
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelectivity over coverage in de novo sequencing of IgGs†
Maurits A.
den Boer‡
 ab,
Jean-Francois
Greisch‡
ab,
Sem
Tamara‡
ab,
Albert
Bondt
ab and
Albert J. R.
Heck
*ab
ab,
Jean-Francois
Greisch‡
ab,
Sem
Tamara‡
ab,
Albert
Bondt
ab and
Albert J. R.
Heck
*ab
aBiomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research, Utrecht Institute of Pharmaceutical Sciences, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands. E-mail: a.j.r.heck@uu.nl
bNetherlands Proteomics Center, Padualaan 8, 3584 CH Utrecht, The Netherlands
First published on 6th October 2020
Abstract
Although incredibly diverse in specificity, millions of unique Immunoglobulin G (IgG) molecules in the human antibody repertoire share most of their amino acid sequence. These constant parts of IgG do not yield any useful information in attempts to sequence antibodies de novo. Therefore, methods focusing solely on the variable regions and providing unambiguous sequence reads are strongly advantageous. We report a mass spectrometry-based method that uses electron capture dissociation (ECD) to provide straightforward-to-read sequence ladders for the variable parts of both the light and heavy chains, with a preference for the functionally important CDR3. We optimized this method on the therapeutic antibody Trastuzumab and demonstrate its applicability on two monoclonal quartets of the four IgG subclasses, IgG1, IgG2, IgG3 and IgG4. The method is based on proteolytically separating the variable F(ab′)2 part from the conserved Fc part, whereafter the F(ab′)2 portions are mass-analyzed and fragmented by ECD. Pure ECD, without additional collisional activation, leads to straightforward-to-read sequence tags covering the CDR3 of both the light and heavy chains. Using molecular modelling and structural analysis, we discuss and explain this selective fragmentation behavior and describe how structural features of the different IgG subclasses lead to distinct fragmentation patterns. Overall, we foresee that pure ECD on F(ab′)2 or Fab molecules can become a valuable tool for the de novo sequencing of serum antibodies.
Introduction
Antibodies are key molecules of our immune system and therefore of great interest to many researchers, both in academia and industry.1–4 The high selectivity of immunoglobulins G (IgGs) for a single antigen has made this class of antibodies of great value in basic research, but even more important in molecular medicine.5 Therapeutic antibodies reflecting those of an individual having survived a severe infection, e.g. by the SARS-CoV-2, Dengue6 or Ebola virus,7 can lead to a substantial reduction in the mortality rate of newly infected patients. Shortening the route from the discovery of an antibody to the production of a functional equivalent is therefore of utmost importance, certainly in present times.8Structurally, IgGs are made of four disulfide-bridged polypeptide chains: two identical light chains (LC) of 25 kDa and two identical heavy chains (HC) of approximately 50 kDa (Fig. 1A). The LC constant domain can be of two types, namely kappa (κ) or lambda (λ). The HC constant domain, on the other hand, is divided into four subclasses, namely IgG1, IgG2, IgG3, and IgG4, having between 83% and 96% sequence similarity. Only specific stretches of the HC regions differ between the subclasses: mainly the hinge region and the N-terminal side of the HC region below the hinge (Fc). Additionally, IgG subclasses exhibit distinctive disulfide bridging with a different number of disulfide bonds in the hinge region and different positioning of disulfides between LC and HC.9,10
 | ||
| Fig. 1 Schematic overview of IgGs structures and the here used sequencing workflow. (A) Distinctive structures of IgG1 and IgG2-4, all consisting of two variable regions (colored) with three antigen-binding Complementarity Determining Regions (CDRs) each, and one constant region (black). The first step in the workflow involves the separation by proteolytic cleavage of the IgG CDR-containing F(ab′)2 from the glycosylated constant Fc using the IdeS enzyme, which cleaves (scissors) just below the disulfides bridges (blue) between the heavy chains (HCs). Besides different numbers of HC-bridging disulfides, IgG 1–4 subclasses also differ by the disulfide bond between the light chain (LC) and the HC: directly above the hinge for IgG1, mediated by a more N-terminal HC cysteine for IgG 2–4. Additionally, the IgG3 hinge region is longer owing to an insertion that is repeated three times, as indicated with a black line above the sequence. (B) The purified F(ab′)2 portions of all IgG subclasses are subjected to ECD-MS, which preferentially fragments the LC and HC between their disulfide-bridged loops. Clearly separated low m/z c-ions and complementary high m/z bridged z-ions are formed, enabling the straightforward interpretation of fragment ion ladders beneficial for de novo IgG sequencing. | ||
Antigen recognition by IgG is mainly determined by three hypervariable regions located in the N-terminal domains of both the LC and HC. Of these so-called complementarity determining regions (CDR1, CDR2 and CDR3), the two CDR3s are often assumed to be major determinants of antibody-binding specificity.11,12 Genetic recombination and hypermutation in these CDR regions lead to a plethora of possible human IgG antibodies, whereby it has been postulated that humans can generate about 10 billion unique antibodies, each potentially capable of binding an antigenic determinant (epitope) with high selectivity.13
This amazingly large repertoire is possible because the CDR sequences are only partly genome-derived. After recombination of multiple genome segments into one antibody sequence, so-called V(D)J-recombination, random mutations are introduced in the process of somatic hypermutation. While initially used to increase the spectrum of recognized antigens, these processes subsequently lead to affinity and selectivity enhancements.
A common approach to elucidate the antibody repertoire in a human is to sequence the immunoglobulin mRNAs from memory B cells present in the donor's circulation.14 The majority of antibodies present in the circulation is however not produced by these circulating B cells, but by long-lived plasma cells that reside in the bone marrow, which are much harder to harvest. The most accessible sequence-containing unit of relevant circulating antibodies is, therefore, the protein itself.15
Sequencing proteins is nowadays generally performed by mass spectrometry. Characterization of peptides of 5 to 20 amino acids produced by proteolytic cleavage results into correct annotations of the full polypeptide backbone, even for de novo approaches when no DNA or protein sequence is available.16–20 However, when it comes to antibodies from mixtures such as present in serum, sample complexity rapidly increases, preventing high-confidence assignments.15 One way to overcome this hurdle is to apply top-down proteomics since the hypervariable region of interest – the paired N-terminal domains of the LC and HC – is then left intact and can be isolated.21 The relevant information density increases even further when the constant regions, or at least most of the constant parts containing PTMs such as lysine clipping or glycosylation, are removed.
Here, we hypothesized that the ideal approach for de novo sequencing of antibodies from complex mixtures is a top-down or middle-down mass spectrometry-based method that results in long stretches of straightforward-to-read sequence ladders covering as much of the variable domains as possible, but at least the CDR3. This would resemble a modern, mass spectrometry-based, version of Edman degradation capable of dealing with impurities or complex mixtures and allowing for longer sequence reads. The work presented here is aimed at such a methodology.
