
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic engineering of single-atom nanozymes for enzyme-like catalysis
Weiwei
Wu†
ab,
Liang
Huang†
ab,
Erkang
Wang
 *ab and
Shaojun
Dong
*ab
*ab and
Shaojun
Dong
*ab
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: ekwang@ciac.ac.cn; dongsj@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, Anhui 230026, China
First published on 11th August 2020
Abstract
Enzyme mimics, especially nanozymes, play a crucial role in replacing natural enzymes for diverse applications related to bioanalysis, therapeutics and other enzyme-like catalysis. Nanozymes are catalytic nanomaterials with enzyme-like properties, which currently face formidable challenges with respect to their intricate structure, properties and mechanism in comparison with enzymes. The latest emergence of single-atom nanozymes (SAzymes) undoubtedly promoted the nanozyme technologies to the atomic level and provided new opportunities to break through their inherent limitations. In this perspective, we discuss key aspects of SAzymes, including the advantages of the single-site structure, and the derived synergetic enhancements of enzyme-like activity, catalytic selectivity and the mechanism, as well as the superiority in biological and catalytic applications, and then highlight challenges that SAzymes face and provide relevant guidelines from our point of view for the rational design and extensive applications of SAzymes, so that SAzyme may achieve its full potential as the next-generation nanozyme.
Introduction
Natural enzymes are powerful biocatalysts and widely used in different fields for their prominent catalytic activities and specificities.1–3 However enzymes are mostly composed of proteins and bioorganic molecules, and the enzymatic activity and stability are highly dependent upon the mild reaction conditions and environments. These intrinsic drawbacks of enzymes as well as high cost dramatically hinder their practical application.4–6 To address these limitations, enormous efforts have been devoted to exploring artificial enzyme mimics over the past few decades.7–10 Nanozymes, the nanomaterials with enzyme-like properties, possess the advantages of high stability, low cost and mass production compared to natural and artificial enzymes.11–16 This ground-breaking research field originated from the unexpected discovery of the peroxidase mimicking activity of magnetic Fe3O4 nanoparticles by Yan and coworkers in 2007,17 and was then clearly defined by Wei and Wang in the first comprehensive review in 2013.18 Since then, nanozymes have attracted enormous interest from various fields, and related research has been growing exponentially. Up to now, more than 200 research laboratories around the world have been working on nanozymes revealing the great value and significance of this field.19Nanozymes are nanomaterial-based enzyme mimics that catalyze the conversion of substrates to products under physiological conditions following the catalytic pathway of natural enzymes. With the rapid development of nanotechnology, over 50 kinds of diverse nanomaterials have been found with different enzyme-like catalytic activity, including noble-metal nanocrystals, transition metal oxides, sulfides, selenides, nitrides, and phosphides, carbon-based nanomaterials, polymer–metal complexes and metal–organic frameworks (MOFs).20–30 Most of which belong to oxidoreductase- and hydrolases-mimics, such as oxidase, peroxidase, catalase, superoxide dismutase, nuclease, phosphatase and so forth. Moreover, the inherent physicochemical properties (magnetic, optical, thermal and electrical properties) of these nanomaterials integrated with the unique enzyme-like activities will endow them with extensive potential compared to natural enzymes. They have been adequately investigated for diverse applications in bioimaging, biosensing, therapeutics, tissue engineering and environmental toxicology.31–41 However, the progress or barrier of nanozymes is totally due to the nature of the nanomaterial itself. The enzyme-like catalysis for nanozymes essentially is a kind of heterogeneous catalysis between a nanomaterial-based catalyst and substrates. Thus the crucial assessment criteria of the activity and selectivity of nanozymes are mainly affected by the nanostructure and composition of the nanomaterials. These factors give rise to two inevitable challenges. First, the size-, shape-, facet-and coordination-dependent enzyme-like activities of nanozymes are always inconsistent and hard to regulate and control. Meanwhile, only small amounts of active atoms on the surface of nanozymes contribute to the enzyme-like catalysis, which lead to the low active site density and atomic utilization efficiency as well as catalytic activity. Second, due to the synthetic strategies and the solid spatial structures of the nanomaterials, nanozymes commonly exhibit inhomogeneous elemental distribution and various facet structures on the surface. These not only give rise to the complicated catalytic mechanisms, but also result in multiple catalytic pathways and enzyme-like activities.42–48 Thus it is extremely difficult to promote the enzyme-like specificity, distinguish the actual active sites and as well as investigate the origin of enzyme-like activity for nanozymes. It is urgent for nanozyme technologies to get out of the current predicament.49
Fortunately, since the groups of Flytzani-Stephanopoulos, Gates, and Zhang successively demonstrated the unexpected performance of single-atom catalysts (SACs), the nanotechnologies have entered the atomic age.50–52 The emerging SACs can not only reduce the negative effects of the physicochemical parameters such as the local facet effect of nanomaterials, but can also uncover the intrinsic active sites.53–58 Furthermore, the atomically dispersed metal sites with simple coordination structures are quite similar to the active centers of the metalloenzymes, which endows them with ultrahigh activity of each site.59–63 Inspired by the specific spatial structures of SACs and metalloenzymes, we put forward the new concept of single-atom nanozymes (SAzymes), which integrated the state-of-the-art single-atom technologies with inherent active sites of enzyme mimics (Fig. 1). Then we demonstrated the great potential and feasibility of FeN5 SAzymes as highly active and stable oxidase mimics.64 In the meantime, some other SAzymes with superior catalytic activities were also reported with diverse enzyme-like properties, and biological and catalytic applications.65–69 These dramatic results indicate that the emerging SAzymes are important breakthroughs and promising next-generation nanozymes.70,71
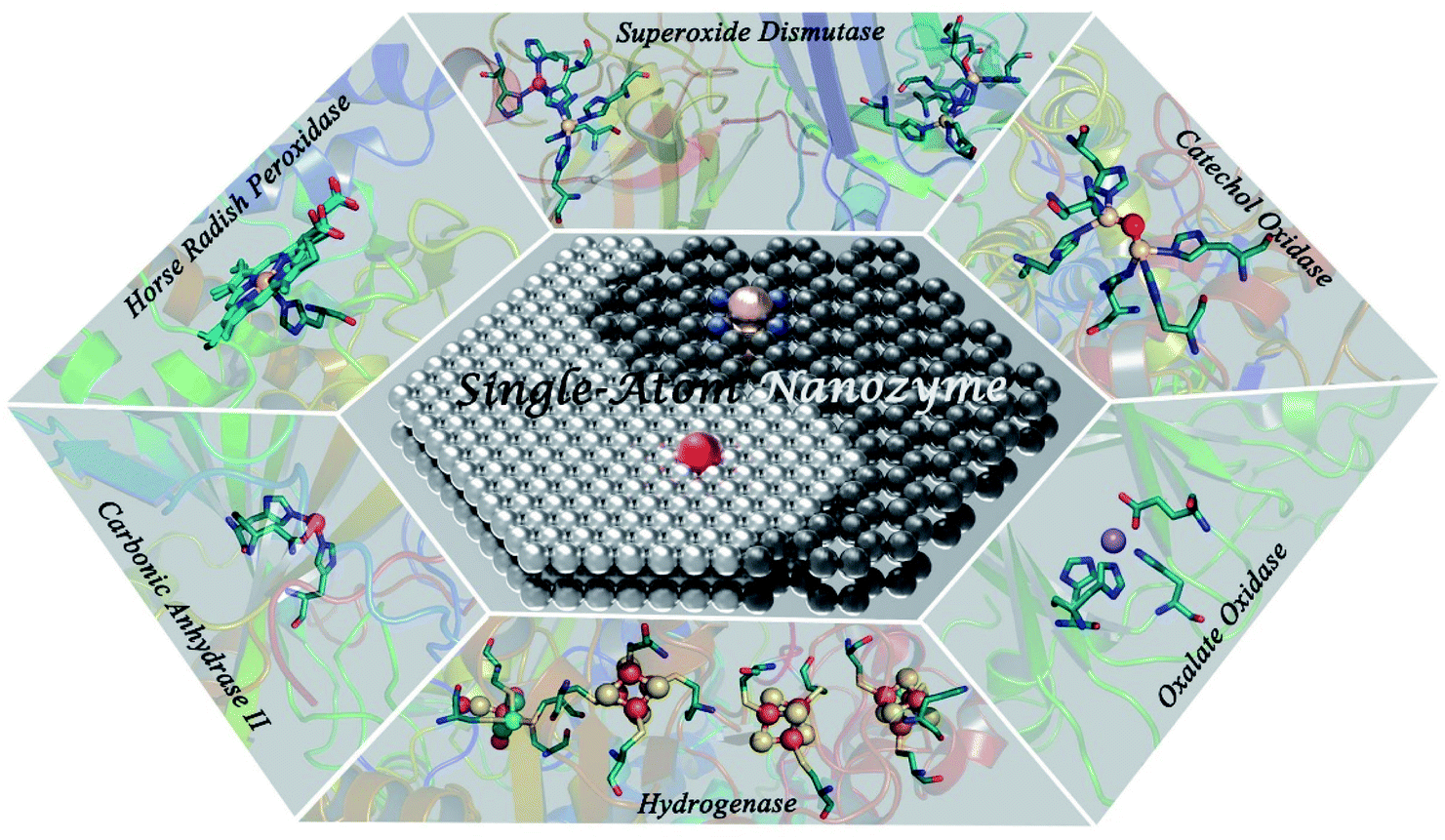 | ||
| Fig. 1 Schematic illustration of the resembling atomically dispersed active centers between SAzymes and various representative types of metalloenzymes. | ||
Therefore, in this perspective, we would like to provide a systematic and timely review of the impressive progress and achievements of SAzymes, which aims to provide deep insight into the unique attributes and great potential of SAzymes, and in turn promote their further development and extensive applications. Firstly, we will highlight the advantages of SAzymes with respect to the specific coordination structure and catalytic mechanism, which are derived from the intrinsic properties of SACs. Then, the synergetic enhancement of the enzyme-like activities and catalytic selectivity of SAzymes, as well as the consequent superiority in biological applications will be discussed in detail. Finally, the current challenges faced by SAzymes are outlined and our viewpoints on boosting this exciting research topic are suggested.
Structural advantages of single-sites
As mentioned above, the enzyme-like catalysis of nanozymes is a special kind of heterogeneous catalytic reaction. Compared to natural enzymes, the activity and selectivity of enzyme mimics are the most important parameters, which are heavily dependent on the structural and atomic properties of the nanomaterials.18,19 However for those nanozymes with inherent enzymatic properties, the activities are primarily derived from the very few highly-active sites on the nanomaterial surface, such as the coordinatively unsaturated atoms in the sites of steps, edges, kinks and defects, and so on.22,72 These factors are difficult to distinguish and quantitatively control, thus resulting in the ultralow enzyme-like catalytic activity and extremely complex mechanism compared with natural enzymes. To further boost the catalytic performance, researchers decreased the size of nanomaterials to increase the number of surface atoms and active sites. When further downsizing the nanoparticles to the atomic level, namely single atoms, the geometric and electronic properties of the SACs are distinctly different from nanomaterials, which only contain isolated metal atoms anchored on substrates and served as the active centers along with the coordination atoms.57 The SACs with a maximum atom utilization and simplest atomic structure always exhibited definite active centers, an unambiguous catalytic mechanism and superior catalytic performance. Meanwhile, SACs also bridged the gaps between homogeneous and heterogeneous catalysis, and broke through the limitations in material design. Therefore, developing SAzymes based on the SAC technology will provide us a promising new perspective in nanozyme research. In this case, SAzymes will have atomically dispersed sites with a definite coordination structure. We can clearly identify the active sites, generally isolated metal atoms coordinated with non-metallic moieties on the supports, and in turn rationally design powerful SAzymes. Furthermore, well-defined configuration of active sites allows us to investigate the catalytic mechanism in depth (Fig. 2a). | ||
| Fig. 2 (a) Fe/Mo centers in natural sulfite oxidase enzyme and simulated active centers in single-atom Fe doped MoS2. Reproduced from ref. 74 with permission from Elsevier. (b) HAADF-STEM image of FeN4/GN-2.7. (c) Low-temperature scanning tunneling microscopy (LS-STM) image of FeN4/GN-2.7, measured at a bias of 1.0 V and a current (I) of 0.3 nA (2 nm × 2 nm). (d) Simulated STM image for (c). Reproduced from ref. 76 with permission from the American Association for the Advancement of Science. | ||
Benefiting from the booming SAC technology, it is possible for us to synthesize SAzymes with a desired center metal atom and coordination structure controllably as well as to optimize the catalytic performance. And the density of active sites is also controllable and adjustable. Furthermore, based on the advanced electron microscope and spectrum technologies with atomic resolution, such as spherical aberration corrected transmission electron microscopy (ACTEM), scanning tunneling microscopy (STM), X-ray absorption spectroscopy (XAS) and so on, we can directly observe the atomic distribution and clearly characterize the spatial structure of active centers (Fig. 2b–d).73–76 These technologies are important guarantees for SAzyme research. Size-controllable SAzymes with exceptional oxidase-mimic activity by pyrolysis of Fe–Zn bimetallic ZIFs were prepared by Chen et al.73 The oxidase-mimic activity could be regulated by changing the ratio of methanol to metal precursors and the calcination temperature. Based on the synergistic effects of the degree of graphitization, particle size-dependent surface areas and exposure level of active sites, an optimal SAzyme was prepared with a molar ratio of methanol to Zn2+ of 1320![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and pyrolysis at 900 °C. In natural sulfite oxidase enzyme, Fe and Mo active sites work synergistically for sulfite oxidation. Inspired by this, Huang et al. prepared single Fe atoms confined in MoS2 nanosheets, denoted as FexMo1−xS2, to simulate the active sites in natural sulfite oxidase enzyme for sulfite activation and oxidative degradation of propranolol (PPA).74 The activity initially was enhanced with increasing Fe content, while a decrease was observed if the Fe content was excessive, indicating that both Fe and Mo were crucial for the activity. Acting like natural sulfite oxidase enzyme, the Fe atoms confined in two-dimensional nanosheets worked collaboratively with Mo atoms in the MoS2 nanosheets for sulfite activation via Fe2+/Fe3+ or Mo4+/Mo5+/Mo6+ redox cycles. They are efficient for PPA degradation with ∼90% degradation efficiency within only 30 min at an optimal pH 4.0, proving to be an efficient enzyme counterpart.
