
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of cyclic hexamers of γ-cyclodextrin in a metallosupramolecular framework with D-penicillamine†
Supattra
Somsri
 ,
Naoto
Kuwamura
,
Tatsuhiro
Kojima
,
Nobuto
Yoshinari
and
Takumi
Konno
*
,
Naoto
Kuwamura
,
Tatsuhiro
Kojima
,
Nobuto
Yoshinari
and
Takumi
Konno
*
Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan. E-mail: konno@chem.sci.osaka-u.ac.jp
First published on 17th August 2020
Abstract
Cyclodextrins are widely used cyclic oligosaccharides of D-glucose whose hydrophilic exterior is covered by hydroxyl groups and whose hydrophobic interior is surrounded by lipophilic moieties. Because of this structural feature, cyclodextrin molecules commonly aggregate into dimensional structures via intermolecular hydrogen bonds, and their aggregation into closed oligomeric architectures has been achieved only via the attachment of functional substituent groups to the cyclodextrin rings. Here, we report the first structurally characterized self-assembly of non-substituted γ-cyclodextrin molecules into cyclic hexamers, which was realized in a chiral coordination framework composed of  complex-anions with D-penicillamine rather than L- or DL-penicillamine. The self-assembly is accompanied by the 3D-to-2D structural transformation of porous coordination frameworks to form helical hexagonal cavities that accommodate helical γ-cyclodextrin hexamers. This finding provides new insight into the development of cyclodextrin chemistry and host–guest chemistry based on chiral recognition and crystal engineering processes.
complex-anions with D-penicillamine rather than L- or DL-penicillamine. The self-assembly is accompanied by the 3D-to-2D structural transformation of porous coordination frameworks to form helical hexagonal cavities that accommodate helical γ-cyclodextrin hexamers. This finding provides new insight into the development of cyclodextrin chemistry and host–guest chemistry based on chiral recognition and crystal engineering processes.
Introduction
Cyclodextrins (CDs) are well-known oligosaccharides in which several molecules of D-glucose are connected via α-1,4-glycosidic bonds in a cyclic form with a truncated cone shape.1 Typical CDs exist in α, β, and γ forms that consist of 6, 7, and 8 D-glucose units, respectively.1 Since the smaller and larger openings of the truncated cone are covered by primary and secondary hydroxyl groups, respectively, the exterior of a CD is hydrophilic, resulting in an appreciable solubility in aqueous media. On the other hand, the interior of a CD is rather hydrophobic, being surrounded by lipophilic moieties, which allows it to accommodate functional, hydrophobic organic molecules with appropriate sizes in the chiral cavity via noncovalent interactions.2–6 These characteristic properties of CDs have widely been utilized for a variety of applications, such as optical resolution,7–9 drug delivery,10–13 and molecular machine systems.14–17 Apart from the conventional assembly of CDs assisted by guest molecules,15–20 the self-assembly of CDs themselves has been proposed based on NMR, TEM, and scattering techniques, in combination with molecular model calculations, since the initial report on dimer formation of CDs in solution.21–26 However, no direct structural evidence for the self-assembly of CDs themselves has yet been obtained by means of X-ray crystallography, although a large number of crystal structures of CD molecules have been reported.27,28 The structures of CDs so far characterized by single-crystal X-ray (SCXR) studies are categorized into cage, channel, and layer types of packing,27 and it has been shown that the difference in the packing modes arises from the different inclusion patterns of guest molecules.28 In X-ray structures, the cavities of CD molecules are commonly occupied by guest molecules, thus preventing the self-assembly of CD molecules.As part of our on-going study on the construction of chiral ionic solids with unusual arrangement of ionic species based on multinuclear complexes with D-penicillamine (D-H2pen; Chart 1),29–31 we have recently reported that treatment of an S-bridged  pentanuclear complex with free carboxylate groups, ΛΛ-[Au3Co2(D-pen)6]3− ([1D]3−), with Co2+ affords a 3D ionic solid, [Co(H2O)6]3[1D]2, with an extremely high porosity of 80%.32 In this compound, [Co(H2O)6]2+ cations and [1D]3− anions are alternately hydrogen bonded to form large, superhydrophilic cavities that can accommodate a large number of water molecules. While the accommodation of γ-CD molecules in [Co(H2O)6]3[1D]2 by soaking the crystals in a mother liquor containing γ-CD was confirmed by 1H NMR spectroscopy (Fig. S1, ESI†), attempts to characterize the inclusion compound by SCXR analysis failed because the crystallinity of [Co(H2O)6]3[1D]2 was entirely lost during the reaction due to its very weak 3D framework sustained only by hydrogen bonds between [Co(H2O)6]2+ and [1D]3−.
pentanuclear complex with free carboxylate groups, ΛΛ-[Au3Co2(D-pen)6]3− ([1D]3−), with Co2+ affords a 3D ionic solid, [Co(H2O)6]3[1D]2, with an extremely high porosity of 80%.32 In this compound, [Co(H2O)6]2+ cations and [1D]3− anions are alternately hydrogen bonded to form large, superhydrophilic cavities that can accommodate a large number of water molecules. While the accommodation of γ-CD molecules in [Co(H2O)6]3[1D]2 by soaking the crystals in a mother liquor containing γ-CD was confirmed by 1H NMR spectroscopy (Fig. S1, ESI†), attempts to characterize the inclusion compound by SCXR analysis failed because the crystallinity of [Co(H2O)6]3[1D]2 was entirely lost during the reaction due to its very weak 3D framework sustained only by hydrogen bonds between [Co(H2O)6]2+ and [1D]3−.
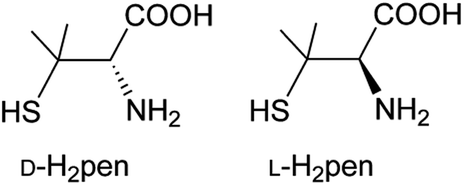 | ||
| Chart 1 Chemical structures of D- and L-penicillamine (H2pen). | ||
In the course of this study, we found that the crystallization of Na3[1D] from an aqueous buffer solution of NaOAc/HOAc produced a coordination compound (2D) composed of [Na4(H2O)15]4+ and [Na(H2O)6]+ cations and [1D]3− anions, which possessed a 3D porous framework comparable with that of [Co(H2O)6]3[1D]2, although the [1D]3− anions were directly connected by [Na4(H2O)15]4+ cations through coordination bonds in 2D (Scheme 1). While single crystals of 2D remained unchanged in the mother liquor, the addition of γ-CD resulted in its transformation to another single-crystalline compound (3D) with a 2D porous framework, accompanied by the inclusion of γ-CD molecules. Remarkably, the γ-CD molecules in 3D were found to self-assemble into cyclic hexamers. To our knowledge, such a structural transformation of the coordination framework induced by the inclusion of CD molecules, as well as the self-assembly of non-substituted CD molecules into cyclic hexamers, is unprecedented. The importance of the homochirality of 2D with D-pen to the appearance of this unique phenomenon was evidenced by the same inclusion experiments using 2L with L-pen or 2DL with a mixture of D-pen and L-pen.
