration of the formation of diagnostic ions for the identification of CC isomers in oleic acid (blue) and cis-vaccenic acid (green). (c) Tandem MS analysis of oleic acid epoxide at m/z 297 yields five major fragments, namely ions at m/z 155, 171, 183, 253 and 279, highlighting the diagnostic ions at m/z 155 and 171 (red) due to shattering of the epoxide ring. (d) Tandem MS analysis of cis-vaccenic acid epoxide at m/z 297 yields four major fragments, namely ions at m/z 199, 183, 253 and 279; diagnostic ions are highlighted at m/z 185 and 199 (orange). (e) Tandem MS analysis of a 1:1 mixture of isomeric oleic and vaccenic fatty acid, showing expected diagnostic ion pairs at m/z (171, 155) and (183, 199), respectively, from CID of the epoxidation reaction product using a Thermo Velos Pro linear ion trap mass spectrometer in negative ion mode. | ||
cis-Vaccenic acid (MW 282 g mol−1) FA 18:1 (11Z) is a positional isomer of oleic acid, and so the two compounds cannot be differentiated based on mass measurements alone. However, the position of the CC bond in FA 18:1 (11Z) is two more carbons away from the carboxylic head group compared with that in oleic acid FA 18:1 (9Z). Therefore, the diagnostic ions of FA 18:1 (11Z) are expected to be 28 Da higher in mass than the diagnostic ions derived from FA 18:1 (9Z) (m/z 155/171). As expected, CID of the epoxide product (m/z 297) from cis-vaccenic acid yielded two major fragments at m/z 183 and 199 (diagnostic ions) that are due to the shattering of the epoxide ring (Fig. 2b and d), as well as two other product ions formed via H2O and CO2 neutral losses to give ions at m/z 253 and 279, respectively (see Scheme S5 and Fig. S14†). With the diagnostic ions for the specific FA isomer identified, we evaluated the ability of our platform to identify the location of double bonds when the two FA isomers (oleic/vaccenic acids) are present together in a single sample mixture (see Fig. S15†). The result from the CID MS/MS analysis of the epoxide products is provided in Fig. 2e, which clearly showed all four expected diagnostic fragment ions at m/z 155/171 (for oleic acid) and m/z 183/199 (for vaccenic acid).
To further examine the robustness of our in situ epoxidation strategy, we explored the analysis of FAs containing two CC bonds such as linoleic acid (MW 280 g mol−1), FA 18:2 (9Z, 12Z). There are three possible epoxidation products from linoleic acid (Fig. 3a): two isomeric products containing a single epoxide functional group separately at C9 and C12 (MW 296 g mol−1 each); these species are accompanied by a reaction in which both double bonds have undergone epoxidation within a single linoleic acid molecule (MW 312 g mol−1). These products were confirmed in an experiment where nESI-MS analysis (using Ir electrode, −1.5 kV spray voltage) of linoleic acid showed three distinct peaks at m/z 279, 295, and 311 for [M − H]−, [M + O–H]−, and [M + 2O–H]−, respectively (Fig. 3b). Upon collisional activation, the ions at m/z 295 (suspected to consist of two isomeric epoxide products) produced two pairs of diagnostic ions at m/z 155/171 (red) and m/z 195/211 (purple) (Fig. 3c). Identification of each pair of diagnostic ions was straightforward because of the characteristic 16 Da mass difference. Hence, the diagnostic ion pair at m/z 155/171 is assigned to the cleavage of the CC bond at C9–C10 while those at m/z 195/211 are thought to be generated after C12–C13 bond cleavage. This suggests that our electrocatalytic epoxidation reaction is non-selective, providing a useful methodology to identify the locations of multiple CC bonds in a poly-unsaturated fatty acid (PUFA) using the signal from the mono-epoxide product (i.e., M + O). In this case, sufficient analytical data are obtained without considering the high order di-epoxide product (i.e., M + 2O), which tends to give complicated MS/MS spectra (See Fig. S16, Scheme S6, and Table S2† for details). We note, however, that when optimized, the appearance of such higher order reaction products occurring at 16 Da from the expected [M + O–H]− ion can signify the presence of a PUFA species. Clearly, such peaks can assist in the MS/MS data interpretation for [M + nO–H]− ions, where n denotes the number of double bonds in the PUFA.