:1 and pyrolysis at 900 °C. In natural sulfite oxidase enzyme, Fe and Mo active sites work synergistically for sulfite oxidation. Inspired by this, Huang et al. prepared single Fe atoms confined in MoS2 nanosheets, denoted as FexMo1−xS2, to simulate the active sites in natural sulfite oxidase enzyme for sulfite activation and oxidative degradation of propranolol (PPA).74 The activity initially was enhanced with increasing Fe content, while a decrease was observed if the Fe content was excessive, indicating that both Fe and Mo were crucial for the activity. Acting like natural sulfite oxidase enzyme, the Fe atoms confined in two-dimensional nanosheets worked collaboratively with Mo atoms in the MoS2 nanosheets for sulfite activation via Fe2+/Fe3+ or Mo4+/Mo5+/Mo6+ redox cycles. They are efficient for PPA degradation with ∼90% degradation efficiency within only 30 min at an optimal pH 4.0, proving to be an efficient enzyme counterpart.
Taking advantage of the definite structure of SAzymes, reasonable catalytic models were built, and the mechanism of simulating enzyme activity could be investigated clearly by combining density functional theory (DFT) calculations. This is of great significance for understanding the structure–activity relationship between the active centers and enzymatic properties, which is crucial for the rational design of multifarious SAzymes and improving the activity and selectivity.75,76 A comprehensive study was performed by Wang et al. to elucidate the mechanism of the structure-dependent oxidase-like activity of Fe–N/C.77 They synthesized a series of single atom nanomaterials and investigated the catalytic mechanism systematically. The results revealed that Fe–N3/C was a preferable oxidase mimic compared with the Fe–C3 model and other Fe–N/C models with different nitrogen coordination numbers. And they further made a comparison between the Fe–N3/C model and other M–N3/C models (M = Co, Ni) to illuminate the mechanism. They explored end-on and side-on O2 adsorption configurations on Fe atoms and revealed that the O2 molecule preferred to adsorb on the Fe–C3 and Fe–N3/C by side-on adsorption configuration while the end-on configuration tended to occur on the four- or five-coordinated Fe atom. Fe–N3/C had a minimum adsorption energy and showed the highest chemical reactivity towards O2. They investigated the relationship between oxidase-like activity, the Fe–N site content and the single Fe content, and found that the activity was related to the Fe–N site content rather than the Fe content, indicating that Fe–N sites were the active sites instead of the isolated Fe atoms. The Fe–N sites exhibited oxidase-like activity via a 4e− reduction pathway and free radical capture experiment confirmed that O2˙− was the main active species in the reaction. The reaction path was deciphered as follows: O2 was adsorbed on the surface of the catalysts and dissociated into two O* atoms, which attracted H+ from solvent to form OH* species. The N atom around the Fe site also attracted H+ from the solvent and generated H* species, which transferred to OH* species to generate H2O subsequently. The H2O finally desorbed from the catalytic site and revived the catalysts. They elucidated that the oxidase-like activity was closely related to the reaction energies of 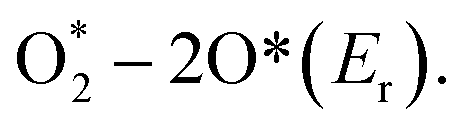 Compared with other models and other M–N3/C models, Fe–N3/C had minimum Er, coincided well with the highest activity. At this point, the catalytic mechanism was investigated thoroughly at the atomic-level benefitting from the definite construction of SAzymes, which is hard to be realized in nanoparticles. Zhao, Xiong and Liu prepared single iron SACs with high peroxidase, oxidase and catalase mimic activity like nature heme-containing enzymes.78 In spite of the low Fe content of 1.2 wt%, the Fe–N–C nanozymes exhibit high enzyme-like activity due to the structural similarity with natural enzymes. And they unveiled the mechanisms of enzyme simulation by operando XAFS spectroscopy, time-resolved quick-scanning XAFS (QXAFS) and DFT calculations. The energy profiles of catalysts and reaction intermediates in peroxidase-like catalysis are depicted in Fig. 3c; H2O2 can form an Fe
Compared with other models and other M–N3/C models, Fe–N3/C had minimum Er, coincided well with the highest activity. At this point, the catalytic mechanism was investigated thoroughly at the atomic-level benefitting from the definite construction of SAzymes, which is hard to be realized in nanoparticles. Zhao, Xiong and Liu prepared single iron SACs with high peroxidase, oxidase and catalase mimic activity like nature heme-containing enzymes.78 In spite of the low Fe content of 1.2 wt%, the Fe–N–C nanozymes exhibit high enzyme-like activity due to the structural similarity with natural enzymes. And they unveiled the mechanisms of enzyme simulation by operando XAFS spectroscopy, time-resolved quick-scanning XAFS (QXAFS) and DFT calculations. The energy profiles of catalysts and reaction intermediates in peroxidase-like catalysis are depicted in Fig. 3c; H2O2 can form an Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate on the Fe–N4 site with releasing a H2O molecule. Subsequently, another H2O2 dissociates on the other side of Fe–N4 and formed an OFeO intermediate. An O species of the OFeO intermediate attracted the H atom from TMB by forming an O–H bond and turned OFeO into OFeOH and oxidized TMB to oxTMB. OFeOH also could attract a H atom from TMB and generated a H2O molecule and another oxTMB. In Fig. 3c we can observe that the total change of energy is −2.80 eV, meaning a thermodynamically favorable reaction. Guo et al. developed a single Mn atom catalyst dispersed on graphitic carbon nitride (g-C3N4) exhibiting superior catalytic activity for the peroxone reaction under acidic conditions.79 X-ray absorption fine structure (EXAFS) and DFT calculations confirmed that Mn atoms worked as efficient sites in the form of Mn–N4 configurations, showing superior catalytic activity even better than that of the homogeneous MnCl2 catalyst. Different catalytic mechanisms from the conventional peroxone reaction were uncovered. The ˙OH was generated from the breakage of the HOO–Mn–N4 bond rather than HO2− anion relevant free radical chain reactions. The HOO–Mn–N4 was formed by the absorption of H2O2 by Mn–N4 sites with the release of a proton, and the breaking to generate HO2˙ and MnI–N4was an endothermal reaction. The addition of O3 helped the breaking of the HOO–Mn–N4 bond and then generated HO2˙, O3˙− and MnII–N4, which is an exothermal reaction (Fig. 3d). O3˙− combined with H+ from the acidic solvent and converted to ˙OH subsequently. O2˙− originated from HO2˙ would generate O3˙− by reacting with O3. The proposed catalytic mechanism is shown in Fig. 3e.
O intermediate on the Fe–N4 site with releasing a H2O molecule. Subsequently, another H2O2 dissociates on the other side of Fe–N4 and formed an OFeO intermediate. An O species of the OFeO intermediate attracted the H atom from TMB by forming an O–H bond and turned OFeO into OFeOH and oxidized TMB to oxTMB. OFeOH also could attract a H atom from TMB and generated a H2O molecule and another oxTMB. In Fig. 3c we can observe that the total change of energy is −2.80 eV, meaning a thermodynamically favorable reaction. Guo et al. developed a single Mn atom catalyst dispersed on graphitic carbon nitride (g-C3N4) exhibiting superior catalytic activity for the peroxone reaction under acidic conditions.79 X-ray absorption fine structure (EXAFS) and DFT calculations confirmed that Mn atoms worked as efficient sites in the form of Mn–N4 configurations, showing superior catalytic activity even better than that of the homogeneous MnCl2 catalyst. Different catalytic mechanisms from the conventional peroxone reaction were uncovered. The ˙OH was generated from the breakage of the HOO–Mn–N4 bond rather than HO2− anion relevant free radical chain reactions. The HOO–Mn–N4 was formed by the absorption of H2O2 by Mn–N4 sites with the release of a proton, and the breaking to generate HO2˙ and MnI–N4was an endothermal reaction. The addition of O3 helped the breaking of the HOO–Mn–N4 bond and then generated HO2˙, O3˙− and MnII–N4, which is an exothermal reaction (Fig. 3d). O3˙− combined with H+ from the acidic solvent and converted to ˙OH subsequently. O2˙− originated from HO2˙ would generate O3˙− by reacting with O3. The proposed catalytic mechanism is shown in Fig. 3e.
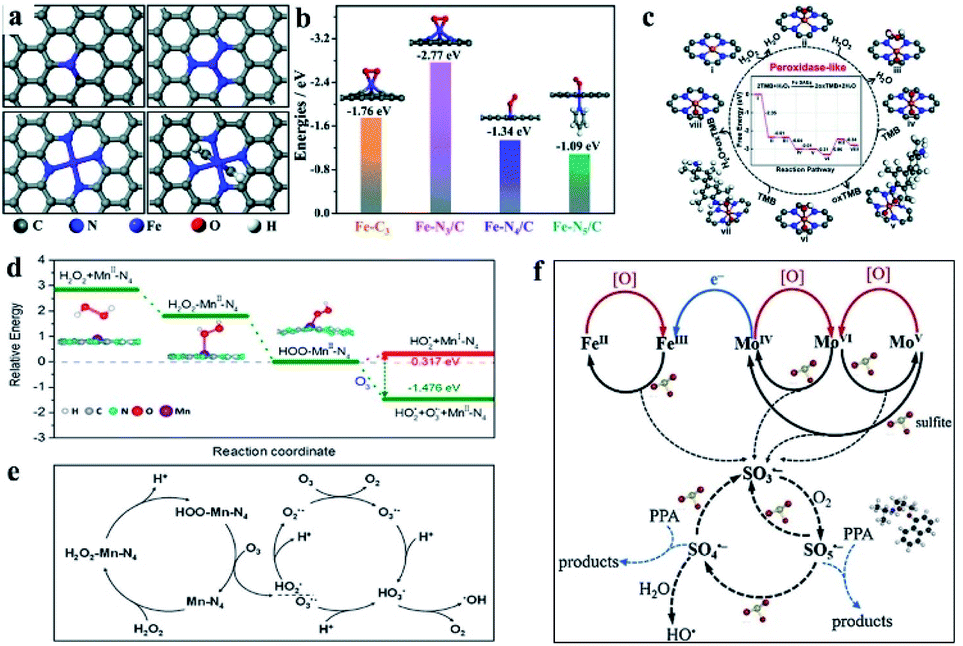 | ||
| Fig. 3 (a) The potential oxidase-like model of Fe–N/C (top left: Fe–C3, top right: Fe–N3/C, bottom left: Fe–N4/C, and bottom right: Fe–N5/C). (b) Adsorption energies of O2 adsorption on Fe–C3, Fe–N3/C, Fe–N4/C, and Fe–N5/C with optimized structures. (c) Optimized geometric structures and free energy profiles of various intermediates in the oxidase-like reaction. (a and b) and (c) reproduced from ref. 77 and 78 with permission from the Royal Society of Chemistry, respectively. (d) Energy change plot for different steps and (e) the reaction mechanism in the Mn–N4 site-catalyzed peroxone reaction. Reproduced from ref. 79 with permission from the American Chemical Society. (f) Sulfite activation mechanism on FexMo1−xS2. Reproduced from ref. 74 with permission from Elsevier. | ||
Considering single-atom dual-site catalysts, the atomically dispersed SACs were also favorable for understanding the mechanism in enzyme-like catalytic reactions. In the Mo–Fe single atom dual-site nanozyme mentioned above, it was confirmed that the catalytic activation of sulfite was on account of the loop of Fe2+/Fe3+ in the Fe center and Mo4+/Mo5+/Mo6+ in the Mo center. SO5˙− is the major ROS in the reactions proved by contrast experiments of free radical scavenging and ESR spectra. Combining the facts observed in the experiments with the DFT calculations, a synergistic mechanism of sulfite activation catalyzed by Mo–Fe was proposed as shown in Fig. 3f. The electron transport between Mo and Fe in MoS2 facilitated the respective loop of Fe and Mo, resulting in a better catalytic performance of FexMo1−xS2.74 In non-carbon support anchored SAzymes, the catalytic mechanism was also investigated clearly for both single atoms and the supports by Zhao and Cui's groups.80 The heterogeneous single-atom Co–MoS2 was adopted as a proof-of-concept nanozyme, and the electron transfer mechanism favored by the single Co atom and Fenton-like reaction dependent mechanisms favored by MoS2 supports were uncovered unambiguously.