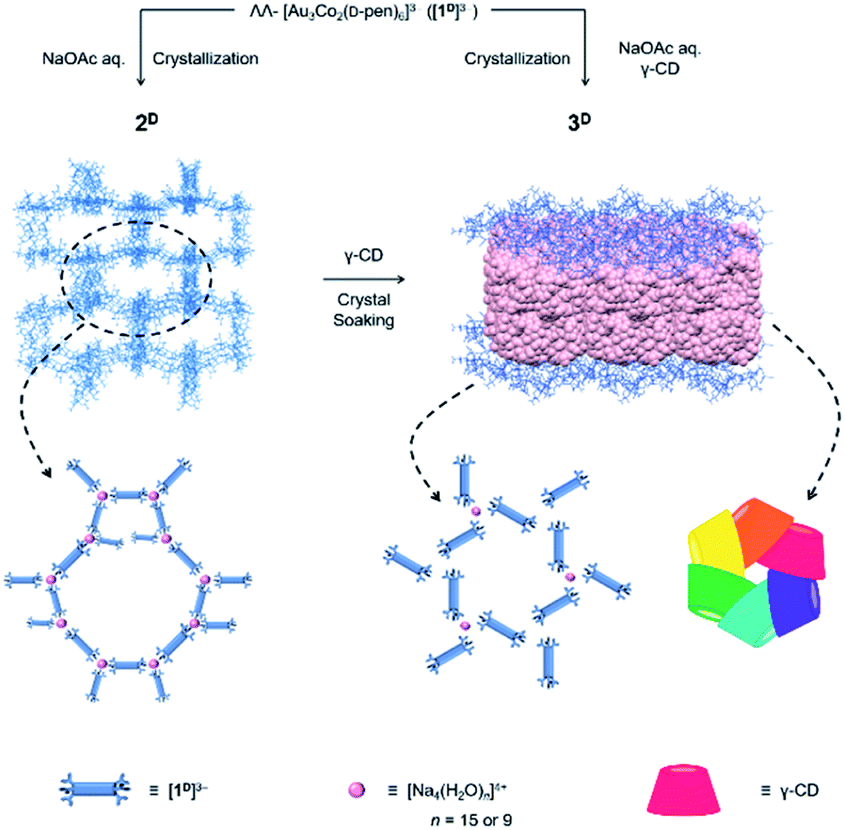 | ||
| Scheme 1 Synthetic route of 2D and 3D from ΛΛ-[Au3Co2(D-pen)6]3− ([1D]3−). | ||
Experimental section
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Na3[1D]·13H2O and Na3[1L]·13H2O instead of Na3[1D]·13H2O. Yield: 45 mg (37%). Anal. found: C, 18.18; H, 4.53; N, 4.17%. Calcd for Na3[Au3Co2(pen)6](NaCH3COO)0.3·18H2O = C30.6H90.9Au3Co2N6Na3.3O30.6S6: C, 18.29; H, 4.56; N, 4.18%. IR spectrum (cm−1, KBr disk): 1638 (νCOO−).
:1 mixture of Na3[1D]·13H2O and Na3[1L]·13H2O instead of Na3[1D]·13H2O. Yield: 45 mg (37%). Anal. found: C, 18.18; H, 4.53; N, 4.17%. Calcd for Na3[Au3Co2(pen)6](NaCH3COO)0.3·18H2O = C30.6H90.9Au3Co2N6Na3.3O30.6S6: C, 18.29; H, 4.56; N, 4.18%. IR spectrum (cm−1, KBr disk): 1638 (νCOO−).
Method A. Compound Na3[1D]·13H2O (100 mg, 0.060 mmol) was dissolved in 5 mL of a 0.5 M aqueous solution of NaOAc/HOAc (pH = 6.0). The dark purple solution was allowed to stand at room temperature for 20 days to give dark purple crystals with a hexagonal pyramidal shape. γ-CD (233 mg, 0.18 mmol) was dissolved in the mother liquor, and the block crystals were immersed in it. Within a day, the block crystals disappeared with the appearance of dark purple crystals with a hexagonal plate shape, which were collected by filtration after 14 days. Yield: 65 mg (22%). Anal. found: C, 30.24; H, 5.96; N, 1.65%. Calcd for Na3[Au3Co2(D-pen)6](NaCH3COO)0.3·2(γ-CD)·41H2O = C126.6H296.9Au3Co2N6Na3.3O133.6S6: C, 30.30; H, 5.96; N, 1.67%. IR spectrum (cm−1, KBr disk): 2921 (νCH2), 1617 (νCOO−), 1158 (νC–C), and 1025 (δOH).
Method B. To a colourless solution containing γ-CD (389 mg, 0.300 mmol) in 5 mL of a 0.5 M aqueous solution of NaOAc/HOAc (pH = 6) was added a solution containing Na3[1D]·13H2O (100 mg, 0.060 mmol) in 5 mL of a 0.5 M aqueous solution of NaOAc/HOAc (pH = 6). The mixture solution was allowed to stand at room temperature for 22 days to give dark purple crystals with a hexagonal plate shape, which were collected by filtration. Yield: 53 mg (17%).
Physical measurements
The IR spectra were collected on a JASCO FT/IR-4100 infrared spectrophotometer by using the KBr method at room temperature. The circular dichroism spectra were recorded on a JASCO J-820 spectrometer at room temperature. The diffuse reflection spectra were measured on a JASCO V-670 UV/Vis/NIR spectrometer. X-ray fluorescence spectrometry was performed on a Shimadzu Model EDX-7000 spectrometer. Elemental analyses (C, H, N) were performed at Osaka University using a Yanaco CHN Corder MT-6. The 1H NMR spectra were recorded on a JEOL ECS400 (400 MHz) spectrometer in D2O. Sodium 4,4′-dimethyl-4-silapentane-1-sulfonate (DSS) was used as the internal standard. High-quality powder X-ray diffraction (PXRD) patterns were recorded at room temperature in transmission mode [synchrotron radiation λ = 1.0 Å; 2θ range = 2°–78°; step width = 0.01°; data collection time 1 min] on a diffractometer equipped with a MYTHEN microstrip X-ray detector (Dectris Ltd) at the SPring-8 BL02B2 beamline. The crystals in the mother liquor were loaded into a glass capillary tube (diameter = 0.3 mm), which was rotated during the measurements. The powder simulation patterns were generated from the SCXR structures using Mercury 3.10.Crystallographic analysis
SCXR diffraction analysis of 2D and 3D was performed on a Rayonix MX225HS CCD area detector at the BL2D SMC beamline in the Pohang Accelerator Laboratory, Korea. Diffraction data were collected from the PAL BL2D-SMDC program,34 and cell refinement, reduction, and absorption correction were performed using HKL3000.35 The synchrotron X-ray diffraction study of 2L and 2DL was carried out at the BL02B1 beamline in SPring-8 with a diffractometer equipped with a PILATUS3 X CdTe 1M. Data collection of all samples was performed at 100 K. The crystal structures were solved and refined by SHELX.36 The least-squares refinement of the structural model was performed under displacement parameter restraints such as DFIX, ISOR, RIGU, and SIMU. The final refinement was performed with the modification of the structure factors for the electron densities of the disordered solvents using the SQUEEZE option of PLATON.37 The crystallographic data are summarized in Table S1 (ESI†).The asymmetric unit of 2D consists of one-third of [Na4(H2O)15]4+, one-third of [Na(H2O)6]+, and half of [Au3Co2(D-pen)6]3− in addition to several water molecules of crystallization, consistent with its chemical formula of Na3[1D]·1/3NaOAc·nH2O. An acetate anion, which might exist with a site occupancy of 1/6 in the asymmetric unit, could not be observed in the Fourier map due to its positional disorder and its low occupancy.