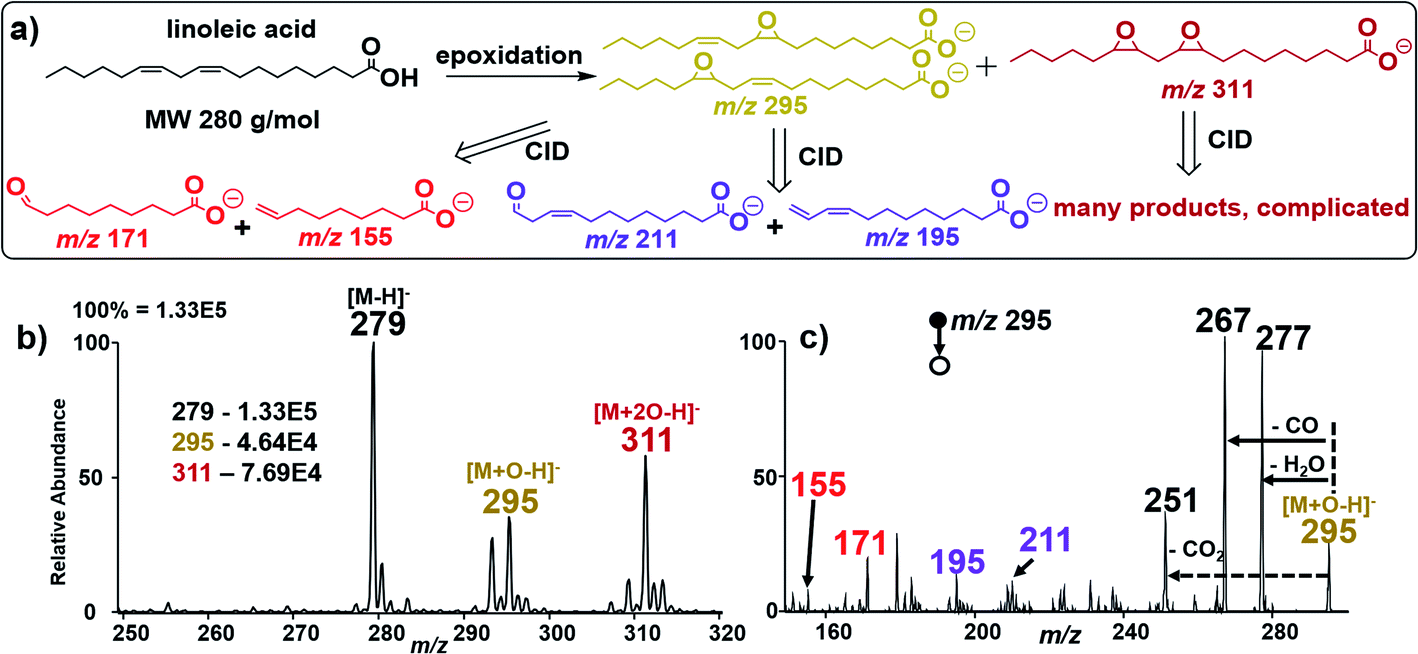 | ||
| Fig. 3 (a) Schematic illustration of the CID fragmentation pattern of deprotonated linoleic acid after epoxidation in negative ion mode. CID pathways show major diagnostic fragment ion pairs at m/z (171, 155) and (211, 195) for the two double bonds in linoleic acid. (b) Negative-ion mode full nESI MS spectrum showing relative ion intensities of linoleic acid epoxides at m/z 295 and 311 on the Ir electrode in the presence of 1% NH4OH in MeOH/H2O (1:1). (c) Tandem MS analysis of the mono-epoxide reaction product at m/z 295 from linoleic acid, yielding four major fragments due to shattering of the epoxide ring into 2 sets of diagnostic ions (m/z 171/155) and (m/z 211/195) using a Thermo Velos Pro linear ion trap mass spectrometer in negative ion mode. | ||
Non-contact (native) spray versus contact (oxidative) spray conditions
The reactive nature of the current nESI MS platform utilizing the Ir electrode means that information on the original chemical content of the analyte solution can be lost. To address this issue, we employed the non-contact nESI method34 recently developed in our laboratory to provide “native-state” mass spectra without oxidation. This nESI setup enables electrospray without physical contact between the reactive metal electrode and analyte solution. We compared the ion signal from three separate experiments: analysis of 200 μM oleic acid in MeOH/H2O (1:1, v/v), with and without the presence of 1% NH4OH, and the analysis of isosafrole solution (MeOH/H2O; 1:1, v/v) in the presence of n-butylamine. Each sample (2 μL present at the tip of the glass capillary) was analysed using non-contact (native) nESI MS and the contact-mode (oxidative) spray condition (using the Ir electrode in all cases). Application of DC voltage (±1.5 kV) to the Ir electrode generated stable spray in both experiments. The mass spectra recorded from these experiments are summarized in Fig. S17.† We observed no traces of the epoxide product when the oleic acid solution was analysed without physical contact between the Ir electrode and analyte solution (Fig. S17a†), providing a means to evaluate the “native-state” chemical species originally present in the analyte solution before the catalytic oxidative process is initiated via the contact spray mode (Fig. S17b†). Upon the addition of 1% NH4OH to the oleic acid solution (MeOH/H2O, 1:1, v/v), the intensity of the epoxide peak at m/z 297 was observed to increase from 10% to 50% relative abundance, under the contact oxidative spray condition (Fig. S17d†) while only a small amount of the oxidized oleic acid was recorded under the non-contact spray condition (Fig. S17c†). In a similar manner, oxidative cleavage of isosafrole was not observed when the non-contact spray mode was used as indicated