In general, the SAzymes belong to single-site heterogeneous catalysts, and their single sites resemble the active centers in homogeneous molecular catalysts, which are spatially isolated, structurally well-characterized, and exert the same interaction energy for each reactant or zymolyte. Therefore, SAzymes will possess the integrated structual advantages of both homogeneous and heterogeneous catalysts, which are far beyond nanomaterial-based nanozymes.54 For example, even uniform or mono-crystalline nanozymes with unified surface facets still have multiple adsorption and reaction sites, which result in diverse enzyme-like activities and catalytic pathways, in turn deeply blurring the actual active sites and catalytic mechanisms. However for SAzymes with the definite and simple single-site, the enzyme-like properties and mechanism are much easier to deduce and confirm.
However, the SAzyme research and even SAC research are in the startup phase; there are still many difficulties to break through before fully utilizing the crucial advantages of the single-atom structure. The following two aspects should be studied emphatically in future research. Firstly, more effective and controllable synthetic strategies for SAzymes need to be developed to further improve the uniformity of the single-atom structure and distribution. Currently, SAzymes are mainly synthesized through bottom-up strategies, and the adsorption and coordination of metal atoms were uncontrollable, which tended to aggregate rather than atomically disperse. Hence, we should lower the non-selective dispersion and enhance the controllability of the single-atom sites to improve the structural and dispersion homogeneity. For instance, complexing the single-atoms to functional organic ligands (such as porphyrin, pyridine, imidazole and sulfydryl groups) and coordinatively unsaturated metal nodes of MOFs, or confining the single-atoms to metal defect sites and non-metallic vacancies (such as N, O, S, and C vacancies) of transition-metal compounds to guarantee the stabilization and definition of the atomic active sites of SAzymes can be carried out.81–83 Secondly, the SAzyme categories with different coordination structures and enzyme-like properties need to be extended, which largely depend on the central metal atoms and neighboring atoms.61,84 However, most reported SAzymes were metal–nitrogen active sites derived from the uncontrollable pyrolysis of metal–organic complexes. Although these SAzymes exhibited high redox activities, the enzyme-like properties were bound in peroxidase or oxidase-like mimics. Through imitating the precise synthesis of nanocrystals, divided into top-down and bottom-up methods, further development of more novel and controllable synthetic strategies to expand the diversity of SAzymes, as well as the enzyme-like mechanisms and applications, can be achieved.
High catalytic activities
Apart from the specific spatial structures and the derived advantages in mechanism investigation, the enzyme-like activity is also a crucial factor for a type of SAzyme. As proved in numerous reports of single-atom catalysis, the SACs were much superior to nanomaterials in both atomic coordination and catalytic activity when the metal sites presented an atomically dispersed state. This is not only because of the maximum atom utilization and active site density, but is also attributed to the high intrinsic activity of each individual metal center. Similarly, the apparent activities of SAC-based SAzymes were far beyond those of nanomaterial-based nanozymes, especially in oxidoreductase-like catalysis, although the metal loading of SAzymes was quite low. For instance, Dong's group designed oxidase-like SAzymes with carbon nanoframe-confined Fe–N–C active centers through a bottom-up strategy.64 With the structural characterization by Fe K-edge X-ray absorption near edge structure (XANES) and Mössbauer spectroscopy, they deduced the atomic structure of the synthesized SAzymes to be FeN5 (Fig. 4a). The Fe–N–C active centers resemble the active sites of natural cytochrome P450, an axial N containing ligand-coordinated heme. The definite structure of synthesized SAzymes (FeN5 SA/CNF) equipped it with an electron push-effect and crucial synergistic effects, creating ultrahigh oxidase-mimic activity (Fig. 4b). In spite of the extremely low Fe content of 1.2 wt%, it's activity is higher than that of other FeNx SACs, FePc and most reported nanoparticles with oxidase-like characteristics, such as CeO2, Fe3O4, MnO2, CuO, Au, Pd, Pt, and Prussian blue. The oxidase-mimic activity was 70 times higher than that of commercial Pt/C and the oxidase-like catalytic rate constant was more than 30 to 1000 times greater than that of nanomaterial-based nanozymes. The definite structure, high density of active sites and maximal atom utilization efficiency ensured the highest oxidase-like activity. In addition, the oxidase-mimic activity of FeN5 SA/CNF maintained at the lowest 90% after being exposed to 21 hour treatment of a strong acid (alkali), indicating the stability of FeN5 species stabilized by the coordination of N atoms on the supports. Kim et al. fabricated single-atom iron sites embedde graphene (Fe–N-rGO) by heat treatment of the mixture of GO and an Fe precursor in an NH3 atmosphere at 750 °C.85 The sites were confirmed to be Fe–N4; the four N atoms form a square-planar structure with the Fe atom at the center, resembling the heme cofactor present in natural horseradish peroxidases (HRP) (Fig. 4c). When Fe or N is doped alone, or Fe was replaced by another transition metal, there is almost no increase in activity compared with rGO, showing the necessity of mimicking the essential cofactor structure of natural enzymes (Fig. 4d). The peroxidase-like activity of the as prepared Fe–N-rGO SAyzme was measured by the oxidation of 3,3,5,5-tetramethylbenzidine (TMB) in the presence H2O2, which was up to ≈700 fold higher than that of undoped rGO even with the weight percentage of Fe being only 1.8 wt%. When it comes to relative activity per iron atom, the Fe–N-rGO SAzyme exhibits 5 million times higher activity compared with Fe3O4 NPs, and even higher than that of the Pt atom in Pt NPs. Moreover, Fe–N-rGO showed effective peroxidase-mimic activity with higher kcat/Km compared with HRP. The unprecedented activity of Fe–N-rGO was attributed to its single-atom dispersed Fe–N4 steric configuration similar to nature enzymes. Wang and Liang's groups reported that single Pt atoms anchored on Te nanowires were used to catalyze the dehydrogenation of formic acid.86 Platinum with 1.1% mass loading on Te nanowires was proved to be atomically dispersed, while increasing the Pt mass loading to 4.6% and 32.0% led to the coexistence of Pt single atoms and Pt nanoclusters on nanowires (4.6% Pt/Te) and Pt nanocrystals supported Te nanowires (32.0% Pt/Te), respectively. At room temperature, the dehydrogenation of HCOOH was enhanced by 1.1% Pt/Te via a plasmon-enhanced catalytic process. The turnover frequency number was 3070 h−1, which is 2.5 and 5.4 times higher than that of 4.6% Pt/Te and 32.0% Pt/Te, respectively, even 8.5 times higher than that of Pt/C. The single Fe atom contained Fe SAE SAzymes reported by Zhao et al. possessed peroxidase, oxidase and catalase enzyme-like activities simultaneously. The peroxidase-like activity was about 40 times higher than that of Fe3O4, even when the content of Fe was 1.2 wt%.78 Similar work has been reported by Chen and Shi's groups. The Vmax of the as prepared Fe SAzymes used in the reaction of TMB and H2O2 was 76.4 fold higher than that of Fe3O4 at the same mass concentration.68 Another study revealed that the turnover number of SAzymes was 4500 times more than that of Fe3O4.87 Zhang's group reported single Fe atom catalysts with FeNx (x = 4–6) species by using MgO as the sacrificial template and an Fe(phen)x (phen = 1,10-phenanthroline) complex as the precursor.61 The relative concentration of FeNx species is related to the pyrolysis temperature, which resulted in different catalytic performances for each sample. For the oxidation of C–H bonds with ethylbenzene as the substrate, the Fe–N–C-700 (pyrolysis at 700 °C) sample had the highest turnover frequency among the three catalysts, attributed to the highest content of FeN5 species. Poisoning experiments by potassium thiocyanate (KSCN) demonstrated that FeN5 was the most active moiety as well, followed by FeN4, and FeN6 was the least active one. The activity of FeN5 was at least 1 order higher than that of Fe–N6 and Fe–N4 species. Among these three FeNx species, FeN5 configuration is most similar to that of natural enzymes with coordination of four nitrogen atoms in the plane and axial coordination of a nitrogen-containing ligand. The unique construction endows it with high intrinsic activity compared to others. For the oxidation of ethylbenzene, 98% conversion was achieved within 5 h. Compared with other catalysts reported previously, Fe–N–C has the highest activity in the oxidation of hydrocarbons at room temperature. | ||
| Fig. 4 (a) Illustration of FeN5 SA/CNF-catalyzed TMB oxidation. (b) Ultrahigh oxidase-like activity of FeN5 SA/CNF. Reproduced from ref. 64 with permission from the American Association for the Advancement of Science. (c) Structural similarity of Fe–N4 sites in Fe–N-rGO and the cofactor of HRP. (d) Comparison of the peroxidase-like activity of Fe–N-rGO and control groups. Reproduced from ref. 85 with permission from John Wiley & Sons. | ||
Furthermore, substrate specificity (Km), the maximal reaction rate (Vm), the catalytic rate constant (kcat) and the catalytic efficiency (kcat/Km) were usually used to compare the catalytic characteristics of nanozymes quantitatively. Yan et al. presented a protocol for measuring and defining the catalytic activity units and kinetics for peroxidase nanozymes.72 The kinetics parameters Vm and Km were calculated by fitting the reaction velocity values and the substrate concentrations to the Michaelis–Menten equation as follows: V = (Vm × [S])/(Km + [S]). kcat was obtained from the equation kcat = Vm/[E]. [E] is the nanozyme concentration (M). We took the most researched peroxidase-like SAzymes and TMB to compare the catalytic activity of single-atom nanozymes and natural enzymes and listed them in the tables above (Tables 1 and 2). As we can see, the Km values of SAzymes on TMB were at the same level as that of HRP, indicating similar affinity toward TMB. But the Km values on H2O2 were much higher than that of HRP, meaning that a higher concentration of H2O2 was required to obtain maximum reaction rates, and SAzymes could tolerate high concentration H2O2. When taking [E] as the molar concentration of single-atom nanozymes, the catalytic efficiency was much higher than that of HRP. However, when we calculate the kinetic constants with [E] as the molar concentration of the metal in the SAzymes, the catalytic efficiency of some SAzymes was inferior compared with HRP.