The asymmetric unit of 2DL consists of one-third of [Na3(H2O)7]3+, one-third of [Na(H2O)6]+, one-third of [Na(H2O)3]+, and half of [Au3Co2(D-pen)6]3−, in addition to several water molecules of crystallization, consistent with the chemical formula of Na3[1D]0.5[1L]0.5·1/3NaOAc·nH2O. An acetate anion that might exist with a site occupancy of 1/6 in the asymmetric unit could not be observed in the Fourier map due to its positional disorder, as well as its low occupancy.
The asymmetric unit of 3D consists of [Na4(H2O)9]4+ and six [Na(H2O)3]+ cations, 3 [Au3Co2(D-pen)6]3− anions, and 6 γ-CD molecules, in addition to several water molecules of crystallization, consistent with its chemical formula of Na3[1D]·1/3NaOAc·2(γ-CD)·nH2O. An acetate anion in the asymmetric unit was not observed in the Fourier map due to its positional disorder and low occupancy.
Results
Synthesis and characterization of 2D
Previously, we reported that crystallizing a powder sample of ΛΛ-Na3[Au3Co2(D-pen)6] (Na3[1D]) from water yielded dark purple crystals with a hexagonal plate shape. It has been shown from the SCXR crystallography that this product possesses a 3D dense coordination framework, in which all carboxylate groups in each [1D]3− anion are involved in the coordination with Na+ cations (Fig. S2, ESI†).33 When crystallization was carried out using an aqueous buffer solution of NaOAc/HOAc at pH 6.0, dark purple crystals with a truncated hexagonal pyramid shape (2D) were obtained.‡ Compound 2D was confirmed to be a sodium salt of [1D]3− based on the solid-state diffuse reflection, circular dichroism, and IR spectra, which are essentially the same as those of Na3[1D] (Fig. S3–S5, ESI†). However, the 1H NMR spectrum of 2D in D2O indicated the presence of acetate (Fig. S6, ESI†), and the elemental analysis data of 2D were consistent with a formula of Na3[1D]·1/3NaOAc·nH2O.Crystal structure of 2D
The structure of 2D was determined by SCXR crystallography. Crystalline 2D contains sodium cations that exist as the isolated aqua cations of [Na(H2O)6]+ and the cluster cations of [Na4(H2O)15]4+, in addition to [1D]3− anions. Each [Na(H2O)6]+ cation is surrounded by 3 [1D]3− anions in a skewed right-handed arrangement by forming OH2⋯OOC hydrogen bonds (av. O⋯O = 2.81 Å), while each [1D]3− anion spans 2 [Na(H2O)6]+ cations through hydrogen bonds (Fig. 1a). As a result, [1D]3− anions are alternately hydrogen-bonded with [Na(H2O)6]+ cations in a 3D porous structure (Fig. S7, ESI†). This structure of 2D corresponds well with that found in [Co(H2O)6]3[1D]2, in which [1D]3− anions are alternately linked by [CoII(H2O)6]2+ cations through hydrogen bonds (Fig. S7, ESI†).32 In 2D, however, 3 [1D]3− anions are connected by a [Na4(H2O)15]4+ cation through coordination bonds (Na–O = 2.40 Å) in a 3-connected mode (Fig. 1b–d), together with a [Na(H2O)6]+ cation through hydrogen bonds, forming a 3D coordination polymer with a porosity of 78% (Fig. 1e). The alternate binding of [Na4(H2O)15]4+ cations and [1D]3− anions affords large, hydrophilic open channels in 2D, each of which consists of 10 [Na4(H2O)15]4+ cations and 10 [1D]3− anions, with a maximum opening of 35 Å (Fig. 1f). It has been shown that [Co(H2O)6]3[1D]2 is transformed into a dense structure in [Co(H2O)4]3[1D]2 by way of the 1D porous structure in [Co(H2O)4][Co(H2O)6]2[1D]2, when its crystals are stored in a mother liquor for one week.32 On the other hand, such a structurally transformation was not observed for 2D under the same conditions for at least one month.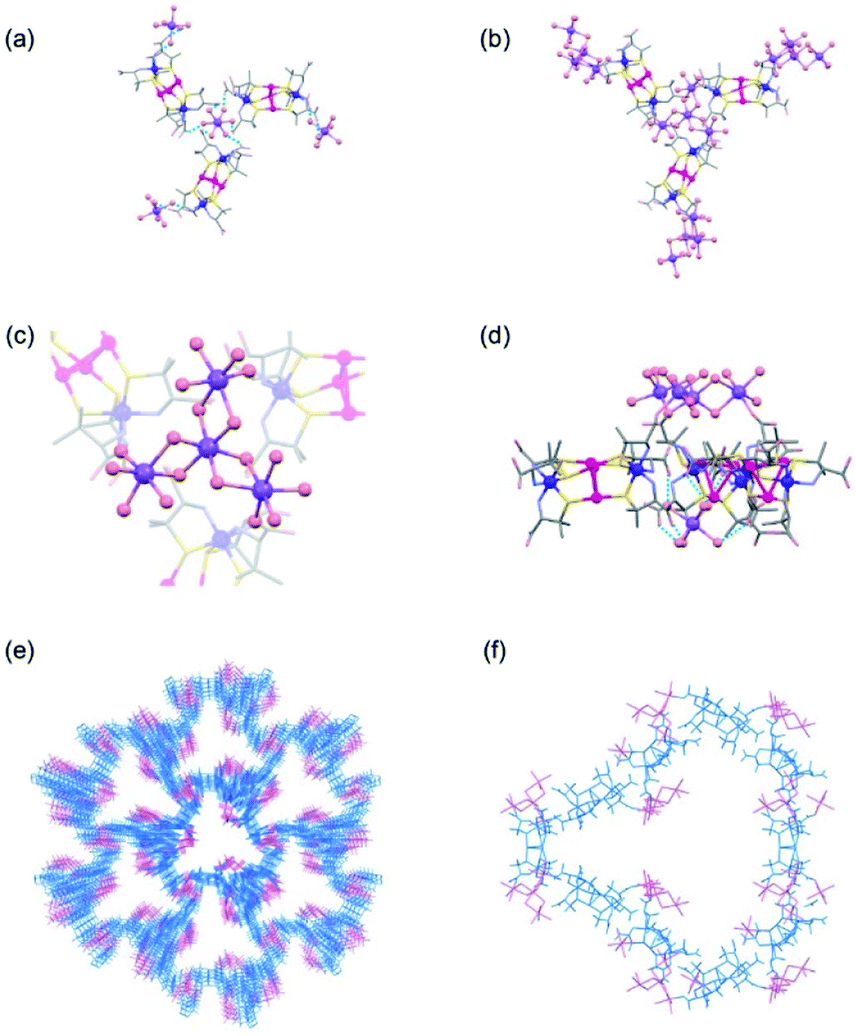 | ||