by the absence of the peak at m/z 206 (Fig. S17e†) compared with the contact-mode oxidative spray (Fig. S17f†), which yields high abundance of this Schiff base (m/z 206) due to the reaction between butylamine and the oxidized aldehyde product. These results are important as they illustrate a two-tiered spray mechanisms for the catalytic nESI-MS platform, allowing unreacted native analytes to be detected before initiating the contact-mode oxidative spray process, all using a single nESI source.
Direct quantification of free fatty acids (FFAs) in untreated serum
As an application, we used the developed electrocatalytic nESI-MS platform to analyze FFAs in complex biofluid. An established protocol exists to extract FFAs from biofluid before MS analysis,34,36 but we sought to develop a two-tiered integrated system where we not only perform in situ catalytic epoxidation, but also enable in-capillary liquid/liquid extraction prior to MS analysis. Therefore, we proposed the integrated experimental process illustrated in Fig. 4 where FFAs present in the complex biofluid sample are first enriched into an ethyl acetate extraction solvent (refer to the experimental methods section in the ESI† for more details). Being immiscible with the biofluids, this organic solvent selectively extracts organic compounds from the biofluid, while leaving behind the aqueous and hydrophilic cellular components and thus minimizing matrix effects during MS analysis. The shaking process (3–5 strokes) results in the disintegration of the biofluid into smaller compartments facilitating highly efficient extraction via increased interfacial contact with the extracting organic solvent. We estimated approximately 34% extraction efficiency for oleic acid spiked in serum (Fig. S18†). The clean organic extract present at the tip of the glass capillary was then placed in contact with the Ir electrode (Fig. 4b). With the application of electrical potential, extra peaks that are shifted by 16 Da are expected to signify the presence of unsaturated FAs, due to the occurrence of the epoxidation reaction in our nESI-MS platform. An example is shown for serum in Fig. 5a where the most prevalent FAs such as FA 18:1 (oleic, m/z 281), FA 18:2 (linoleic, m/z 279), FA 18:3 (linolenic, m/z 277), and FA 16:1 (palmitoleic, m/z 253) were all observed accompanied by the expected epoxide products at m/z 297, 295, 293, and 269, respectively. As anticipated, FA 18:0 (stearic) and FA 16:0 (palmitic) having no CC bond did not show the corresponding epoxide peaks. Structures of all fatty acids were confirmed in tandem MS analysis (see Fig. S19 and S20†). These FAs were characterized directly from a complex (blank) serum sample without any dilution or pretreatment processes, except for the in-capillary liquid/liquid extraction.