| Sample | [E] (M) | Substrate | K m (mM) | V m (μM s−1) | k cat (s−1) | k cat/Km (mM−1 s−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a [E] is the concentration of nanozymes. b [E] is the concentration of the metal in nanozymes. | |||||||
| PMCSa | 2.147 × 10−14 | TMB | 0.224 | 0.1066 | 5.66 × 106 | 2.5268 × 107 | 66 |
| H2O2 | 40.160 | 0.1215 | 4.97 × 106 | 1.2376 × 105 | |||
| Fe–N-rGO sheetsa | 1.2 × 10−11 | TMB | 0.074 | 1.74 | 1.45 × 105 | 1.9595 × 106 | 85 |
| H2O2 | 43 | 1.44 | 1.20 × 105 | 2.7907 × 103 | |||
| Fe–N–C SANa | 1.6 × 10−11 | TMB | 0.08 | 0.7452 | 4.66 × 104 | 5.8219 × 105 | 67 |
| H2O2 | 28.3 | 0.4285 | 2.68 × 104 | 9.4633 × 102 | |||
| CNT/FeNCb | 1.7 × 10−7 | TMB | 0.1294 | 0.156 | 0.9176 | 7.0912 | 87 |
| H2O2 | — | — | — | — | |||
| SAF NCsb | 6.07 × 10−6 | TMB | — | — | — | — | 68 |
| H2O2 | 0.0120 | 0.223 | 0.0367 | 3.0711 | |||
| Fe SAEsb | 1.07 × 10−6 | TMB | 3.92 | 0.588 | 0.5495 | 0.1402 | 78 |
| H2O2 | 0.243 | 0.0825 | 0.0771 | 0.3173 | |||
| Fe–N–Cb | 1.48 × 10−6 | TMB | 3.6 | 1.16 | 0.7838 | 0.2177 | 103 |
| H2O2 | 12.2 | 0.356 | 0.2405 | 0.0197 | |||
| Cu–N–Cb | 2.68 × 10−6 | TMB | 3.76 | 0.7505 | 0.2803 | 0.0746 | 113 |
| H2O2 | 19.94 | 0.2007 | 0.0750 | 0.0038 | |||
| Fe–N–C SAzymesb | — | TMB | 5.20 | 1.49 | — | — | 65 |
| H2O2 | 4.31 | 0.62 | — | — | |||
| Co–N–C SAzymesb | — | TMB | 5.06 | 0.190 | — | — | |
| H2O2 | 16.26 | 0.165 | — | — | |||
| Zn–N–C SAzymesb | — | TMB | 0.28 | 0.043 | — | — | |
| H2O2 | 6.27 | 0.048 | — | — | |||
| HRP | 2.5 × 10−11 | TMB | 0.434 | 0.100 | 4.00 × 103 | 9.2166 × 103 | 17 |
| H2O2 | 3.7 | 0.087 | 3.50 × 103 | 9.4595 × 102 | |||
The superior enzyme-like activity of SAzymes should be ascribed to not only the maximum atom utilization of the active centers, but also the high intrinsic activity of each single-site. Although the apparent activities of SAzymes have exceeded those of nanomaterial-based nanozymes, they still far below those of the corresponding natural enzymes.71,88 Therefore, we should greatly improve the enzyme-like activity of SAzymes through the above two aspects to meet the requirements of practical applications. Since the single-site loading rates of most SAzymes were lower than 1 wt%, there is still room for activity improvements. Through more moderate synthetic conditions and stronger metal-support interactions to dramatically increase the density and stabilize the single-sites, the fold increase of the enzyme-like activity can be realized. In addition, regulating the coordination structure of the active center, such as the coordination atom, coordination number and spatial structure, will essentially alter the catalytic properties of SAzymes, and even exhibit orders of magnitude enhancement of the enzyme-like activity. Furthermore, the definite and atomically dispersed active sites of SAzymes afford us with new opportunities to establish more suitable evaluation methods to reflect the intrinsic enzyme-like activity of each single-atom. Whether SAzymes or nanozymes, if one nanoparticle contains a number of active sites, it is inappropriate to consider the nanoparticle as the enzyme unit while ignoring the size, surface area and active site density.72 Different from the uncountable nanozymes, for the SAzymes with definite active centers and atomic density it is much easier to uniformize and quantify the enzyme-like activity, which is crucial for the activity evaluation of different SAzymes.
Enzyme-like selectivities
Enzymatic specificity is the most important feature that distinguishes natural enzymes from other chemical catalysts. They would exhibit remarkable chemo-, regio-, and stereo-selectivities for specific biochemical processes, which are dependent not only on the uniform and simple active centers, but also on the intricate and complete hierarchical structure of the protein dimensional folding configurations.89,90 Therefore, it is nearly impossible to achieve the enzyme-like specificity or selectivity by nanomaterial-based nanozymes, although that is the ultimate goal for enzyme mimics research. On the one hand, it is difficult to mimic the spatial structure of enzymes for heterogeneous nanoparticles to attain the selective adsorption, activation and desorption of the substrate molecules. On the other hand, the inhomogeneous distribution of elements and facets of the nanozymes leads to a distinctly different coordination environment and atomic structure for metal active sites, and then inevitably brings about distinguishing enzyme-like activity intensity, different catalytic mechanisms, and even multiple enzyme properties. While these atomic structures of nanozymes become simpler, such as SAzymes, things might be changed. For SAzymes with a uniform coordination structure, the metal active centers are equivalent, so the enzyme-like catalytic process and mechanism on each active site are the same owing to these reactions being inclined to the reaction paths with the lowest potential energy barrier and free energy. As a consequence, it is possible for SAzymes to obtain the catalytic selectivity for a specific substrate through altering the coordination environment to divide their reactive activations.71 For example, He et al. synthesized Pt single-atom catalysts on a N-doped porous carbon support via a precursor-dilution strategy.91 Tetraphenylporphyrin (TPP) with chelated metal cations (TPP–M) was used as the precursor and excess amount of free TPP as the diluent. By changing the concentration of TPP–M and free TPP, they could tune the surface Pt atom density in Pt/N–C. And they prepared single atoms, nanoclusters and nanoparticles of Pt catalysts (denoted as Pt/N–C, Pt-NCs/N–C and Pt-NPs/N–C respectively) at different pyrolysis temperatures. The Pt/N–C exhibited superior chemoselectivity and regioselectivity in hydrogenation reactions (Fig. 5a and b). Pt/N–C showed 99% chemoselectivity in hydrogenation of 1-nitro-4-ethynylbenzene and 1-ethynyl-4-vinylbenzene at a 20% conversion level, and commendable chemoselectivity even at a 100% conversion level. It only transformed alkyne groups into alkenyl groups and kept –NO2 and –CCH2 intact, with 99% selectivity to 1-nitro-4-vinylbenzene in the hydrogenation of 1-nitro-4-ethynylbenzene and 99% selectivity to 1,4-divinylbenzene in the hydrogenation of 1-ethynyl-4-vinylbenzene at the 20% conversion level. As expected, multiple products were produced in the hydrogenation reactions catalyzed by Pt-NPs/N–C. What is more, Pt/N–C exhibited regioselectivity in the hydrogenation of 1-ethynyl-4-(phenylethynyl)benzene and 1-(dec-1-yn-1-yl)-3-ethynylbenzene. 99% selectivity to 1-(phenylethynyl)-4-vinylbenzene and 99% selectivity to 1-(dec-1-yn-1-yl)-3-vinylbenzene at the ∼20% conversion level were realized, indicating that Pt/N–C only converted terminal alkyne to alkenyl while keeping internal alkyne intact. However, both terminal and internal alkynes were hydrogenated in the reactions catalyzed by Pt-NPs/N–C, indicating the lack of regioselectivity of nanoparticle-catalysts. The chemoselectivity was explained as the good match between the relatively low catalytic activity of Pt SACs and high reactivity of terminal alkynes, while the regioselectivity was deciphered as the possible steric hindrance effect of Pt/N–C, neither of which could be achieved by nanoparticles. The hydrogenation of unsaturated aldehydes to unsaturated alcohols with high selectivity is challenging since the hydrogenation of CC bonds is much easier than that of the CO bonds. Homogeneous catalysts could catalyze these reactions effectively but suffer from the difficulty of separation. And 100% selectivity was hard to realize by other heterogeneous catalysts. SAzymes were expected to realize selective catalysis due to their well-defined active sites. Steric hindrance around the active sites may further improve the selectivity. Lou et al. reported single atoms Rh anchored to the edges of 2D MoS2 sheets with Rh–S4 configuration (Fig. 5c and d), showing excellent selectivity in the conversion of crotonaldehyde to crotyl alcohol.92 Hydrogen molecules dissociate on the anchored Rh atoms from MoS2 edges and formed OH moieties. The OH moieties bonded to the edge Mo atoms later and formed pocket-like HO–Mo–Rh–Mo–OH configuration, with unique geometric and electronic structures like enzymes. The single Rh atoms catalysts realized 100% selectivity even at a high conversion level for the hydrogenation of crotonaldehyde to crotyl alcohol under mild reaction conditions (Fig. 5e and f). In contrast, nano-Rh/MoS2 with nanoparticles dispersed onto 2D MoS2 nanosheets showed only 47% selectivity with the main product of 1-butanol. The perfect selectivity could be attributed to the special Rh–S4 configuration formed by the edge-anchored Rh atom and S atoms in MoS2. The unique adsorption properties of positive charged Rh atoms facilitated the adsorption of the strongly polarized CO bond rather than the CC bond due to the steric effect. Back-bonding interactions between the Rh and CO π* orbital prompted the electron transfer from Rh atoms to the CO π* orbital and the weak CO bond, leading to the preferential hydrogenation. Conventional catalysts coexisting with single atoms and nanoparticles have an inherent defect for realizing the goal of 100% selectivity. Selective oxidation of the C–H bond is challenging because of the high dissociation energy of the C–H bond and probable overoxidation side reactions. The groups of Zhang and Wang demonstrated that Fe SACs were capable of selective oxidation of the C–H bond.61 Fe–N–C converted ethylbenzene to acetophenone with 97% selectivity at a 98% conversion level, and showed excellent selectivity for other aromatic hydrocarbons with electron-donating groups or electron-withdrawing groups. Lee and colleagues reported that single-atom Pt catalysts supported on titanium nitride (TiN) nanoparticles showed high selectivity for the 2e− process in electrochemical oxygen reduction.93 And lower Pt loading with higher portion of single-atom Pt showed higher selectivity, reaching 90%. Similarly, the formation of Pt–S4 configuration through sulfur-doped carbon stabilized Pt single atoms has realized 96% selectivity for producing H2O2 through the 2e− O2 reduction process, while Pt loaded on low S-content supports with clusters or nanoparticles showed selectivity far below that of single-atom Pt catalysts.94 These facts indicate that SACs have commendable selectivity comparable to that of homogeneous catalysts and enzymes.
 | ||
| Fig. 5 Catalytic performance of Pt/N–C and Pt-NPs/N–C in the hydrogenation of (a) 1-nitro-4-ethynylbenzene and (b) 1-ethynyl-4-vinylbenzene. Reproduced from ref. 91 with permission from the Nature Publishing Group. (c) HAADF-STEM images of Rh/MoS2 SAC. (d) Rh–S4 configuration of the Rh active site simulated by DFT calculations. (e) Catalytic performance and (f) stability of a Rh/MoS2 SAC. Reproduced from ref. 92 with permission from the American Chemical Society. | ||
As enzyme mimics, the performance of nanomaterial-based nanozymes was not satisfactory in specific recognition of substrates, which is a primary feature of enzymes. They always have multiple enzyme activities, lacking selectivity for the target substrate. Although molecular imprinting and chiral molecular modification were developed to improve the selectivity of nanozymes, the selectivity did not originate from the material itself and the actual selectivity was low. The Fe–N-rGO mentioned above showed excellent peroxidase-like activity without any oxidase activity.85 The sole enzyme activity of Fe–N-rGO means that it could selectively activate hydrogen peroxide rather than O2. DFT calculations indicated that the O2 dissociation reaction is endothermic on Fe–N-rGO, hindering the oxidase activity (Fig. 6b). Based on the high and selective peroxidase-mimic activity of Fe–N-rGO, trace amounts of H2O2 would be successfully quantified. Wei's group uncovered that doping the nitrogen element into graphene can dramatically enhance the peroxidase-like activity of reduced graphene oxide (rGO) by 105-fold while barely affecting oxidase, SOD and catalase activity (Fig. 6c).95 This strategy is proved to be adaptable for mesoporous carbon with more than 60-fold enhancement in peroxidase-like activity, which means that N-doping could be a general strategy for designing highly active nanozymes with specific peroxidase-like activity. It can be seen that the sole enzyme activity, which is the precondition for substrate-specificity, could be realized by N-doped tailoring. In addition to nitrogen, other elements could also be doped to improve the activity or allow them to show specific enzyme activity. Well-defined SACs, generally metal atoms with coordination of N or S, could be regulated more easily than other nanomaterials.
