
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring modular reengineering strategies to redesign the teicoplanin non-ribosomal peptide synthetase†
Milda
Kaniusaite
 abc,
Robert J. A.
Goode
ad,
Julien
Tailhades
abc,
Ralf B.
Schittenhelm
ad and
Max J.
Cryle
*abc
abc,
Robert J. A.
Goode
ad,
Julien
Tailhades
abc,
Ralf B.
Schittenhelm
ad and
Max J.
Cryle
*abc
aThe Monash Biomedicine Discovery Institute, Department of Biochemistry and Molecular Biology, Monash University, Clayton, Victoria 3800, Australia. E-mail: max.cryle@monash.edu
bEMBL Australia, Monash University, Clayton, Victoria 3800, Australia
cThe Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, Monash University, Clayton, Victoria 3800, Australia
dMonash Proteomics and Metabolomics Facility, Monash University, Clayton, Victoria 3800, Australia
First published on 24th August 2020
Abstract
Non-ribosomal peptide synthesis is an important biosynthesis pathway in secondary metabolism. In this study we have investigated modularisation and redesign strategies for the glycopeptide antibiotic teicoplanin. Using the relocation or exchange of domains within the NRPS modules, we have identified how to initiate peptide biosynthesis and explored the requirements for the functional reengineering of both the condensation/adenylation domain and epimerisation/condensation domain interfaces. We have also demonstrated strategies that ensure communication between isolated NRPS modules, leading to new peptide assembly pathways. This provides important insights into NRPS reengineering of glycopeptide antibiotic biosynthesis and has broad implications for the redesign of other NRPS systems.
Introduction
Non-ribosomal peptide synthetases (NRPSs) are mega-enzyme assembly lines that are responsible for the biosynthesis of many clinically important compounds, including anticancer agents, immunosuppressants and antibiotics.1 Unlike ribosomal peptide synthesis, peptide synthesis mediated by NRPSs relies on the activity and interplay of multi-functional catalytic units known as modules, where each module consists of several domains that each have a specific function in peptide biosynthesis.1,2 In this process, adenylation (A)-domains are responsible for the selection and activation of specific monomers for subsequent incorporation into the growing peptide chain, and without the constraints of the ribosome these domains are able to utilise a greatly expanded array of substrates for peptide synthesis.3 Following substrate activation, monomers are covalently tethered to the phosphopantetheinyl cofactor linked to an adjacent peptidyl carrier protein (PCP) domain, which serves to retain the monomer as an activated, yet enzyme bound thioester.4 Finally, peptide bond formation between two PCP-bound substrates is formed by the condensation (C)-domain, which leads to the transfer of the upstream peptide fragment onto the downstream aminoacyl-PCP, thus extending the peptide by one residue.5 C-domains play central roles as gatekeepers within NRPS biosynthesis, where they act to not only control the stereochemistry of the peptide fragment together with the action of neighbouring epimerisation (E)-domains – additional tailoring domains that supplement the core C–A–PCP domains found within a minimal extension module – but can also allow modification of the PCP-bound amino acid either directly or via controlling substrate availability for trans-interacting domains such as hydroxylases or halogenases.6,7 Module expansion by the addition of tailoring domains can extend beyond E-domains,8,9 with terminal modules typically containing a thioesterase (TE)-domain that then releases the mature peptide product through hydrolysis, dimerisation or cyclisation by attack of an internal nucleophile.10 Further complex modifications are also common within NRPS-biosynthesis, with one important example being the recruitment of multiple external oxygenase enzymes during the cyclisation cascade found in glycopeptide antibiotic (GPA) biosynthesis (Fig. 1).11,12 This process, which is performed on the final module of the GPA-producing NRPS and that is mediated by a specialised recruitment (X)-domain,13,14 serves to highlight the impressive range of chemical transformations that are naturally found within NRPS biosynthesis. The complex structures of the peptide products of NRPS-biosynthesis often leads to the requirement to use in vivo biosynthesis for production of such compounds at commercial scale, which in turn can prevent the effective production of optimised derivatives of these natural agents.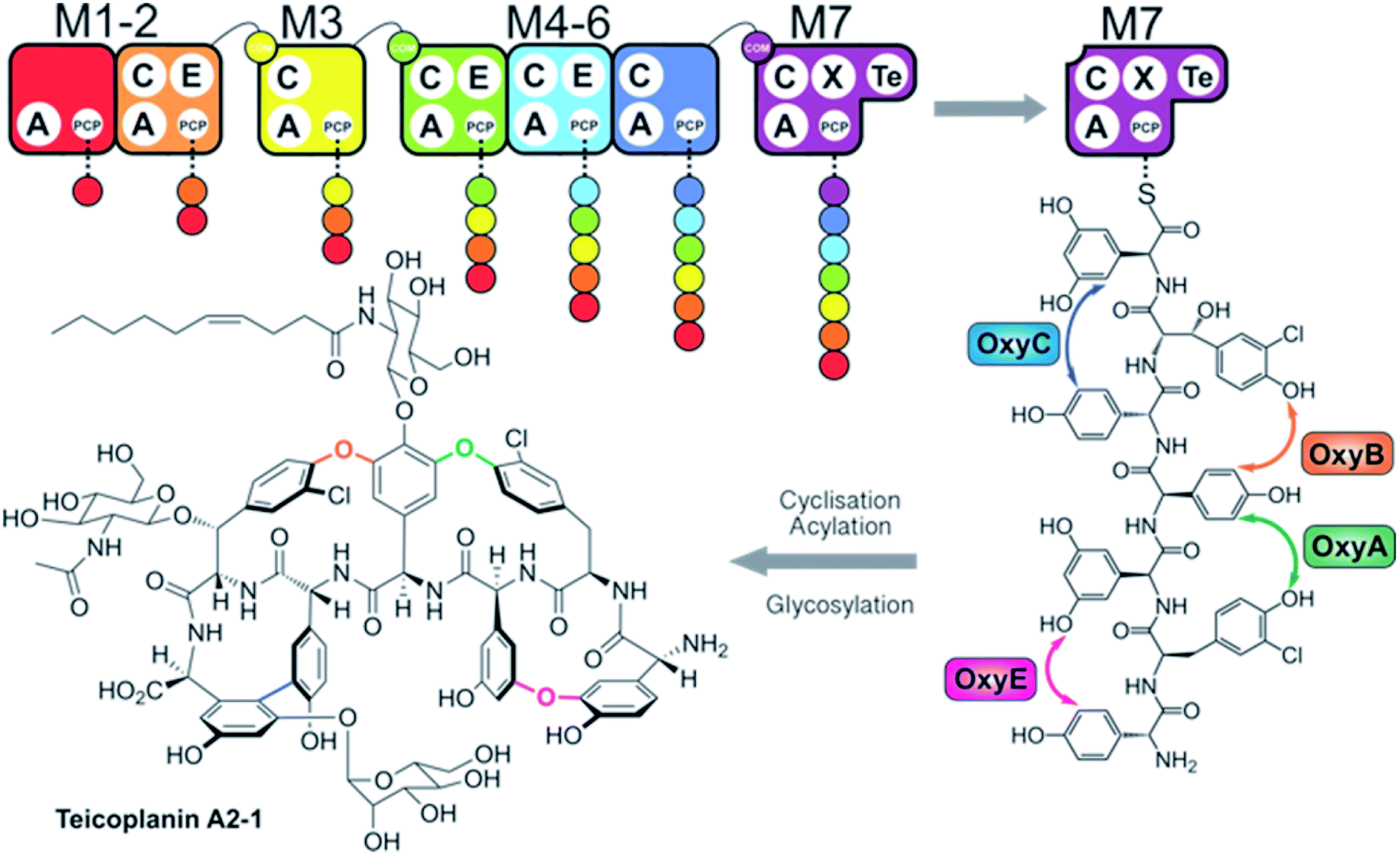 | ||
| Fig. 1 Overview of teicoplanin biosynthesis. The heptapeptide core of teicoplanin is assembled in a stepwise fashion by a seven module NRPS machinery (M1–M7, shown from red to violet) divided across four separate proteins. The linear heptapeptide is then cyclised on the final NRPS module by the actions of four cytochrome P450 (oxy) enzymes (B–E–A–C) before the cleavage of the tetracyclised aglycone by the M7 TE domain. The free aglycone is subsequently modified by several enzymes to generate the final structure of teicoplanin. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, X-P450-recruitment, TE – thioesterase, COM – communication. | ||
Given the importance of the products of natural megaenzyme synthases, an ability to alter such biosynthetic pathways to engineer the production of desired compounds would be of great value.15 In such endeavours, the modularity of NRPS machineries would appear to make reengineering such assembly lines highly feasible, given the shared enzymology and stepwise nature of peptide biosynthesis.1,2 However, the reality of reengineering these large and complex proteins has often shown that there are significant challenges yet to solve if we are to be able to perform such biosynthetic reengineering in a reliable and efficient way.15 Given the challenges of working with large proteins in vitro and the use of in vivo biosynthesis for eventual scale up and production of non-ribosomal peptides, it is unsurprising that the majority of efforts have been performed in vivo.16–23 Recent efforts in this regard have focused on C-domains as crucial junctions in modular redesign, either via module division within C-domains for linear systems from strains of Xenorhabdus and Photorhabdus19 or alternate module architecture for iterative fungal systems.24 Whilst in vivo approaches have made valuable contributions to NRPS redesign, few have been explored in vitro, which leads to difficulties in fully characterising these approaches especially in situations that are only partially successful or that show unexpected outcomes. Given this, our approach to NRPS redesign has focussed on the in vitro reconstitution of GPA biosynthesis from teicoplanin and related molecules (Fig. 1).6,7,14,25–28 Now, we explore the ability to generate alternate NRPS assembly lines from teicoplanin biosynthesis using a combination of approaches including module hybridisation, re-purposing extension modules as initiation modules and redesigning modules to control intermodule communication through the use of specific domain interaction interfaces. In this way, we present a set of instructions to tackle the reconstitution and reengineering of complex NRPS assembly lines in vitro.
Results
Repurposed elongation modules initiate peptide biosynthesis
To understand the potential for modular reorganisation of the teicoplanin NRPS, we explored the ability of elongation modules to initiate peptide biosynthesis using our established NRPS peptide reconstitution assay (Fig. 2 and 3). In such assays, the PCP-domains within the NRPS constructs under investigation were first converted into their holo-form using coenzyme A and the phosphopantetheinyl transferase Sfp.29,30 The holo-NRPS proteins were then incubated with their amino acid substrates and ATP, before reactions were terminated by addition of methylamine. This results in the offloading of any PCP-bound species as methylamide peptides, allowing PCP-bound species to be identified using MS analysis (labelled “a”) as opposed to those resulting from hydrolysis (labelled “b”). Methylamine cleavage can result in the epimerisation of phenylglycine residues at the peptide C-terminus, leading to double peaks in such cases (this is indicated in figure captions).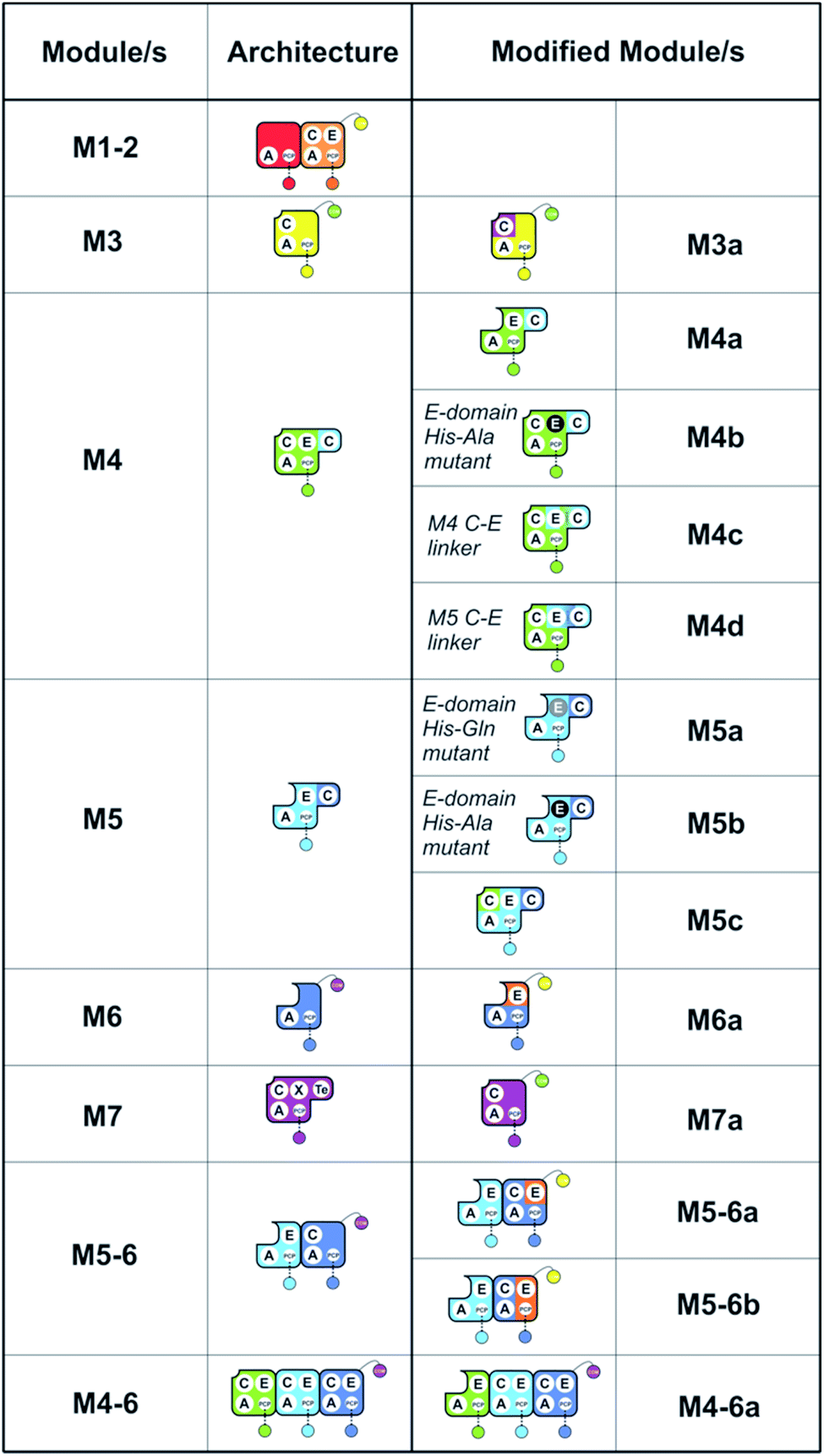 | ||
| Fig. 2 Summary of the modules and their domain composition in the teicoplanin NRPS together with the alternate constructs designed and tested in this study. | ||
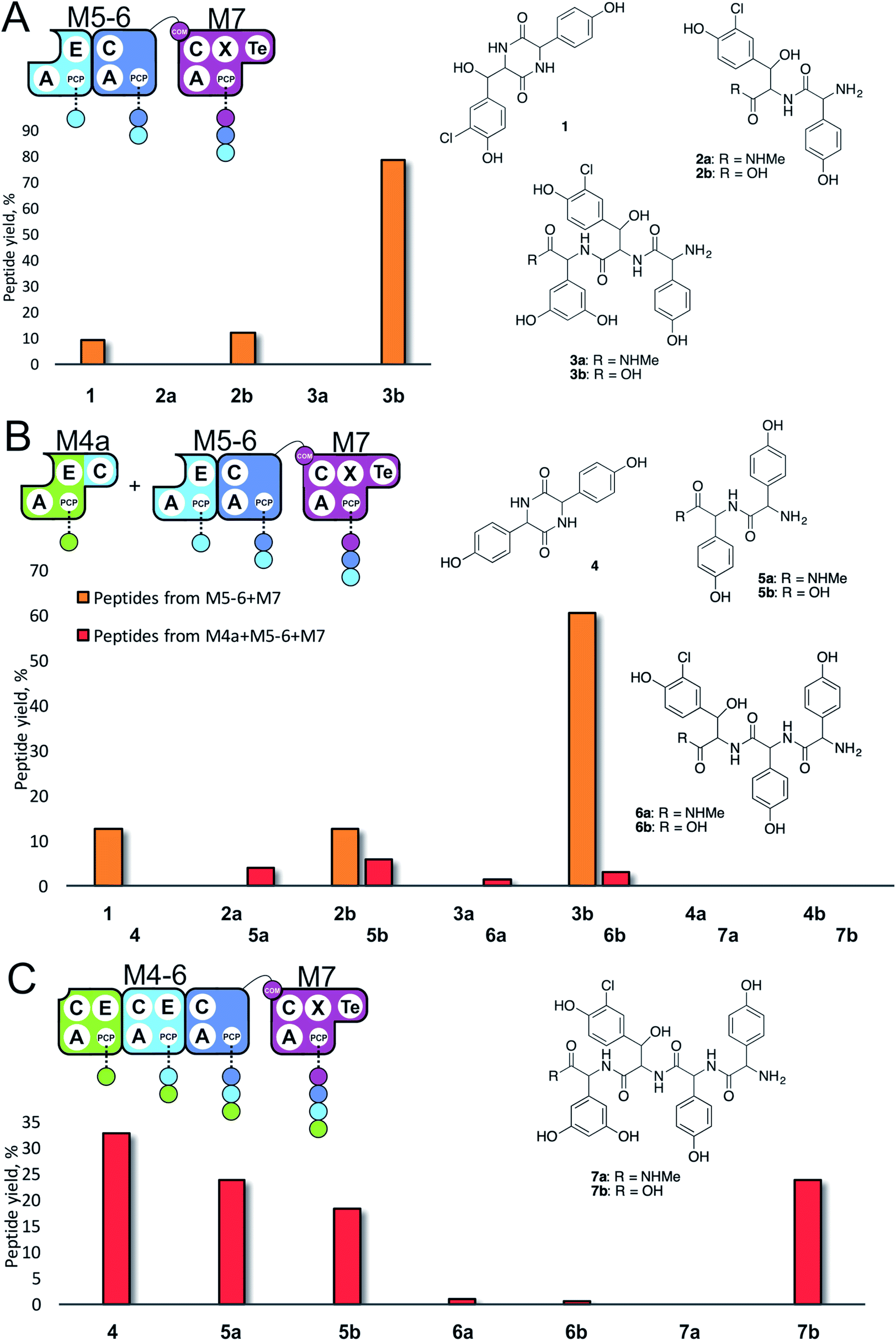 | ||
| Fig. 3 Initiation of peptide biosynthesis from elongation modules of the teicoplanin NRPS. Peptide biosynthesis assays included either 3 (M5–M7, A) or 4 (M4–M7, B and C) modules of the teicoplanin NRPS in various configurations together with ATP and the substrates of the A-domains of each module (M4, M5 – Hpg; M6 – ClBht; M7 – 3,5-Dpg). Peptides detected for M5–M7 are indicated in orange, for M4–M7 in red; yield is calculated for each species as a percentage of the total ion current determined by LCMS analysis. Peptide species indicated; for m/z data see ESI Table S3.† Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, X-P450-recruitment, TE – thioesterase, COM – communication. Module colour codes: M4 – green, M5 – pale blue, M6 – dark blue, M7 – violet. | ||
Given that the majority of the NRPS initiation modules begin with an A-domain (except modules that contain a C-domain to load acyl groups, for example) we first generated an M5-6 dimodule construct in which the initial C-domain had been deleted. When M5-6 was used in a peptide reconstitution assay together with M7, biosynthesis of tripeptide 3 (Hpg–ClBht–Dpg) was observed (Fig. 3A). Next, we attempted to extend 3 into tetrapeptide 7 (Hpg–Hpg–ClBht–Dpg) via the addition of module 4 (Fig. 3B and C). This was not achieved by adding module 4 constructs (M4, M4a) into the M5-6 + M7 assay (Fig. 3B), but was possible when replacing M4 + M5-6 with complete trimodule constructs (M4-6, M4-6a; Fig. 3C).
These experiments revealed three key findings: firstly, that initiation of NRPS biosynthesis is achievable using re-purposed extension modules. The second finding stems from the inability of a single module (M4) to compete for initiation with the M5-6 dimodule. This indicates that the affinity of the C/A domain interface between M4 and M5-6 is insufficient to allow peptide extension from a single amino acid, but that it is possible when these modules are fused in the M4-6 constructs. Thus, module fusion allows peptide synthesis to be initiated as they are restrained in the same construct, which dramatically increases the effective local concentration. The third finding stems from the fact that formation of 7 is obtained using both M4-6 and M4-6a, although M4-6a lacks the natural N-terminal C-domain. This indicates that the presence of such C-domains in an NRPS module is insufficient to prevent the initiation of peptide synthesis. Rather, it appears that interactions with neighbouring modules control this process. This provides clues that the interactions between modules split into separate proteins must be sufficiently high to prevent unwanted peptide chain initiation during normal peptide biosynthesis, which is clearly of interest for NRPS module redesign.
Engineering modular interactions across separated E- and C-domains
With extension modules able to initiate peptide synthesis, we now focused on M5-6 redesign to alter this into a dimodule resembling the natural M1-2 protein (Tcp9). To generate the same peptide stereochemistry with a reengineered M5-6 construct as occurs with M1-2, we needed to relocate the E-domain from M1-2 to the C-terminus of M5-6 (Fig. 2 and 4). Two different constructs were designed: M5-6a, adding the E-domain from M2 onto the C-terminus of the PCP-domain in M6, and M5-6b, exchanging the M6 PCP domain with the complete PCP–E didomain from M2. Peptide reconstitution assays were then performed using M5-6a together with M3, which led to production of tripeptide 3 albeit without epimerisation of the Tyr-residue (Fig. 4 and ESI Fig. S10†). Results obtained with M5-6b in these experiments matched those obtained for M5-6a (ESI Fig. S10†), whilst control experiments using M5-6 + M3 and M5-6a + M7 showed no formation of 3 in either case (data not shown). | ||
| Fig. 4 Engineering modular interactions across E- and C-domains for modules M6 and M3. Peptide biosynthesis assay using the engineered dimodule M5-6a + M3 together with ATP and the substrates of the A-domains of each module (M3 – 3,5-Dpg; M5 – Hpg; M6 – ClBht); HRMS analysis shows the formation of 3a; for m/z data see ESI Table S3.† Double peak caused by epimerisation of C-terminal Dpg residue during methylamine offloading. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, COM – communication. Module colour codes: M2 – orange, M3 – yellow, M5 – pale blue, M6 – dark blue. | ||
These experiments show that natural communication between M2 and M3 requires the M2 E-domain. The ability of M5-6a + M3 to generate tripeptide 3 demonstrates effective communication between reengineered M6 and M3, which are not adjacent within the natural NRPS assembly line (Fig. 4). As the same results were obtained with M5-6a and M5-6b, this shows that there was no need to transplant the PCP–E didomain in this case. It is important to note, however, that the activity of the transplanted E-domain appears unable to compete with peptide extension by M3. This is consistent with previous results that have showed the M3 C-domain is able to extend incorrectly configured peptides with little effect on extension rate.25
Engineering modular interactions across fused E- and C-domains
Next, we wanted to further explore how to hybridise modules across the interface between E- and C-domains within multi-modular proteins. We concentrated on the M4/M5 interface, as significant differences in peptide hydrolysis in M4 (high) vs.M5 (low) had previously been published.7 Our hypothesis was that the M4 E-domain was responsible for the significant hydrolysis seen in in vitro assays, and we wanted to test if E-domain exchange could overcome this.First, we explored the role of the individual M4 and M5 E-domains on peptide hydrolysis. As constructs, we generated active site mutants of the catalytic histidine residue for the E-domains in M4 (His to Ala, M4b) and M5 (His to Gln, M5a; His to Ala, M5b). Next, we prepared synthetic tetra- and pentapeptides (8, 9 respectively, see ESI†) matching the natural teicoplanin peptide sequence but in which the C-terminal residue of each peptide was as either in the L- or D-configuration. These peptides were converted into peptidyl-CoAs (ESI Fig. S5–S8†) and loaded enzymatically onto the apo-PCP domains in these M4 and M5 constructs using Sfp. Hydrolysis and epimerisation were then measured via LCMS by comparing the retention times of the product peptides with synthetic standards.
Using this assay setup, we first compared the effect of the E-domain on peptide epimerisation in M4 and mutant M4b constructs using synthetic tetrapeptides D/L-8 loaded onto these modules (ESI Fig. S9A–H†). After overnight incubation, M4 led to hydrolysis of both D- and L-8, whilst M4b displayed no hydrolysis in either case (ESI Fig. S9C–H†). Here, we were unable to assay epimerisation activity due to the co-elution of the tetrapeptides 4 L-8 and 4 D-8 (ESI Fig. S9A and B†). Next, we tested the activity of M5 constructs using synthetic pentapeptides D/L-9 loaded onto these modules (ESI Fig. S9I–P†). After overnight incubation, all of 5 L-9 loaded on M5 had been converted into the 5 D-form, whilst the mutants showed that epimerization was either suppressed (M5a, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 L:D) or abolished (M5b) (ESI Fig. S9M–P†). The hydrolysis of 9 was low in all cases. These experiments reveal that the M4 E-domain displays significant hydrolytic activity that is not seen for M5.
:2 L:D) or abolished (M5b) (ESI Fig. S9M–P†). The hydrolysis of 9 was low in all cases. These experiments reveal that the M4 E-domain displays significant hydrolytic activity that is not seen for M5.
With evidence of role of the M4 E-domain in peptide hydrolysis, we undertook the design of modified M4 modules in which the E-domain was replaced with the corresponding E-domain from M5. As the M4 construct contains the downstream C-domain from module 5 fused with the E-domain of module 4, it was necessary to find an appropriate non-cognate E-domain accommodation site in these constructs (Fig. 5). We therefore designed two constructs in which the interdomain linker between the C- and E-domains was either retained from E4-and C5-domains (M4c) or matched the E5-and C6-domains (M4d).
 | ||
| Fig. 5 Importance of the E–C interdomain linker for activity in hybrid modules with transplanted E-domains (A). Within the M4–M6 protein there are two linkers between E- and C-domains, shown in green (M4 to M5) and red (M5 to M6). Peptide biosynthesis assays analysed by LCMS commencing from synthetic tripeptide 10 loaded on M3 with M4 and M5 shows effective biosynthesis of pentapeptide 12 (B) when incubated together with ATP and the specific substrates of the A-domains of each extension module (M4, M5 – Hpg). Incorporation of the M5 E-domain in M4 either retaining the M4 linker (M4c, C) or incorporating the M5 linker (M4d, D) into comparable assays shows that biosynthesis of 12 is only maintained for the M4 linker. Peptide species indicated; for m/z data see ESI Table S3.† Double peaks caused by epimerisation of C-terminal Hpg/Dpg residues during methylamine offloading. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, COM – communication. Module colour codes: M3 – yellow, M4 – green, M5 – pale blue, M6 – dark blue. | ||
To explore the activity of these modified modules, we pre-loaded M3 with synthetic tripeptide 10 (as performed above) before using this together with M4c/d and M5 in peptide reconstitution assays. Reconstitution using M4c provided excellent conversion of 10 into pentapeptide 12. In contrast, reconstitution using M4d revealed formation of tetrapeptide 11 without hydrolysis, although there was little extension to 12. These results indicate that the existing E–C linker found within an NRPS module should be retained when transplanting E-domains into modules (Fig. 5). This also offers an explanation for the lack of activity of the transplanted E-domain in the M5-6a and M5-6b constructs given the lack of native E–C linker in the M6 module.
Relocating COM domains to engineer interactions across A- and C-domains
Having seen the results of M5-6 reengineering and the possibilities for E/C-domain mediated communication across separated modules, we next wanted to assay the effects of intermodule communication that is naturally mediated between A- and C-domains across different proteins As M3/M7 and M4/M5 display the same substrate specificities (Dpg and Hpg, respectively) we now undertook to explore C/A compatibility by designing hybrid modules for both of these module sets (Fig. 2).First, we wanted to test if communication between M7 and M4 could be enabled solely by relocating the small COM domain that is found in M3. Relocation of the M3 COM domain onto M7 generated the M7a construct and we explored if this protein was active in peptide reconstitution assays. Firstly, M5-6 was incubated together with M7a as a control, and afforded tripeptide 3 as anticipated (Fig. 6). Next, we attempted to extend the M5-6 + M7a assay by adding M4. This did not afford tetrapeptide formation (ESI Fig. S11†), which showed a lack of communication between M7a and M4.
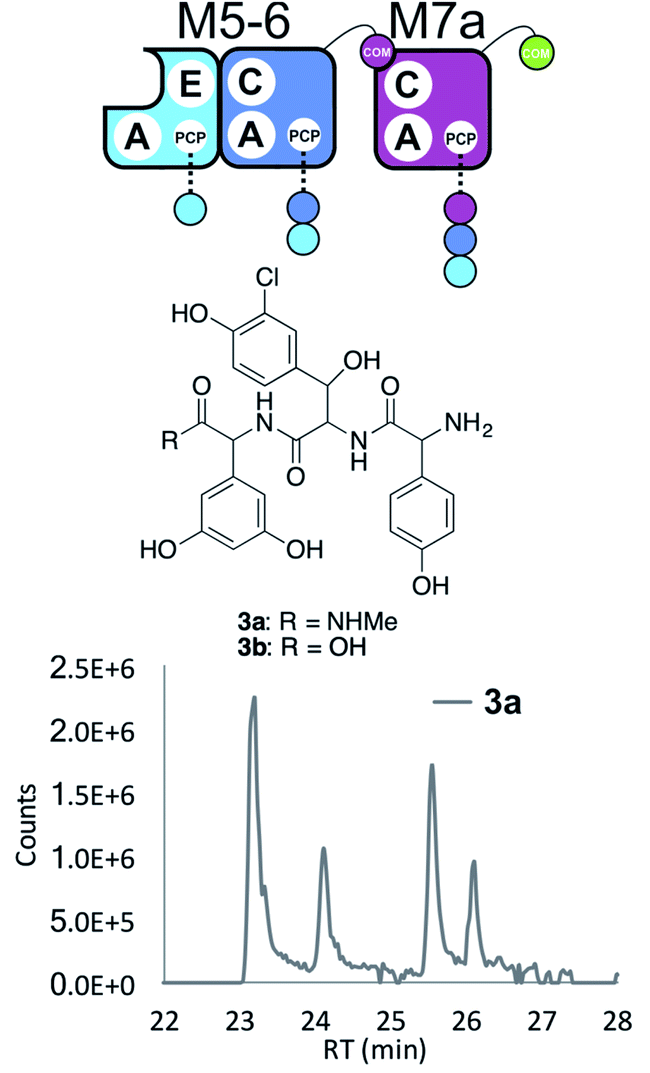 | ||
| Fig. 6 COM-domain transplantation is insufficient to allow interaction across modules M7 and M4. Peptide biosynthesis assays using dimodule M5-6 and an engineered M7 module M7a bearing the M3 COM-domain (A) forms 3, but addition of M4 has no effect (LCMS, ESI Fig. S12†). HRMS analysis shows formation of 3a when M5-6 + M7a or M5-6 + M7a + M4 was incubated together with ATP and the substrates of the A-domains of each module (M4, M5 – Hpg; M6 – ClBht; M7a – 3,5-Dpg). Peptide species indicated; for m/z data see ESI Table S3.† Double peaks caused by epimerisation of C-terminal Dpg residue during methylamine offloading. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, COM – communication. Module colour codes: M4 – green, M5 – pale blue, M6 – dark blue, M7 – violet. | ||
Engineering modular interactions across A- and C-domains
Having seen that COM domain transplantation was insufficient to allow M7a communication with M4, we next explored the concept of generating hybrid modules that contain C-domains from alternate modules in their architecture. The hypothesis here was that transplanting C-domains would allow hybrid modules to communicate via the A/C interface natural to the transplanted C-domain. To test this, we prepared a hybrid M3a construct in which we fused C7 with A–PCP3 within the artificial module M3a (Fig. 7A).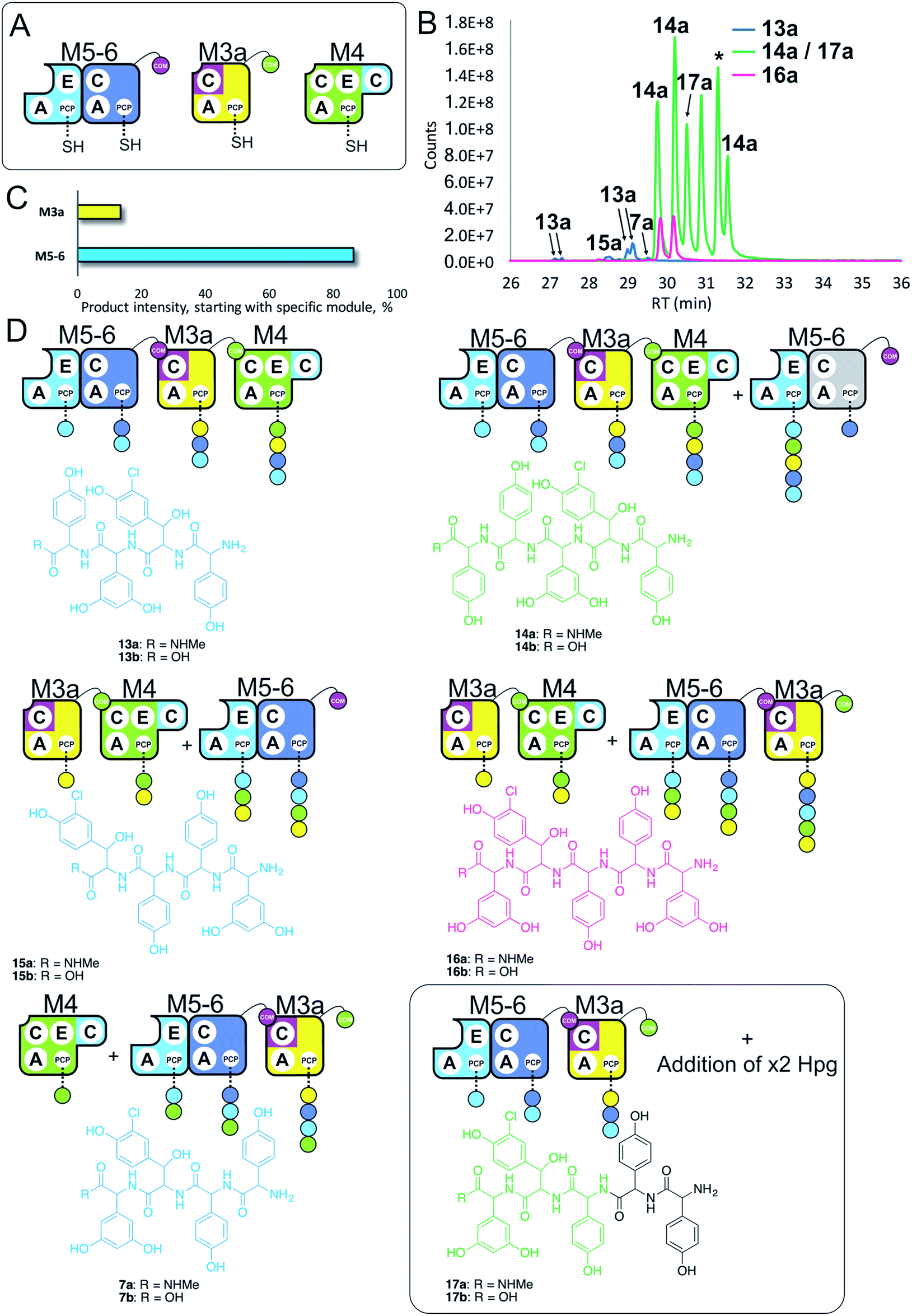 | ||
| Fig. 7 Engineering modular interactions across A- and C-domains for modules M7 and M4. Peptide biosynthesis assays using dimodule M5-6, an engineered M7 replacement module M3a bearing the M7 C-domain and M4 (A) shows communication between M3a and M4 and affords two starting points for NRPS-mediated peptide assembly (B). Peptide biosynthesis assays including ATP and the substrates of the A-domains of each extension module (M3a – 3,5-Dpg; M4, M5 – Hpg; M6 – ClBht). HRMS analysis (C and D) shows the formation of tetrapeptides 7a, 13a & 15a and pentapeptides 14a, 16a & 17a. 17a contains a sequence that can be rationalised through dipeptide acting as an acceptor substrate during peptide biosynthesis. Apparent pentapeptide peak indicated with an asterisk (*) did not provide an MS2 spectrum to allow structural analysis. Peptide species indicated; for m/z data see ESI Table S3.† Colour code indicates the amino acids added during the assays and matches the colours used in the LCMS traces. Double peaks caused by epimerisation of C-terminal Hpg/Dpg residues during methylamine offloading. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, COM – communication. Module colour codes: M3 – yellow, M4 – green, M5 – pale blue, M6 – dark blue, M7 – violet. | ||
We tested the functionality of M3a in the reengineered NRPS assembly line M5-6 + M3a + M4 using our peptide reconstitution assay. This array of modules also offered the possibility for several cycles of peptide synthesis due to the potential interaction of M4 with M5-6. Analysis of the results of this assay showed a complex series of products was present (Fig. 7B). Firstly, a small amount of tetrapeptide 13 was detected, which corresponds to the anticipated M5-6 + M3a + M4 pathway (Fig. 7). Tetrapeptides 15 and 7 were also detected at very low level, and result from M3a + M4 + M5-6 and M4 + M5-6 + M3a pathways, respectively (Fig. 7C and D). However, far more significant production of several pentapeptide species was detected, with smaller amounts of 16 and larger amounts of 14 (Fig. 7D). Production of these pentapeptides can be described by the same cyclic “set” of NRPS interactions 〈-M5-6 + M3a + M4-〉 albeit with different initiation points. Peptide 16 can be rationalised as being formed by M3a + M4 + M5-6 + M3a, whilst formation of 14 is supported by M5-6 + M3a + M4 + M5-6 activity, with no final M6 activity in this case.
Further analysis of the MS2 fragmentation of the pentapeptide products of this assay revealed the presence of pentapeptide sequence 17, which cannot be formed through the biosynthesis pathway discussed above (Fig. 7D). To determine the pathway responsible for formation of 17, we first hypothesised that the module reengineering undertaken to produce M3a could have affected the amino acid specificity of this module, specifically here allowing activation of Hpg. We explored the Hpg vs. Dpg activation properties of M3a and compared them to M3 using a spectroscopic activity assay, in which an enzymatic cascade couples the formation of pyrophosphate during amino acid activation with the oxidation of NADH.27 These assays showed no appreciable difference in the activity of M3a for Hpg versusM3, which is in agreement with A-domain mutagenesis data that shows how selective M3 is for Dpg vs. Hpg (ESI Fig. S13†).28 This makes the incorporation of Hpg by M3a highly unlikely to explain the formation of 17.
In these experiments, it is clear that engineering hybrid modules to enable alternate module interactions through A/C interfaces is possible. Formation of 17 is unexpected: one explanation would be the unusual extension of the M4 + M5-6 + M3a assembly line at the N-terminus by an additional round of either M4 or M5 activity. Results obtained using synthetic peptides loaded on M3 together with M5 (see below and ESI Fig. S12†) support the ability of M5 to perform such extensions. However, the inability of M4 to compete with M5-6 initiation would argue against this pathway. Instead, an alternative that is supported by other experiments (see ESI Fig. S12†) is the formation of tripeptide 3 by M5-6 + M3a, followed by two rounds of N-terminal extension by M5.
Results obtained in these experiments indicate that the pathway of NRPS-mediated peptide synthesis is maintained throughout synthesis of these peptides, with alterations in sequence occurring either through alternate start modules within the pathway (M5 or M3; a general inability to start at M4) or unusual extension of the peptide due to effects on module interactions because of the modular division of the assembly line. In the case of M3a, this retains the naturally split A/C interface between M3 and M4, and shows that these interfaces must be result in higher affinity than the artificially split M4 and M5 modules. This also indicates that such interaction interfaces between divided modules extends beyond isolated COM domain pairs, and suggests that further interactions – presumably mediated between A- and C-domains – are required for effective intermodule interaction.
NRPS modularisation reveals unexpected biosynthesis pathways
Having seen that hybridising modules can enable effective peptide biosynthesis pathways to be generated through dynamic module exchange, we next tested a similar NRPS assembly line comprising M5-6a + M3 + M4 to compare the activity of this artificial assembly line (Fig. 8A and B). The results of this peptide reconstitution assay support the majority of initiation from the M5-6a dimodule (Fig. 8C). The expected M5 initiation products include the tetrapeptide 13, formed by M5-6a + M3 + M4 and a hexapeptide extension product 19 formed by the additional activity of M5-6a at the end of the tetrapeptide assembly line (Fig. 8D). Unexpectedly, pentapeptide 20 was the dominant product of these assays. This sequence can be rationalised through the assembly line M5-6a + M3 + M4 with subsequent acceptance of the M4 loaded tetrapeptide by M3 (Fig. 8D). Whilst we have noted M5 interactions with M3 (see Fig. 7 and ESI Fig. S12†), these interactions appear to favour the N-terminal extension of the M3-loaded peptide rather than this more typical NRPS-mediated peptide elongation. Having seen that the M4/M5 interaction is too weak to initiate peptide biosynthesis in an efficient manner, we postulate that this unusual extension activity is due to the strong interaction mediated by the M3/M4 interface, which in turn is due to the separated nature of these modules in the assembly line. This could then allow an unusual pathway to occur by virtue of increased local concentration. It is clear, however, that modularisation of the teicoplanin NRPS assembly line can enable non-standard peptide assembly pathways to proceed if the interactions of these separated modules are more favourable than those seen in a standard pathway with modules physically linked within larger proteins.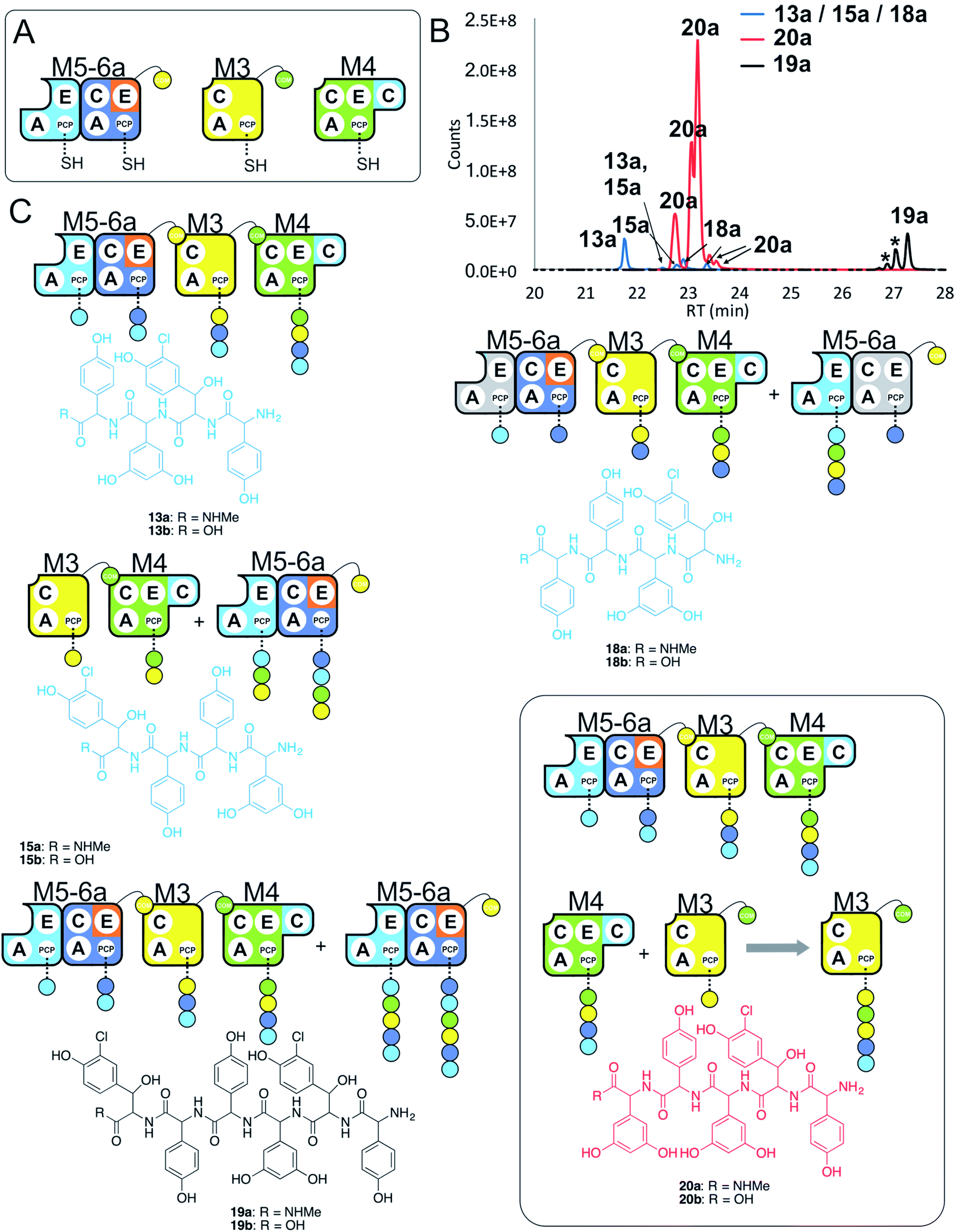 | ||
| Fig. 8 Comparative four module NRPS biosynthesis using engineered M5-6a didomain. Peptide biosynthesis assays using engineered dimodule M5-6a, M3 and M4 (A) shows communication between M5-6a and M3, and reveals competition for NRPS-mediated peptide elongation from M4 that favours M3 over M5 (B). Peptide biosynthesis assays including ATP and the substrates of the A-domains of each extension module (M3a – 3,5-Dpg; M4, M5 – Hpg; M6 – ClBht). HRMS analysis (C) shows tetrapeptides 13a, 15a & 18a, pentapeptide 20a and hexapeptide 19a; 19a is the result of initiation from M5 but with M4 tetrapeptide extension mediated by M3 in this case (box). Peptide species indicated; for m/z data see ESI Table S3.† Double peaks caused by epimerisation of C-terminal Hpg/Dpg residues during methylamine offloading. Colour code indicates the amino acids added during the assays and matches the colours used in the LCMS traces. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, COM – communication. Module colour codes: M2 – orange, M3 – yellow, M4 – green, M5 – pale blue, M6 – dark blue. | ||
Given these unusual findings, we further investigated hybrid modules through C-domain incorporation. We generated a novel M5 in a similar manner to that used above, where we now fused the M4 C-domain into M5 to generate M5c (C–A–PCP–E–C; Fig. 2 and 9A). To test the functionality of M5c, we utilised this module in peptide reconstitution assays together with synthetic tripeptide 10-loaded M3 and M6 (Fig. 9B). This demonstrated the successful extension of 10 to pentapeptide 21. When M3 loaded with 10 was used together only with M5c, this led to the anticipated tetrapeptide 11 as the major product. In this assay we also identified pentapeptides bearing an additional Hpg residue at the peptide N- or C-termini (ESI Fig. S12†). A control assay using M3 with M5 also showed tetrapeptide and pentapeptide formation, although in this case both were exclusively found at the N-terminus of the synthetic tripeptide (ESI Fig. S12†). Whilst such peptide extension is unexpected, it is clearly able to be an effective process under these conditions as opposed to traditional peptide biosynthesis. These results support those obtained for the M5-6 + M3a + M4 reconstitution experiments, which indicate that unusual N-terminal peptide extension is likely also proceeding in these pathways.
 | ||
| Fig. 9 Engineering modular interactions across A- and C-domains for modules M3 and M5. Peptide biosynthesis assays commencing from synthetic tripeptide 10-loaded on M3 with engineered M5c and M6 (A) shows effective biosynthesis of pentapeptide 21 when incubated together with ATP and the substrates of the A-domains of each extension module (M5 – Hpg; M6 – ClBht). HRMS analyses (B) show no residual 10, with conversion to tetrapeptide 11a and pentapeptide 21. Peptide species indicated; for m/z data see ESI Table S3.† Colour code indicates the amino acids added during the assays and matches the colours used in the LCMS traces. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, COM – communication. Module colour codes: M3 – yellow, M4 – green, M5 – pale blue, M6 – dark blue, M7 – violet. | ||
These results show that the effective redesign of naturally separated modules within an NRPS can be obtained through the use of C-domain replacement, which we have demonstrated here by retaining the M3/M4 and M6/M7 interfaces across reengineered modules. This supports other studies that highlight the value of modular redesign by altering C-domains, and also shows how unexpected module interactions can be identified by the modularisation of larger NRPS multimodular proteins.
NRPS assembly lines comprised of isolated modules show decreased peptide assembly efficiency
In a final experiment, we explored the effect of mixing single NRPS modules together to see how selective the assembly line was for reassembly (Fig. 10A). To this end, we included M6a + M3 + M4 + M5 + M6 + M7 in a peptide reconstitution assay. This removed the fusion M5/M6 to determine how selective M6a and M6 were for M3 and M7, respectively. Reconstitution of this assembly line showed that this module assembly order was mostly conserved (Fig. 10B and C). The major peptides produced were 22 from M6a + M3 + M4 + M5 and 24 from M5 + M6 + M7 (Fig. 10). Small amounts of pentapeptide 25 formed from M3 + M4 + M5 + M6 + M7 was detected, and there was further evidence for M5–M5 interactions with the formation of the pentapeptide 23. Low levels of the complete hexapeptide 26 was present, indicating that it is possible to regenerate activity of the complete assembly line M6a + M3 + M4 + M5 + M6 + M7, although the generation of shorter peptides was clearly favoured when each isolated module was present. This experiment shows that the interactions present when using isolated modules can support peptide biosynthesis that follows the desired biosynthesis pathway, but that the efficiency of NRPS assembly lines using isolated modules is optimal for shorter assembly lines of 3–5 modules.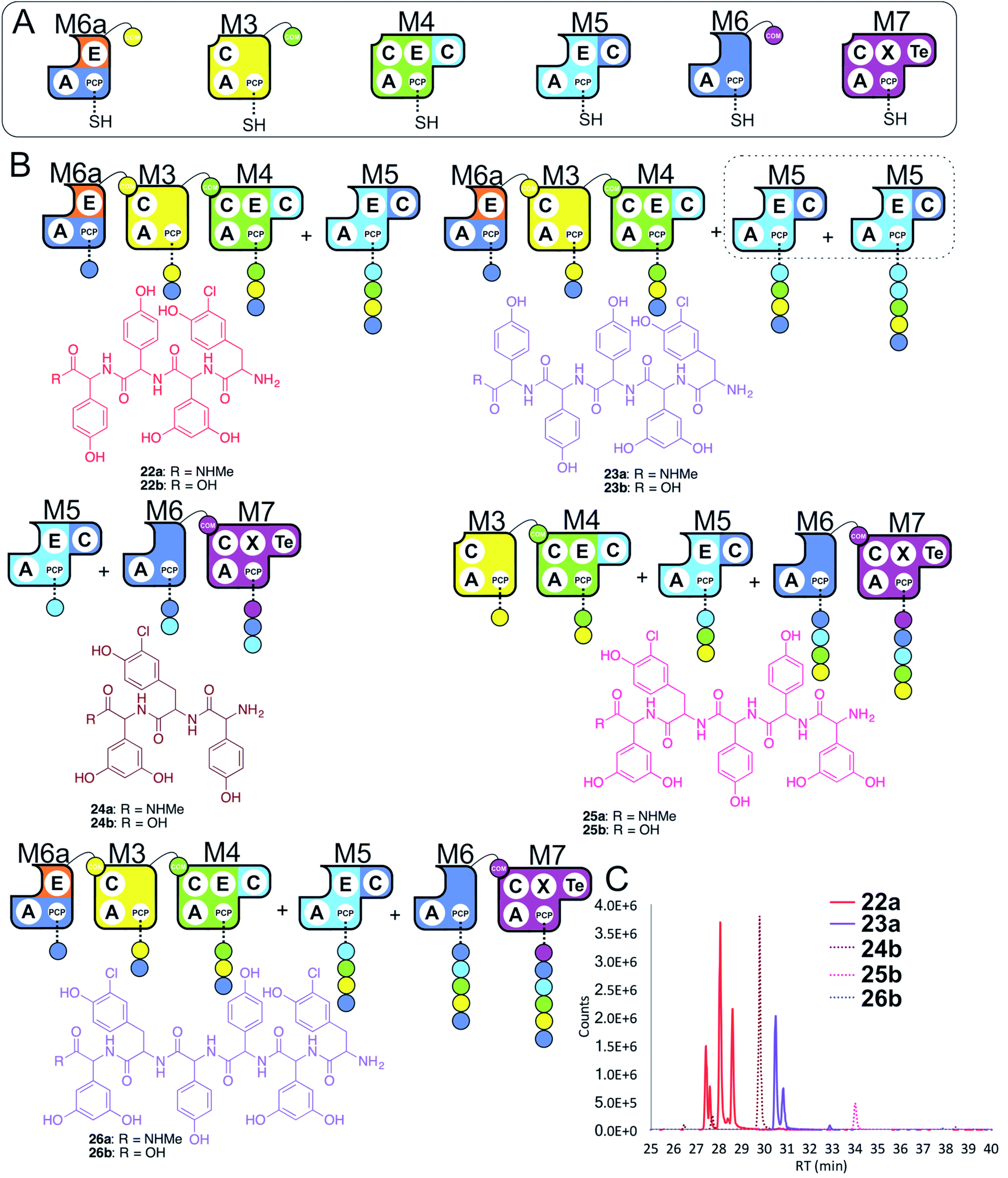 | ||
| Fig. 10 Exploring the reconstitution of hybrid NRPS assembly lines from individual modules. Peptide biosynthesis assays using engineered module M6a, M3, M4, M5, M6 and M7 (A) shows a preference for the biosynthesis of peptides of 3–5 residues, and evidence for M5–M5 interactions (B). Peptide biosynthesis assays including ATP and the specific substrates of the A-domains of each extension module (M3, M7 – 3,5-Dpg; M4, M5 – Hpg; M6, M6a – ClTyr, chosen here to favour M3 activity). HRMS analysis (C) shows tripeptide 24, tetrapeptide 22, pentapeptides 23 & 25 and hexapeptide 26. Peptide species indicated; for m/z data see ESI Table S3.† Double peaks caused by epimerisation of C-terminal Hpg/Dpg residues during methylamine offloading. Colour code indicates the amino acids added during the assays and matches the colours used in the LCMS traces. Domain key: A – adenylation, C – condensation, PCP – peptidyl carrier protein, E – epimerisation, X-P450-recruitment, TE – thioesterase, COM – communication. Module colour codes: M2 – orange, M3 – yellow, M4 – green, M5 – pale blue, M6 – dark blue, M7 – violet. | ||
Discussion
NRPS reengineering to produce modified peptide sequences has been intensively studied using in vivo approaches, largely centred on altering A-domain selectivity, C-domain division and mutasynthesis approaches. Here, we present an NRPS reconstitution platform from teicoplanin biosynthesis to study reengineering strategies in vitro. We focussed on four approaches to enable redesign of peptide biosynthesis: (1) construction of novel NRPS initiation modules, (2) reengineering modules across the C/A-domain and C/E-domain interfaces, (3) domain relocation and fusion with non-cognate modules, and (4) optimisation of interdomain linkers to allow effective non-native domain accommodation in redesigned NRPS modules.Firstly, we tested the hypothesis that elongation modules, as well as dimodular and trimodular NRPS proteins, can be converted into initiation modules. Investigating constructs derived from the M4-6 protein Tcp11 protein showed that this was indeed possible from M4, M5 and M6, although the abilities of dimodular constructs from Tcp11 to initiate peptide biosynthesis were generally higher than single modules. In contrast to other reported results,31 we identified that elongation modules containing an N-terminal C-domain were not inhibited from peptide initiation, indicating that the affinity of the C-domain acceptor site in such cases is insufficient to prevent an aminoacyl-PCP from acting as the donor substrate for a downstream C-domain.
We next endeavoured to understand how to artificially induce intermodule communication between separate protein constructs. Given the similarities in the peptide formed by M5-6 and M1-2, we explored reengineering to allow M5-6 to replace M1-2 in its interaction with M3. Here, we tested two new approaches: (1) using C/A interface reengineering between distinct modules and (2) the relocation of an E-domain to ensure communication with the downstream module. The success of these experiments, taken together with the results of our previous M6 PCP exchange experiments perform in the M6 A-PCP construct,26 indicates that both C/A and A/PCP regions are flexible in terms domain exchange. The critical factor in these bio-combinatorial experiments is the selectivity of the acceptor site of the upstream C-domain, which in teicoplanin M6 is high due to the need for this domain to gate aminoacyl-PCP modifications by trans-acting enzymes.7,26 In order to generate communication between non-adjacent modules in the teicoplanin NPRS we identified that, in contrast to other NRPS systems,32–34 it is not sufficient to relocate/exchange compatible COM domains at the end of module of interest. Whilst recent strategies that divide fused modules have shown compatibility with COM domain relocation,35 our results clearly show that larger adjacent domain–domain interaction surfaces are also required to ensure module–module recognition and ensure communication between M5-6 and M3. This maintenance of E- and C-domain interactions is likely the result of the tight coupling of activity of these two domains, where the E-domain is required to act prior to the acceptance of the modified peptide by the subsequent C-domain. It also helps to explain why inactive E-domains are retained within some NRPS assembly lines, exemplified in GPAs by M3/M4 interactions in A47934 biosynthesis36 and M6/M7 interactions in complestatin biosynthesis.37,38 The utility of this approach is not limited by the requirement to maintain E-domain activity, as C-domains are not always exclusively active on the correct peptide stereochemistry as seen previously for teicoplanin M3 and M7, for example.14,25
Having seen the importance of E-domains within modular exchange strategies, we also tested domain exchange experiments investigating the linkers with the E-domains found in the M4 and M5 modules from the Tcp11 protein. Here, we demonstrated that the construct possessing the C–E domain inter-modular linker (IML) connecting M4 with M5 retains activity but not one with the IML connecting M5 with M6. This finding is in agreement with a published IML compatibility analysis, which found successful domain exchanges requires compatible linkers connecting the upstream and downstream modules of interest.39 We identified that the 26 amino acid length linkers connecting E- and C-domains – displaying more than 80% sequence identity in this case – are key to ensuring productive substrate delivery to the E-domain and C-domains. In contrast, we did not observe that E-domain activity was linked to the presence of its partner PCP domain (M5-6avs.M5-6b constructs, ESI Fig. S10†). This disagrees with a previous study based on structural observations that PCP–E di-domains should act as a functional and conservative unit.40 In our case, an inappropriate E/C interface in these reengineered constructs, combined with the lack of stereoselectivity exhibited by the downstream module, would appear to explain these results. The lack of structural data for large NRPS biosynthetic protein constructs limits our understanding the structural role of different NRPS inter-modular linkers that connect different modules. Recent dimodular NRPS protein X-ray structures41 as well as photocrosslinking studies42 provides us with an understanding of the flexibility of the NRPS biosynthetic machinery, which suggests that linker exchange in multi-modular NRPS proteins could well alter domain–domain motion and potentially prevent productive substrate delivery to downstream peptide processing domains. However, further structural investigations of large multi-modular NRPS constructs are needed in order to deliver the molecular insights into the roles that such linkers play in NRPS-mediated peptide assembly.
Given that E-domains are optional in NRPS modules, the exploration of redesigning modules by fusion at the C/A interface shows the most general promise in redesign. In exploring this interface, we again noted the importance of maintaining the catalytic terminal domains (i.e. beyond the COM-domain) between modules split into separate proteins. In redesigning the assembly line to allow M7 to communicate with M4, we showed that the M3a construct allowed communication with M6via the M7 C-domain and M4via the A-domain of M3. We also note that adding the COM-domain of M3 to M7 was insufficient for communication with M4. The suitability of this redesign strategy was further supported by the ability to append the M4 C-domain directly onto M5 and produce M5a. M5a retained the interaction with M3via the M4 C-domain and also retained M6 interaction via the natural M5/6 A/C interface. The A/C interface also provides an explanation why the excision of modules through the division of C-domains provides a path to successful modular redesign for NRPS systems.19
Whilst most of the peptide synthesis pathways established in this work conform to those anticipated based on the natural assembly line, we did identify that modularisation of the teicoplanin NRPS led to unanticipated modular interactions in some cases. Within the M5-6a + M3 + M4 system, for example, the acceptance of the M4 loaded tetrapeptide by M3 was the major pathway present, and shows an unusual M4/M3 interaction (Fig. 8). Here, it is important to note that this interaction only occurs after the activity of M3 within the anticipated M5-6a + M3 + M4 pathway, possibly due to the lack of affinity between the M4 and M5 modules that are normally fused within one protein. Perhaps most curiously of all, we have also noted the apparent N-terminal extension of M3-loaded tripeptides by M5 in a number of assays. This extension must be occurring on the PCP-bound peptide, as all these unusual peptide products were detected in their methylamide forms. Whilst in the context of the M5-6 + M3a + M4 pathway this could possibly be explained by other as yet unidentified intermodule interactions, in the experiments where synthetic tripeptide was loaded on M3 and incubated with M5 the evidence for N-terminal extension appears unambiguous. Whilst unexpected, it should be noted that there is a general lack of structural information concerning the presentation of acceptor substrates within C-domains,5,41,43–45 and that coupled with the reported flexibility of the NRPS assembly line (even within fused modules),25,41,42,46 there is no evidence that the attack of an acceptor peptide onto a donor amino acid is explicitly prevented. This intriguing result highlights the importance of obtaining further structural snapshots of the NRPS C-domain in relevant catalytic states, and is further underlined by the diverse range of catalytic activities performed by domains derived from C-domains.5,9,13,47,48 It also raises further questions as to origins of the replacement of the teicoplanin M3 domain in vancomycin/pekiskomycin-type GPAs,49–51 given that the M3 module appears to be the major source of atypical module interactions in our experiments with the teicoplanin NRPS. The unexpected interactions uncovered in these experiments (M4 with M3, and M3 with M5) also shows further promise for NRPS redesign, for this indicates that the alteration of the fused state of modules within an assembly line can then lead to alterations in the assembly pathway, and hence the formation of new peptide products.
Experimental
Construct cloning
All protein constructs were cloned into a pET-GB1-1d vector, which encodes an N-terminal 6xHis-Tag followed by the IgG-binding B1 domain of Streptococcus (GB1) with a TEV cleavage site under the control of a T7 promoter and a Strep-Tag II at the C-terminus, using In-Fusion® HD Cloning kit. DNA fragments required for hybrid proteins construction were amplified by PCR from previously constructed protein expression plasmids. PCR primers were designed to share 15 bases of homology with adjacent DNA fragments. These primers were then used to amplify both the insert/(-s) and plasmid DNA. The plasmid DNA, containing the gene encoding the NRPS module of interest, was used as the template DNA for a PCR reaction. Fragments were amplified using Phusion® Hot Start Flex Master Mix (NEB) and the appropriate forward and reverse primers (ESI Table S1†). The PCR products were analysed on a 0.8% agarose gel in TAE buffer and the DNA subsequently gel-extracted and purified using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific). The extracted PCR products (insert/(-s) and vector) were combined in an In-Fusion® cloning reaction as per the manufacturer's instructions. In-Fusion® cloning reactions were incubated for 15 min at 50 °C, then placed on ice and 2.5 μL of the reaction mixture was used for transformation of NEB 10-beta competent E. coli cells. The next day, plasmid DNA from individual colonies was isolated and sequenced to confirm that the appropriate module fragment was correctly inserted into the plasmid.Protein expression
All proteins analysed in this study were co-expressed with the teicoplanin MbtH-like protein Tcp17. For co-expression of the proteins, E. coli BL21 (DE3) (NEB) competent cells possessing the plasmid encoding Tcp17 were co-transformed together with a plasmid encoding the NRPS module of interest. For selection, two antibiotics (kanamycin and streptomycin) were used; expression of all modules were performed in 10 L of TB media, supplemented with 50 μg mL−1 kanamycin and 50 μg mL−1 streptomycin. Cells were grown and protein expression was induced according to the previously described procedure.7Protein purification
The majority of proteins constructs analysed in this study were purified as previously described,7 with the exception of Tcp12 that was purified using a modified procedure.14 Purified construct yields from 10 L of bacterial growth media: 26 mg of M3a, 3 mg of M4a, 1 mg of M4b, 1.5 mg of M4c, 2.4 mg of M4d, 12 mg of M5a, 8 mg of M5b, 5 mg of M5c, 7 mg of M6a, 55 mg of M7a, 9 mg of M5-6, 9 mg of M5-6a, 1 mg of M5-6b and 1 mg of M4-6a. For SDS-PAGE gels see ESI (Fig. S1–S4†).Peptidyl-CoA synthesis
10-CoA was synthesised as described previously.7 (4 D/L)-8- and (5 D/L)-9-CoAs were synthesised manually on solid phase at 0.05 mmol scale using hydrazide activation and displacement to generate the desired CoA thioesters.52 2-Chlorotrityl chloride resin (200 mg) was swelled in DCM (8 mL, 30 min), washed with DMF (3×), and incubated with a 5% hydrazine solution in DMF (6 mL, 2 × 30 min). The resin was washed with DMF (3×), and a solution of DMF/TEA/MeOH (7:2:1) (4 mL, 15 min) added. The first Fmoc-protected amino acid (0.06 mmol) was coupled to the resin using COMU (0.06 mmol) and 2,6-lutidine (0.06 mmol, 0.12 M) overnight. In the second step, unreacted hydrazine groups were capped with Boc-glycine (0.15 mmol) that had been activated using COMU (0.15 mmol) and 2,6-lutidine (0.15 mmol, 0.12 M) for 1 h. Subsequent Fmoc removal was performed using a 1% DBU solution (3 mL, 3 × 30 s) in DMF followed by coupling of the subsequent Fmoc- or Boc-protected amino acid (0.15 mmol) after activation with COMU (0.15 mmol) and 2,6-lutidine (0.15 mmol, 0.12 M) for 40 min; the final amino acid added to the peptide was always Boc-protected. Cleavage of the hydrazide peptide from resin and removal of protecting groups (tBu, Boc, Pbf) was accomplished using TFA/TIS/H2O (95:2.5:2.5 v/v′/v′′, 5 mL) with shaking at room temperature for 1 h. The resin was removed by filtration and washed with TFA (2×). Subsequently, the filtrate was concentrated under a N2 stream to ∼1 mL and the peptide precipitated with ice cold diethyl ether (∼8 mL) and collected by centrifugation in a flame-resistant centrifuge. Crude peptide was purified using preparative RP-HPLC (gradient 10–40% ACN or 15–45% ACN over 30 min). Purified hydrazide peptides were then dissolved in buffer 1 containing urea (6 M) and NaH2PO4 (0.2 M), pH 3 (obtained via addition of HCl) to a final concentration of 5 mM. The solution was cooled to −15 °C using a salt/ice bath. Subsequently, 0.5 M NaNO2 (0.95 equivalents) was added and the mixture was stirred for 10 min. Coenzyme A (1.2 equivalents dissolved in buffer 1) was then added to the reaction. After 15 minutes, the pH was slowly adjusted to 6.5 using KH2PO4/K2HPO4 buffer (6:94 v/v 1 M, pH 8.0). The reaction mixture was stirred on ice for 2 hours, before the final peptidyl-CoA product was purified using preparative RP-HPLC, gradient 10–40% ACN over 30 min. All purifications were performed using a Shimadzu high performance liquid chromatography system equipped with a SPD-M20A Prominence photo diode array detector and two LC-20AP pumps. Preparative separations were performed using a Waters XBridge BEH300 Prep C18 column (5 μm, 19 × 150 mm) with a flow rate of 10 mL min−1. The solvents used were water + 0.1% TFA (solvent A) and HPLC-grade ACN + 0.1% TFA (solvent B). For compound analysis by LCMS (see ESI Fig. S5–S8†).
PCP loading
Following purification, PCP-containing proteins were converted from their apo to holo form by loading their PPE linker in a reaction catalysed by the phosphopantetheinyl transferase Sfp (R4-4 mutant). Depending on the desired reaction two different substrate types were used to transform NRPS proteins: (1) either loading CoA to generate holo-PCP constructs or (2) peptidyl-CoA conjugates to generate peptidyl-PCP constructs. PCPs loading were performed according to the same procedure as described previously.7A-domain activity assay
Analysis of A-domain activation for various substrates were performed as previously reported.27E-domain activity assay
In order to evaluate both wild type E-domains in M4 and M5 as well as the activity of active site mutants, proteins were converted into their peptidyl form by loading 8-CoA (ESI Fig. S5 and S6†) or 9-CoA (ESI Fig. S7 and S8†) onto the corresponding PCP domains. Thus generated, these peptidyl-PCP constructs (5–10 μM) were immediately used for E-domain activity assays. Peptidyl-NRPS protein was mixed with 1 mM ATP and 10 mM MgCl2 in reconstitution assay buffer (50 mM HEPES, pH 7.0; 50 mM NaCl) and incubated overnight at 30 °C with shaking at 300 rpm. Peptide cleavage with methylamine, purification and analysis by LC-MS was performed as described previously.7 Epimerisation activity (conversion of L-peptide to D-peptide) was evaluated by the comparison of LC-MS traces of the control peptidyl-CoA compounds in the reconstitution reaction buffer (non-loaded, direct cleavage with methylamine).In vitro reconstitution of non-ribosomal peptide biosynthesis
Experimental procedures, sample preparation and analysis were performed using the assay conditions described previously.7 2–10 μM of holo NRPS-proteins were used for in vitro reconstitution assays. In each reaction, NRPS proteins were mixed with 1 mM ATP, 10 mM MgCl2 and amino acid substrates for each A-domain (1 mM) in reconstitution assay buffer (50 mM HEPES, pH 7.0; 50 mM NaCl) to a final volume of 200 μL. These were incubated overnight at 30 °C with shaking at 300 rpm, with peptides attached to any PCP domain then chemically cleaved by the addition of methylamine (15 μL) to liberate the methylamide peptides. Reaction mixtures were incubated for 15 min at room temperature. To neutralise the mixture, 4 mL of reconstitution assay buffer was added. The peptide products were then purified from the reaction mixture using solid phase extraction (Strata™-X-33 μm Polymeric Reversed Phase Tubes; 30 mg mL; Phenomenex). Before sample loading, the cartridges were activated with methanol (1 mL) and subsequently equilibrated with water (1 mL). Neutralised samples were then loaded onto equilibrated cartridges and the solution allowed to pass through the column bed under gravity. When the entire sample was loaded, the cartridge was washed with 0.1% aqueous formic acid (1 mL) before the peptides were eluted with 1% formic acid in methanol (500 μL). After removal of the methanol using a centrifugal concentrator (Concentrator plus; Eppendorf; 45 °C) the residue was dissolved in 6 μL of 50% MeCN in water (Optima® LC/MS Grade) and diluted to a final volume of 36 μL using aqueous 0.1% formic acid (Optima® LC/MS Grade). 14 μL of the sample was subsequently injected onto a XBridge® Peptide BEH C18 column (5 × 300 mm, Waters) and analysed by LCMS (LCMS-2020, ESI, Shimadzu) in positive ionisation mode, using a 5–45% acetonitrile in water gradient over 40 min and a 1 mL min−1 flow rate.HRMS and MS2 measurements
HRMS was performed using Orbitrap-based mass spectrometers (QExactive HF, QExactive Plus and Fusion tribrid, Thermo Scientific) coupled to nanoflow high performance liquid chromatography (Ultimate 3000 RSLCnano, Dionex/Thermo scientific) via a nanospray source. Chromatographic separation was performed by trap-elution using an Acclaim PepMap 100 trap column (100 mm × 2 cm, nanoViper, C18, 5 mm, 100 Å; Thermo Scientific) and an Acclaim PepMap RSLC column (75 mm × 50 cm, nanoViper, C18, 2 mm, 100 Å; Thermo Scientific). Loading onto the trap column was performed at 15 μL min−1 in 2% acetonitrile, 0.1% TFA. Elution was performed at 250 nL min−1 using 0.1% formic acid (buffer A) and 80% acetonitrile, 0.1% formic acid (buffer B). After equilibration at 2.5% buffer B for 2 minutes, peptides were eluted with a gradient from 2.5% to 37.5% buffer B over 30 min, followed by a ramp to 42.5% buffer B over 3 min. Full scan MS was performed in the Orbitrap at either 70000 (Plus) or 60000 (HF and Fusion) nominal resolution, with 5 data dependent MS2 scans acquired using 1.4–1.6 isolation width and 15000 to 30000 nominal resolution in the Orbitrap using HCD with 27% normalised collision energy (32% on Fusion). Up to 20 predicted peptides were also targeted for MS2 in each cycle using the same resolution and stepped collision energies between 21 and 30%. Extracted ion chromatograms at 6 ppm were performed for all possible sequence compositions (based on input amino acids) with significant peaks inspected for appropriate MS isotope distributions and manual assignment of MS2 fragments when available (ESI Table S2†).
Conclusions
Non-ribosomal peptide synthetases have long been viewed as highly amenable to redesign due to their modular nature. In this work, we have explored a wide range of modular redesign strategies for the teicoplanin NRPS, which have shown that modules within an NRPS are indeed highly versatile units, whose interactions can be controlled through modification of their composite domains. These experiments show that elongation domains can be effective at initiating biosynthesis, and thus assembly lines are not limited to commencing from a small subset of modules. Furthermore, our results indicate that the interactions of NRPS modules that are found on separate proteins does not rely solely on smaller communication domains, and rather implies larger interactions mediated through C-domains with either A- or E-domains. The ability to reconstitute hybrid modules by transplantation of C- and E-domains appears to be facilitated by such interactions, whilst the limits of C-domain gating for peptide stereochemistry also can be used to allow the generation of novel assembly lines across unwanted E-domains. Division of fused modules within an NRPS indicates that there is the potential for alternate module interactions to occur, which shows that alternate assembly pathways can be generated even through minor alterations to the NRPS machinery. Taken together, our results show the versatility of teicoplanin NRPS modules for alternate redesign strategies and provides new insights into the molecular interactions of these fascinating peptide assembly lines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. Stier (BZH-Heidelberg) for fusion protein vectors; J. Yin (University of Chicago) for the R4-4 Sfp expression plasmid and N. Ziemert, E. Stegmann (University of Tübingen) and G. Challis (Monash University) for helpful discussions. This work was supported by Monash University, EMBL Australia and the National Health and Medical Research Council (APP1140619 to M. J. C.) and further supported under the Australian Research Council's Discovery Projects funding scheme (project number DP190101272 to M. J. C.). This research was supported by the Monash University Postgraduate Publications award (to M. K.). This research was conducted by the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012) and funded by the Australian Government.References
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- T. Izoré and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 1120–1139 RSC.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- T. Kittilä, A. Mollo, L. K. Charkoudian and M. J. Cryle, Angew. Chem., Int. Ed., 2016, 55, 9834–9840 CrossRef PubMed.
- K. Bloudoff and T. M. Schmeing, Biochim. Biophys. Acta, 2017, 1865, 1587–1604 CrossRef CAS.
- T. Kittilä, C. Kittel, J. Tailhades, D. Butz, M. Schoppet, A. Büttner, R. J. A. Goode, R. B. Schittenhelm, K.-H. van Pee, R. D. Süssmuth, W. Wohlleben, M. J. Cryle and E. Stegmann, Chem. Sci., 2017, 8, 5992–6004 RSC.
- M. Kaniusaite, J. Tailhades, E. A. Marschall, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, Chem. Sci., 2019, 10, 9466–9482 RSC.
- J. M. Reimer, I. Harb, O. G. Ovchinnikova, J. Jiang, C. Whitfield and T. M. Schmeing, ACS Chem. Biol., 2018, 13, 3161–3172 CrossRef CAS PubMed.
- K. Bloudoff, C. D. Fage, M. A. Marahiel and T. M. Schmeing, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 95–100 CrossRef CAS PubMed.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- M. Peschke, K. Haslinger, C. Brieke, J. Reinstein and M. Cryle, J. Am. Chem. Soc., 2016, 138, 6746–6753 CrossRef CAS PubMed.
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31–41 CrossRef CAS.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- M. Schoppet, M. Peschke, A. Kirchberg, V. Wiebach, R. D. Süssmuth, E. Stegmann and M. J. Cryle, Chem. Sci., 2019, 10, 118–133 RSC.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- T. Stachelhaus, A. Schneider and M. Marahiel, Science, 1995, 269, 69–72 CrossRef CAS PubMed.
- M. J. Calcott and D. F. Ackerley, Biotechnol. Lett., 2014, 36, 2407–2416 CrossRef CAS PubMed.
- J. W. Han, E. Y. Kim, J. M. Lee, Y. S. Kim, E. Bang and B. S. Kim, Biotechnol. Lett., 2012, 34, 1327–1334 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y.-N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 11, 653–661 CrossRef.
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. D. Süssmuth, J. Am. Chem. Soc., 2004, 126, 5942–5943 CrossRef CAS PubMed.
- S. Weist, B. Bister, O. Puk, D. Bischoff, S. Pelzer, G. J. Nicholson, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed., 2002, 41, 3383–3385 CrossRef CAS PubMed.
- J. Thirlway, R. Lewis, L. Nunns, M. Al Nakeeb, M. Styles, A.-W. Struck, C. P. Smith and J. Micklefield, Angew. Chem., Int. Ed., 2012, 51, 7181–7184 CrossRef CAS PubMed.
- G. C. Uguru, C. Milne, M. Borg, F. Flett, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2004, 126, 5032–5033 CrossRef CAS PubMed.
- C. Steiniger, S. Hoffmann and R. D. Süssmuth, Cell Chem. Biol., 2019, 26, 1526–1534 CrossRef CAS PubMed , e1522.
- M. Kaniusaite, J. Tailhades, T. Kittilä, C. D. Fage, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, FEBS J., 2020 DOI:10.1111/febs.15350.
- M. Kaniusaite, R. J. A. Goode, R. B. Schittenhelm, T. M. Makris and M. J. Cryle, ACS Chem. Biol., 2019, 14, 2932–2941 CrossRef CAS PubMed.
- T. Kittilä, M. Schoppet and M. J. Cryle, ChemBioChem, 2016, 17, 576–584 CrossRef PubMed.
- M. Kaniusaite, T. Kittila, R. J. A. Goode, R. B. Schittenhelm and M. J. Cryle, ACS Chem. Biol., 2020 DOI:10.1021/acschembio.0c00435.
- J. Beld, E. C. Sonnenschein, C. R. Vickery, J. P. Noel and M. D. Burkart, Nat. Prod. Rep., 2014, 31, 61–108 RSC.
- M. Sunbul, N. J. Marshall, Y. Zou, K. Zhang and J. Yin, J. Mol. Biol., 2009, 387, 883–898 CrossRef CAS PubMed.
- U. Linne and M. A. Marahiel, Biochemistry, 2000, 39, 10439–10447 CrossRef CAS PubMed.
- A. Tanovic, S. A. Samel, L.-O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS PubMed.
- M. Hahn and T. Stachelhaus, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 275–280 CrossRef CAS PubMed.
- M. Hahn and T. Stachelhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15585–15590 CrossRef CAS PubMed.
- C. Kegler and H. B. Bode, Angew. Chem., Int. Ed., 2020, 59(32), 13463–13467 CrossRef CAS PubMed.
- J. Pootoolal, M. G. Thomas, C. G. Marshall, J. M. Neu, B. K. Hubbard, C. T. Walsh and G. D. Wright, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8962–8967 CrossRef CAS PubMed.
- A. Mollo, A. N. von Krusenstiern, J. A. Bulos, V. Ulrich, K. S. Åkerfeldt, M. J. Cryle and L. K. Charkoudian, RSC Adv., 2017, 7, 35376–35384 RSC.
- S. B. Singh, H. Jayasuriya, G. M. Salituro, D. L. Zink, A. Shafiee, B. Heimbuch, K. C. Silverman, R. B. Lingham, O. Genilloud, A. Teran, D. Vilella, P. Felock and D. Hazuda, J. Nat. Prod., 2001, 64, 874–882 CrossRef CAS PubMed.
- S. Farag, R. M. Bleich, E. A. Shank, O. Isayev, A. A. Bowers and A. Tropsha, Bioinformatics, 2019, 35, 3584–3591 CrossRef CAS PubMed.
- W.-H. Chen, K. Li, N. S. Guntaka and S. D. Bruner, ACS Chem. Biol., 2016, 11, 2293–2303 CrossRef CAS PubMed.
- J. M. Reimer, M. Eivaskhani, I. Harb, A. Guarné, M. Weigt and T. M. Schmeing, Science, 2019, 366, eaaw4388 CrossRef CAS PubMed.
- E. Dehling, J. Rüschenbaum, J. Diecker, W. Dörner and H. D. Mootz, Chem. Sci., 2020 10.1039/D0sc01969k.
- K. Bloudoff, D. A. Alonzo and T. M. Schmeing, Cell Chem. Biol., 2016, 23, 331–339 CrossRef CAS PubMed.
- S. Kosol, A. Gallo, D. Griffiths, T. R. Valentic, J. Masschelein, M. Jenner, E. L. C. de los Santos, L. Manzi, P. K. Sydor, D. Rea, S. Zhou, V. Fülöp, N. J. Oldham, S.-C. Tsai, G. L. Challis and J. R. Lewandowski, Nat. Chem., 2019, 11, 913–923 CrossRef CAS PubMed.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Mol. Biol., 2002, 9, 522–526 CAS.
- M. J. Tarry, A. S. Haque, K. H. Bui and T. M. Schmeing, Structure, 2017, 25, 783–793 CrossRef CAS PubMed , e784.
- J. B. Patteson, Z. D. Dunn and B. Li, Angew. Chem., Int. Ed., 2018, 57, 6780–6785 CrossRef CAS PubMed.
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383–387 CrossRef CAS PubMed.
- E. J. Culp, N. Waglechner, W. Wang, A. A. Fiebig-Comyn, Y.-P. Hsu, K. Koteva, D. Sychantha, B. K. Coombes, M. S. Van Nieuwenhze, Y. V. Brun and G. D. Wright, Nature, 2020, 578, 582–587 CrossRef CAS PubMed.
- Y. Zhao, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades and M. J. Cryle, J. Org. Chem., 2019, 85, 1537–1547 CrossRef PubMed.
- N. Waglechner, A. G. McArthur and G. D. Wright, Nat. Microbiol., 2019, 4, 1862–1871 CrossRef CAS PubMed.
- J. Tailhades, M. Schoppet, A. Greule, M. Peschke, C. Brieke and M. J. Cryle, Chem. Commun., 2018, 54, 2146–2149 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Primer sequences and template DNA for construct design; analysis of protein purification; peptidyl-CoA analysis; analysis of biochemical assays including E-domain epimerisation experiments. See DOI: 10.1039/d0sc03483e |
| This journal is © The Royal Society of Chemistry 2020 |