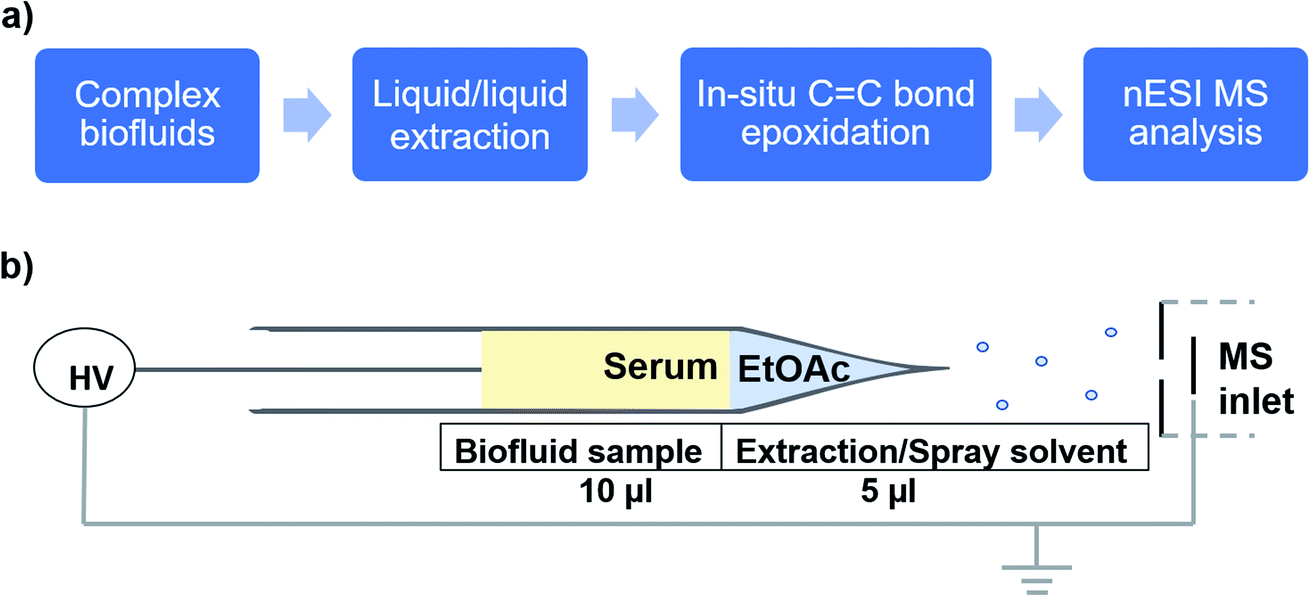 | ||
| Fig. 4 (a) Sequence of the experimental approach for complex biofluid analysis involving in-capillary extraction, in situ oxide creation and epoxidation, and real-time analysis by nESI MS. (b) Schematic illustration of the platform for in-capillary liquid/liquid extraction of FAs from serum using ethyl acetate and subsequent MS analysis with nESI using Ir where in situ fatty acid epoxidation occurs. | ||
 | ||
| Fig. 5 (a) Negative-ion mode nESI MS analysis of serum after in-capillary liquid/liquid extraction of FAs, showing the presence of unsaturated FAs at m/z 253, 277, 279, and 281 with concomitant appearance of epoxide reaction products at m/z 269, 293, 295, and 297, respectively, as indicated by the 16 Da shift. Saturated FAs lacking the epoxide peaks were observed at m/z 255 and 283. (b) Calibration curve showing the quantification of oleic acid (25–300 μM) spiked in serum using 100 μM oleic-d17 as the internal standard (IS); the intensities of [M − H]− peaks at m/z 281 and m/z 298 for the analyte and IS, respectively, were used following the in-capillary liquid/liquid extraction process. Error bars indicate SD for 3 replicates. A Thermo Velos Pro linear ion trap mass spectrometer was used in negative ion mode. | ||
As exemplified above, a variety of FAs naturally exist in serum, a substantial amount of which are unsaturated. We sought to use our platform to quantify selected FAs, including oleic acid (FA 18:1), cis-vaccenic acid (FA 18:1) and linoleic acid (FA 18:2 (9Z, 12Z)). We spiked each of the FAs in serum separately and calibration functions (concentration range 25–300 μM) were constructed. By doping standards of specific fatty acids into the serum sample, we expected the corresponding [M − H]− peak to increase with increasing concentration.