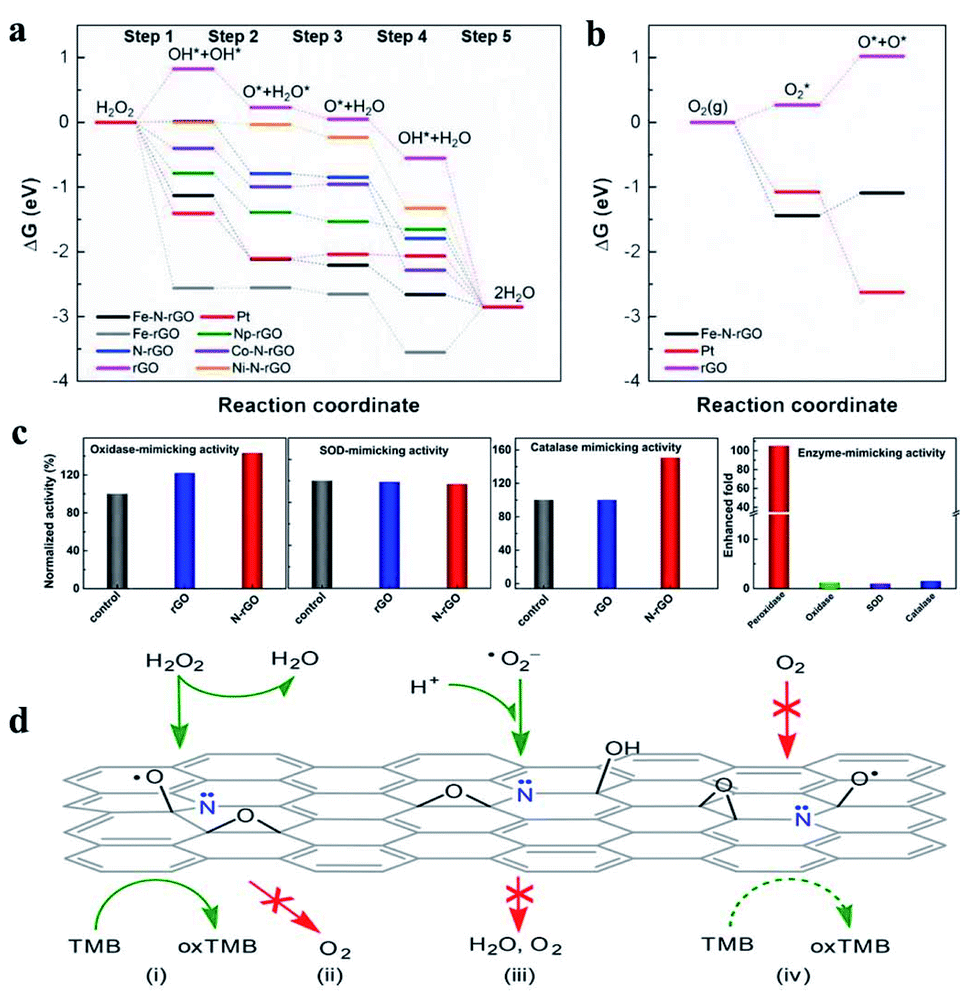 | ||
| Fig. 6 Gibbs free energy diagram of (a) peroxidase-like and (b) oxidase-like reactions on different enzyme mimics. Reproduced from ref. 85 with permission from John Wiley & Sons. (c) Specific enhancement of the peroxidase-mimicking activity of rGO by N-doping and (d) the possible mechanism obtained by DFT calculations. Reproduced from ref. 95 with permission from the American Chemical Society. | ||
The above examples have demonstrated some catalytic selectivity of SAzymes under certain conditions for heterogeneous and enzyme-like catalysis. These could be attributed to the concise single-site structures of SAzymes that resemble the active centers of enzymes and homogeneous catalysts. However, the selectivity of these SAzymes was still far less than that of natural enzymes, and most of the SAzymes even have no selectivity at all. Based on the best of our knowledge and the reported results, the ab initio prediction and rational design of highly selective single-sites, as well as an effective post-processing strategy to endow the SAzymes with tunable catalytic selectivity will be a crucial approach to promote the future nanozyme research. Firstly, bioinspired design has been proved to be an effective approach for research on both artificial enzyme mimics and SAzymes. By mimicking the active center of enzymes and constructing the derived single-site structures on SACs, the SAzymes might possess a similar enzyme-like mechanism and selectivity to natural enzymes.96 Secondly, due to the enzyme-like properties of SAzymes depending mainly upon the steric configuration of the single-sites, the features of the simple composition, definite structure and few atoms make it feasible to predict and simulate the corresponding enzyme-like selectivity of different active sites through theoretical calculations, such as first-principles, the ab initio method and DFT.97 Thirdly, the enzyme-like catalysis on SAzymes in essence is the heterocatalysis process, the selectivity arises from the differences of interaction force between single-sites and reactants, which includes the adsorption, desorption, and electron and proton transfer processes. Based on the well-established single-atom electrocatalyst and electrochemical research systems, the reaction kinetics and electrocatalytic properties of SACs could be extended to the analogous enzyme-like catalysis to filtrate highly selective SAzymes,98 for instance, the SACs with highly selectivity in four-electron or two-electron oxygen reduction reaction electrocatalysis respectively corresponding to the half-reaction of oxidase-like or glucose oxidase-like SAzyme catalysis.99,100 Shen et al. reported the catalyst of single-atom Pt with a CuSx support (h-Pt1–CuSx), which can consistently reduce O2 to H2O2 with high selectivity.99 Due to the stronger affinity between Pt–S than between Pt–O or Pt–N, the H2O2 selectivity and activity of h-Pt1–CuSx are much higher than that of other Pt single-atom catalysts. In addition, the DFT calculations show that the *O is preferentially adsorbed on the 3-fold hollow sites of Pt clusters rather than single Pt sites, and thus the O2 reduction is limited to yielding H2O2 through a 2e− process. Therefore, through altering the coordination number and single-atom support will be potential and effective strategies for regulating the inherent properties of single-atom nanozymes, which will tailor the intrinsic electronic and geometric structures of the active sites and the nanosupports, and then the regulation of the binding energy between different organic substrates can further enhance the enzyme-like selectivity of SAzymes. The above train of thoughts are put forward to avoid trial-and-error attempts and guide the rational and efficient design of SAzymes with tunable specificity and selectivity in future research.
Applications of SAzymes
For nanozymes, the inherent enzyme-like properties, which were essentially determined by nanostructures, definitively determined their diverse applications in bioanalysis, therapeutics, tissue engineering, heterocatalysis and even environmental toxicology. As discussed above, SAzymes consist of atomically dispersed metal sites, with an ideal atomic structure, superior catalytic activity and uniform catalytic properties. These advantages endow SAzymes with highly enzyme-like activity and possible specificity which are more similar to those of natural enzymes. Integrated with the intrinsic structural and physicochemical properties simultaneously, the emerging SAzymes will undoubtedly boost the crucial applications of nanozyme technologies.67,69,101 For instance, in vivo and in vitro bioassays, such as H2O2, glucose, ascorbic acid, and glutathione, are the most extensive and essential applications of nanozymes. The detection sensitivity could be significantly improved benefitting from the ultrahigh activity of SAzymes, and specific detections could be realized by the SAzymes with sole enzyme activity. Single-Fe-site SAzymes, Fe atoms anchored on N-doped carbons supported on carbon nanotubes, were applied for ultrasensitive bioassays of H2O2, glucose and ascorbic acid.87 Ultra-low detection limits were realized, which were much lower than the allowance level. This strategy was generic for the detection of H2O2, biomolecules that can produce H2O2, small molecules that can reduce oxTMB and other peroxidase substrates or enhance/inhibit enzyme activity (Fig. 7a and b).102,103 Fe–N-rGO reported by Kim et al. exhibits sole peroxidase-like activity without any oxidase-like activity, and could be applied for the detection of trace amounts of H2O2. Previously reported peroxidase-like nanozymes always had oxidase-like activity as well, which could interfere the with detection of H2O2 because of the reaction of O2 in the atmosphere, especially at a low H2O2 content. The specific SAzyme Fe–N-rGO was used to detect trace amounts of H2O2 produced from enzymatic reactions and cancerous cells. When Fe–N-rGO was applied to detect choline and acetylcholine, limit of detection values of ≈10 × 10−9 M and 20 × 10−9 M were obtained, respectively, indicating the sensitivity of Fe–N-rGO in H2O2 detection.85 | ||
| Fig. 7 (a) Synthesis of CNT/FeNC SAzyme and colorimetric biosensing of H2O2via various chromogenic substrates. Reproduced from ref. 87 with permission from John Wiley & Sons. (b) Schematic diagram of four different types of bioassays. | ||
In environmental pollution treatment, homogeneous catalysts and enzymes faced difficulties in separation and were easy to degrade and the complex preparation process hinders their application. Heterogeneous catalysts such as metallic oxide, and metal sulfide nanoparticles used in the treatment of pollutants face the risk of metal ion leaching, causing secondary pollution probability. Single-atom catalysts with a minimal metal content but high catalytic efficiency have inherent advantages over other catalysts in pollutant degradation. Single iron atom SAzyme with 40 times higher peroxidase-like activity than Fe3O4 was applied to degrade phenol in polluted water.78 Fe SAzymes showed effective pollutant removal ability, which removed 83% phenol within 30 min. After ten cycles, Fe SAzymes maintained almost unaltered activity, demonstrating the durability of Fe–N4 SAzymes under harsh conditions. Single-atom Mn–N4 sites supported on graphitic carbon nitride (g-C3N4) could catalyze a peroxone reaction efficiently in acid solution (Fig. 8a), which could be applied to degrade stubborn organic pollutants by the generation of hydroxyl radicals (˙OH).79 In the solution with O3 and H2O2, the addition of Mn–N4 catalysts leads to complete degradation of oxalic acid within 40 min, while only 28% degradation efficiency was obtained after 60 min without Mn–N4 catalysts (Fig. 8b). Mn–N4 mediated a special mechanism to catalyze the peroxone reaction and generated ˙OH effectively, and the catalytic activity had no obvious decrease after 5 cycles. Single-Co-atom anchored on N-doped graphene was developed as an efficient Fenton-like catalyst and used for catalytic oxidation of recalcitrant organics via activation of peroxymonosulfate (PMS).104 In the catalytic oxidation of bisphenol A (BPA), Co–N4 was confirmed to be the sites for PMS activation while the pyrrolic N served as the site for organics adsorption. The dual sites in catalysts reduced the migration distance for singlet oxygen (1O2), and were able to recognize and activate the respective substrates independently, making active species generation and substrate activation occur simultaneously (Fig. 8c). As a result, the catalytic activity was improved dramatically. The catalysts were able to completely remove BPA in 4 min with minimum leaching of Co.