Straightforward-to-read sequence ladders require highly optimized mass spectrometric methods preferentially yielding only one type of fragment pairs (a/x, b/y, or c/z) to avoid spectral congestion and dilution of signal intensity. To this end, electron capture dissociation (ECD),22 which recently became available for benchtop mass spectrometers, has been shown to offer significant advantages in obtaining straightforward-to-read sequence ladders.23,24 Since, ECD has been used to query antibody sequences. It resulted in significant backbone cleavages in both the LC and HC variable regions of both denatured and native intact antibodies. It also yielded mainly (c/z·) fragment ions.25,26 While the presence of intrachain disulfide bridges complicates the detection of several variable domain regions, it enhances the detection of CDR3 fragments.25,27
In the present work, we build upon these exciting results and further optimize ECD for the de novo sequencing of IgG CDR3s and their downstream regions, generating very clean, straightforward-to-read sequence-ladders comprised solely of c-ions (Fig. 1B). Using native conditions in electrospray to ionize the IgG antibodies, we achieve maximal separation between the precursor and the informative fragment ions. We show that removal of the Fc region also reduces the complexity of the spectrum and simplifies the precursor ion selection. We optimize and apply the method using the IgG1 Trastuzumab. Next, in order to investigate in detail whether the method is generically applicable, we apply it to two quartets of IgG covering all human IgG subclasses (i.e. IgG1, IgG2, IgG3 and IgG4). We show that the straightforward sequence reads are obtained for all IgG subclasses. Through structural modeling, we aim to explain and predict the observed fragmentation patterns.
Experimental
Antibody samples
Monoclonal antibodies against CD20 (7D8)28 and DNP (DNP-G2a2)29,30 were recombinantly expressed as IgG1, IgG2, IgG3 and IgG4, and provided by Genmab (Utrecht, the Netherlands). The IgG1 antibody Trastuzumab was provided by Roche (Penzberg, Germany).Preparation of intact IgG, F(ab′)2 and Fab samples for native top-down MS analysis
F(ab′)2 molecules were generated by overnight digestion at 37 °C of the IgGs bound to a CaptureSelect FcXL affinity matrix (Thermo Fisher Scientific) using 0.25 U μg−1 FabRICATOR® IdeS (Genovis AB, Llund, Sweden) as described earlier.31 IgG1 Fab molecules were similarly prepared using 1 U μg−1 FabALACTICA® IgdE (Genovis AB, Llund, Sweden). Prior to native top-down analysis, buffers were exchanged to aqueous 150 mM ammonium acetate (pH 7.5) through six consecutive dilution and concentration steps at 4 °C using Amicon® Ultra centrifugal filters with a 10 kDa molecular weight cutoff (Merck KGaA, Darmstadt, Germany). IgG concentrations used for native electrospray ionization mass spectrometry were around 4 μM.Native top-down ECD MS
Top-down MS of native IgG (sub)complexes was performed on an ultra-high mass range (UHMR) Q-Exactive Orbitrap (Thermo Fisher Scientific, Bremen, Germany) using an ECD cell developed by e-MSion.32 First, IgG, F(ab′)2 or Fab precursor ions were mass-selected by the quadrupole. The in-source trapping desolvation voltage displays an optimum in terms of ECD at −100 V, likely a consequence of small structural changes and cleavage enhancement occurring as the effective temperature of the ion is raised.33 The ions were then transmitted into the ECD cell where they were subjected to electron capture dissociation. The small permanent magnets ensure the radial confinement of electrons emitted by a cathode while additional electrodes ensure longitudinal confinement and efficient ion transfer to the HCD cell. Second, upon transfer from the ECD cell to the HCD cell, post-ECD collisional activation was kept to a minimum (HCD direct eV setting = 1) to avoid (b, y) ion formation and concentrate the fragment signal intensity into the c- and z-ions of interest. Overall, the HCD cell trapping and extraction parameters were optimized for low nitrogen collision gas pressures to achieve efficient detection of the subunits and fragments. All spectra were acquired with the noise threshold parameter set to 3.64. A single charge state of the precursor (21+ for F(ab′)2, 13+ for F(ab), and 24+ for intact IgG) was isolated to simplify interpretation of the fragment ion spectra. All the spectra were acquired at a set resolution of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 @ m/z 400.
000 @ m/z 400.
Data analysis
Processing of the fragmentation spectra involved the conversion of raw files to mzML format by Proteowizard.34 We used the MSDeisotope python library (Joshua Klein, Boston University CBMS)35,36 with a minimum_score = 10.0 and mass_error_tolerance = 0.02 to generate a charge deconvoluted spectrum with all the isotopic peaks retained.35,36 Fragments were assigned by applying LcMsSpectator (Pacific Northwest National Laboratory)37,38 to the charge deconvoluted spectra generated by MSDeisotope. The accuracy threshold was set to 3 ppm for all assignments following recalibration of the fragment's m/z via shifting by the average error on assignable c-ion fragments. The results were exported as tsv files for further analysis. Sequence assignment accommodated the major ECD ion types (c, z, z·) without considering H2O and NH3 neutral losses, except when explicitly mentioned.Modelling
The structures of the F(ab′)2 fragments studied are not available in structural databases. Consequently, homology models of the F(ab′)2 were built using MODELLER39,40 similarly to the protocol introduced by Politis and co-workers.41 For that, we used crystallographic structures with PDB IDs IgG1 1HZH (human),42 IgG2 1IGT (mouse),43 IgG3 5A16 (mouse)44 and IgG4 5DK3 (human)45 as a template. Refinement of the obtained models was also performed in MODELLER using a combination of variable target function method and molecular dynamics with simulated annealing; optimization was repeated for three cycles. All models were assessed using the (negative) Discrete Optimized Protein Energy score.39,40 Since IgG3 lacks an available F(ab′)2 structure, the hinge region was built in several steps to avoid knots introduced by MODELLER when more than approximately 15 residues are missing from the template structure. All disulfide bridges were checked and manually defined when they were not modelled automatically.To explore the conformational space of the Fab arm movement, each F(ab′)2 model was subjected to a rapidly exploring random tree based sampling, which is part of the Integrative Modelling Platform.46 For that, residues in the upper hinge region were set as flexible linker residues, namely: D226-T230 for anti-CD20 IgG1 and D225-T229 for anti-DNP IgG1, E221-K223 for anti-CD20 IgG2 and E220-K222 for anti-DNP IgG2, E221-T232 for anti-CD20 IgG3 and E220-T231 for anti-DNP IgG3, E221-P227 for anti-CD20 IgG4 and E220-P226 for anti-DNP IgG4. The rest of the molecule was connected through inter-chain disulfides and set as a rigid body. Next, the algorithm randomly tested the possibility of rotating an atom of the linker residues in agreement with the torsional space of the corresponding residue. The sampling was performed with the number of iterations and nodes set to 10000. Subsequently, conformations without steric clashes and overlaps were used as output. High-throughput collision-cross section (CCS) calculations were done using Impact software47 using the standard parameters. The obtained CCS values were used for selecting the appropriate set of F(ab′)2 conformations. Calculation of the center-of-mass (COM) of the light chain, heavy chain and the F(ab′)2 was performed in Pymol48 on a sample of 2500 random and 500 most compact conformations. For COM calculations of the IgG3 F(ab′)2, we used HC sequences down to the first two hinge disulfides to avoid the COM offset introduced by the larger hinge region of IgG3, when compared to other IgG subclasses. Next, we calculated the average distances between the COM of the LC or HC and the COM of the F(ab′)2 for a subset of conformations to assess the degree to which each chain is either inward- or outward-facing. Finally, we used this metric to estimate which chain is likely to be more exposed to the environment when the collapsed gas-phase structures are formed.