Thus, after characterization by MS/MS using diagnostic peaks from [M + O–H]−, we were able to reliably use the [M − H]− signal for quantification. The [M − H]− ion signal from C18:1-d17 (MW 299 g mol; 100 μM) was used as the internal standard (IS).37 For oleic acid quantification in serum, we observed that the ratio of the analyte (A, m/z 281) to IS (m/z 298) increased linearly with added oleic acid concentration (Fig. 5b). Table 1 shows the limit of detection (LOD) and limit of quantification (LOQ) data obtained for FA 18:1 (both Δ9 and Δ11 isomers) and FA 18:2, in only 10 μL of serum. The data are consistent with those reported in the literature.38 Relative standard deviations <10% were obtained for each FA indicating high accuracy. The method showed excellent precision and good reproducibility was also demonstrated as indicated by low standard deviation (SD, shown by the error bars) as well as high linearity (R2 > 0.99; Fig. S21† for details of the calibration data).
| Analyte | LOD (μM/ppm) | LOQ (μM/ppm) |
|---|---|---|
| FA 18:1 (9Z) | 1.18/0.34 | 3.95/1.12 |
| FA 18:1 (11Z) | 8.00/2.26 | 26.66/7.53 |
| FA 18:2 (9Z, 12Z) | 2.81/0.79 | 9.38/2.63 |
Conclusions
In conclusion, we have successfully developed an integrated nESI MS platform fitted with reactive electrodes that enables structural identification of positional isomers in FAs. The platform is capable of complex mixture analysis through on-line liquid/liquid extraction, in situ electrooxidation of CC bonds to form epoxides and ionization of product/analytes, all in a single experimental setup. The entire process can be completed in under 2 min. The use of lipid profiles to “fingerprint” biofluids may find significant future applications in diagnostic pathology. Using serum samples from healthy patients we established a baseline level of the selected FAs using the Ir electrode. This should enable quantification of FAs in biofluids to construct a spectral database for a large variety of CC isomeric lipids. While the epoxide reaction product [M + O–H]− exhibited exceptional qualitative abilities, its use for quantification purposes, based on diagnostic ion intensities, may require further optimization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation (award number CHE-1900271). The authors thank Dr Lisa Alexander for her support with the utilization of the XPS facility at The Ohio State University.Notes and references
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef.
- M. P. Wymann and R. Schneiter, Nat. Rev. Mol. Cell Biol., 2008, 9, 162–176 CrossRef CAS.
- M. R. Wenk, Cell, 2010, 143, 888–895 CrossRef CAS.
- B. J. Arsenault, S. M. Boekholdt and J. J. P. Kastelein, Nat. Rev. Cardiol., 2011, 8, 197–206 CrossRef CAS.
- J. E. Chavarro, S. A. Kenfield, M. J. Stampfer, M. Loda, H. Campos, H. D. Sesso and J. Ma, Am. J. Epidemiol., 2013, 178, 1246–1255 CrossRef.
- A. R. Lima, M. de L. Bastos, M. Carvalho and P. Guedes de Pinho, Transl. Oncol., 2016, 9, 357–370 CrossRef.
- S. A. Abdelmagid, S. E. Clarke, D. E. Nielsen, A. Badawi, A. El-Sohemy, D. M. Mutch and D. W. L. Ma, PLoS One, 2015, 10(2), e0116195 CrossRef.
- W. Zhang, S. Chiang, Z. Li, Q. Chen, Y. Xia and Z. Ouyang, Angew. Chem., Int. Ed., 2019, 58, 6064–6069 CrossRef CAS.
- B. Burla, M. Arita, M. Arita, A. K. Bendt, A. Cazenave-Gassiot, E. A. Dennis, K. Ekroos, X. Han, K. Ikeda, G. Liebisch, M. K. Lin, T. P. Loh, P. J. Meikle, M. Orešič, O. Quehenberger, A. Shevchenko, F. Torta, M. J. O. Wakelam, C. E. Wheelock and M. R. Wenk, J. Lipid Res., 2018, 59, 2001–2017 CrossRef CAS.
- S. J. Blanksby and T. W. Mitchell, Annu. Rev. Anal. Chem., 2010, 3, 433–465 CrossRef CAS.
- K. Yang, B. G. Dilthey and R. W. Gross, Anal. Chem., 2013, 85, 9742–9750 CrossRef CAS.
- M. M. Mahfouz, A. J. Valicenti and R. T. Holman, Biochim. Biophys. Acta, Lipids Lipid Metab., 1980, 618, 1–12 CrossRef CAS.