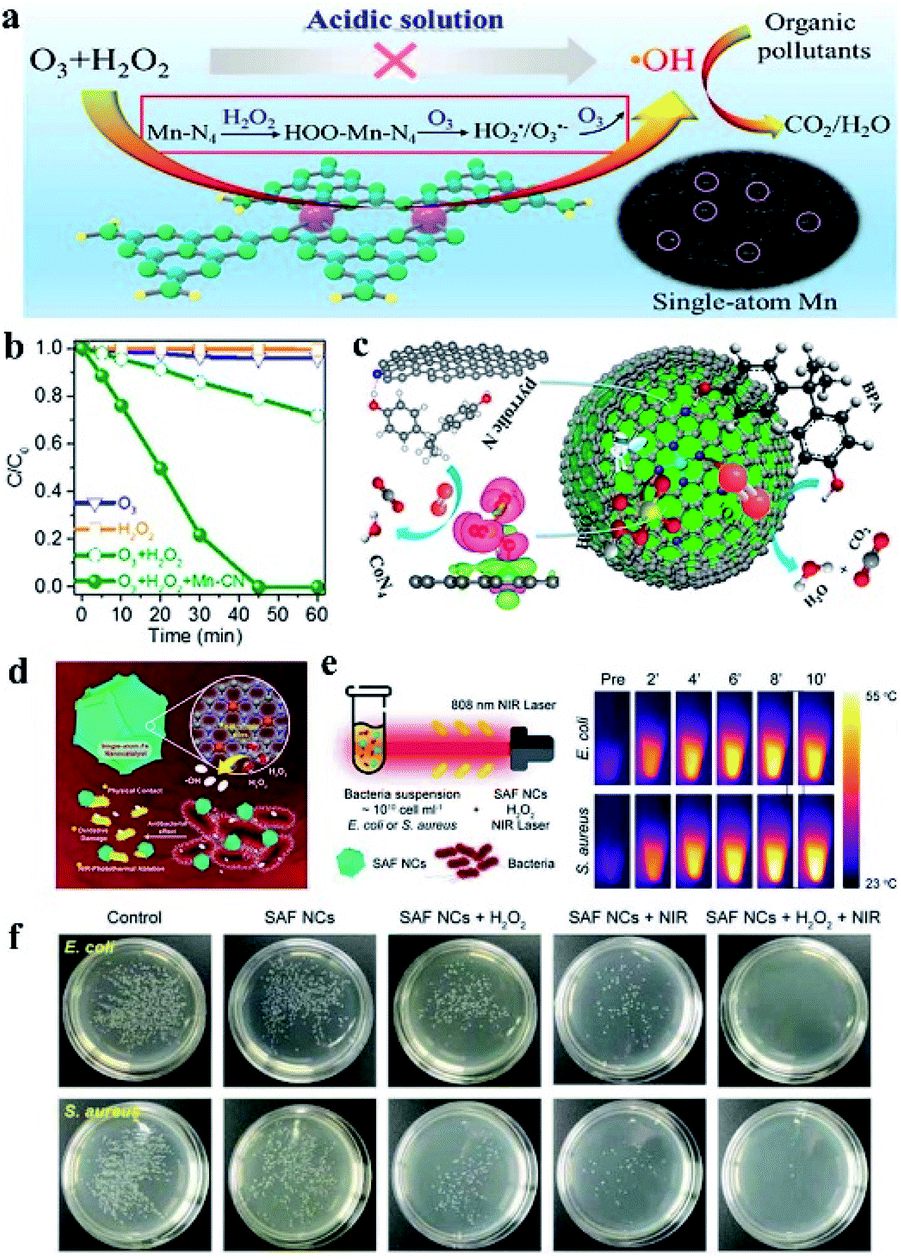 | ||
| Fig. 8 (a) Single-atom Mn–N4 site-catalyzed peroxone reaction for efficient production of hydroxyl radicals and organic pollutant degradation in acidic solution. (b) Degradation curves of oxalic acid in ozonation, the H2O2 process, and the peroxone process with or without Mn–CN. (c) Single-Co-atom anchored on N-doped graphene for degradation of bisphenol A. (a–c) reproduced from ref. 79 and 104 with permission from the American Chemical Society, respectively. (d) Schematic illustration of ˙OH generation and antibacterial applications of single-iron-atom nanozyme with peroxidase-like activity. (e) NIR-involved antibacterial experiment and corresponding IR images of E. coli or S. aureus bacterial suspension. (f) Digital photographs of remaining bacteria-inoculated agar plates after treatment of respective conditions. Reproduced from ref. 68 with permission from John Wiley & Sons. | ||
SAzymes could also be used for antibacterial and cancer treatment, for their superiority in biosecurity with little leakage of metal ions. Huo et al. prepared single iron atom catalysts fabricated in N-doped amorphous carbon.68 The as prepared SACs showed high peroxidase-like activity, generating abundant hydroxyl radicals in the presence of H2O2. The SAzymes with free radical production capacity could kill both Gram positive (Staphylococcus aureus) and Gram negative (Escherichia coli) bacteria at the physiological level of H2O2, which benefited from the synergetic effect of physical contact damages and ˙OH toxicity to bacteria (Fig. 8d). In the group of bacteria treated with SAzymes and H2O2, the remaining activities were only 28.9% and 17.40% for E. coli and S. aureus respectively. In addition, the N-doped amorphous carbon supported single iron atom catalysts showed optical absorption and 19.37% of the high photothermal conversion effects in the near infrared region (NIR) (Fig. 8e). Under NIR irradiation conditions, the antibacterial efficiency was improved to almost 100% for both E. coli and S. aureus (Fig. 8f). The in vivo wound healing experiment revealed that SAzymes and H2O2 treated groups showed a shorter wound healing time compared with the contrast groups, and SAzymes + H2O2 + NIR treated groups reduced the time further, showing a better antibacterial effect with NIR irradiation. The antibacterial mechanism was explained by the membrane destruction, leading to the breaking of cell biointegrity. These results were consistent with the findings of Yan's group. They reported a Zn–N4 SAzyme with efficient peroxidase activity derived from pyrolysis of zinc-based zeolitic-imidazolate-frameworks (ZIF-8).66 The Zn–N4 SAzyme was used for antimicrobial treatment and showed a 99.87% inhibition rate of P. aeruginosa. The in vivo bactericidal efficacy was revealed by the fact that the mice infected by P. aeruginosa needed more than 11 days for wound-healing, while infected mice treated with Zn–N4 SAzyme achieved complete wound-healing in only 6 days. And toxicological analysis of the main organs showed that no obvious toxicity was detected, indicating the good biosafety of the Zn–N4 SAzyme. Dong's group found that SAzymes of FeN5 SA/CNF with oxidase-like activity would generate reactive oxygen species or oxidative stress during the catalytic reduction of oxygen; it was used to destroy the integrity of bacterial cell membranes.64 When E. coli and S. aureus cells were exposed to FeN5 SA/CNF, the bacterial survival rates were markedly reduced, indicating a high antibacterial efficiency. When used in vivo, the clear remission of ulceration and accelerated wound healing in the mice were observed compared with the control group.
The group of Mao prepared SAzyme through the pyrolysis process after encapsulating FePc molecules into the cages of ZIF-8. The obtained single iron sites were confirmed to be Fe–N4 resembling heme-contained enzymes, and exhibited bifunctional enzyme-like activity of catalase and SOD. Fe–N4 SAzymes were used for cytoprotection by reducing oxidative stress. SOD activity could convert O2− to H2O2 and O2 while catalase-like activity can decompose hydrogen peroxide into oxygen and water, and therefore scavenging ROS in cells and protecting cells from oxidative stress. When HeLa cells were treated with 10 μM β-lapachone (β-Lap), which could be bioactivated by NAD(P)H quinone oxidoreductase 1 (NQO1) to generate ROS swiftly, cell viability was reduced to 20%. However, cell viability could be increased to 85% if the HeLa cells were pre-incubated with SAzymes before being treated with β-Lap, while N-doped carbon did not change the cell viability, showing the excellent cell protection ability of the as prepared Fe–N4 SAzymes.105 Similar work was reported by Lu et al.106 Besides, Ming and Zhang's groups developed single-atom Pt/CeO2 based bandage for treatment of brain trauma.88 The single-atom Pt/CeO2 showed excellent peroxidase (POD)-, catalase (CAT)-, superoxide dismutase (SOD)- and glutathione peroxidase (GPx)-like activity, much higher than that of CeO2 clusters (Fig. 9a). Attributed to the multi-antioxidant activities, the SAzymes could scavenge active oxygen and nitrogen species (RONS) induced by traumatic brain injury, preventing subsequent neuronal damage. In addition, single-atom Pt/CeO2 showed great anti-inflammation responses to inflammation caused by H2O2 and lipopolysaccharides, and improved cell viability significantly. Treatment of mice with traumatic brain injury (TBI) demonstrated that the single-atom Pt/CeO2 based bandage showed a remarkable wound healing effect, reducing the wound size to healthy levels while the untreated mice only recovered 50% (Fig. 9b and c). Compared with previously reported Pt-based materials, single-atom Pt/CeO2 had persistent activity and remained unchanged after 30 days. Single Co atoms supported on nitrogen-doped carbon was shown to have SOD, catalase and GPx activity under physiological conditions, which would be used to eliminate RONS as well.107 O2˙− could be eliminated by SOD-like activity, while H2O2 was consumed by mimicking catalase and GPx catalysis. ˙NO was scavenged through the formation of a nitrosyl–metal complex with Co-porphyrin centers. Based on the ability to eliminate RONS, the SAzyme was used for sepsis management. In vivo treatment demonstrated that the SAzyme could eliminate RONS effectively, which protected cells from DNA damage and inhibited lipid peroxidation, leading to remarkable therapeutic effects of sepsis.
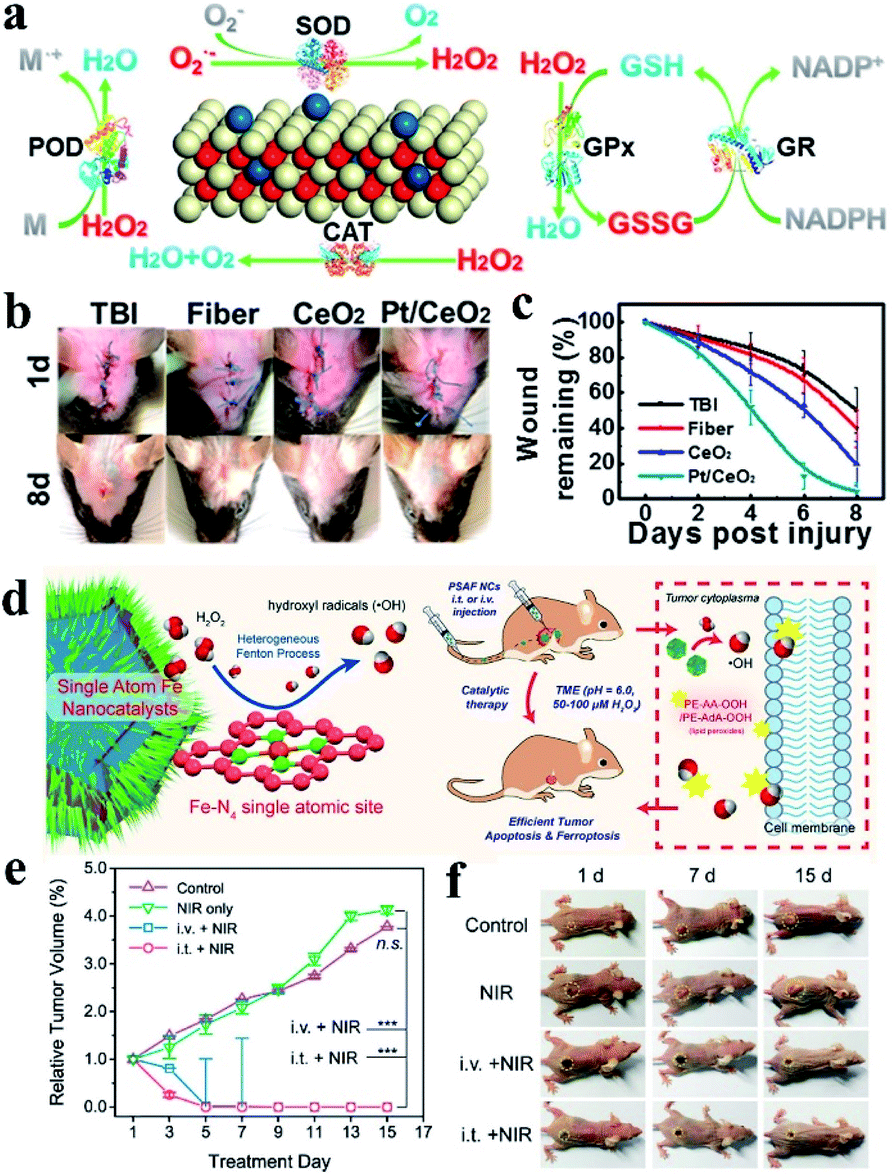 | ||
| Fig. 9 (a) Multiple enzyme activities of single-atom Pt/CeO2. (b) Digital photographs of the healing effect and (c) residual wound over time with or without the treatment of Pt/CeO2 based bandage. (d) Single atom Fe nanocatalysts with the Fe–N4 site for tumor therapy. (e) In vivo 4T1 tumor proliferation curves and (f) corresponding digital photos of mice treated under different conditions. (a–c) and (d–f) reproduced from ref. 88 and 108 with permission from the American Chemical Society, respectively. | ||
Huo and Wang prepared single iron atom catalysts for tumor therapy through an “isolation-pyrolysis” method.108 The tumor therapy was realized by toxic hydroxyl radicals (˙OH) generated from the Fenton reaction in a faintly acid tumor microenvironment, leading to cell apoptosis and ferroptosis caused by accumulation of lipid peroxides (Fig. 9d). The therapy was tumor-selective because of the acidity-accelerated heterogeneous Fenton reactions and reduced ˙OH generation under neutral conditions. Pegylation endowed the SAzymes with biocompatibility, combining the high-performance peroxidase-like activity of Fe-SAzyme with the pegylation endowed biocompatibility, it was used for in vivo tumor therapy. The in vitro antiproliferation effects were evaluated by treating 4T1 mammary tumor cells with Fe-SAzymes and investigated the cytotoxic impacts. They had a substantial proliferation inhibition effect under mild acid conditions while much weaker in a neutral medium, indicating the target of the tumor microenvironment. By introducing ascorbic acid and α-Tocopherol to the system, the cell death was reduced adequately. Therefore, they elucidated the mechanism of cell death to be the combined action of ˙OH induced oxidative stress and lipid peroxidation. In vivo therapeutic experiments showed that Fe-SAzymes had sustained catalytic performance in inhibiting tumor cell proliferation. Benefiting from the high photothermal conversion efficiency of N-doped carbon, the SAzymes exhibited better therapeutic effects under NIR irradiation, with more than 150% tumor inhibition rates compared with the control group (Fig. 9e and f). Single-atom Ru assembled with the chlorin e6 photosensitizer was used as a catalase-like nanozyme for photodynamic therapy treatment of tumors.69 Local generation of O2 by Ru SAzyme using the highly expressed intracellular H2O2 in tumors could relieve the hypoxia in the tumor microenvironment and enhance the therapeutic efficiency. Au SACs were used for cancer treatment by breaking the mitochondrial redox homeostasis,109 which was maintained by the balance of reactive oxygen species and antioxidants. The homeostasis imbalance always leads to apoptosis. Carbon-dot supported atomically dispersed gold modified with triphenylphosphine and cinnamaldehyde (MitoCAT-g) was used as a mitochondrial oxidative stress amplifier reported by Liang and Li's groups. MitoCAT-g could deplete intracellular antioxidants GSH though the formation of Au–S bonds, and then amplify the oxidative stress caused by cinnamaldehyde, as well as the cancer cell death. The atomically dispersed Au atoms in MitoCAT-g had the highest atom utilization and more active sites. These inspired the development of single-atom catalysts with GPx-like activity or other antioxidant enzyme-like activity for cancer treatment.