Results and discussion
Benchmarking ECD-only on intact IgG1, F(ab′)2, and Fab molecules of Trastuzumab
At the start of our experiments, we tested the performance of ECD on different preparations of Trastuzumab. A cross-comparison between ECD fragment ion spectra obtained for the 148 kDa intact IgG1, the 98 kDa F(ab′)2 and the 48 kDa Fab molecules revealed a striking similarity (Fig. S1 and Table S1†). In line with previously reported data,27 we observed that most of the abundant fragment ions have masses between 9000 and 16000 Da, regardless of the Trastuzumab moiety that was used.
After careful consideration and examination of the ECD fragmentation spectra, we chose to focus on F(ab′)2 molecules for the rest of our study for the reasons described below. The ECD fragment ion spectra of intact Trastuzumab were notably more complex to analyze due to spectral congestion by z-ions from the highly conserved Fc region, which overlap with the c-ions from the region of interest. Additionally, the glycosylation on the Fc dilutes the precursor ion signal over a broader range of m/z values, hampering the use of narrow m/z windows for precursor isolation. Therefore, we chose to disfavor the analysis of intact mAbs.
Although both Fab and F(ab′)2 preparations yield straightforward sequence ladders corresponding to c-ions from the LC and HC regions of interest, the F(ab′)2 molecules provide some key advantages. While the IgdE enzyme can reliably produce Fab molecules of IgG1,49 other IgG subclasses require the use of less specific enzymes such as papain, which result in higher sample heterogeneity. The IdeS enzyme on the other hand, which generates F(ab′)2 molecules by cleaving at a specific position below the hinge of all IgG subclasses,50 provides both a reproducible and universal method irrespective of the subclass investigated. The resulting low-heterogeneity samples derived for the F(ab′)2 preparations enable strict precursor isolation without impacting sensitivity. Additionally, for denatured samples as well as native Fabs, the relatively low m/z of the precursor ions place them in the same m/z window where the ECD fragments are observed. The use of native F(ab′)2 molecules concentrates the precursor ion intensity on a small number of charge states outside the fragment ion regions, thereby simplifying data analysis. For these reasons, we chose the native F(ab′)2 molecules as precursors in the remainder of our analysis, although our data confirm that ECD also works reasonably well for both intact mAbs as well as Fab fragments.
Supplemental activation reduces the selectivity of ECD
Combining ECD with supplemental collisional activation can enhance precursor fragmentation. Although this has been demonstrated by Shaw et al.27 to extend sequence coverage, collisional activation also generates additional b/y-ion series. We argue that the generation of these extra fragment ion series could be disadvantageous for straightforward and confident de novo sequencing of IgGs.We investigated the effects of supplemental activation by recording EChcD-MS spectra of F(ab′)2 molecules using different levels of collisional activation. The higher collision voltages used in EChcD indeed introduced other ion types, which increase spectral complexity and make it more difficult to extract unambiguous sequence reads (Fig. S2 and Table S2†). Moreover, the slightly improved sequence coverage obtained by EChcD primarily provided information on the highly conserved parts of the antibody sequence, notably the FR3 and constant regions.
Since de novo sequencing benefits from non-ambiguous sequence reads, we opted for the use of “pure” ECD (i.e. without HCD) to reduce the fragmentation pattern to the single c-ion type covering the regions of interest. Notably, after careful analysis of all fragment ions with masses in between 9000 and 16000 Da, we could conclude that almost all fragment ion intensity originated exclusively from c-ions, with the low-abundant remainder originating from a-ions. Thus, conditions can be employed wherein ECD of F(ab′)2 molecules purely generates c-ions in this mass window.
Pure ECD produces clean CDR3-spanning c-ion ladders
Following our encouraging benchmark data obtained for the F(ab′)2 molecules of Trastuzumab, we sought to determine whether the observed fragmentation patterns were generic and thus useful for a range of different IgG mAbs. To this end, we characterized two quartets of IgG mAbs, covering the subclasses IgG1, IgG2, IgG3 and IgG4. One of these mAbs targets CD20, the other 2,4-dinitrophenol (DNP). Sequences, as well as theoretical and observed masses, are provided at the end of the ESI in Tables S5 and S6†.Application of pure ECD to F(ab′)2 molecules of all subclasses of the two IgG families consistently yielded ECD fragment ion mass spectra containing pure and easily interpretable c-ion sequence ladders covering CDR3 and FR4 of both the LC and HC (Fig. 2, S3, and Table S3†). We noticed that the fragment ions originate nearly exclusively from backbone cleavages in a part of the LC and HC sequence that is framed by intramolecular disulfide loops, and which span from the beginning of the CDR3 to the constant region. For both LC and HC, the observed backbone cleavages start immediately after the cysteine residue preceding the CDR3 (LC: Cys-88, HC: Cys-96). This resulted in near-complete sequence coverage of this very important region, with the exception that we observed no c-ion cleavages at the N-terminal-side of proline residues, in line with earlier reports.51–53 Notably, several of these gaps are covered by the corresponding a-ions, although at relatively lower intensities than the c-ion series.
 | ||
| Fig. 2 (A) Charge-deconvoluted ECD-MS spectra of IgG1, IgG2, IgG3 and IgG4 F(ab′)2 molecules of anti-DNP showing exclusively c-ion ladders covering the LC (purple) and HC (red) CDR3 regions. Cysteine residues involved in disulfide bonds are highlighted on the color-coded sequences by a solid line. Residue X, in …PLAPXS… in the heavy chain, represents a Ser for IgG1, and a Cys for IgG2-4. (B) Bar-chart displaying normalized c- and a-ion fragment intensities for each residue of IgG1, IgG2, IgG3 and IgG4 LC and HC. Although all ECD spectra for a given mAb are very similar, IgG1 HC c-ion fragments further extend towards the C-terminus for both investigated mAb families (Fig. S3†). For some cleavage sites, a-ions complement missing c-ions. | ||
Ion ladders covering the CDR3 of the LC do not overlap with HC fragment ions in the charge-deconvoluted spectra, making them suitable for direct interpretation. Ion ladders covering the CDR3 of the HC, on the other hand, overlap with fragment ions covering FR4 and the constant region of the LC. Because these regions are highly conserved, a comparison with theoretical spectra from these regions, accounting for the unknown N-terminal part by a variable mass shift, could help disentangle LC and HC fragment ions, making these mass spectra very suitable for de novo sequence assignment.