- X. Ma, L. Chong, R. Tian, R. Shi, T. Y. Hu, Z. Ouyang and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2573–2578 CrossRef.
- M. R. L. Paine, B. L. J. Poad, G. B. Eijkel, D. L. Marshall, S. J. Blanksby, R. M. A. Heeren and S. R. Ellis, Angew. Chem., 2018, 130, 10690–10694 CrossRef.
- K. A. Harrison and R. C. Murphy, Anal. Chem., 1996, 68, 3224–3230 CrossRef.
- M. C. Thomas, T. W. Mitchell, D. G. Harman, J. M. Deeley, R. C. Murphy and S. J. Blanksby, Anal. Chem., 2007, 79, 5013–5022 CrossRef.
- S. Torii, K. Uneyama and K. Ueda, J. Org. Chem., 1984, 49, 1830–1832 CrossRef.
- X. Ma and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 2592–2596 CrossRef CAS.
- P. E. Williams, D. R. Klein, S. M. Greer and J. S. Brodbelt, J. Am. Chem. Soc., 2017, 139, 15681–15690 CrossRef CAS.
- C. L. Feider, L. A. Macias, J. S. Brodbelt and L. S. Eberlin, Anal. Chem., 2020, 92, 8386–8395 CrossRef CAS.
- H.-Y. Hsieh, L.-H. Li, R.-Y. Hsu, W.-F. Kao, Y.-C. Huang and C.-C. Hsu, Anal. Chem., 2017, 89, 6146–6152 CrossRef CAS.
- Y. Zhao, H. Zhao, X. Zhao, J. Jia, Q. Ma, S. Zhang, X. Zhang, H. Chiba, S.-P. Hui and X. Ma, Anal. Chem., 2017, 89, 10270–10278 CrossRef CAS.
- Y. Feng, B. Chen, Q. Yu and L. Li, Anal. Chem., 2019, 91, 1791–1795 CrossRef CAS.
- S. Tang, H. Cheng and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 209–214 CrossRef CAS.
- N. Danilovic, R. Subbaraman, K.-C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y.-T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS.
- S. Cherevko, J. Electroanal. Chem., 2017, 787, 11–13 CrossRef CAS.
- R. Kötz, H. Neff and S. Stucki, J. Electrochem. Soc., 1984, 131, 72–77 CrossRef.
- O. Simond and Ch. Comninellis, Electrochim. Acta, 1997, 42, 2013–2018 CrossRef CAS.
- A. Li, Q. Luo, S.-J. Park and R. G. Cooks, Angew. Chem., Int. Ed., 2014, 53, 3147–3150 CrossRef CAS.
- Q. Wan, S. Chen and A. K. Badu-Tawiah, Chem. Sci., 2018, 9, 5724–5729 RSC.
- H. Zhang, K. Yu, N. Li, J. He, L. Qiao, M. Li, Y. Wang, D. Zhang, J. Jiang and R. N. Zare, Analyst, 2018, 143, 4247–4250 RSC.
- T. A. Brown, H. Chen and R. N. Zare, J. Am. Chem. Soc., 2015, 137, 7274–7277 CrossRef CAS.
- C. L. P. S. Zanta, A. R. de Andrade and J. F. C. Boodts, Electrochim. Acta, 1999, 44, 3333–3340 CrossRef CAS.
- D. S. Kulyk, D. J. Swiner, T. Sahraeian and A. K. Badu-Tawiah, Anal. Chem., 2019, 91, 11562–11568 CrossRef CAS.
- W. Li, J. Electrochem. Soc., 1999, 146, 592 CrossRef CAS.
- Y. Ren, M. N. McLuckey, J. Liu and Z. Ouyang, Angew. Chem., Int. Ed. Engl., 2014, 53, 14124–14127 CrossRef CAS.
- X. Ma, X. Zhao, J. Li, W. Zhang, J.-X. Cheng, Z. Ouyang and Y. Xia, Anal. Chem., 2016, 88, 8931–8935 CrossRef CAS.
- E. Kish-Trier, E. L. Schwarz, M. Pasquali and T. Yuzyuk, Clin. Mass. Spectrom., 2016, 2, 11–17 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03403g |
| This journal is © The Royal Society of Chemistry 2020 |