The high catalytic activity and selectivity of SAzymes also made them capable for the application in organic synthesis and conversion. Fe–N–C SAzyme was applied to biomimetic catalysis and industrial oxidation processes reported by He et al.110 Dehydrogenation of 1,4-dihydropyridine (1,4-DHP) into diethyl 2,6-dimethyl-3,5-pyridine-dicarboxylate (DDPD) was realized using O2 as the terminal oxidant with comparable activation energy as nature enzymes. In addition to biomimetic catalysts like the cytochrome P450 enzymes, Fe–N–C SAzyme could also be used in dehydrogenation of a series of N-heterocycles and monoxygenation of triphenylphosphine with 100% selectivity. Due to their robust structure, they can remain active in a variety of organic solvents. The catalytic mechanism was investigated and it was uncovered that Fe–N–C catalyzed the activation of O2 with formation of 1O2, O2˙−, and H2O2 rather than strongly aggressive OH˙−, ensuring the selective oxidation of substrates. Meanwhile, Fe–N–C exhibited both accelerated and inhibited cytochrome P450-like behaviors in the drug metabolization based on the enzyme-like central structures. Fe–N–C obtained at 400 °C could catalyze the oxidation of 1,4-DPH and showed similar inhibiting interactions with other drugs like cytochrome P450,111 indicating that Fe–N–C SAzyme could replace cytochrome P450 in the evaluation of potential chemicals, studies of drug–drug interactions, dosage guidance and outcome prediction.
Enzymes were employed in the electrochemical field frequently, such as enzyme-modified electrodes used for sensing and enzymatic biofuel cells. Nanozyme modified electrodes were fabricated for detection of a wide range of species, such as small molecules, proteins and cells. In addition, Mu et al. recently reported that the addition of Zn–TCPP(Fe) nanozymes to Li–O2 batteries would scavenge superoxide radicals, reduce the damage caused by aggressive superoxide intermediates, and improve the cycling stability.112 The Zn–TCPP(Fe) nanozymes performed as an ORR/OER bifunctional catalyst and served as a molecular shuttle of superoxide species and electrons between cathodes and products in both discharge and recharge processes, leading to enlarged discharge capacity and elevated energy efficiency.
In short, the emerging SAzymes have been used in many fields. However, their scope of application is still limited because of some key issues being unresolved. For example, sensitivity and selectivity are crucial in sensing applications. SAzymes with higher activity and selectivity are desired to be prepared for practical application. Besides, more convenient and sensitive detection means should be developed by combining the properties of nanomaterial-based supports and excellent enzyme activity of single-atom active sites. In the removal of pollutants, more stable SAzymes are desired to remain active under harsh conditions and reduce metal ion leakage to avoid secondary contamination. In the treatment of disease, disease diagnosis and treatment could be combined based on the ability of SAzymes to detect various biological substances. Thermal conversion capacity of N-doped carbon supports was integrated with the enzyme-mimic activity, and more types of support loaded single-atom active sites could be developed for a synergistic effect in disease treatment. Moreover, although SAzymes with good biosafety were demonstrated by some researchers, more in vivo experiments should be conducted before clinical therapeutics and effective SAzymes with low toxicity are needed.
In addition to the applications mentioned above, a wider range of applications should be taken into consideration based on the superiority of SAzymes. For example, SAzymes are expected to replace natural enzymes in industrial catalysis for their satisfactory selectivity and high organic conversion capacity. And in consideration of the high enzyme-like activity of SAzymes, we except them to be used in enzymatic biofuel cells to substitute natural enzymes. The development of SAzymes is still in its infancy, and only oxidoreductase-mimics are developed. More types of SAzymes are required to be used in more fields.
Conclusion and outlook
In this perspective, we have detailedly described the developments of the emerging SAzymes, which were defined as single-site heterogeneous catalysts with inherent enzyme-like characteristics. Because of the indivisible correlations between the structure, properties and application of SAzymes, we successively discussed the features and advantages of SAzymes with respect to the single-site structure, enzyme-like activity, catalytic selectivity and extensive applications based on the reported results. Meanwhile, we also gave our perspective on the current challenges facing SAzyme research and the corresponding possible directions for future research to the best of our knowledge. Firstly, attributed to the atomically dispersed sites, SAzymes have gained outstanding structural advantages in comparison with nanozymes. The atomized moieties of SAzymes made the active centers much simple, more controllable and definite, which in consequence promote the comprehension of the catalytic reaction process, enzyme-like mechanism and rational design of SAzymes. However, the current outcomes were unable to fulfill the potential applications, thus calling for development of more effective strategies to increase the structural uniformity and expand the enzyme-like categories. Secondly, SAzymes always feature high enzyme-like activity, which was mainly derived from the structural advantages: the maximum atom utilization and high intrinsic activity of single-sites. Therefore, for the insufficient activity of SAzymes compared to natural enzymes, the above two aspects should be taken into full consideration. On the one hand, even though SAzymes maximized the atom utilization of metal sites, the metal loading rate and active site density were still relatively low. Developing novel synthetic strategies to increase the active site density can effectively promote the catalytic activity. On the other hand, the intrinsic enzyme-like activity originates from the central metal atoms and the neighboring coordination atoms. The enzyme-like activity can also be radically boosted by tailoring the electronic and geometric structures of the single-sites. Furthermore, the definite and atomically dispersed active sites of SAzymes afford us with new opportunities to establish more suitable evaluation methods for reflecting the intrinsic enzyme-like activity of each single-atom. Thirdly, the catalytic selectivity, including substrate selectivity and product selectivity, was the most important property for enzymes and enzyme mimics, which promotes the corresponding applications in analysis and catalysis, respectively. Although the current SAzymes exhibited few enzyme-like selectivity or specificity, the superior structural advantages of the single-sites make it more easy to regulate the inherent properties through the central metal atom and coordination structure tailoring. The purposive adjustment and rational design of SAzymes require the collaborative interplay of SAC catalysis and theoretical calculations to indicate how single metal atoms coordinated with adjacent atoms act as the active sites and affect the kinetic process. Finally, the extensive applications of SAzymes were derived from not only the advantages of high enzyme-like properties but also the physicochemical features. The future research of SAzymes applications need to fully explore the irreplaceable advantages that are far beyond those of natural enzymes, and develop novel and multiple SAzyme-combined catalytic systems to carry forward their significant role in the enzyme mimics field.In conclusion, the emergence of SAzymes do promote the nanozyme research entering a new period of the atomic level. Through the summary and outlook of the endeavor and progress of SAzymes from the structure and properties to applications, we believe that the SAzyme research will usher in a bright prospect in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21675151) and the Ministry of Science and Technology of China (no. 2016YFA0203203).References
- R. Wolfenden and M. J. Snider, Acc. Chem. Res., 2001, 34, 938–945 CrossRef CAS.
- M. Garcia-Viloca, J. Gao, M. Karplus and D. G. Truhlar, Science, 2004, 303, 186–195 CrossRef CAS.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC.
- X. Zhang, H. P. Xu, Z. Y. Dong, Y. P. Wang, J. Q. Liu and J. C. Shen, J. Am. Chem. Soc., 2004, 126, 10556–10557 CrossRef CAS.
- V. T. D'Souza, Supramol. Chem., 2003, 15, 221–229 CrossRef.
- Y. Aiba, J. Sumaoka and M. Komiyama, Chem. Soc. Rev., 2011, 40, 5657–5668 RSC.
- J. H. Dawson, Science, 1988, 240, 433–439 CrossRef CAS.
- E. L. Onderko, A. Silakov, T. H. Yosca and M. T. Green, Nat. Chem., 2017, 9, 623–628 CrossRef CAS.
- R. Breslow, Acc. Chem. Res., 1995, 28, 146–153 CrossRef CAS.
- Q. Wang, H. Wei, Z. Zhang, E. Wang and S. Dong, TrAC, Trends Anal. Chem., 2018, 105, 218–224 CrossRef CAS.
- J. Golchin, K. Golchin, N. Alidadian, S. Ghaderi, S. Eslamkhah, M. Eslamkhah and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2017, 45, 1069–1076 CrossRef CAS.
- L. Gao and X. Yan, Sci. China: Life Sci., 2016, 59, 400–402 CrossRef.
- R. Ragg, M. N. Tahir and W. Tremel, Eur. J. Inorg. Chem., 2016, 1906–1915 CrossRef CAS.
- Y. Lin, J. Ren and X. Qu, Acc. Chem. Res., 2014, 47, 1097–1105 CrossRef CAS.
- X. Wang, Y. Hu and H. Wei, Inorg. Chem. Front., 2016, 3, 41–60 RSC.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- R. Zhang, K. Fan and X. Yan, Sci. China: Life Sci., 2020, 63, 1183–1200 CrossRef.
- I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127–4170 RSC.
- S. Ghosh, P. Roy, N. Karmodak, E. D. Jemmis and G. Mugesh, Angew. Chem., Int. Ed., 2018, 57, 4510–4515 CrossRef CAS.
- A. Asati, S. Santra, C. Kaittanis, S. Nath and J. M. Perez, Angew. Chem., Int. Ed., 2009, 48, 2308–2312 CrossRef CAS.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS.
- X. Shen, W. Liu, X. Gao, Z. Lu, X. Wu and X. Gao, J. Am. Chem. Soc., 2015, 137, 15882–15891 CrossRef CAS.
- K. Lu, T. Aung, N. Guo, R. Weichselbaum and W. Lin, Adv. Mater., 2018, 30, 1707634 CrossRef.
- Y. Zhang, S. Li, H. Liu, W. Long and X.-D. Zhang, Front. Chem., 2020, 8, 219 CrossRef.
- H. Wang, K. Wan and X. Shi, Adv. Mater., 2019, 31, 1805368 CrossRef CAS.
- T. M. Benedetti, C. Andronescu, S. Cheong, P. Wilde, J. Wordsworth, M. Kientz, R. D. Tilley, W. Schuhmann and J. J. Gooding, J. Am. Chem. Soc., 2018, 140, 13449–13455 CrossRef CAS.
- W. Zhang, S. Hu, J.-J. Yin, W. He, W. Lu, M. Ma, N. Gu and Y. Zhang, J. Am. Chem. Soc., 2016, 138, 5860–5865 CrossRef CAS.
- X. Zhang, D. Wu, X. Zhou, Y. Yu, J. Liu, N. Hu, H. Wang, G. Li and Y. Wu, TrAC, Trends Anal. Chem., 2019, 121, 115668 CrossRef CAS.
- Y. Guan, M. Li, K. Dong, N. Gao, J. Ren, Y. Zheng and X. Qu, Biomaterials, 2016, 98, 92–102 CrossRef CAS.
- Y. Huang, Z. Liu, C. Liu, E. Ju, Y. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2016, 55, 6646–6650 CrossRef CAS.
- L. Rizzello and P. P. Pompa, Chem. Soc. Rev., 2014, 43, 1501–1518 RSC.
- Z. Wang, K. Dong, Z. Liu, Y. Zhang, Z. Chen, H. Sun, J. Ren and X. Qu, Biomaterials, 2017, 113, 145–157 CrossRef CAS.
- W. Yin, J. Yu, F. Lv, L. Yan, L. R. Zheng, Z. Gu and Y. Zhao, ACS Nano, 2016, 10, 11000–11011 CrossRef CAS.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS.
- Y. Lin, Z. Li, Z. Chen, J. Ren and X. Qu, Biomaterials, 2013, 34, 2600–2610 CrossRef CAS.