| Fig. 1 Crystal structures of 2D, crystallized in the cubic space group of I213. (a) The linkage of [1D]3− anions with [Na(H2O)6]+ cations in a 3-connected mode. (b) The linkage of [1D]3− anions with [Na4(H2O)15]4+ cations in a 3-connected mode. (c) The tetranuclear structure of [Na4(H2O)15]4+ that connects 3 [1D]3− anions. (d) The linkage of [1D]3− anions with [Na(H2O)6]+ and [Na4(H2O)15]4+ cations. (e) The 3D porous framework. (f) The open channel consisting of 10 [1D]3− anions that are connected by 10 [Na(H2O)6]+ and 10 [Na4(H2O)15]4+ cations. | ||
Synthesis and crystal structures of 2L and 2DL
A similar crystallization of ΔΔ-Na3[Au3Co2(L-pen)6] (Na3[1L]), instead of Na3[1D], from aqueous NaOAc/HOAc buffer (pH 6.0) produced purple crystals with the same truncated hexagonal pyramid shape (2L). The crystal structure of 2L, which is an enantiomer of 2D, was confirmed by SCXR crystallography (Fig. S9, ESI†), as well as solid-state circular dichroism spectroscopy (Fig. S4, ESI†). A noticeable structural feature in 2L is the linkage of 3 [1D]3− anions with [Na4(H2O)15]4+ and [Na(H2O)6]+ cations in a skewed left-handed arrangement, opposite to the right-handed arrangement in 2D.When a 1:1 mixture of Na3[1D] and Na3[1L] was crystallized under the same conditions, purple crystals with a tetrahedron shape (2DL) were produced.‡ Based on the solid-state diffuse reflection, circular dichroism, IR, and 1H NMR spectra (Fig. S3–S6, ESI†), together with the elemental analysis, 2DL was determined to have a formula of Na3[1D]0.5[1L]0.5·1/3NaOAc)·nH2O, which corresponds to the formula of Na3[1D]·1/3NaOAc·nH2O for 2D. The SCXR analysis revealed that 2DL contains a 3D porous framework composed of [1D]3− anions and a 3D porous framework composed of [1L]3− anions, which are essentially the same as the frameworks of 2D and 2L, respectively (Fig. 2a, b, and S10, ESI†). In 2DL, the two enantiomeric frameworks penetrate each other by forming O–H⋯O hydrogen bonds (av. O⋯O = 2.75 Å) between water molecules coordinated to NaI centres (Fig. 2c and S10, ESI†). The porosity of 2DL is 41%, and the largest opening is 7 Å, which is much lower than that of 2D.
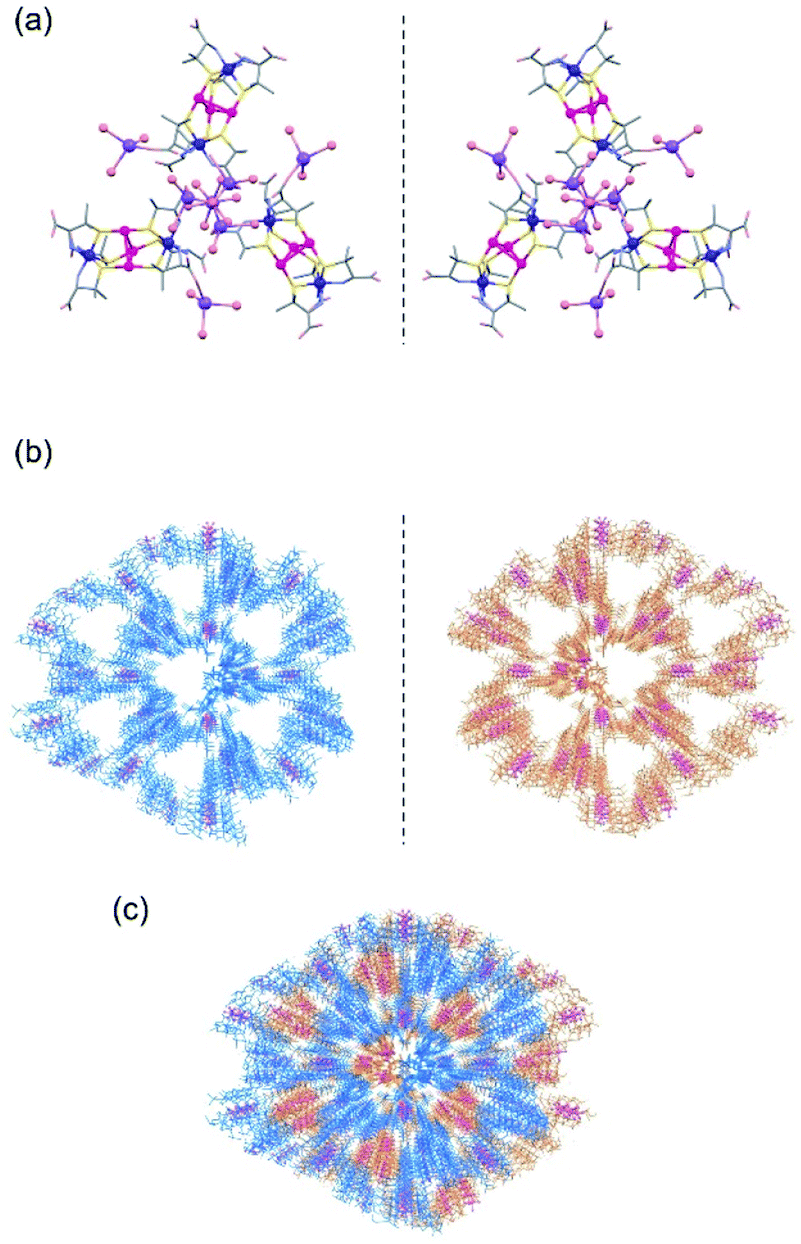 | ||
Fig. 2 Crystal structures of 2DL crystallized in the cubic space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d. (a) The linkage of [1D]3− anions and the linkage of [1L]3− anions with [Na(H2O)6]+ and [Na3(H2O)7]3+ cations in a 3-connected mode. A [Na(H2O)3]+ moiety is attached to each anion through a carboxylate group. (b) The 3D porous framework composed of [1D]3− anions (blue) and that composed of [1L]3− anions (orange). (c) Packing structure of 2DL shows the interpenetrating framework of the ΛΛ-[Au3Co2(D-pen)6]3− complex (blue) and the ΔΔ-[Au3Co2(L-pen)6]3− complex (orange). 3d. (a) The linkage of [1D]3− anions and the linkage of [1L]3− anions with [Na(H2O)6]+ and [Na3(H2O)7]3+ cations in a 3-connected mode. A [Na(H2O)3]+ moiety is attached to each anion through a carboxylate group. (b) The 3D porous framework composed of [1D]3− anions (blue) and that composed of [1L]3− anions (orange). (c) Packing structure of 2DL shows the interpenetrating framework of the ΛΛ-[Au3Co2(D-pen)6]3− complex (blue) and the ΔΔ-[Au3Co2(L-pen)6]3− complex (orange). | ||
Formation and characterization of 3D
Soaking truncated hexagonal pyramid crystals of 2D in a mother liquor containing excess γ-CD resulted in the disappearance of these crystals and the appearance of different crystals with a thin hexagonal plate shape (3D) within one day.‡ Crystals of 3D were also obtained when Na3[1D] was crystallized from aqueous NaOAc/HOAc buffer in the presence of γ-CD (Scheme 1). While the solid-state diffuse reflection and CD spectra of 3D (Fig. S3 and S4, ESI†) are very similar to those of 2D, the 1H NMR spectrum shows characteristic multiplet signals due to γ-CD (Fig. S6, ESI†). 3D was assigned as a 1:2 adduct of 2D and γ-CD with a chemical formula of Na3[1D]·1/3NaOAc·2(γ-CD)·nH2O based on the 1:2 integration ratio of the 1H NMR signals due to [1D]3− and γ-CD, together with elemental analysis. Consistent with the difference in crystal shapes between 2D and 3D, the PXRD pattern of 3D differs significantly from that of 2D (Fig. S8, ESI†).
Crystal structure of 3D
The SCXR analysis revealed that 3D contains Na+ cations and [1D]3− anions in a 10:3 ratio, as does 2D. However, Na+ cations in 3D exist as [Na4(H2O)9]4+ and [Na(H2O)3]+ cations in a 1:6 ratio, with 2 [Na(H2O)3]+ cations being attached to each [1D]3− anion (Fig. 3a). This is different from the presence of [Na4(H2O)15]4+ and [Na(H2O)6]+ cations in a 1:1 ratio in 2D. The structural difference between 2D and 3D is also observed in the sodium(I) cluster cations; the central NaI atom bridges the 3 terminal NaI atoms through 6 water molecules to form [Na4(μ-H2O)6(H2O)9]4+ in 2D, while 3 of the 6 water molecules are involved in bridging to form [Na4(μ-H2O)3(H2O)6]4+ in 3D (Fig. 3a). As in 2D, the [1D]3− anions in 3D are linked by sodium(I) cluster cations in a 3-connected mode, forming a trimer subunit composed of 3 [1]3− anions linked by the [Na4(H2O)9]4+ cation (Fig. 3a). In 3D, however, the trimer subunits are connected in a planar fashion by 6 adjacent subunits though H2N⋯OOC hydrogen bonds (av. N⋯O = 2.90 Å) in a 2D layer structure, having hexagonal open channels with a dimeter of 20 Å surrounded by 6 [1]3− anions in a helical fashion (Fig. 3b).
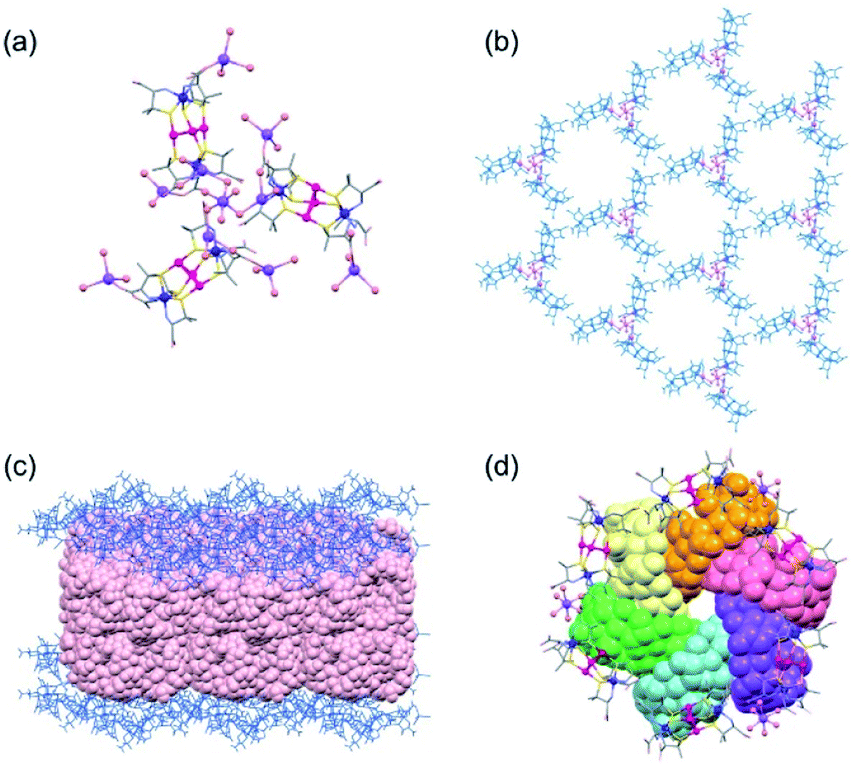 | ||
| Fig. 3 Crystal structures of 3D crystallized in the monoclinic space group C2. (a) The linkage of [1D]3− anions with [Na4(H2O)9]4+ cations in a 3-connected mode. (b) The 2D porous framework. (c) The double layer of γ-CD between the 2D porous frameworks. (d) The cyclic hexamer of γ-CD located in the hexagonal cavity. | ||
In 3D, γ-CD molecules are accommodated between the 2D porous layers (Fig. 3c), such that 6 γ-CD molecules self-assemble into a helical cyclic hexamer with a diameter of 34 Å by forming intermolecular OH⋯O hydrogen bonds (av. O⋯O = 2.90 Å). Part of each γ-CD molecule in the hexamer is included in the cavity of its adjacent molecule, retaining the original truncated cone shape (Fig. 3d and S11, ESI†). From the comparison of the volume of the hexamer of γ-CD molecules (8561 Å3) with that of each γ-CD molecule (1532 Å3), the inclusion ratio of γ-CD was estimated to be 7% in the hexamer. Each helical hexamer of γ-CD in 3D is hydrogen-bonded to 6 adjacent hexamers (av. OH⋯O = 2.77 Å) to form a single layer, which is stacked with the other layer through OH⋯O hydrogen bonds (av. OH⋯O = 2.81 Å), forming a double layer structure of γ-CD (Fig. 3c and S11, ESI†). Note that each helical hexamer of γ-CD sits on a helical hexagon composed of 6 [1]3− anions sharing a 6-fold axis (Fig. 3d), such that carboxylate groups of each [1]3− anion are tightly hydrogen-bonded (av. O⋯O = 2.62 Å) with the secondary hydroxyl groups of γ-CD molecules (Fig. S12, ESI†).
As in the case of 2D, soaking crystals of 2L in a mother liquor after adding γ-CD resulted in the disappearance of the crystals. However, no crystalline product appeared for at least 2 weeks. On the other hand, the crystals of 2DL remained unchanged for at least 2 weeks without dissolution when the crystals were soaked in a mother liquor containing γ-CD under the same conditions.
Discussion
In this study, we found that the crystallization of a powder sample of ΛΛ-Na3[Au3Co2(D-pen)6] (Na3[1D]) from an aqueous NaOAc/HOAc buffer leads to the production of purple crystals with a truncated hexagonal pyramid shape (2D), which possesses a 3D structure with a high porosity of 78%, comparable to that of the previously reported [Co(H2O)6]3[1D]2.32 This structure contains [Na(H2O)6]+ and [Na4(H2O)15]4+ as cationic species and is entirely different from the dense 3D structure that was formed by the crystallization of Na3[1D] from water. Since the formation of the dense structure is a result of the coordination of all the carboxylate groups in [1D]3− to Na+ centres, the presence of excess acetate ions in the aqueous buffer, which cover Na+ ions to prevent carboxylate coordination, is key to the construction of the highly porous structure in 2D. The presence of excess sodium cations is also crucial to the generation of 2D because they are needed for the formation of the [Na4(H2O)15]4+ cluster cations. The most characteristic structural feature of 2D is the linkage of 3 [1D]3− anions in a skewed arrangement not only via [Na(H2O)6]+ through hydrogen bonds but also via [Na4(μ-H2O)6(H2O)9]4+ through coordination bonds; this linkage leads to the generation of nanometre-sized open channels consisting of 10 [1D]3− anions connected by 10 [Na(H2O)6]+ and 10 [Na4(μ-H2O)6(H2O)9]4+ cations. Despite the presence of large open channels, crystal 2D is quite stable in a mother liquor for a long period of time, which is in contrast to the facile structural conversion of the 3D porous [Co(H2O)6]3[1D]2 under the same conditions. This is due to the linkage of [1D]3− anions by [Na4(μ-H2O)6(H2O)9]4+ cations through coordination bonds in 2D, together with [Na(H2O)6]+ through hydrogen bonds, while [1D]3− anions are linked only by [Co(H2O)6]2+ through hydrogen bonds in [Co(H2O)6]3[1D]2.As expected, a crystallization procedure similar to that of ΔΔ-Na3[Au3Co2(L-pen)6] (Na3[1L]) yielded truncated hexagonal pyramid crystals (2L), which have a 3D porous structure enantiomeric to that of 2D. Notably, the crystallization of a 1:1 mixture of Na3[1D] and Na3[1L] yielded different shaped crystals (2DL) rather than a mixture of 2D and 2L crystals. While 2DL contains [1D]3− and [1L]3− anions in a 1:1 ratio, [1D]3− and [1L]3− anions are independently connected by [Na3(H2O)7]3+ cations to form a pair of enantiomeric 3D frameworks that are essentially the same as those of 2D and 2L. In 2DL, the frameworks are interpenetrated in each other to form a denser structure with a much lower porosity than that of 2D or 2L. Such an enantioselective linkage of a pair of enantiomeric ionic species to a pair of enantiomeric coordination frameworks that are interpenetrated with each other in a crystalline lattice is unprecedented, although the interpenetration of a pair of enantiomeric metal–organic frameworks generated from metal ions and organic ligands has been reported.38–42
The high robustness of the 3D coordination framework with large, hydrophilic open channels in 2D prompted us to investigate the inclusion of γ-CD molecules in its cavities. Contrary to our expectation, soaking crystals of 2D in a mother liquor containing γ-CD resulted in transformation of the crystals into another crystalline phase (3D) within one day, incorporating γ-CD molecules in the crystal. While 3D contains [1D]3− and Na+ in a 3:10 ratio, as does 2D, 3D adopts a 2D layer structure rather than the 3D structure observed in 2D. The structural transformation of the 3D framework in 2D to the 2D framework in 3D is accompanied by the alternation of [Na4(μ-H2O)6(H2O)9]4+ in 2D to [Na4(μ-H2O)3(H2O)6]4+ in 3D, which allows the linkage of 3 [1D]3− anions in a planar arrangement in 3D rather than the skewed arrangement in 2D. In addition, each [1D]3− anion bears pendent [Na(H2O)3]+ cations, which appears to prevent the linkage of the trimer units with additional [Na4(H2O)9]4+ ions. Thus, the trimer units are hydrogen-bonded to each other in a planar fashion, forming a 2D layer structure with hexagonal cavities, each of which is surrounded by 6 [1D]3− anions in a helical arrangement. In 3D, γ-CD molecules are accommodated between the 2D layers to form a unique double layer of γ-CD. Of particular note is that the γ-CD molecules in the double layer exist as cyclic hexamers with a diameter of 34 Å, in which part of one γ-CD ring is included in an adjacent ring in a helical fashion. It has been proposed that γ-CD can be self-assembled to form molecular aggregates,21,23 but their exact structures have remained unknown. This is the first structurally characterized self-assembly of γ-CD molecules, the structure of which is reminiscent of hexamers found in natural proteins such as glutamine synthetase.43,44 The self-assembly of γ-CD with the partial inclusion of one molecule within another molecule is itself remarkable because the hydrophilic nature of the exterior of γ-CD covered by hydroxyl groups is quite suitable for 1D and 2D arrangements through intermolecular hydrogen bonds, as reported in previous studies;45–52 the assembly of CD molecules into discrete oligomers has been achieved only by the modification of the CD ring via organic syntheses such that a hydrophobic substituent group attached to one CD ring can be inserted into another CD ring via hydrophobic interactions.15,53
An additional note is that crystals of 2L and 2DL do not permit such incorporation of γ-CD molecules accompanied by the structural transformation. This implies that the chirality of the host framework in 2D composed of [1D]3− anions with D-pen ligands, as well as its highly porous structure with open channels larger than the size of γ-CD, is essential for the appearance of this unique phenomenon. Molecular model examinations revealed that nonbonding interactions, rather than hydrogen bonding, exist between each [1L]3− anion and γ-CD molecule when the [1D]3− anions in 3D are replaced by [1L]3− anions (Fig. S13, ESI†). Thus, γ-CD molecules and [1D]3− anions mutually recognize their chiralities, leading to the 3D-to-2D structural transformation to generate helical hexagonal cavities surrounded by [1D]3− anions, with the concomitant self-assembly of γ-CD molecules into helical cyclic hexamers that are best fitted for the hexagonal cavities.
Conclusion
We showed that the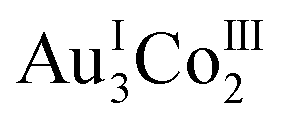 complex-anions with D-penicillamine, ΛΛ-[Au3Co2(D-pen)6]3− ([1D]3−), are organized into the 3D porous framework in 2D with a very high porosity of 78%, assisted by aqua sodium(I) species via hydrogen bonds and coordination bonds, generating nanometre-sized open channels that allow the inclusion of γ-CD molecules inside the crystal. Such an inclusion of γ-CD molecules was not observed for the corresponding porous framework in 2L with L-penicillamine and the dense framework in 2DL with mixed D-penicillamine and L-penicillamine. This event was found to be accompanied by the transformation of the 3D framework in 2D to the 2D layer framework in 3D so as to form hexagonal cavities that can accept γ-CD molecules. Remarkably, the γ-CD molecules in 3D existed as huge cyclic hexamers, which is the first self-assembly of non-substituted γ-CD molecules so far structurally characterized. Thus, the present study realized the unprecedented self-assembly of γ-CD using the 3D chiral coordination framework, which was transformed to the 2D framework induced by the chiral-selective inclusion of γ-CD molecules. The chiral, highly porous, guest-responsible framework in 2D appears to be applicable for the chiral-selective inclusion and the structural determination of other huge molecules/aggregates such as natural proteins, which cannot be realized via the recently developed ‘crystal sponge method’ that employs rigid MOFs with limited void spaces.54–56
complex-anions with D-penicillamine, ΛΛ-[Au3Co2(D-pen)6]3− ([1D]3−), are organized into the 3D porous framework in 2D with a very high porosity of 78%, assisted by aqua sodium(I) species via hydrogen bonds and coordination bonds, generating nanometre-sized open channels that allow the inclusion of γ-CD molecules inside the crystal. Such an inclusion of γ-CD molecules was not observed for the corresponding porous framework in 2L with L-penicillamine and the dense framework in 2DL with mixed D-penicillamine and L-penicillamine. This event was found to be accompanied by the transformation of the 3D framework in 2D to the 2D layer framework in 3D so as to form hexagonal cavities that can accept γ-CD molecules. Remarkably, the γ-CD molecules in 3D existed as huge cyclic hexamers, which is the first self-assembly of non-substituted γ-CD molecules so far structurally characterized. Thus, the present study realized the unprecedented self-assembly of γ-CD using the 3D chiral coordination framework, which was transformed to the 2D framework induced by the chiral-selective inclusion of γ-CD molecules. The chiral, highly porous, guest-responsible framework in 2D appears to be applicable for the chiral-selective inclusion and the structural determination of other huge molecules/aggregates such as natural proteins, which cannot be realized via the recently developed ‘crystal sponge method’ that employs rigid MOFs with limited void spaces.54–56
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST CREST (Grant No. JPMJCR13L3) and JSPS KAKENHI (Grant No. 18H05344, 19K05667 and 19K05496). The synchrotron radiation experiments were performed at the BL02B1 and BL02B2 beamlines of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2019A1350, 2018B1296, 2019A1279 and 2019B1107) and at 2D-SMC of the Pohang Accelerator Laboratory.Notes and references
- G. Crini, Chem. Rev., 2014, 114, 10940 CrossRef CAS PubMed.
- W. Saenger, Angew. Chem., Int. Ed. Engl., 1980, 19, 344 CrossRef.
- G. Wenz, Angew. Chem., Int. Ed., 1994, 33, 803 CrossRef.
- J. Szejtli, Chem. Rev., 1998, 98, 1743 CrossRef CAS PubMed.
- E. M. M. del Valle, Process Biochem., 2004, 39, 1033 CrossRef CAS.
- F. Hapiot, S. Tilloy and E. Monflier, Chem. Rev., 2006, 106, 767 CrossRef CAS PubMed.
- T. J. Ward and D. W. Armstrong, J. Liq. Chromatogr., 1986, 9, 407 CrossRef CAS.
- K. Kano, J. Phys. Org. Chem., 1997, 10, 286 CrossRef CAS.
- B. Chankvetadze, Chem. Soc. Rev., 2004, 33, 337 RSC.
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045 CrossRef CAS PubMed.
- Y. Chen and Y. Liu, Chem. Soc. Rev., 2010, 39, 495 RSC.
- C. O. Mellet, J. M. G. Fernández and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586 RSC.
- J. Zhang and P. X. Ma, Adv. Drug Delivery Rev., 2013, 65, 1215 CrossRef CAS PubMed.
- A. Harada, Acc. Chem. Res., 2001, 34, 456 CrossRef CAS PubMed.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875 RSC.
- F. Niess, V. Duplan and J.-P. Sauvage, Chem. Lett., 2014, 43, 964 CrossRef CAS.
- A. Hashidzume, H. Yamaguchi and A. Harada, Eur. J. Org. Chem., 2019, 3344 CrossRef CAS.
- D. Armspach, P. R. Ashton, C. P. Moore, N. Spencer, J. F. Stoddart, T. J. Wear and D. J. Williams, Angew. Chem., Int. Ed., 1993, 32, 854 CrossRef.
- K. A. Udachin, L. D. Wilson and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 12375 CrossRef CAS.
- S. Kamitori, O. Matsuzaka, S. Kondo, S. Muraoka, K. Okuyama, K. Noguchi, M. Okada and A. Harada, Macromolecules, 2000, 33, 1500 CrossRef CAS.
- K. Miyajima, M. Sawada and M. Nakagaki, Bull. Chem. Soc. Jpn., 1983, 56, 3556 CrossRef CAS.
- M. Bonini, S. Rossi, G. Karlsson, M. Almgren, P. L. Nostro and P. Baglioni, Langmuir, 2006, 22, 1478 CrossRef CAS PubMed.
- M. Messner, S. V. Kurkov, M. E. Brewster, P. Jansook and P. Loftsson, Int. J. Pharm., 2011, 407, 174 CrossRef CAS PubMed.
- S. Yang, Y. Yan, J. Huang, A. V. Petukhov, L. M. J. Kroon-Batenburg, M. Drechsler, C. Zhou, M. Tu, S. Granick and L. Jiang, Nat. Commun., 2017, 8, 15856 CrossRef CAS PubMed.
- A. J. M. Valente, R. A. Carvalho, D. Murtinho and O. Söderman, Langmuir, 2017, 33, 8233 CrossRef CAS PubMed.
- H. Shigemitsu and T. Kida, Polym. J., 2018, 50, 541 CrossRef CAS.
- W. Saenger and T. Steiner, Acta Crystallogr., Sect. A: Found. Crystallogr., 1998, 54, 798 CrossRef.
- K. Harata, Crystallographic Study of Cyclodextrins and Their Inclusion Complexes, in Cyclodextrins and Their Complexes: Chemistry, Analytical Methods, Applications, ed. H. Dpdzoik, Willey-VCH, 2006, ch. 7, pp. 147–198 Search PubMed.
- (a) R. Lee, A. Igashira-Kamiyama, H. Motoyoshi and T. Konno, CrystEngComm, 2012, 14, 1936 RSC; (b) R. Lee, A. Igashira-Kamiyama, M. Okumura and T. Konno, Bull. Chem. Soc. Jpn., 2013, 86, 908 CrossRef CAS; (c) S. Yamashita, Y. Nakazawa, S. Yamanaka, M. Okumura, T. Kojima, N. Yoshinari and T. Konno, Sci. Rep., 2018, 8, 2606 CrossRef PubMed.
- N. Yoshinari and T. Konno, Chem. Rec., 2016, 16, 1647 CrossRef CAS PubMed.
- N. Yoshinari and T. Konno, Coordination Molecular Technology, in Molecular Technology, Volume 4: Synthesis Innovation, ed. H. Yamamoto and T. Kato, Wiley-VCH, Weinheim, Germany, 2019, pp. 199–230 Search PubMed.
- S. Surinwong, N. Yoshinari, B. Yotnoi and T. Konno, Chem.–Asian J., 2016, 11, 486 CrossRef CAS PubMed.
- T. Konno, A. Toyota and A. Igashira-Kamiyama, J. Chin. Chem. Soc., 2009, 56, 26 CrossRef CAS.
- J. W. Shin, K. Eom and D. Moon, J. Synchrotron Radiat., 2016, 23, 369 CrossRef PubMed.
- W. Minor, M. Cymborowski, Z. Otwinowski and M. Chruszcz, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 859 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- B. F. Abrahams, S. R. Batten, H. Hamit, B. F. Hoskins and R. Robson, Chem. Commun., 1996, 1313 RSC.
- X.-H. Bu, K. Biradha, T. Yamaguchi, M. Nishimura, T. Ito, K. Tanaka and M. Shionoya, Chem. Commun., 2000, 1953 RSC.
- T. J. Prior and M. J. Rosseinsky, Inorg. Chem., 2003, 42, 1564 CrossRef CAS PubMed.
- Y. Bai, C. Duan, P. Cai, D. Dang and Q. Meng, Dalton Trans., 2005, 2678 RSC.
- Q. Sun, M. Wei, Y. Bai, C. He, Q. Meng and C. Duan, Dalton Trans., 2007, 4089 RSC.
- R. J. Almassy, C. A. Janson, R. Hamlin, N.-H. Xuong and D. Eisenberg, Nature, 1986, 323, 304 CrossRef CAS PubMed.
- M. M. Yamashita, R. J. Almassy, C. A. Janson, D. Cascio and D. Eisenberg, J. Biol. Chem., 1989, 264, 17681 CAS.
- R. A. Smaldone, R. S. Forgan, H. Furukawa, J. J. Gassensmith, A. M. Z. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8630 CrossRef CAS PubMed.
- Z. Liu, M. Frasconi, J. Lei, Z. J. Brown, Z. Zhu, D. Cao, J. Iehl, G. Liu, A. C. Fahrenbach, Y. Y. Botros, O. K. Farha, J. T. Hupp, C. A. Mirkin and J. F. Stoddart, Nat. Commun., 2013, 4, 1855 CrossRef PubMed.
- K. I. Assaf, M. S. Ural, F. Pan, T. Georgiev, S. Simova, K. Rissanen, D. Gabel and W. M. Nau, Angew. Chem., Int. Ed., 2015, 54, 6852 CrossRef CAS PubMed.
- M. A. Moussawi, N. Leclerc-Laronze, S. Floquet, P. A. Abramov, M. N. Sokolov, S. Cordier, A. Ponchel, E. Monflier, H. Bricout, D. Landy, M. Haouas, J. Marrot and E. Cadot, J. Am. Chem. Soc., 2017, 139, 12793 CrossRef CAS PubMed.
- E. Patyk-Kaźmierczak, M. R. Warren, D. R. Allan and A. Katrusiak, Phys. Chem. Chem. Phys., 2017, 19, 9086 RSC.
- A. A. Ivanov, C. Falaise, P. A. Abramov, M. A. Shestopalov, K. Kirakci, K. Lang, M. A. Moussawi, M. N. Sokolov, N. G. Naumov, S. Floquet, D. Landy, M. Haouas, K. A. Brylev, Y. V. Mironov, Y. Molard, S. Cordier and E. Cadot, Chem.–Eur. J., 2018, 24, 13467 CrossRef CAS PubMed.
- C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, J. Am. Chem. Soc., 2018, 140, 11198 CrossRef CAS PubMed.
- P. Yang, W. Zhao, A. Shkurenko, Y. Belmabkhout, M. Eddaoudi, X. Dong, H. N. Alshareef and N. M. Khashab, J. Am. Chem. Soc., 2019, 141, 1847 CrossRef CAS PubMed.
- A. Kanaya, Y. Takashima and A. Harada, J. Org. Chem., 2011, 76, 492 CrossRef CAS PubMed.
- Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780 CrossRef CAS PubMed.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 ( Nature , 2013 , 501 , 262 ) CrossRef CAS PubMed.
- M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, IUCrJ, 2016, 3, 139 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR (Fig. S1 and S6), crystal structure (Fig. S2, S7, S9–S12 and Table S1), diffuse reflection (Fig. S3), circular dichroism (Fig. S4), IR (Fig. S5), PXRD (Fig. S8), modelling study (Fig. S13). CCDC 2006554–2006557. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03925j |
| ‡ The homogeneity of the bulk samples of 2D, 2DL, and 3D was confirmed by the PXRD patterns, which were consistent with the patterns simulated from the single-crystal X-ray data (Fig. S8†). |
| This journal is © The Royal Society of Chemistry 2020 |