Exploring fragment complementarity in pure ECD of IgG F(ab′)2 molecules
By using low pressure in the HCD cell and avoiding collisional activation, the ECD fragments of the two IgG quartets nearly exclusively correspond to c-ions (Table S4†), similarly to our data for Trastuzumab, without the need to invoke small neutral losses such as ammonia or water, or sidechain losses. Apart from c-ions, a minor contribution is being made by a-ions, often when there is no c-ion fragment.Although C-terminal z-ions are not apparent within the shown deconvoluted analysis window (9000 < Mw < 16000 Da) of Fig. 2, they were detected completely separated in m/z from the concomitant c-ions (Fig. 3A). This is because the formed z-ions remain in complex with the remaining F(ab′)2 precursor, as they are connected through disulfide bridges at or near their C-termini. Charge and mass determination support this conclusion. While the 9–16 kDa c-ion fragments carry approximately 3–8 charges, their concomitant 83–90 kDa bridged z-ion fragments carry approximately 8–17 charges (Fig. 3B). Overall, pure ECD of F(ab′)2 molecules results specifically into a small subset of low-mass c-ions and corresponding high-mass bridged z-ions.
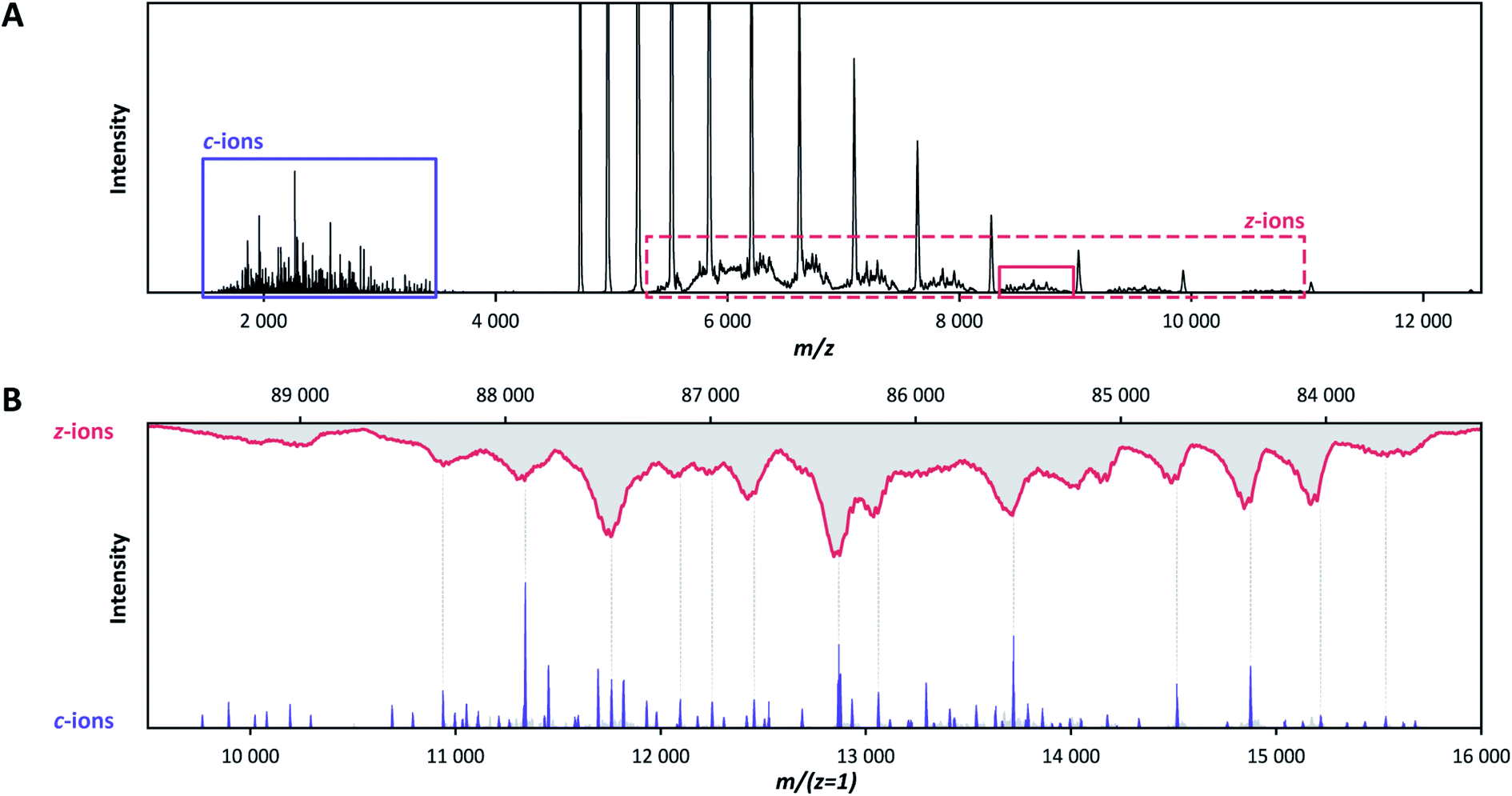 | ||
| Fig. 3 Concomitant N-terminal c-ions and C-terminal bridged z-ions produced in the ECD spectra of the anti-DNP IgG1 F(ab′)2 precursor, revealing that the disulfide bonds remain intact. (A) The c-ions (purple box) are well separated in the m/z window from their concomitant bridged z-ions (red dotted box) in the native ECD-MS spectrum of the IgG1 anti-DNP F(ab′)2 molecule. Because they are minimally affected by signal overlapping, bridged z-ions in the solid red box were selected for further analysis. (B) Alignment of the c-ions (purple) with the bridged z-ions (red) after charge deconvolution reveals that their mass adds up to that of the intact 99.2 kDa F(ab′)2 precursor. | ||
The disulfide links present in the IgG subclasses direct specific ECD fragmentation
The intact IgGs, but also the F(ab′)2 and Fab molecules, contain hundreds of amino acid residues, which could all theoretically be cleaved by ECD, potentially leading to hundreds of c- and z-ions. Still, we observe that, very selectively and reproducibly, only a few dozen specific fragment ions are preferentially formed. To understand this distinct fragmentation pattern, observed in all ECD spectra of F(ab′)2 molecules from the different IgG subclasses, we next looked into their (distinct) structural features. Since exact structural models of the studied F(ab′)2 molecules are not available, we performed homology modelling, whereby available IgG structures were used as templates for the structural reconstruction of the target F(ab′)2 molecules. The high quality of the resulting F(ab′)2 structural models, as displayed in Fig. 4 and S5,† can be assessed from their Discrete Optimized Protein Energy (DOPE) profiles (Fig. S4†). An overview of the tools used for the structural analysis of the here studied IgG F(ab′)2 molecules is provided in Fig. S6.† | ||
| Fig. 4 Influence of structural features on ECD fragmentation of F(ab′)2 from the four subclasses of IgGs (anti-CD20). (A) Structures of F(ab′)2 comparatively modelled against available structures of IgG subclasses, IgG1: 1HZH, IgG2: 1IGT, IgG3: 5A16, IgG4: 5DK3. The disulfide bridges are displayed as paired yellow spheres. (B and C) The number of disulfide bonds that span each sequence position of (B) the LC and (C) the HC in different subclasses of IgG anti-CD20 overlaid with detected ECD fragments. (D) Fractions of ECD fragment ion intensities originating from the LC or HC for the different subclasses of IgG anti-CD20. (E) Fractions of the 500 most compact conformations of each subclass in which either the LC or HC is most solvent-exposed following collapse.31 See Fig. S7† for a more detailed description of the estimated chain relative exposure based on center-of-mass calculations. (F) Sampling of Fab arm movements displayed as spheres representing the center-of-mass of (purple) LCs and (red) HCs, showing remarkable differences in between the different subclasses. | ||
The disulfide bridges are among the most distinctive structural features that differentiate one IgG subclass from another, and not surprisingly these linkages affect the observed ECD cleavages in intact IgG and F(ab′)2 molecules most notably.25 In Fig. 4A (anti-CD20) and Fig. S5A† (anti-DNP), the distinctive morphologies of the IgG subclasses are readily distinguishable. Differences are primarily in the distribution of the disulfide bridges and the length of the hinge regions, a notable distinctive feature in between the IgG subclasses.
To assess the impact of the disulfide linkages on the ECD, we first assessed the number of disulfide bond cleavages required to produce N-terminal fragments of either the LC or HC. Overlaid with the c-ion patterns produced by ECD of F(ab′)2 molecules, the resulting disulfide profiles nicely explain nearly all detected ECD fragments for both the LCs and HCs (Fig. 4B and C for anti-CD20 IgGs and Fig. S5B and C† for anti-DNP IgGs). In other words, only backbone cleavages in the regions that are not part of an intra- or interchain disulfide loop result in separation and detection of free N-terminal ions.
Since the LCs of all subclasses within a given IgG quartet are identical, it is not surprising that they yield identical ECD fragments with comparable abundances (Fig. 2 and S5B†). The ECD patterns of HCs differ between the IgG subclasses (Fig. 2 and S5C†), but this is readily explained by the location of the LC-HC interchain disulfide bridges. In the HC sequences of IgG2-4, the interchain link is mediated by Cys-136, which is in the middle part of the sequence. For the IgG1 HC on the other hand, the interchain link is mediated by the more C-terminal Cys-225. As a result, a larger part of the IgG1 HC sequence is free from disulfide looping, and therefore susceptible to ECD.
Probing distinctive structural dynamics of F(ab′)2 originating from different IgG subclasses
In addition to the striking disulfide-directed fragmentation, we noticed that the total ion intensity of HC fragments is higher for IgG1 compared to other subclasses, with IgG2 displaying the lowest fraction of HC fragment ion intensity (Fig. 4D and S5D†). We hypothesized that a possible explanation could involve differences in the conformations and structural dynamics of the IgG molecules.33,41,54,55In the absence of extensive collisional activation, low energy (<1 eV) electron capture dissociation is known to result in the preferential release of fragments from the surface-exposed regions of native proteins.17,45–47 The rationale is that exposed domains and chains have fewer non-covalent interactions upon transition to the gas phase, and therefore more readily release backbone fragments upon electron attachment. Thus, we set out to probe the structural features of F(ab′)2 molecules using modelling.
It has recently been reported that IgG molecules undergo a substantial structural collapse upon transfer to the gas phase during electrospray ionization.41,54 In this structural rearrangement, the high flexibility of the hinge region enables one or more Fab arms to clasp the IgG Fc region.41 When processed by the IdeS enzyme, the remaining F(ab′)2 portion of IgG retains its hinge region and, hence, F(ab′)2 should also be able to adopt a similar more compact structure upon transition to the gas phase. Although we are not able to directly predict the exact collapsed state of IgG F(ab′)2 molecules, we can assess their conformational space prior to the collapse for which we used rapidly exploring random tree based conformational sampling from the Integrative Modelling Platform.46 First, we sampled the motions of the Fab arms (n = 2500, Fig. 4F and S5F†). The differences in conformational dynamics between the different IgG subclasses can be assessed by displaying the center-of-mass (COM) of both chains of a Fab arm for a conformation sample.55 In order to infer the relative position of LC and HC in the putative F(ab′)2 gas-phase conformations, we calculated the average distances between the COM of each chain and the COM of the molecule (Fig. S7†). Second, we considered the 500 most compact F(ab′)2 conformations as determined from their collision cross-sections. We then assumed that the most exposed chains, as inferred from the COMs, are less prone to end up buried within the collapsed structures and will, therefore, be more likely to capture electrons and release backbone fragments. Finally, we compared all subclasses for anti-CD20 IgG (Fig. 4E and F) and anti-DNP IgG (Fig. S5E and F†).
Our modelling data suggest that the LCs and HCs are nearly equally solvent-exposed in the collapsed structures of IgG1 F(ab′)2, resulting in comparable abundances of the LC and HC ECD fragments. IgG4 F(ab′)2 exhibit slightly less exposed HCs which seemingly result in a higher fraction of LC fragments. Notably, the relative rigidity55 and predominant presence of LCs on the surface of collapsed IgG2 F(ab′)2 structures correlate pleasingly well with the prevalence of LC fragments over HC fragments, which explains the lower abundance of HC fragments compared to the other subclasses. Owing to its larger and more flexible hinge region, F(ab′)2 of IgG3 shows the widest range of Fab arm motions and the broadest distribution of collision cross-sections (Fig. S8†). It is likely that the high flexibility of IgG3 F(ab′)2 leads to dramatic structural rearrangements upon transfer to the gas phase, which can affect fragment separation upon ECD. Summarizing the modelling data, in IgG2, IgG3 and IgG4, the LC is more exposed than the HC while, in contrast, IgG1 displays nearly equal surface occupation for the LC and HC when sampling the 500 most compact F(ab′)2 conformations. Notably, our analysis supports that the predominant presence of the LC on the surface of the collapsed IgG2 F(ab′)2 structures and relative rigidity of the overall structure hamper the formation of abundant HC fragments compared to the other subclasses of both anti-CD20 and anti-DNP IgG.
Conclusions
Here, we explored the benefits of electron capture dissociation in attempting to sequence antibodies. Differentiating our approach from earlier attempts,21,27,56 we aimed deliberately for a very restricted, albeit targeted, sequence coverage, spanning just the important hypervariable CDR3 of both the light and heavy chain (LC and HC) of the mAbs. We optimized our method by first getting rid of the sequence-wise less informative constant regions (Fc) by using the IdeS protease, to obtain F(ab′)2 fragments. Next, we conducted ECD at minimal collisional activation and low pressure, allowing us to get exclusively straightforward-to-read amino acid sequence ladders of c-ions spanning the CDR3s. The ladders we obtained provide easily interpretable sequence reads covering the CDR3 of both chains, whereby we seemingly only observe a gap in the sequence when there is a proline present. We argue that this optimized method can be very powerful and beneficial for the de novo sequencing of human antibodies.Next, we sought to explain the observed highly specific fragmentation behavior by studying whether it was generic. Therefore, we investigated two quartets of mAbs (one directed against CD20 and one against DNP) being expressed in all their subclass variants; IgG1, IgG2, IgG3 and IgG4. On the one hand, the ECD data of these eight mAbs showed extremely conserved fragmentation of their CDR3s with the exclusive formation of c-ions in the low mass region (9000–16000 Da). On the other hand, specific features were observed in between the different subclasses, which could be largely explained by differences in the disulfide bridges within these molecules, and by their distinctive structural features in the context of the gas-phase transition.
Through this work, we improve our understanding of the observed very specific fragmentation in the CDR3 regions of all different subclasses of the IgGs. The models generated based on this data provide a first step in predicting the fragmentation patterns observed in ECD of IgGs. Such a spectrum prediction tool would greatly facilitate de novo sequencing of antibodies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the members of the Heck laboratory for general support, especially Arjan Barendregt. JFG thanks Joshua Klein (Boston University CBMS) for modifications of the MSDeisotope python library. This research received funding through the Netherlands Organization for Scientific Research (NWO) TTW project 15575 (Structural analysis and position-resolved imaging of macromolecular structures using novel mass spectrometry-based approaches), the ENPPS.LIFT.019.001 project (AJRH and JFG), the NACTAR project 16442 (AJRH and MAdB) and the Spinoza Award SPI.2017.028 to AJRH. This project received additional funding from the European Union's Horizon 2020 research and innovation program under the grant agreement 686547 (EPIC-XS) for AJRH. We kindly acknowledge the team of Janine Schuurman and Frank Beurskens of Genmab, Utrecht, NL for continuous support over the years and the gift of the two quartets of IgG subclass antibodies, and Dietmar Reusch and Markus Haberger from Roche, Penzberg, Germany for the gift of Trastuzumab.Notes and references
- I. Schwab and F. Nimmerjahn, Intravenous immunoglobulin therapy: how does IgG modulate the immune system?, Nat. Rev. Immunol., 2013, 13, 176–189, DOI:10.1038/nri3401.
- H. W. Schroeder and L. Cavacini, Structure and Function of Immunoglobulins, J. Allergy Clin. Immunol., 2010, 125, S41–S52, DOI:10.1016/j.jaci.2009.09.046.
- E. Wagner, O. Colas, S. Chenu, A. Goyon, A. Murisier, S. Cianferani, Y. François, S. Fekete, D. Guillarme, V. D'Atri and A. Beck, Determination of size variants by CE-SDS for approved therapeutic antibodies: Key implications of subclasses and light chain specificities, J. Pharm. Biomed. Anal., 2020, 184, 113166, DOI:10.1016/j.jpba.2020.113166.
- J. Giorgetti, A. Beck, E. Leize-Wagner and Y.-N. François, Combination of intact, middle-up and bottom-up levels to characterize 7 therapeutic monoclonal antibodies by capillary electrophoresis – Mass spectrometry, J. Pharm. Biomed. Anal., 2020, 182, 113107, DOI:10.1016/j.jpba.2020.113107.
- O. Leavy, Therapeutic antibodies: past, present and future, Nat. Rev. Immunol., 2010, 10, 297, DOI:10.1038/nri2763.
- W. Dejnirattisai, W. Wongwiwat, S. Supasa, X. Zhang, X. Dai, A. Rouvinski, A. Jumnainsong, C. Edwards, N. T. H. Quyen, T. Duangchinda, J. M. Grimes, W.-Y. Tsai, C.-Y. Lai, W.-K. Wang, P. Malasit, J. Farrar, C. P. Simmons, Z. H. Zhou, F. A. Rey, J. Mongkolsapaya and G. R. Screaton, A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus, Nat. Immunol., 2015, 16, 170–177, DOI:10.1038/ni.3058.
- S. Mulangu, L. E. Dodd, R. T. Davey, O. Tshiani Mbaya, M. Proschan, D. Mukadi, M. Lusakibanza Manzo, D. Nzolo, A. Tshomba Oloma, A. Ibanda, R. Ali, S. Coulibaly, A. C. Levine, R. Grais, J. Diaz, H. C. Lane, J.-J. Muyembe-Tamfum and The PALM Writing Group, A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics, N. Engl. J. Med., 2019, 381, 2293–2303, DOI:10.1056/NEJMoa1910993.
- A. Iwasaki and Y. Yang, The potential danger of suboptimal antibody responses in COVID-19, Nat. Rev. Immunol., 2020, 1–3, DOI:10.1038/s41577-020-0321-6.
- G. Vidarsson, G. Dekkers and T. Rispens, IgG Subclasses and Allotypes: From Structure to Effector Functions, Front. Immunol., 2014, 5, 1–17, DOI:10.3389/fimmu.2014.00520.
- H. Liu and K. May, Disulfide bond structures of IgG molecules, mAbs, 2012, 4, 17–23, DOI:10.4161/mabs.4.1.18347.
- Y. Barrios, P. Jirholt and M. Ohlin, Length of the antibody heavy chain complementarity determining region 3 as a specificity-determining factor, J. Mol. Recognit., 2004, 17, 332–338, DOI:10.1002/jmr.679.
- D. Kuroda, H. Shirai, M. Kobori and H. Nakamura, Structural classification of CDR-H3 revisited: A lesson in antibody modeling, Proteins, 2008, 73, 608–620, DOI:10.1002/prot.22087.
- L. J. Fanning, A. M. Connor and G. E. Wu, Development of the Immunoglobulin Repertoire, Clin. Immunol. Immunopathol., 1996, 79, 1–14, DOI:10.1006/clin.1996.0044.
- G. Georgiou, G. C. Ippolito, J. Beausang, C. E. Busse, H. Wardemann and S. R. Quake, The promise and challenge of high-throughput sequencing of the antibody repertoire, Nat. Biotechnol., 2014, 32, 158–168, DOI:10.1038/nbt.2782.
- J. J. Lavinder, A. P. Horton, G. Georgiou and G. C. Ippolito, Next-generation sequencing and protein mass spectrometry for the comprehensive analysis of human cellular and serum antibody repertoires, Curr. Opin. Chem. Biol., 2015, 24, 112–120, DOI:10.1016/j.cbpa.2014.11.007.
- A. F. M. Altelaar, J. Munoz and A. J. R. Heck, Next-generation proteomics: towards an integrative view of proteome dynamics, Nat. Rev. Genet., 2013, 14, 35–48, DOI:10.1038/nrg3356.
- N. Bandeira, V. Pham, P. Pevzner, D. Arnott and J. R. Lill, Automated de novo protein sequencing of monoclonal antibodies, Nat. Biotechnol., 2008, 26, 1336–1338, DOI:10.1038/nbt1208-1336.
- A. Guthals, Y. Gan, L. Murray, Y. Chen, J. Stinson, G. Nakamura, J. R. Lill, W. Sandoval and N. Bandeira, De Novo MS/MS Sequencing of Native Human Antibodies, J. Proteome Res., 2017, 16, 45–54, DOI:10.1021/acs.jproteome.6b00608.
- N. H. Tran, M. Z. Rahman, L. He, L. Xin, B. Shan and M. Li, Complete De Novo Assembly of Monoclonal Antibody Sequences, Sci. Rep., 2016, 6, 31730, DOI:10.1038/srep31730.
- K. I. Sen, W. H. Tang, S. Nayak, Y. J. Kil, M. Bern, B. Ozoglu, B. Ueberheide, D. Davis and C. Becker, Automated Antibody De Novo Sequencing and Its Utility in Biopharmaceutical Discovery, J. Am. Soc. Mass Spectrom., 2017, 28, 803–810, DOI:10.1007/s13361-016-1580-0.
- L. Fornelli, K. Srzentić, R. Huguet, C. Mullen, S. Sharma, V. Zabrouskov, R. T. Fellers, K. R. Durbin, P. D. Compton and N. L. Kelleher, Accurate Sequence Analysis of a Monoclonal Antibody by Top-Down and Middle-Down Orbitrap Mass Spectrometry Applying Multiple Ion Activation Techniques, Anal. Chem., 2018, 90, 8421–8429, DOI:10.1021/acs.analchem.8b00984.
- R. A. Zubarev, Electron-capture dissociation tandem mass spectrometry, Curr. Opin. Biotechnol., 2004, 15, 12–16, DOI:10.1016/j.copbio.2003.12.002.
- N. Taouatas, M. M. Drugan, A. J. R. Heck and S. Mohammed, Straightforward ladder sequencing of peptides using a Lys-N metalloendopeptidase, Nat. Methods, 2008, 5, 405–407, DOI:10.1038/nmeth.1204.
- F. Lermyte, D. Valkenborg, J. A. Loo and F. Sobott, Radical solutions: Principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure, Mass Spectrom. Rev., 2018, 37, 750–771, DOI:10.1002/mas.21560.
- Y. Mao, S. G. Valeja, J. C. Rouse, C. L. Hendrickson and A. G. Marshall, Top-Down Structural Analysis of an Intact Monoclonal Antibody by Electron Capture Dissociation-Fourier Transform Ion Cyclotron Resonance-Mass Spectrometry, Anal. Chem., 2013, 85, 4239–4246, DOI:10.1021/ac303525n.
- J. B. Shaw, N. Malhan, Y. V. Vasil'ev, N. I. Lopez, A. Makarov, J. S. Beckman and V. G. Voinov, Sequencing Grade Tandem Mass Spectrometry for Top–Down Proteomics Using Hybrid Electron Capture Dissociation Methods in a Benchtop Orbitrap Mass Spectrometer, Anal. Chem., 2018, 90, 10819–10827, DOI:10.1021/acs.analchem.8b01901.
- J. B. Shaw, W. Liu, Y. V. Vasil'ev, C. C. Bracken, N. Malhan, A. Guthals, J. S. Beckman and V. G. Voinov, Direct Determination of Antibody Chain Pairing by Top-down and Middle-down Mass Spectrometry Using Electron Capture Dissociation and Ultraviolet Photodissociation, Anal. Chem., 2020, 92, 766–773, DOI:10.1021/acs.analchem.9b03129.
- J. L. Teeling, Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas, Blood, 2004, 104, 1793–1800, DOI:10.1182/blood-2004-01-0039.
- K. D. White, M. B. Frank, S. Foundling and F. J. Waxman, Effect of immunoglobulin variable region structure on C3b and C4b deposition, Mol. Immunol., 1996, 33, 759–768, DOI:10.1016/0161-5890(96)00031-4.
- D. Ugurlar, S. C. Howes, B.-J. de Kreuk, R. I. Koning, R. N. de Jong, F. J. Beurskens, J. Schuurman, A. J. Koster, T. H. Sharp, P. W. H. I. Parren and P. Gros, Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement, Science, 2018, 359, 794–797, DOI:10.1126/science.aao4988.
- A. Bondt, Y. Rombouts, M. H. J. Selman, P. J. Hensbergen, K. R. Reiding, J. M. W. Hazes, R. J. E. M. Dolhain and M. Wuhrer, Immunoglobulin G (IgG) Fab Glycosylation Analysis Using a New Mass Spectrometric High-throughput Profiling Method Reveals Pregnancy-associated Changes, Mol. Cell. Proteomics, 2014, 13, 3029–3039, DOI:10.1074/mcp.M114.039537.
- K. L. Fort, C. N. Cramer, V. G. Voinov, Y. V. Vasil'ev, N. I. Lopez, J. S. Beckman and A. J. R. Heck, Exploring ECD on a Benchtop Q Exactive Orbitrap Mass Spectrometer, J. Proteome Res., 2018, 17, 926–933, DOI:10.1021/acs.jproteome.7b00622.
- K. J. Pacholarz, S. J. Peters, R. A. Garlish, A. J. Henry, R. J. Taylor, D. P. Humphreys and P. E. Barran, Molecular Insights into the Thermal Stability of mAbs with Variable-Temperature Ion-Mobility Mass Spectrometry, ChemBioChem, 2016, 17, 46–51, DOI:10.1002/cbic.201500574.
- M. C. Chambers, B. Maclean, R. Burke, D. Amodei, D. L. Ruderman, S. Neumann, L. Gatto, B. Fischer, B. Pratt, J. Egertson, K. Hoff, D. Kessner, N. Tasman, N. Shulman, B. Frewen, T. A. Baker, M.-Y. Brusniak, C. Paulse, D. Creasy, L. Flashner, K. Kani, C. Moulding, S. L. Seymour, L. M. Nuwaysir, B. Lefebvre, F. Kuhlmann, J. Roark, P. Rainer, S. Detlev, T. Hemenway, A. Huhmer, J. Langridge, B. Connolly, T. Chadick, K. Holly, J. Eckels, E. W. Deutsch, R. L. Moritz, J. E. Katz, D. B. Agus, M. MacCoss, D. L. Tabb and P. Mallick, A cross-platform toolkit for mass spectrometry and proteomics, Nat. Biotechnol., 2012, 30, 918–920, DOI:10.1038/nbt.2377.
- J. Klein, L. Carvalho and J. Zaia, Application of network smoothing to glycan LC-MS profiling, Bioinformatics, 2018, 34, 3511–3518, DOI:10.1093/bioinformatics/bty397.
- J. Klein, heckendorfc, S. Lukauskas, mobiusklein/ms_deisotope: Release v0.0.10, Zenodo, 2019, https://zenodo.org/record/3475687#.XnNv8HLTVPY Search PubMed.
- PNNL-Comp-Mass-Spec/LCMS-Spectator, Computational Mass Spectrometry @ Pacific Northwest National Laboratory, 2020, https://github.com/PNNL-Comp-Mass-Spec/LCMS-Spectator Search PubMed.
- J. Park, P. D. Piehowski, C. Wilkins, M. Zhou, J. Mendoza, G. M. Fujimoto, B. C. Gibbons, J. B. Shaw, Y. Shen, A. K. Shukla, R. J. Moore, T. Liu, V. A. Petyuk, N. Tolić, L. Paša-Tolić, R. D. Smith, S. H. Payne and S. Kim, Informed-Proteomics: open-source software package for top-down proteomics, Nat. Methods, 2017, 14, 909–914, DOI:10.1038/nmeth.4388.
- M. A. Martı-Renom, A. C. Stuart, A. Fiser, R. Sanchez and F. Melo, Comparative Protein Structure Modeling of Genes and Genomes, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325, DOI:10.1146/annurev.biophys.29.1.291.
- A. Fiser and A. Šali, in Methods in Enzymology, Elsevier, 2003, vol. 374, pp. 461–491, https://linkinghub.elsevier.com/retrieve/pii/S0076687903740208 Search PubMed.
- K. Hansen, A. M. Lau, K. Giles, J. M. McDonnell, W. B. Struwe, B. J. Sutton and A. Politis, A Mass-Spectrometry-Based Modelling Workflow for Accurate Prediction of IgG Antibody Conformations in the Gas Phase, Angew. Chem., Int. Ed., 2018, 57, 17194–17199, DOI:10.1002/anie.201812018.
- E. O. Saphire, P. W. H. I. Parren, R. Pantophlet, M. B. Zwick, G. M. Morris, P. M. Rudd, R. A. Dwek, R. L. Stanfield, D. R. Burton and I. A. Wilson, Crystal Structure of a Neutralizing Human IgG Against HIV-1: A Template for Vaccine Design, Science, 2001, 293, 1155–1159, DOI:10.1126/science.1061692.
- L. J. Harris, S. B. Larson, K. W. Hasel and A. McPherson, Refined Structure of an Intact IgG2a Monoclonal Antibody, Biochemistry, 1997, 36, 1581–1597 CrossRef CAS.
- T. Arakawa, T. Kobayashi-Yurugi, Y. Alguel, H. Iwanari, H. Hatae, M. Iwata, Y. Abe, T. Hino, C. Ikeda-Suno, H. Kuma, D. Kang, T. Murata, T. Hamakubo, A. D. Cameron, T. Kobayashi, N. Hamasaki and S. Iwata, Crystal structure of the anion exchanger domain of human erythrocyte band 3, Science, 2015, 350, 680–684, DOI:10.1126/science.aaa4335.
- G. Scapin, X. Yang, W. W. Prosise, M. McCoy, P. Reichert, J. M. Johnston, R. S. Kashi and C. Strickland, Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab, Nat. Struct. Mol. Biol., 2015, 22, 953–958, DOI:10.1038/nsmb.3129.
- D. Russel, K. Lasker, B. Webb, J. Velázquez-Muriel, E. Tjioe, D. Schneidman-Duhovny, B. Peterson and A. Sali, Putting the Pieces Together: Integrative Modeling Platform Software for Structure Determination of Macromolecular Assemblies, PLoS Biol., 2012, 10, e1001244, DOI:10.1371/journal.pbio.1001244.
- E. G. Marklund, M. T. Degiacomi, C. V. Robinson, A. J. Baldwin and J. L. P. Benesch, Collision cross sections for structural proteomics, Structure, 2015, 23, 791–799, DOI:10.1016/j.str.2015.02.010.
- W. L. DeLano, Pymol: An open-source molecular graphics tool, CCP4 Newsletter on protein crystallography, 2002, vol. 40, pp. 82–92 Search PubMed.
- C. Spoerry, J. Seele, P. Valentin-Weigand, C. G. Baums and U. von Pawel-Rammingen, Identification and Characterization of IgdE, a Novel IgG-degrading Protease of Streptococcus suis with Unique Specificity for Porcine IgG, J. Biol. Chem., 2016, 291, 7915–7925, DOI:10.1074/jbc.M115.711440.
- U. von Pawel-Rammingen, B. P. Johansson and L. Björck, IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G, EMBO J., 2002, 21, 1607–1615, DOI:10.1093/emboj/21.7.1607.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process, J. Am. Chem. Soc., 1998, 120, 3265–3266, DOI:10.1021/ja973478k.
- R. A. Zubarev, D. M. Horn, E. K. Fridriksson, N. L. Kelleher, N. A. Kruger, M. A. Lewis, B. K. Carpenter and F. W. McLafferty, Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations, Anal. Chem., 2000, 72, 563–573, DOI:10.1021/ac990811p.
- E. A. Syrstad and F. Turecček, Toward a general mechanism of electron capture dissociation, J. Am. Soc. Mass Spectrom., 2005, 16, 208–224, DOI:10.1021/jasms.8b02352.
- K. J. Pacholarz, M. Porrini, R. A. Garlish, R. J. Burnley, R. J. Taylor, A. J. Henry and P. E. Barran, Dynamics of Intact Immunoglobulin G Explored by Drift-Tube Ion-Mobility Mass Spectrometry and Molecular Modeling, Angew. Chem., Int. Ed., 2014, 53, 7765–7769, DOI:10.1002/anie.201402863.
- G. K. Hui, A. D. Gardener, H. Begum, C. Eldrid, K. Thalassinos, J. Gor and S. J. Perkins, The solution structure of the human IgG2 subclass is distinct from those for human IgG1 and IgG4 providing an explanation for their discrete functions, J. Biol. Chem., 2019, 294, 10789–10806, DOI:10.1074/jbc.RA118.007134.
- L. He, L. C. Anderson, D. R. Barnidge, D. L. Murray, C. L. Hendrickson and A. G. Marshall, Analysis of Monoclonal Antibodies in Human Serum as a Model for Clinical Monoclonal Gammopathy by Use of 21 Tesla FT-ICR Top-Down and Middle-Down MS/MS, J. Am. Soc. Mass Spectrom., 2017, 28, 827–838, DOI:10.1007/s13361-017-1602-6.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03438j |
| ‡ Equal contributing first authors. |
| This journal is © The Royal Society of Chemistry 2020 |