- S. Li, L. Shang, B. Xu, S. Wang, K. Gu, Q. Wu, Y. Sun, Q. Zhang, H. Yang, F. Zhang, L. Gu, T. Zhang and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 12624–12631 CrossRef CAS.
- X. Zhang, G. Li, D. Wu, X. Li, N. Hu, J. Chen, G. Chen and Y. Wu, Biosens. Bioelectron., 2019, 137, 178–198 CrossRef CAS.
- T. Mahmoudi, M. de la Guardia, B. Shirdel, A. Mokhtarzadeh and B. Baradaran, TrAC, Trends Anal. Chem., 2019, 116, 13–30 CrossRef CAS.
- M. Komiyama and K. Ariga, Mol. Catal., 2019, 475, 110492 CrossRef CAS.
- M. Liang and X. Yan, Acc. Chem. Res., 2019, 52, 2190–2200 CrossRef CAS.
- D. Jiang, D. Ni, Z. T. Rosenkrans, P. Huang, X. Yan and W. Cai, Chem. Soc. Rev., 2019, 48, 3683–3704 RSC.
- N. Chaibakhsh and Z. Moradi-Shoeili, Mater. Sci. Eng., C, 2019, 99, 1424–1447 CrossRef CAS.
- W. Song, B. Zhao, C. Wang, Y. Ozaki and X. Lu, J. Mater. Chem. B, 2019, 7, 850–875 RSC.
- R. Yang, S.-F. Cai and C. Wang, Prog. Biochem. Biophys., 2018, 45, 170–192 Search PubMed.
- X.-M. Shen, X.-J. Gao and X.-F. Gao, Prog. Biochem. Biophys., 2018, 45, 204–217 Search PubMed.
- J. Wu, S. Li and H. Wei, Nanoscale Horiz., 2018, 3, 367–382 RSC.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS.
- J. Guzman and B. C. Gates, J. Catal., 2004, 226, 111–119 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- X. Zhang, H. Shi and B. Q. Xu, Angew. Chem., Int. Ed., 2005, 44, 7132–7135 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2019, 52, 237–247 CrossRef CAS.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- H. Yan, C. Su, J. He and W. Chen, J. Mater. Chem. A, 2018, 6, 8793–8814 RSC.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS.
- T. J. Collins and A. D. Ryabov, Chem. Rev., 2017, 117, 9140–9162 CrossRef CAS.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS.
- U. Tylus, Q. Jia, K. Strickland, N. Ramaswamy, A. Serov, P. Atanassov and S. Mukerjee, J. Phys. Chem. C, 2014, 118, 8999–9008 CrossRef CAS.
- A. T. Gallagher, J. Y. Lee, V. Kathiresan, J. S. Anderson, B. M. Hoffman and T. D. Harris, Chem. Sci., 2018, 9, 1596–1603 RSC.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS.
- L. Jiao, J. Wu, H. Zhong, Y. Zhang, W. Xu, Y. Wu, Y. Chen, H. Yan, Q. Zhang, W. Gu, L. Gu, S. P. Beckman, L. Huang and C. Zhu, ACS Catal., 2020, 10, 6422–6429 CrossRef CAS.
- B. Xu, H. Wang, W. Wang, L. Gao, S. Li, X. Pan, H. Wang, H. Yang, X. Meng, Q. Wu, L. Zheng, S. Chen, X. Shi, K. Fan, X. Yan and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 4911–4916 CrossRef CAS.
- X. Niu, Q. Shi, W. Zhu, D. Liu, H. Tian, S. Fu, N. Cheng, S. Li, J. N. Smith, D. Du and Y. Lin, Biosens. Bioelectron., 2019, 142, 111495 CrossRef CAS.
- M. Huo, L. Wang, H. Zhang, L. Zhang, Y. Chen and J. Shi, Small, 2019, 15, 1901834 CrossRef.
- D. Wang, H. Wu, S. Z. F. Phua, G. Yang, W. Q. Lim, L. Gu, C. Qian, H. Wang, Z. Guo, H. Chen and Y. Zhao, Nat. Commun., 2020, 11, 357 CrossRef CAS.
- X. Zhang, G. Li, G. Chen, D. Wu, X. Zhou and Y. Wu, Coord. Chem. Rev., 2020, 418, 213376 CrossRef CAS.
- L. Jiao, H. Yan, Y. Wu, W. Gu, C. Zhu, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2020, 59, 2565–2576 CrossRef CAS.
- B. Jiang, D. Duan, L. Gao, M. Zhou, K. Fan, Y. Tang, J. Xi, Y. Bi, Z. Tong, G. F. Gao, N. Xie, A. Tango, G. Nie, M. Liang and X. Yan, Nat. Protoc., 2018, 13, 1506–1520 CrossRef CAS.
- Q. Chen, S. Li, Y. Liu, X. Zhang, Y. Tang, H. Chai and Y. Huang, Sens. Actuators, B, 2020, 305, 127511 CrossRef.
- L.-Z. Huang, X. Wei, E. Gao, C. Zhang, X.-M. Hu, Y. Chen, Z. Liu, N. Finck, J. Luetzenkirchen and D. D. Dionysiou, Appl. Catal., B, 2020, 268, 118459 CrossRef.
- G. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu, P. Du, R. Si, J. Wang, X. Cui, H. Li, J. Xiao, T. Xu, J. Deng, F. Yang, P. N. Duchesne, P. Zhang, J. Zhou, L. Sun, J. Li, X. Pan and X. Bao, Sci. Adv., 2015, 1, e150046 Search PubMed.
- Y. Wang, Z. Zhang, G. Jia, L. Zheng, J. Zhao and X. Cui, Chem. Commun., 2019, 55, 5271–5274 RSC.
- C. Zhao, C. Xiong, X. Liu, M. Qiao, Z. Li, T. Yuan, J. Wang, Y. Qu, X. Wang, F. Zhou, Q. Xu, S. Wang, M. Chen, W. Wang, Y. Li, T. Yao, Y. Wu and Y. Li, Chem. Commun., 2019, 55, 2285–2288 RSC.
- Z. Guo, Y. Xie, J. Xiao, Z.-J. Zhao, Y. Wang, Z. Xu, Y. Zhang, L. Yin, H. Cao and J. Gong, J. Am. Chem. Soc., 2019, 141, 12005–12010 CrossRef CAS.
- Y. Wang, K. Qi, S. Yu, G. Jia, Z. Cheng, L. Zheng, Q. Wu, Q. Bao, Q. Wang, J. Zhao, X. Cui and W. Zheng, Nano-Micro Lett., 2019, 11, 102 CrossRef.
- C. Wan, X. Duan and Y. Huang, Adv. Energy Mater., 2020, 10, 1903815 CrossRef CAS.
- J. Li, S. Chen, F. Quan, G. Zhan, F. Jia, Z. Ai and L. Zhang, Chem, 2020, 6, 885–901 CAS.
- F. S. Hage, G. Radtke, D. M. Kepaptsoglou, M. Lazzeri and Q. M. Ramasse, Science, 2020, 367, 1124–1127 CrossRef CAS.
- Y. Ren, Y. Tang, L. Zhang, X. Liu, L. Li, S. Miao, D. S. Su, A. Wang, J. Li and T. Zhang, Nat. Commun., 2019, 10, 4500 CrossRef.
- M. S. Kim, J. Lee, H. S. Kim, A. Cho, K. H. Shim, T. N. Le, S. S. A. An, J. W. Han, M. I. Kim and J. Lee, Adv. Funct. Mater., 2020, 30, 1905410 CrossRef CAS.
- L. Han, L. Zhang, H. Wu, H. Zu, P. Cui, J. Guo, R. Guo, J. Ye, J. Zhu, X. Zheng, L. Yang, Y. Zhong, S. Liang and L. Wang, Adv. Sci., 2019, 6, 1900006 CrossRef.
- N. Cheng, J.-C. Li, D. Liu, Y. Lin and D. Du, Small, 2019, 15, 1901485 CrossRef CAS.
- R. Yan, S. Sun, J. Yang, W. Long, J. Wang, X. Mu, Q. Li, W. Hao, S. Zhang, H. Liu, Y. Gao, L. Ouyang, J. Chen, S. Liu, X.-D. Zhang and D. Ming, ACS Nano, 2019, 13, 11552–11560 CrossRef CAS.
- C. M. Krest, A. Silakov, J. Rittle, T. H. Yosca, E. L. Onderko, J. C. Calixto and M. T. Green, Nat. Chem., 2015, 7, 696–702 CrossRef CAS.
- P. Campomanes, U. Rothlisberger, M. Alfonso-Prieto and C. Rovira, J. Am. Chem. Soc., 2015, 137, 11170–11178 CrossRef CAS.
- X. He, Q. He, Y. Deng, M. Peng, H. Chen, Y. Zhang, S. Yao, M. Zhang, D. Xiao, D. Ma, B. Ge and H. Ji, Nat. Commun., 2019, 10, 3663 CrossRef.
- Y. Lou, Y. Zheng, X. Li, N. Ta, J. Xu, Y. Nie, K. Cho and J. Liu, J. Am. Chem. Soc., 2019, 141, 19289–19295 CrossRef CAS.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS.
- Y. Hu, X. J. Gao, Y. Zhu, F. Muhammad, S. Tan, W. Cao, S. Lin, Z. Jin, X. Gao and H. Wei, Chem. Mater., 2018, 30, 6431–6439 CrossRef CAS.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. G. Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 CrossRef.
- M. Sun, A. W. Dougherty, B. Huang, Y. Li and C.-H. Yan, Adv. Energy Mater., 2020, 10, 1903949 CrossRef CAS.
- J. Gao, H. B. Yang, X. Huang, S.-F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, Chem, 2019, 5, 2099–2110 CAS.
- M. Ledendecker, E. Pizzutilo, G. Malta, G. V. Fortunato, K. J. J. Mayrhofer, G. J. Hutchings and S. J. Freakley, ACS Catal., 2020, 10, 5928–5938 CrossRef CAS.
- H. Xiang, W. Feng and Y. Chen, Adv. Mater., 2020, 32, 1905994 CrossRef CAS.
- Y. Wu, L. Jiao, X. Luo, W. Xu, X. Wei, H. Wang, H. Yan, W. Gu, B. Z. Xu, D. Du, Y. Lin and C. Zhu, Small, 2019, 15, 1903108 CrossRef CAS.
- L. Jiao, W. Xu, H. Yan, Y. Wu, C. Liu, D. Du, Y. Lin and C. Zhu, Anal. Chem., 2019, 91, 11994–11999 CrossRef CAS.
- X. Li, X. Huang, S. Xi, S. Miao, J. Ding, W. Cai, S. Liu, X. Yang, H. Yang, J. Gao, J. Wang, Y. Huang, T. Zhang and B. Liu, J. Am. Chem. Soc., 2018, 140, 12469–12475 CrossRef CAS.
- W. Ma, J. Mao, X. Yang, C. Pan, W. Chen, M. Wang, P. Yu, L. Mao and Y. Li, Chem. Commun., 2019, 55, 159–162 RSC.
- M. Lu, C. Wang, Y. Ding, M. Peng, W. Zhang, K. Li, W. Wei and Y. Lin, Chem. Commun., 2019, 55, 14534–14537 RSC.
- F. Cao, L. Zhang, Y. You, L. Zheng, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2020, 59, 5108–5115 CrossRef CAS.
- M. Huo, L. Wang, Y. Wang, Y. Chen and J. Shi, ACS Nano, 2019, 13, 2643–2653 CAS.
- N. Gong, X. Ma, X. Ye, Q. Zhou, X. Chen, X. Tan, S. Yao, S. Huo, T. Zhang, S. Chen, X. Teng, X. Hu, J. Yu, Y. Gan, H. Jiang, J. Li and X.-J. Liang, Nat. Nanotechnol., 2019, 14, 379–387 CrossRef CAS.
- F. He, L. Mi, Y. Shen, T. Mori, S. Liu and Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 35327–35333 CrossRef CAS.
- Y. Xu, J. Xue, Q. Zhou, Y. Zheng, X. Chen, S. Liu, Y. Shen and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14498–14503 CrossRef CAS.
- X. Mu, Y. Liu, X. Zhang, H. Wei, P. He and H. Zhou, Batteries Supercaps, 2020, 3, 336–340 CrossRef CAS.
- Y. Wu, J. Wu, L. Jiao, W. Xu, H. Wang, X. Wei, W. Gu, G. Ren, N. Zhang, Q. Zhang, L. Huang, L. Gu and C. Zhu, Anal. Chem., 2020, 92, 3373–3379 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |