
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatial confinement alters the ultrafast photoisomerization dynamics of azobenzenes†
Christopher J.
Otolski‡
a,
A. Mohan
Raj
 b,
Vaidhyanathan
Ramamurthy
*b and
Christopher G.
Elles
*a
b,
Vaidhyanathan
Ramamurthy
*b and
Christopher G.
Elles
*a
aDepartment of Chemistry, University of Kansas, Lawrence, Kansas, USA. E-mail: elles@ku.edu
bDepartment of Chemistry, University of Miami, Coral Gables, Florida, USA. E-mail: murthy1@miami.edu
First published on 24th August 2020
Abstract
Ultrafast transient absorption spectroscopy reveals new excited-state dynamics following excitation of trans-azobenzene (t-Az) and several alkyl-substituted t-Az derivatives encapsulated in a water-soluble supramolecular host–guest complex. Encapsulation increases the excited-state lifetimes and alters the yields of the trans → cis photoisomerization reaction compared with solution. Kinetic modeling of the transient spectra for unsubstituted t-Az following nπ* and ππ* excitation reveals steric trapping of excited-state species, as well as an adiabatic excited-state trans → cis isomerization pathway for confined molecules that is not observed in solution. Analysis of the transient spectra following ππ* excitation for a series of 4-alkyl and 4,4′-dialkyl substituted t-Az molecules suggests that additional crowding due to lengthening of the alkyl tails results in deeper trapping of the excited-state species, including distorted trans and cis structures. The variation of the dynamics due to crowding in the confined environment provides new evidence to explain the violation of Kasha's rule for nπ* and ππ* excitation of azobenzenes based on competition between in-plane inversion and out-of-plane rotation channels.
1 Introduction
Many important photoisomerization reactions take place in the confined environment of a protein or solid-state material.1–3 Steric restrictions often play a key role in these reactions by inhibiting large amplitude motions of the atoms and stabilizing, or destabilizing, specific structures along the reaction path. For example, crowding facilitates the primary steps responsible for vision and phototaxy by steering the photoisomerization of protein-bound chromophores toward the desired outcome.4–7 In contrast, confinement in a polymer matrix or nanoporous material often inhibits the reactions of photochromic molecules compared with solution.8–10 Understanding how confinement affects the excited-state dynamics therefore promises new insight on the fundamental details of light-activated biological systems, and provides important guidance for designing new materials that preserve the efficiency of photochromic molecules in solid-state applications.Azobenzenes are prototypical molecular switches that combine the benefits of relatively fast and efficient trans → cis photoisomerization with thermal cis → trans relaxation.11–14 The photoisomerization dynamics of azobenzenes are particularly sensitive to the influence of a confined environment because of the different geometric requirements for the competing out-of-plane rotation and in-plane inversion pathways for isomerization around the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond.15–23 Even in solution, the relative importance of the rotation and inversion channels is not fully understood due to short (few ps or less) excited-state lifetimes and wavelength-dependent isomerization yields.24–26 In fact, the different quantum yields following excitation to the S1(nπ*) and S2(ππ*) excited states is a defining feature of azobenzenes,27,28 and a rare example of a reaction that violates the venerable Kasha–Vavilov rule.29–31
N bond.15–23 Even in solution, the relative importance of the rotation and inversion channels is not fully understood due to short (few ps or less) excited-state lifetimes and wavelength-dependent isomerization yields.24–26 In fact, the different quantum yields following excitation to the S1(nπ*) and S2(ππ*) excited states is a defining feature of azobenzenes,27,28 and a rare example of a reaction that violates the venerable Kasha–Vavilov rule.29–31
Given the influence of steric restrictions on photoisomerization, measuring the excited-state dynamics in a confined environment provides a sensitive probe of the reaction mechanism for azobenzenes. With that goal in mind, this paper examines the excited-state dynamics for a series of azobenzenes encapsulated in a supramolecular host–guest complex that restricts the motion of the phenyl rings around the central NN bond.32–35 Each self-assembled complex consists of a single molecule of azobenzene encapsulated in the hydrophobic cavity formed by two water-soluble ‘octa-acid’ (OA) host molecules (Fig. 1).36–40 The photochemistry of geometric isomerization and bimolecular reactions in the OA2 cavity have been studied extensively using steady-state methods,39 and we recently reported on the ultrafast photoisomerization dynamics of 4-propylstilbene and 4-propylazobenzene,34 as well as a series of alkyl-substituted stilbenes with increasing size.35 Interactions with the capsule directly influence the reaction and relaxation channels by distorting the ground- and excited-state potential energy surfaces of the confined molecule, providing a unique window on the reaction dynamics.
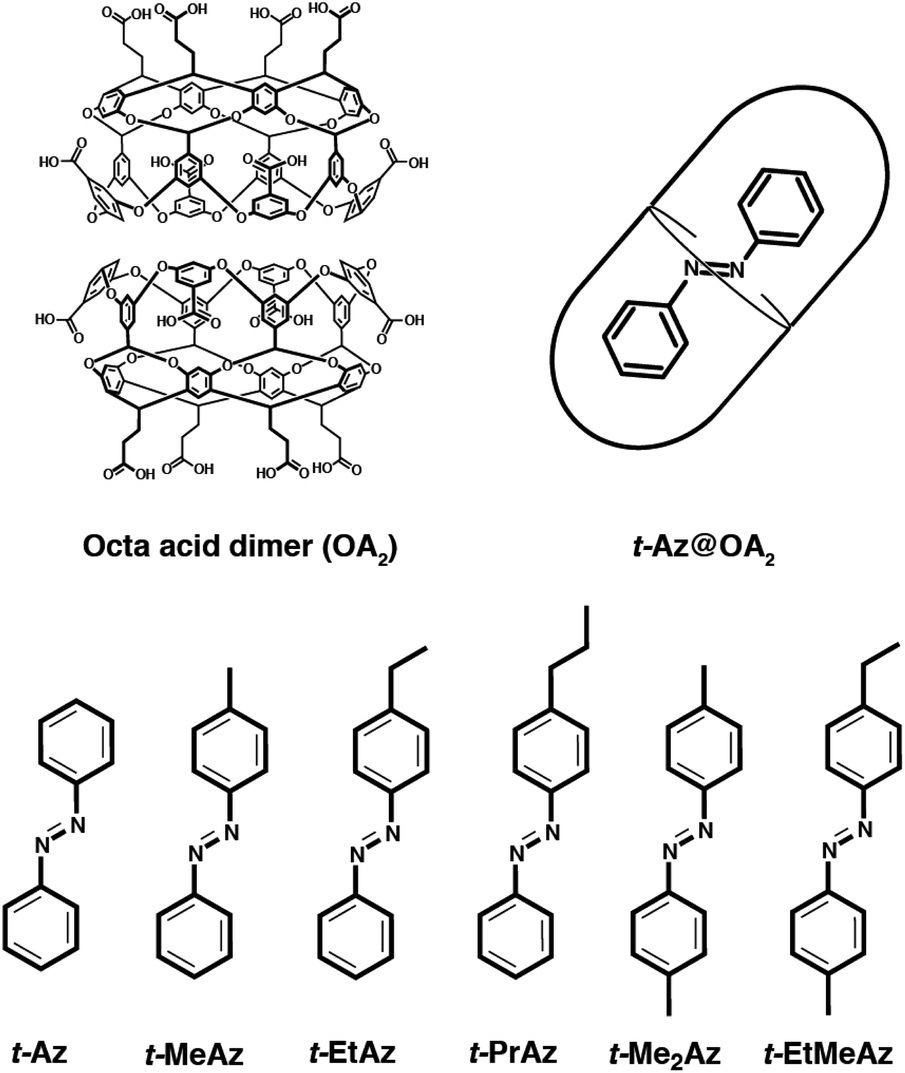 | ||
| Fig. 1 Structures of the octa-acid dimer (OA2) and the series of trans-azobenzenes. From left to right: azobenzene (Az), 4-methylazobenzene (MeAz), 4-ethylazobenzene (EtAz), 4-propylazobenzene (PrAz), 4,4′-dimethylazobenzene (Me2Az), 4,4′-ethylmethylazobenzene (EtMeAz). | ||
In this paper, we examine the role of crowding effects on the azo isomerization by comparing the excited-state dynamics of encapsulated trans-azobenzene (t-Az@OA2) and several 4-alkyl- and 4,4′-dialkyl-substituted derivatives (Fig. 1). For unsubstituted t-Az, we compare the excited-state dynamics following nπ* excitation at 470 nm and ππ* excitation at 320 nm. These wavelength-dependent measurements reveal enhanced violation of Kasha's rule in the capsule compared with solution, including a new adiabatic excited-state isomerization channel that is not observed for the freely solvated molecule. We also compare the dynamics following ππ* excitation across the series of alkyl-substituted compounds in the two environments. Increasing the alkyl-chain length changes the relative stability of the trans and cis isomers inside the capsule, and thus alters the photoisomerization yields compared with solution. Comparing the dynamics across the series of encapsulated compounds provides new insight on the role of steric interactions in tuning the excited-state dynamics, including the “trapping” of excited-state species that are not observed in solution.
2 Experimental details
Unsubstituted t-Az was purchased from Sigma-Aldrich and used as received. Octa acid (OA) and all of the alkyl-substituted trans-azobenzenes were synthesized as previously reported.32,33,37 We form self-assembled complexes (guest@OA2) by sonicating 2 equivalents of OA in pH = 8.9 borate buffer with 1 equivalent of azobenzene until the solution is transparent.35,36,40 The transient absorption (TA) measurements for nπ* excitation use an 8 mM solution of either t-Az in cyclohexane or mixed isomers of Az in OA2 at photostationary state (PSS). We obtain PSS by continuously irradiating the sample of Az@OA2 with a high-intensity 300 nm light-emitting diode to obtain a steady-state equilibrium between the two isomers. Comparing the absorption spectrum of the sample before and after each TA measurement confirms that the relative concentration of isomers remains unchanged throughout the experiment. The TA measurements for ππ* excitation use either a 1 mM solution of the trans isomer of a compound dissolved in cyclohexane or a ∼0.5 mM solution of the trans isomer inside the aqueous OA2 capsule. We measure all TA spectra in a circularly translating quartz cuvette with a path length of 1 mm.The TA measurements use the modified output of a regeneratively amplified Ti:sapphire laser (Legend Elite, Coherent) operating at 1 kHz. A portion of the 800 nm output of the laser is passed into an optical parametric amplifier (OPA) with two additional stages of nonlinear frequency conversion to generate pump pulses at either 470 or 320 nm. A CaF2 prism pair compresses the pump pulses to a duration < 80 fs and a zero-order λ/2 wave plate sets the pump polarization to parallel or perpendicular orientation with respect to the probe. We focus the pump pulses into the sample with an energy of <1.2 μJ per pulse and diameter of ∼200 μm. Broadband probe pulses covering the range 340–990 nm come from white-light continuum generation in a circularly translating CaF2 crystal using the 1200 nm signal from a second, home-built OPA. We use a pair of parabolic mirrors to collimate, then focus the probe beam into the continuously translating sample, where the pump and probe beams overlap at a small crossing angle. After passing through the sample, a prism disperses the probe pulses onto a 256-element photodiode array for shot-to-shot detection. A synchronized chopper blocks every other pump pulse for active background subtraction, and we average 103 laser pulses per time delay for each spectrum. We continuously translate the sample in order to avoid accumulation of photoproduct in the laser focal volume. We measure TA spectra for both parallel and perpendicular polarization, then calculate the isotropic spectra, ΔAiso = (ΔA‖ + 2ΔA⊥)/3, in order to eliminate contributions from rotational reorientation.41
Independently, we measure the trans → cis photoisomerization quantum yields (Φt→c) for ππ* excitation by monitoring the change in transmission of 320 nm laser pulses through a solution of the trans isomer in a static 1 mm cuvette.42,43 In order to account for small shot-to-shot fluctuations of the laser, we use a 50% beam splitter to separate the UV laser light into signal and reference beams before the sample, then simultaneously measure the intensity of the two beams with identical integrating photodiodes operating at 1 kHz. The transmission of the signal beam through the sample (1.5 mm beam diameter, 2.5 nJ per pulse incident energy) increases as a function of time due to the conversion from trans to cis. The change in absorption is proportional to the number of molecules that isomerize, because only the trans isomer has appreciable absorption at 320 nm. Thus, we calculate Φt→c from the relative change in absorption as a function of time compared with the absolute absorption of the sample.
3 Results and analysis
3.1 Ground-state spectra and quantum yields
Fig. 2 shows the ground-state absorption spectra of t-Az in cyclohexane, t-Az@OA2 in borate buffer, and adamantane@OA2 in borate buffer. The t-Az spectrum has a strong ππ* absorption band near 320 nm and a weaker nπ* absorption band near 440 nm. The weaker band is also shown on an expanded scale for clarity. The spectrum of t-Az@OA2 is very similar to that of the freely solvated molecule, except for an overlapping absorption band of OA2 below 300 nm and a shift of the ππ* band to slightly longer wavelength. The spectrum of adamantane@OA2 confirms that the band at 280 nm is due to the OA2 capsule, because adamantane is transparent above 200 nm.44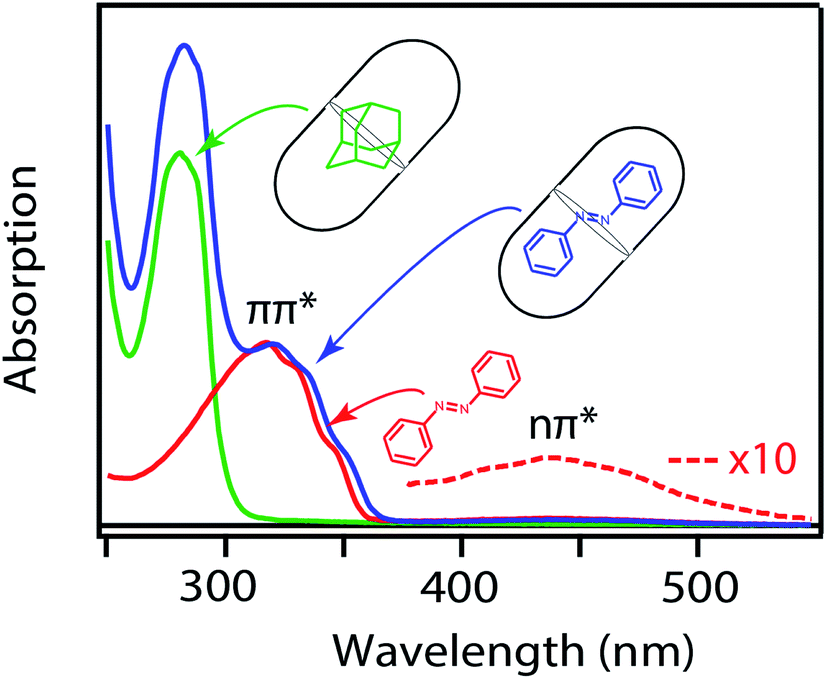 | ||
| Fig. 2 Ground-state absorption spectra of t-Az in cyclohexane (red), t-Az@OA2 (blue), and adamantane@OA2 (green). Adamantane is transparent in this wavelength range. | ||
In Fig. 3, we compare the ππ* absorption bands for the series of alkyl-substituted azobenzenes. All six molecules have similar spectra in cyclohexane, except for a slight shift to longer wavelength with the addition of one or two alkyl substituents. Similar to t-Az, most of the spectra shift further to the red upon encapsulation, but retain the same vibronic structure. However, the spectra for t-EtAz@OA2 and t-PrAz@OA2 clearly deviate from the others. The ππ* absorption bands for these two encapsulated compounds are shifted to higher energy and have significantly less vibronic structure, which is a signature of reduced conjugation and indicates a distorted, non-planar equilibrium geometry inside the OA2 capsule.35 The effect is not surprising for these two molecules, as they have the longest alkyl chains, and therefore experience the most crowding inside the capsule. Interestingly, the di-substituted compounds t-Me2Az and t-EtMeAz retain their vibronic structure in the capsule, even though the total lengths of these two molecules are similar to t-EtAz and t-PrAz, respectively. The difference probably reflects better packing of the alkyl groups into the ends of the OA2 capsule for the di-substituted compounds compared with the packing of the unmodified phenyl ring for the mono-substituted compounds of the same length. Azobenzenes with alkyl tails longer than three carbon atoms do not fit inside the capsule.32,33
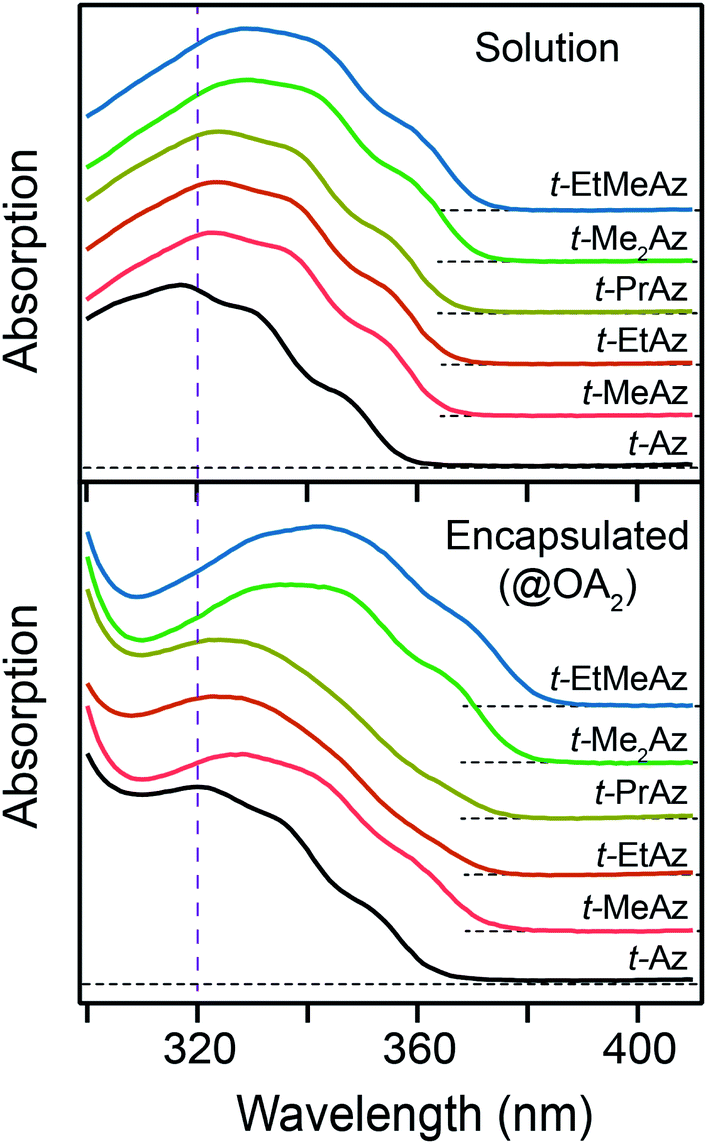 | ||
| Fig. 3 Ground-state ππ* absorption band for the series of t-Az derivatives in cyclohexane (top panel) and in the OA2 capsule (bottom panel). Vertical dashed line indicates the excitation wavelength of 320 nm. | ||
We report the trans → cis photoisomerization quantum yields (Φt→c) for ππ* excitation of all six compounds in Table 1. The estimated 2σ uncertainties in the table are for the absolute quantum yields, but the relative uncertainty between the measurements is much smaller because we made back-to-back measurements using identical conditions. The quantum yields are similar for all of the compounds in solution, ranging from 0.10 to 0.15, but vary considerably for the encapsulated compounds. The quantum yield increases systematically with chain length for the encapsulated mono-substituted compounds, from 0.05 for unsubstituted t-Az@OA2 to 0.17 for the molecule with the longest alkyl chain, t-PrAz@OA2. Notably, the value for t-PrAz@OA2 is even larger than the quantum yield for the same molecule in solution. In contrast with the mono-substituted compounds of similar total length, the quantum yields for the encapsulated dialkyl-substituted azobenzenes t-Me2Az@OA2 and t-EtMeAz@OA2 are only 0.05 and 0.03, respectively, which are much lower than the corresponding value of 0.13 in solution.
| Φ t→c(solution) | Φ t→c(capsule) | |
|---|---|---|
| a Estimated 2σ uncertainties for the absolute quantum yields in parentheses. The relative uncertainties are smaller due to back-to-back measurements under identical conditions. | ||
| t-Az | 0.11(4) | 0.05(3) |
| t-MeAz | 0.10(4) | 0.10(4) |
| t-EtAz | 0.15(4) | 0.12(4) |
| t-PrAz | 0.12(4) | 0.17(4) |
| t-Me2Az | 0.13(4) | 0.05(3) |
| t-EtMeAz | 0.13(4) | 0.03(2) |
3.2 Transient absorption spectroscopy
The top panel of Fig. 4 shows the evolution of the transient absorption (TA) spectrum following nπ* excitation of unsubstituted t-Az in cyclohexane. The transient spectrum has a strong excited-state absorption (ESA) band centered near 395 nm and a weaker band near 540 nm. As reported previously,18,21 the stronger absorption band shifts and narrows slightly due to vibrational relaxation within the first few hundred fs, then both bands decay simultaneously to the baseline within a few ps as excited molecules return to the ground state. A weak absorption feature below 375 nm persists for up to ∼20 ps due to broadening and red-shifting of the ground-state ππ* absorption band for the vibrationally excited trans isomer.45 Consistent with the low quantum yield for trans → cis isomerization of azobenzene (Table 1),46 the TA signal essentially returns to the baseline at longer delay times.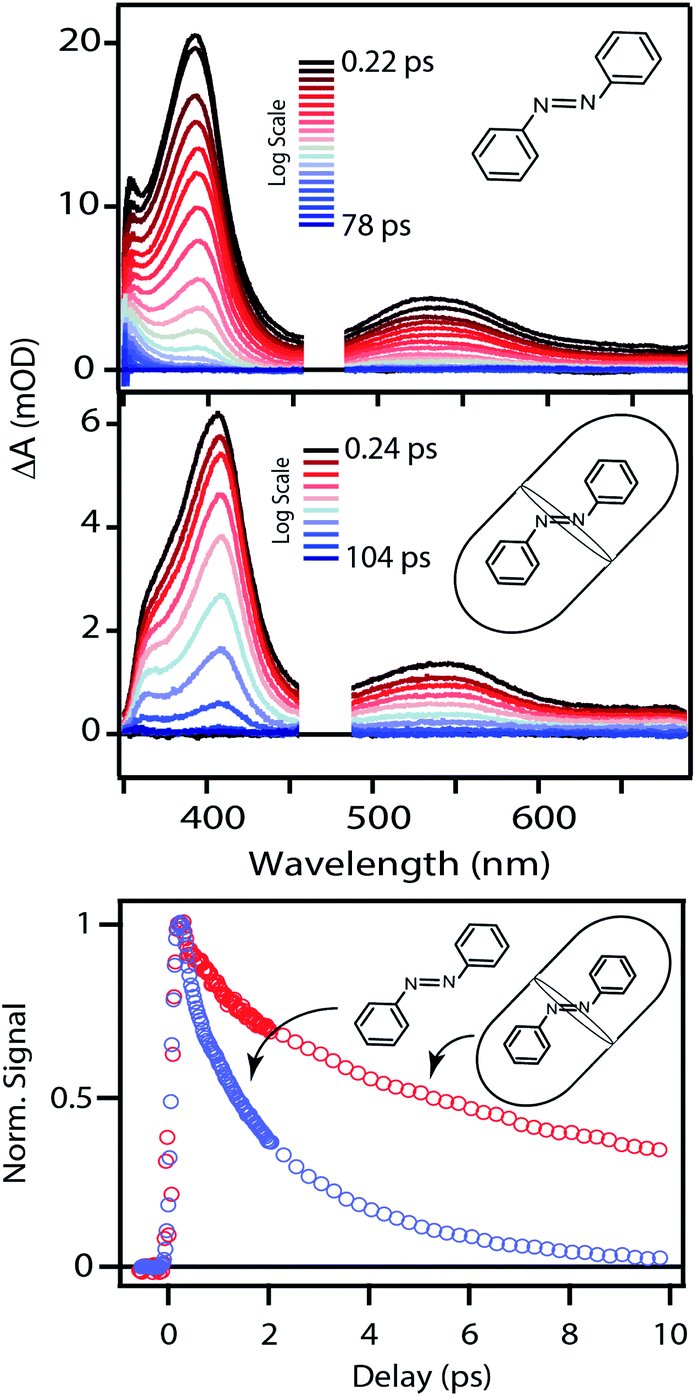 | ||
| Fig. 4 Evolution of the TA spectra following nπ* excitation at 470 nm for t-Az in cyclohexane and Az@OA2 at PSS. The bottom panel compares the decay at the maximum of the excited-state absorption bands. | ||
The middle panel of Fig. 4 shows the evolution of the TA spectrum for nπ* excitation of Az@OA2 at photo-stationary state (PSS). We use PSS in this case in order to avoid variation of the background signal from direct excitation of c-Az@OA2 at 470 nm. Unlike the experiments in solution, the accumulation of c-Az@OA2 is difficult to avoid for nπ* excitation in the small-volume sample due to the very slow thermal cis → trans recovery of the encapsulated molecules.47 Even a small population of c-Az@OA2 contributes to the TA signal because the molar absorbance of the cis isomer is nearly three times larger than trans at this excitation wavelength. Although the measurement at PSS contains contributions from both isomers, the relative concentration remains constant throughout the duration of the measurement, and therefore simplifies the analysis. Based on the ground-state absorption spectra of the two isomers (Fig. S1†), we estimate that ∼23% of encapsulated molecules are in the cis geometry at PSS, giving an initial excited-state population consisting of ∼40% cis and ∼60% trans.
Despite the simultaneous excitation of both c-Az@OA2 and t-Az@OA2, the TA spectrum for the encapsulated sample at PSS is very similar to the spectrum of t-Az in solution, suggesting that the trans isomer provides the dominant contribution to the transient signal. Consistent with this observation, our global fits to the TA data (described below) reveal only a weak contribution from the excited state of c-Az@OA2. However, the ESA band shifts to slightly longer wavelength and decays much slower in the capsule than in solution, as illustrated in the bottom panel of Fig. 4. The ∼10 nm shift of the ESA band is similar to the shift of the ground-state ππ* absorption band in Fig. 2. The longer excited-state lifetime in the capsule prevents accumulation of vibrationally excited molecules in the ground state, which was responsible for the positive absorption feature below ∼375 nm in cyclohexane. At the same time, the shift of the ground-state absorption band results in a transient ground-state bleach, and therefore appears as a steep reduction of the TA signal below ∼360 nm that we do not observe in cyclohexane for the same range of probe wavelengths.
The TA spectra following ππ* excitation of t-Az and t-Az@OA2 are shown in Fig. 5, where we divide the evolution of the spectra into separate panels to better represent the rapid relaxation from S2 to S1 within the first ∼200 fs, and the subsequent relaxation from S1 to S0 on a longer time scale.34 Unlike the nπ* experiments above, PSS is unnecessary in the case of UV excitation, because the significantly weaker absorption of the cis isomer at 320 nm results in a negligible contribution to the transient signal from direct excitation of that isomer. The lower isomerization yield for UV excitation also reduces the amount of cis isomer that forms. Comparing the static absorption spectra before and after each TA measurement confirms that there is minimal accumulation of cis isomer over the duration of the experiment for ππ* excitation.
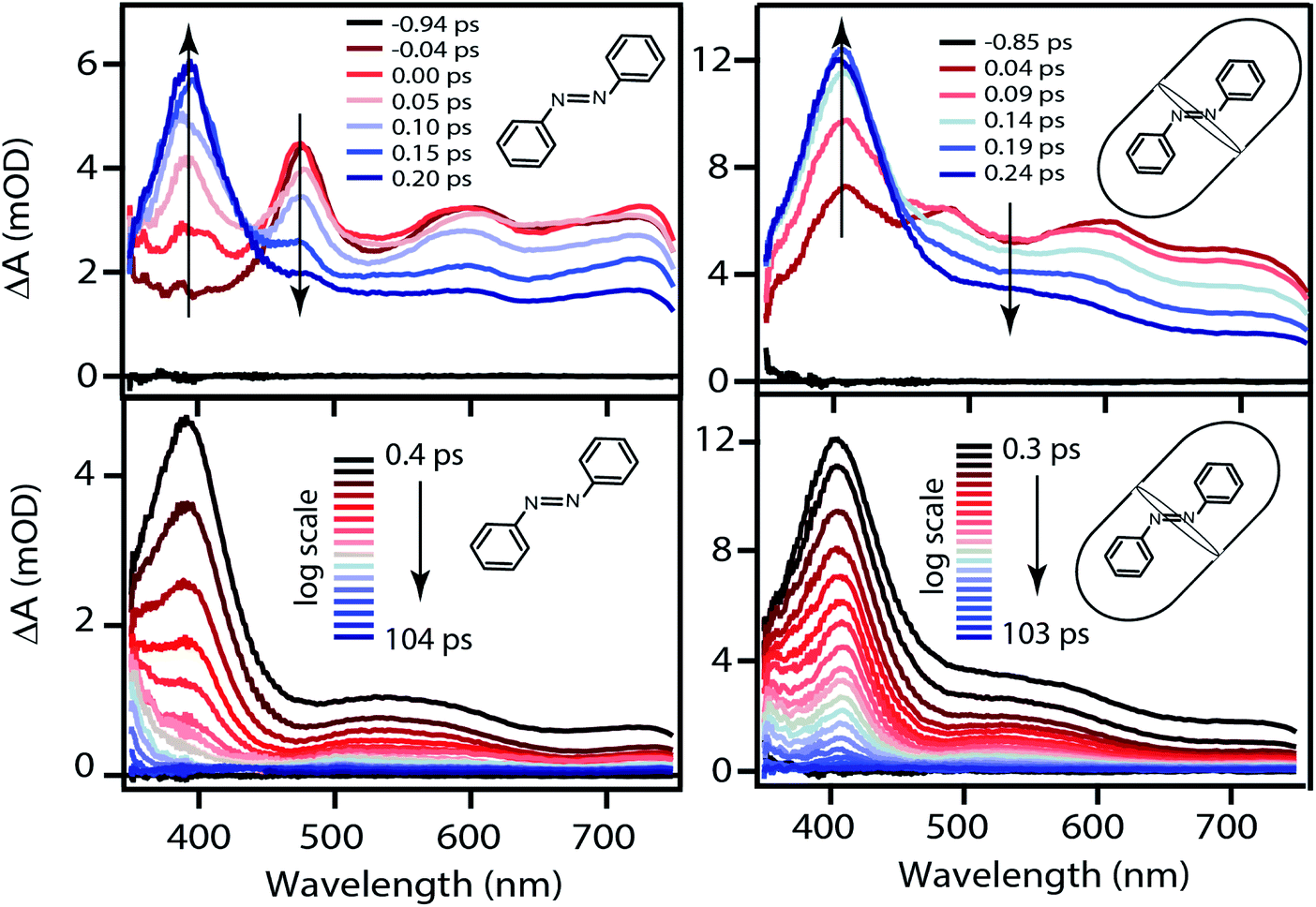 | ||
| Fig. 5 Evolution of the TA spectra following ππ* excitation at 320 nm of t-Az in cyclohexane (left) and t-Az@OA2 (right). The upper panels show only the first few hundred fs, the lower panels show the evolution up to ∼100 ps. | ||
The initial ESA spectrum following ππ* excitation has several broad features, including a prominent absorption band near 485 nm and weaker bands near 600 and 710 nm.45,48 This spectrum is similar in both samples, except for additional broadening of the individual ESA bands for the encapsulated compound. In both cases, the S2 absorption decays within ∼100 fs and is replaced by a spectrum that is very similar to the S1 spectrum that we observe following direct nπ* excitation at 470 nm. An isobestic point near ∼450 nm confirms direct relaxation from S2 to S1 in both environments. The slight broadening of the S1 spectra compared with Fig. 4 probably reflects excess vibrational energy following internal conversion from S2.
3.3 Kinetic models
Global fits to the TA spectra reveal more detailed information about the excited-state dynamics. We use a combination of singular value decomposition (SVD)49 and fits to the TA data with a sum of exponentials (e.g. Fig. S3†) to determine the smallest number of time constants that are necessary to accurately reproduce the evolution of each spectrum. We then use target analysis to apply more restrictive kinetic models in the fits to the data.50 Target analysis gives species-associated spectra (SAS) that represent individual states in the kinetic model, where the time evolution of the spectra are restricted by the specified pathways between states. All of the fits include a convolution of the kinetics with a Gaussian instrument response function (100 fs fwhm) to account for the time-resolution of our measurement. We allow all of the time constants and amplitudes to vary in each of the fits, except where indicated otherwise.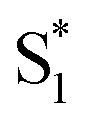 that relaxes through two competing channels, one that returns directly to the ground state in ∼1 ps, and a second that undergoes vibrational relaxation in S1 before returning to the ground state on a timescale of 2.4 ps. This branching in the excited state represents a bifurcation between one pathway that directly accesses a conical intersection with the ground state and a second pathway that relaxes to a minimum-energy structure in the excited state before crossing a barrier to reach either the same or a different conical intersection.20 Both channels lead to the vibrationally hot ground state,
that relaxes through two competing channels, one that returns directly to the ground state in ∼1 ps, and a second that undergoes vibrational relaxation in S1 before returning to the ground state on a timescale of 2.4 ps. This branching in the excited state represents a bifurcation between one pathway that directly accesses a conical intersection with the ground state and a second pathway that relaxes to a minimum-energy structure in the excited state before crossing a barrier to reach either the same or a different conical intersection.20 Both channels lead to the vibrationally hot ground state, 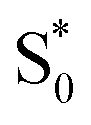 , which then dissipates energy to the solvent to reach the equilibrated S0 on a timescale of 13.0 ps. Importantly, the SAS from the fits are consistent with the kinetic model, including a slight shift and narrowing of the primary ESA band from
, which then dissipates energy to the solvent to reach the equilibrated S0 on a timescale of 13.0 ps. Importantly, the SAS from the fits are consistent with the kinetic model, including a slight shift and narrowing of the primary ESA band from 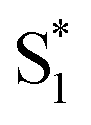 to S1, and a tail below ∼375 nm due to broadening of the ππ* absorption band for
to S1, and a tail below ∼375 nm due to broadening of the ππ* absorption band for 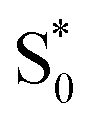 compared with S0.45 The model does not distinguish between cis and trans isomers in the relaxed ground state, because the weak contribution from
compared with S0.45 The model does not distinguish between cis and trans isomers in the relaxed ground state, because the weak contribution from 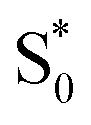 is not sufficient to determine the branching ratio directly from the TA measurements.
is not sufficient to determine the branching ratio directly from the TA measurements.
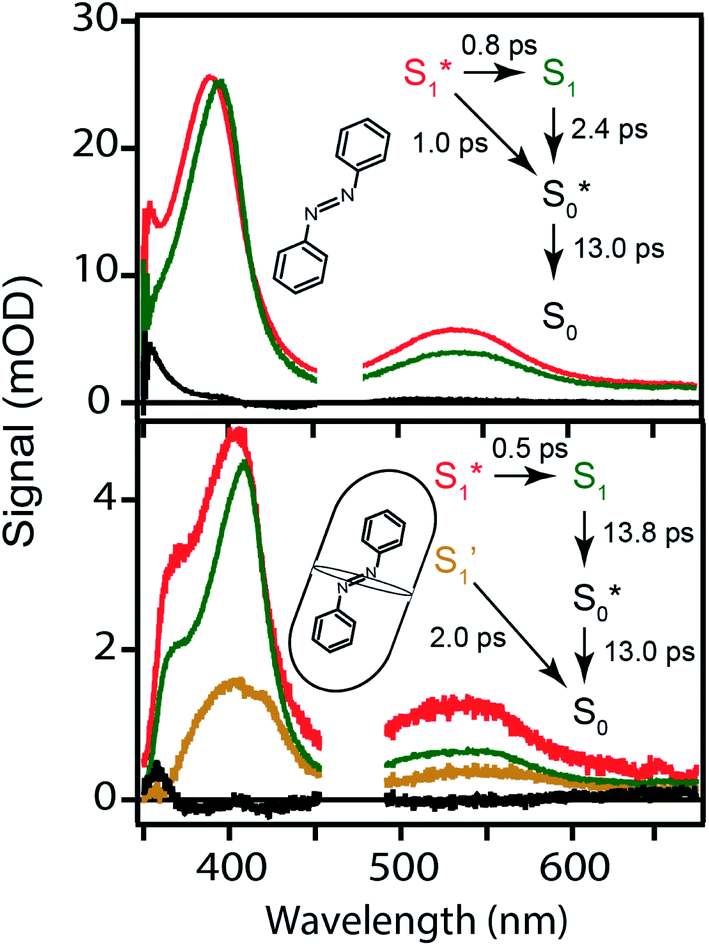 | ||
| Fig. 6 Species-associated spectra for nπ* excitation of t-Az in cyclohexane (top) and mixed t-Az@OA2/c-Az@OA2 at PSS (bottom). The kinetic models and lifetimes from the global fits are shown in the insets. See text for details. | ||
The bottom panel of Fig. 6 shows the SAS that we obtain for nπ* excitation of the mixed isomers of Az@OA2 at PSS. We use a kinetic model with parallel contributions for the cis and trans isomers, as shown in the inset. The model for relaxation of t-Az@OA2 is similar to the model in solution, except that we do not observe a rapid decay of the total TA signal strength within the first ps for the encapsulated molecule, and therefore do not include a direct relaxation channel from 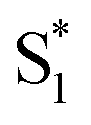 to
to 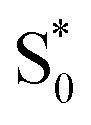 in the capsule. Instead, the ESA band decays on a single timescale of 13.8 ps, compared with the 2.4 ps lifetime in solution. Despite the very different excited-state lifetimes, the SAS for t-Az@OA2 are remarkably similar to the spectra in cyclohexane. It is not necessary to include ground-state vibrational cooling (
in the capsule. Instead, the ESA band decays on a single timescale of 13.8 ps, compared with the 2.4 ps lifetime in solution. Despite the very different excited-state lifetimes, the SAS for t-Az@OA2 are remarkably similar to the spectra in cyclohexane. It is not necessary to include ground-state vibrational cooling (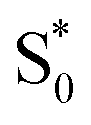 → S0) in order to accurately reproduce the TA spectrum for the encapsulated sample, but we find that adding this component with a fixed lifetime of 13.0 ps recovers a weak signature below 375 nm that is similar to the vibrationally hot
→ S0) in order to accurately reproduce the TA spectrum for the encapsulated sample, but we find that adding this component with a fixed lifetime of 13.0 ps recovers a weak signature below 375 nm that is similar to the vibrationally hot 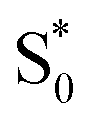 in solution. The spectrum for
in solution. The spectrum for 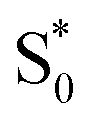 is much noisier in the capsule because the hot ground state cools on the same timescale as the electronic relaxation from S1, and therefore prevents accumulation of highly vibrationally excited molecules.
is much noisier in the capsule because the hot ground state cools on the same timescale as the electronic relaxation from S1, and therefore prevents accumulation of highly vibrationally excited molecules.
The global fits reveal an additional feature in the evolution of the TA spectrum of the mixed encapsulated isomers that decays on a time scale of 2.0 ps. We confidently assign this feature as the excited state of c-Az@OA2, because the SAS (gold spectrum in Fig. 6) has absorption bands near 400 and 550 nm that closely resemble the ESA spectrum for nπ* excitation of c-Az in solution.21 The previously reported spectrum for c-Az has broader and weaker absorption bands than the excited-state spectrum of the trans isomer, just like we observe here. Although the 2.0 ps lifetime that we observe for c-Az@OA2 is significantly longer than the ∼100 fs lifetime previously reported for c-Az in solution,21,45,51 this difference mirrors the order-of-magnitude longer lifetime of the trans isomer in the capsule compared with solution.
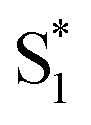 . In order to improve the quality of the fits we fix the S2 →
. In order to improve the quality of the fits we fix the S2 → 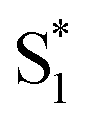 internal conversion time at 50 fs based on measurements with higher time resolution by Nenov, et al.28 The top panel of Fig. 7 shows the SAS. The spectra for
internal conversion time at 50 fs based on measurements with higher time resolution by Nenov, et al.28 The top panel of Fig. 7 shows the SAS. The spectra for 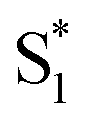 , S1, and
, S1, and 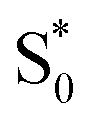 are all very similar to the spectra from nπ* excitation, with some additional broadening of
are all very similar to the spectra from nπ* excitation, with some additional broadening of 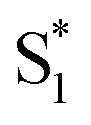 that is probably a signature of excess vibrational energy following internal conversion from S2.42 The spectrum for S2 is distinct from the other states, and includes a negative stimulated emission band near 400 nm that closely matches an S2 → S0 fluorescence band that was previously reported following ππ* excitation of t-Az.22,52
that is probably a signature of excess vibrational energy following internal conversion from S2.42 The spectrum for S2 is distinct from the other states, and includes a negative stimulated emission band near 400 nm that closely matches an S2 → S0 fluorescence band that was previously reported following ππ* excitation of t-Az.22,52
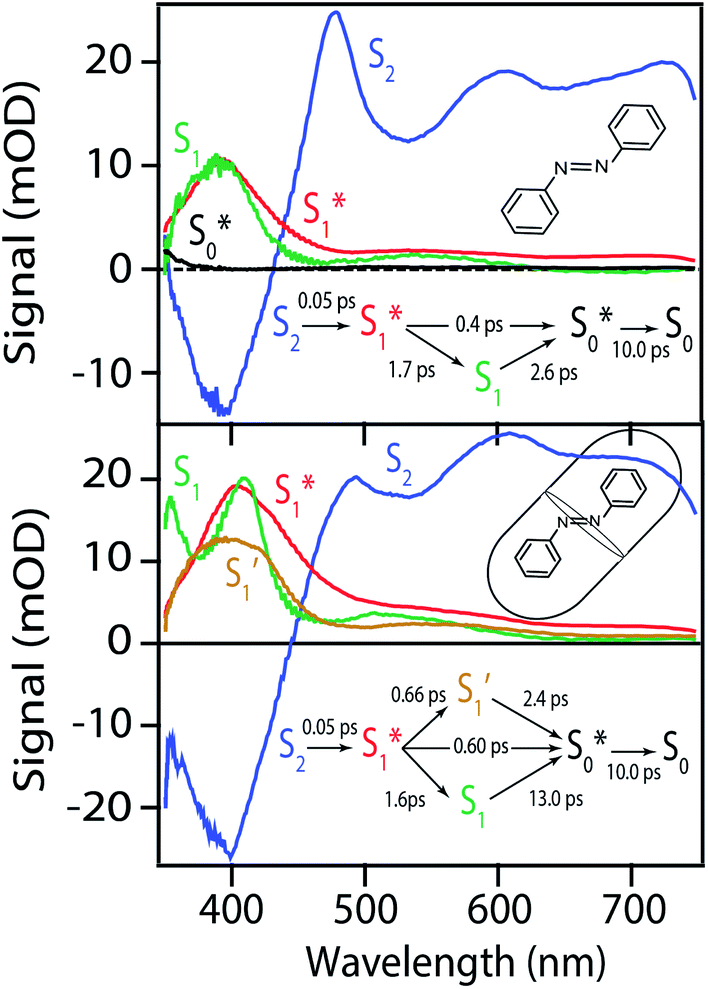 | ||
| Fig. 7 Species associated spectra for ππ* excitation of azobenzene in cyclohexane (top) and the OA2 capsule (bottom). The spectra are from global fits to the transient absorption spectra using the kinetic models shown in the insets. See text for details. | ||
Importantly, the kinetic model for ππ* excitation of t-Az@OA2 requires an additional component compared with t-Az in cyclohexane. The additional lifetime is evident from both SVD and the global fits to the data using a sum of exponentials, each of which suggests that a total of at least five time constants are necessary to accurately reproduce the evolution of the experimental TA spectrum for the encapsulated sample. Thus, in addition to the initially excited S2, we include an extra feature (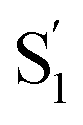 ) for the encapsulated molecule. The kinetic model that gives the best agreement with experiment is shown in the bottom panel of Fig. 7, along with the resulting SAS from target analysis. We tested several different kinetic models, but only this model, which includes three distinct channels for the relaxation of
) for the encapsulated molecule. The kinetic model that gives the best agreement with experiment is shown in the bottom panel of Fig. 7, along with the resulting SAS from target analysis. We tested several different kinetic models, but only this model, which includes three distinct channels for the relaxation of 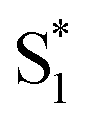 , gives a good fit to the data and also is consistent with the results from above. We exclude other kinetic models based on poor fits to the data or unreasonable results, such as artificially short lifetimes (i.e. <50 fs) for species other than S2, or SAS with negative spectral features above 500 nm. As before, we convoluted the fits with a 100 fs instrument-response function, and used a fixed lifetime of 50 fs for the relaxation of S2.
, gives a good fit to the data and also is consistent with the results from above. We exclude other kinetic models based on poor fits to the data or unreasonable results, such as artificially short lifetimes (i.e. <50 fs) for species other than S2, or SAS with negative spectral features above 500 nm. As before, we convoluted the fits with a 100 fs instrument-response function, and used a fixed lifetime of 50 fs for the relaxation of S2.
The SAS for S2 and 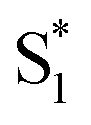 of t-Az@OA2 are similar to the spectra in solution, and decay on roughly the same two time scales (50 fs and ∼300 fs, respectively) in both environments. However, a key result from the model for ππ* excitation of t-Az@OA2 is the presence of two distinct excited-state species (S1 and
of t-Az@OA2 are similar to the spectra in solution, and decay on roughly the same two time scales (50 fs and ∼300 fs, respectively) in both environments. However, a key result from the model for ππ* excitation of t-Az@OA2 is the presence of two distinct excited-state species (S1 and 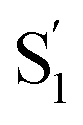 ) with lifetimes of 13 and 2.4 ps, respectively. We assign the species with a lifetime of 13 ps as the relaxed S1 state of t-Az@OA2, based on the similar spectrum and lifetime that we observe following nπ* excitation of the encapsulated molecule. We assign the other feature,
) with lifetimes of 13 and 2.4 ps, respectively. We assign the species with a lifetime of 13 ps as the relaxed S1 state of t-Az@OA2, based on the similar spectrum and lifetime that we observe following nπ* excitation of the encapsulated molecule. We assign the other feature, 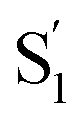 , as the relaxed excited state of the cis isomer, even though we excite only the trans isomer of the encapsulated compound at 320 nm. Importantly, both the spectrum and the 2.4 ps lifetime of
, as the relaxed excited state of the cis isomer, even though we excite only the trans isomer of the encapsulated compound at 320 nm. Importantly, both the spectrum and the 2.4 ps lifetime of 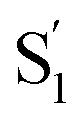 closely match the spectrum and lifetime that we observe following direct nπ* excitation of c-Az@OA2 at 470 nm. Formation of the cis excited state after ππ* excitation of t-Az@OA2 implies adiabatic isomerization in S1, which we observe only at the higher excitation energy and in the confined environment of the capsule.
closely match the spectrum and lifetime that we observe following direct nπ* excitation of c-Az@OA2 at 470 nm. Formation of the cis excited state after ππ* excitation of t-Az@OA2 implies adiabatic isomerization in S1, which we observe only at the higher excitation energy and in the confined environment of the capsule.
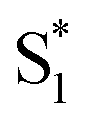 to the ground state. For unsubstituted t-Az@OA2 and the mono-alkyl substituted derivatives, we observe rapid decay of the ESA that requires the direct
to the ground state. For unsubstituted t-Az@OA2 and the mono-alkyl substituted derivatives, we observe rapid decay of the ESA that requires the direct 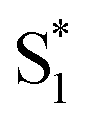 →
→ 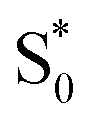 relaxation channel. The same model gives a poor fit to the data for the di-substituted azobenzenes, Me2Az@OA2 and EtMeAz@OA2, where the TA spectra do not rapidly decrease in intensity after the initial electronic relaxation to
relaxation channel. The same model gives a poor fit to the data for the di-substituted azobenzenes, Me2Az@OA2 and EtMeAz@OA2, where the TA spectra do not rapidly decrease in intensity after the initial electronic relaxation to 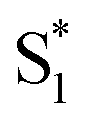 . Instead, we remove the direct
. Instead, we remove the direct 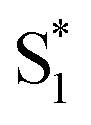 →
→ 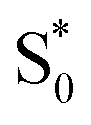 relaxation channel from the model for these two compounds. In all cases, we use a fixed lifetime of 10 ps for the
relaxation channel from the model for these two compounds. In all cases, we use a fixed lifetime of 10 ps for the 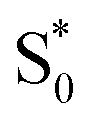 lifetime due to the insensitivity of the fit to this small-amplitude signal. The SAS for the full series of compounds are presented in the ESI† and the lifetimes of S1 and
lifetime due to the insensitivity of the fit to this small-amplitude signal. The SAS for the full series of compounds are presented in the ESI† and the lifetimes of S1 and 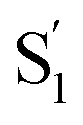 are listed in Table 2.
are listed in Table 2.
| Solution | Capsule | ||
|---|---|---|---|
| τ S1 (ps) | τ S1 (ps) |
|
|
a From global fits using the kinetic models described in the text. For encapsulated molecules, the model includes two distinct excited-state species, S1 and 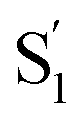 . .
|
|||
| t-Az | 2.6 | 13 | 2.4 |
| t-MeAz | 1.8 | 15 | 2.4 |
| t-EtAz | 2.3 | 21 | 3.8 |
| t-PrAz | 2.5 | 26 | 5.6 |
| t-Me2Az | 2.4 | 35 | 3.3 |
| t-EtMeAz | 2.6 | 55 | 3.5 |
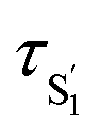
A comparison of the lifetimes of the encapsulated molecules with those in solution is informative. While the S1 lifetimes are essentially the same for all of the compounds in solution (∼2.5 ps), the lifetimes are significantly longer in the capsule, ranging from 13 ps for unsubstituted t-Az@OA2 to 55 ps for t-EtMeAz@OA2. The 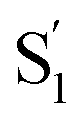 lifetimes in the capsule also depend on the length of the substituent, increasing from 2.4 to 5.6 ps for the mono-substituted compounds, with the di-substituted compounds having intermediate lifetimes.
lifetimes in the capsule also depend on the length of the substituent, increasing from 2.4 to 5.6 ps for the mono-substituted compounds, with the di-substituted compounds having intermediate lifetimes.
Fig. 8 compares the SAS for the S1 state of each compound in solution with the S1 and 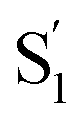 spectra in OA2. For the encapsulated compounds, we shade the S1 spectra below 360 nm due to complications from overlapping spectral features in the target analysis. On one hand, the similar timescales for electronic relaxation of S1 and vibrational cooling of
spectra in OA2. For the encapsulated compounds, we shade the S1 spectra below 360 nm due to complications from overlapping spectral features in the target analysis. On one hand, the similar timescales for electronic relaxation of S1 and vibrational cooling of  results in an artificial increase of the S1 SAS below ∼360 nm for t-Az@OA2, t-MeAz@OA2, and t-EtAz@OA2. On the other hand, the red-shifted ground-state ππ* absorption bands for the two di-substituted compounds result in ground-state bleaching that is not accounted for in our model and therefore gives a steep decrease of the SAS below 360 nm. t-PrAz@OA2 has a long enough S1 lifetime and small enough shift of the ground-state absorption band that it is not susceptible to these issues, but the relative population of S1 is much smaller in that case, giving a poorer signal-to-noise ratio for the SAS.
results in an artificial increase of the S1 SAS below ∼360 nm for t-Az@OA2, t-MeAz@OA2, and t-EtAz@OA2. On the other hand, the red-shifted ground-state ππ* absorption bands for the two di-substituted compounds result in ground-state bleaching that is not accounted for in our model and therefore gives a steep decrease of the SAS below 360 nm. t-PrAz@OA2 has a long enough S1 lifetime and small enough shift of the ground-state absorption band that it is not susceptible to these issues, but the relative population of S1 is much smaller in that case, giving a poorer signal-to-noise ratio for the SAS.
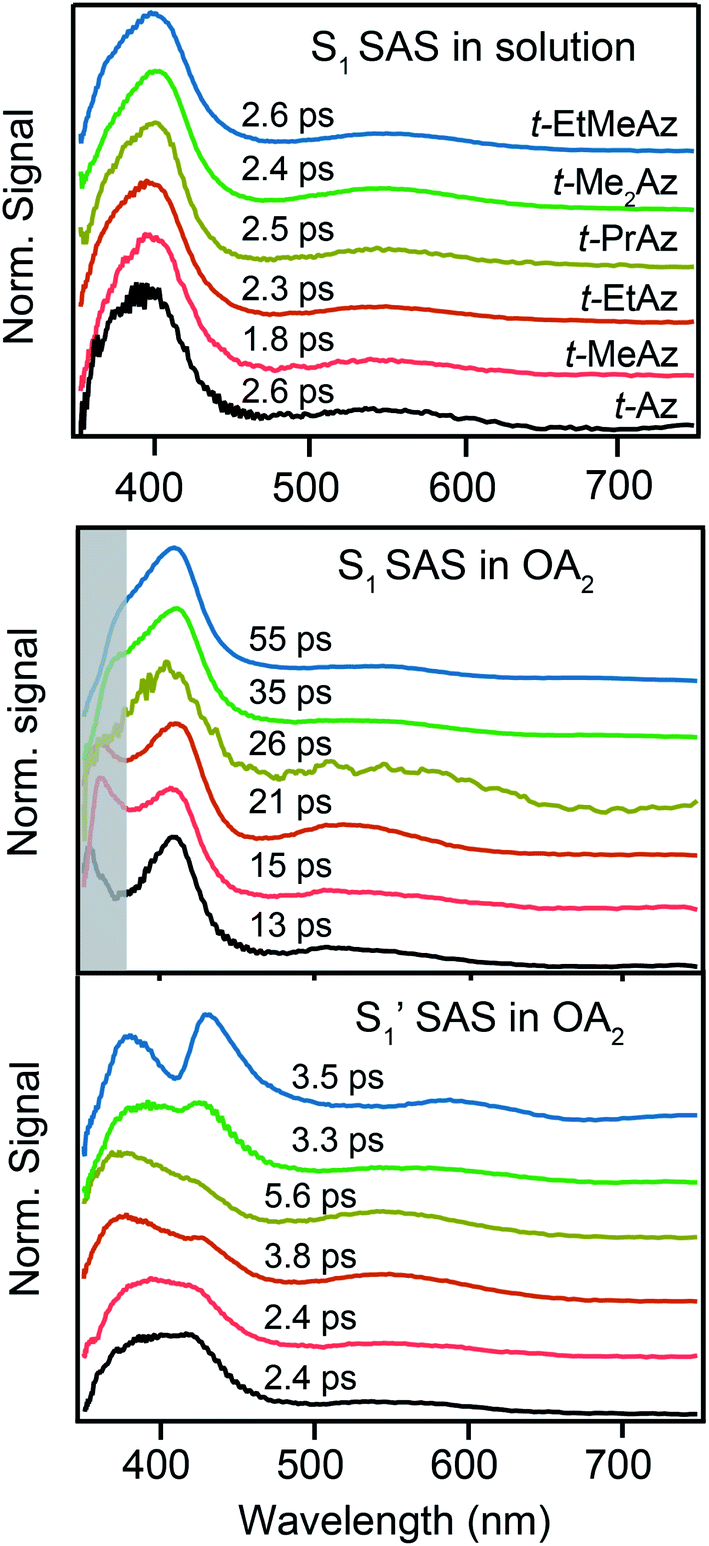 | ||
Fig. 8 The upper panel shows the SAS for S1 in solution. The lower panels show the S1 and  SAS inside the OA2 capsule. The shaded band indicates a region with known artifacts in the S1 SAS of the encapsulated molecules. SAS inside the OA2 capsule. The shaded band indicates a region with known artifacts in the S1 SAS of the encapsulated molecules. | ||
Neglecting the known discrepancies below 360 nm, Fig. 8 shows that the S1 SAS are similar for all of the compounds, both in solution and in the capsule. However, the  SAS are more variable across the series of encapsulated molecules. The main ESA band in the
SAS are more variable across the series of encapsulated molecules. The main ESA band in the 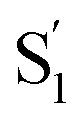 spectrum broadens and splits into two overlapping features with increasing length of the alkyl substituent. Notably, the spectra for
spectrum broadens and splits into two overlapping features with increasing length of the alkyl substituent. Notably, the spectra for 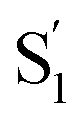 of the di-substituted compounds have a different shape than the spectra for the mono-substituted compounds, including more pronounced splitting of the ESA band and higher intensity of the longer wavelength component near 440 nm. A weaker ESA band near 550 nm for Az@OA2 and the encapsulated mono-substituted compounds also shifts ∼50 nm to longer wavelength for the disubstituted compounds in the capsule.
of the di-substituted compounds have a different shape than the spectra for the mono-substituted compounds, including more pronounced splitting of the ESA band and higher intensity of the longer wavelength component near 440 nm. A weaker ESA band near 550 nm for Az@OA2 and the encapsulated mono-substituted compounds also shifts ∼50 nm to longer wavelength for the disubstituted compounds in the capsule.
4 Discussion
4.1 Isomerization mechanism
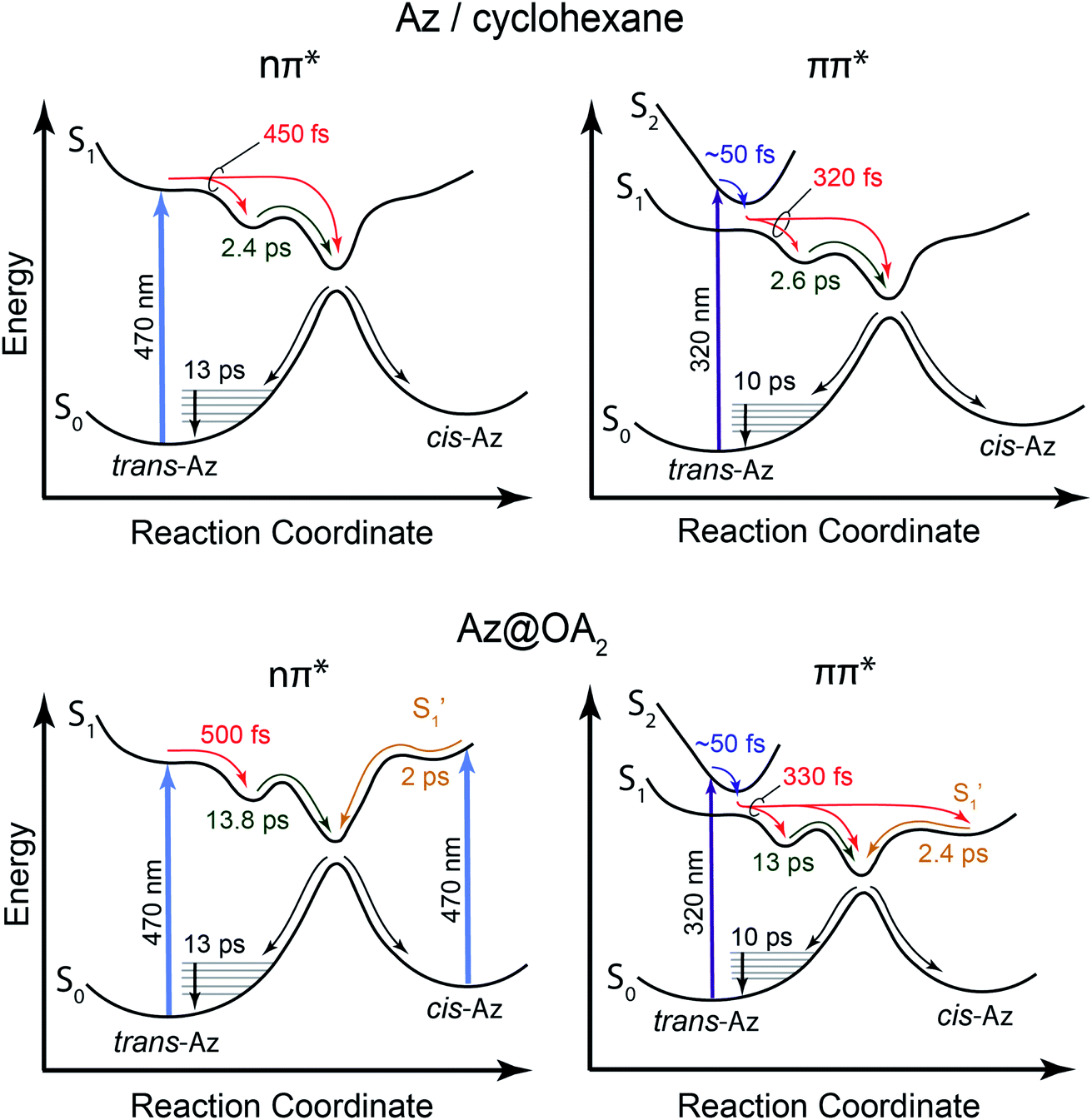
Our kinetic modeling and global fits reveal interesting details about the excited-state dynamics of t-Az in solution and in the capsule. The schematic potential energy diagrams in Fig. 9 illustrate the reaction pathways and lifetimes that we extract from the models for nπ* and ππ* excitation in each environment. An important difference between the models in Fig. 9 is the fast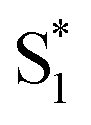 →
→ 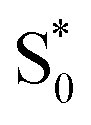 electronic relaxation channel. We observe this prompt relaxation for both nπ* and ππ* excitation in solution, indicating that there is a direct pathway to access a conical intersection (or conical seam) with the ground state. However, not all molecules take this direct pathway, and some relax to a local minimum energy structure on the S1 excited state. The excited-state lifetime of ∼2.5 ps in solution represents the activated barrier crossing to escape the local minimum and access the region of the conical intersection. The topology of the conical intersection determines the branching between trans and cis isomers when molecules return to the ground electronic state.26,53
electronic relaxation channel. We observe this prompt relaxation for both nπ* and ππ* excitation in solution, indicating that there is a direct pathway to access a conical intersection (or conical seam) with the ground state. However, not all molecules take this direct pathway, and some relax to a local minimum energy structure on the S1 excited state. The excited-state lifetime of ∼2.5 ps in solution represents the activated barrier crossing to escape the local minimum and access the region of the conical intersection. The topology of the conical intersection determines the branching between trans and cis isomers when molecules return to the ground electronic state.26,53
 | ||
| Fig. 9 Schematic diagram of the potential energy curves for Az in cyclohexane (top) and Az@OA2 (bottom). The left side shows the reaction pathway for nπ* excitation, and the right side shows ππ* excitation. | ||
The direct pathway to the conical intersection has been identified as a likely explanation for the different quantum yields following nπ* and ππ* excitation.28,34 The trans → cis isomerization yield for ππ* excitation to S2 is lower than it is for direct nπ* excitation to S1.14 Although early reports attributed the different quantum yields to the different electronic character of the initially excited states, more recent explanations point to the role of excess vibrational energy following rapid internal conversion from S2 to 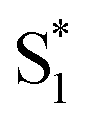 .17,54–56 Internal conversion from S2 to
.17,54–56 Internal conversion from S2 to 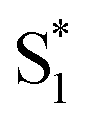 leaves the molecule with sufficient excess vibrational energy to access higher-energy regions of the S1–S0 conical seam, which preferentially leads back to the trans isomer in the ground state. Thus, the ‘non-Kasha’ behavior, i.e. the different reaction yields for excitation to S2 and S1, is a result of competition between rapid (few ps) isomerization and vibrational cooling in the excited state.
leaves the molecule with sufficient excess vibrational energy to access higher-energy regions of the S1–S0 conical seam, which preferentially leads back to the trans isomer in the ground state. Thus, the ‘non-Kasha’ behavior, i.e. the different reaction yields for excitation to S2 and S1, is a result of competition between rapid (few ps) isomerization and vibrational cooling in the excited state.
In contrast with the dynamics in solution, the direct 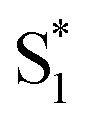 →
→ 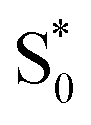 relaxation pathway is absent for nπ* excitation in the capsule, where steric restrictions may inhibit the motions required to access the conical seam. Steric restriction is consistent with the larger barrier to escape the S1 local minimum and therefore the longer excited-state lifetime of 13.8 ps in the capsule compared with only 2.4 ps in solution. However, the fast electronic relaxation pathway becomes accessible again in the capsule for higher-energy excitation into the ππ* band. Excitation of the ππ* band provides enough energy to overcome the barrier (including steric restrictions) after internal conversion from S2.
relaxation pathway is absent for nπ* excitation in the capsule, where steric restrictions may inhibit the motions required to access the conical seam. Steric restriction is consistent with the larger barrier to escape the S1 local minimum and therefore the longer excited-state lifetime of 13.8 ps in the capsule compared with only 2.4 ps in solution. However, the fast electronic relaxation pathway becomes accessible again in the capsule for higher-energy excitation into the ππ* band. Excitation of the ππ* band provides enough energy to overcome the barrier (including steric restrictions) after internal conversion from S2.
Our results are consistent with the proposed reaction mechanism of Nenov et al.,28 who showed that the minimum-energy reaction path for freely solvated t-Az involves inversion-assisted rotation on S1. This reaction path begins with in-plane bending of a phenyl ring along the inversion coordinate, then includes some out-of-plane rotation to access a conical intersection with the ground state. The inversion-assisted rotation mechanism results from mixing of the nπ* and ππ* states along the reaction coordinate.27 In the capsule, the deeper local minimum on the S1 potential inhibits the motion to access the region of the conical intersection, possibly due to distortion of the molecule into a nonplanar geometry that inhibits the proposed initial motion along the in-plane inversion coordinate. For ππ* excitation, rapid internal conversion from S2 provides sufficient excess energy for the molecule to deviate from the minimum-energy path and overcome the larger barrier at nonplanar geometries. The ∼50 fs lifetime of S2 suggests that the motion toward the conical seam probably occurs in the vibrationally excited 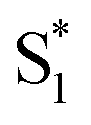 , rather than S2. The timescales for the initial relaxation from S2 to S1 and the vibrational and structural relaxation on S1 are essentially the same in both environments, indicating that the early relaxation processes are largely insensitive to confinement.57
, rather than S2. The timescales for the initial relaxation from S2 to S1 and the vibrational and structural relaxation on S1 are essentially the same in both environments, indicating that the early relaxation processes are largely insensitive to confinement.57
Another key result from our measurements is the observation of the second excited-state species, 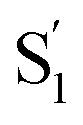 , after ππ* excitation in the capsule. Although we excite c-Az@OA2 directly in the nπ* excitation experiments at PSS, the measurements for ππ* excitation use a pure solution of t-Az@OA2. Nevertheless, we observe the same
, after ππ* excitation in the capsule. Although we excite c-Az@OA2 directly in the nπ* excitation experiments at PSS, the measurements for ππ* excitation use a pure solution of t-Az@OA2. Nevertheless, we observe the same 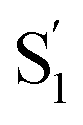 absorption feature in both cases. The two distinct excited-state features indicate the presence of both trans and cis excited-state structures following ππ* excitation of t-Az@OA2. This implies an excited-state isomerization pathway, probably on the S1 potential energy surface. Adiabatic isomerization to populate the trans excited state after excitation of the cis isomer has been observed,45 but the encapsulated azobenzenes in OA2 are the first example to our knowledge showing the reverse process of trans → cis excited-state isomerization.
absorption feature in both cases. The two distinct excited-state features indicate the presence of both trans and cis excited-state structures following ππ* excitation of t-Az@OA2. This implies an excited-state isomerization pathway, probably on the S1 potential energy surface. Adiabatic isomerization to populate the trans excited state after excitation of the cis isomer has been observed,45 but the encapsulated azobenzenes in OA2 are the first example to our knowledge showing the reverse process of trans → cis excited-state isomerization.
Our results support an emerging picture that molecules may explore a wide range of structures in the excited state even though the reaction dynamics are largely determined by the minimum-energy path.58,59 In other words, the excited-state wave packet may sample both cis and trans regions of the potential, even in solution, but we observe the cis isomer only in the capsule due to “trapping” of that structure in the restricted environment of the capsule. Branching between the three channels (i.e., the direct pathway to the ground state, and relaxation to the trans and cis local minima) depends on the relative time scales for vibrational cooling and to explore energetically accessible regions of the excited-state potential energy surface. Vibrational relaxation essentially quenches the excited-state dynamics within a few hundred fs, trapping the molecules in any accessible local minima. We observe the encapsulated cis isomer because of the deeper well in the confined environment, which is evident from the significantly longer lifetime of 2 ps for c-Az@OA2 compared with the sub-100 fs lifetime of c-Az in solution.45 This picture suggests that excitation of the ππ* band not only provides enough energy to overcome the barrier to access the S1–S0 conical intersection directly, but also to reach the cis geometry of the excited state.
4.2 Crowding effects
The variation of the excited-state lifetimes and spectra across the series of encapsulated alkyl-substituted azobenzenes reveals additional changes in the excited-state dynamics due to confinement. In solution, the lifetimes and spectra are essentially independent of alkyl substitution, therefore the differences among the encapsulated compounds are largely due to steric restrictions of the capsule. Increasing the alkyl-chain length probes the role of crowding on the reaction dynamics because longer molecules have less space to rearrange. The similar lifetimes in solution are due to the substituents having little influence on the π-conjugation, and being located at the para position of the phenyl rings, where they do not inhibit the isomerization.For t-Az@OA2 and the encapsulated mono-substituted compounds, the excited-state lifetime increases with chain length, suggesting a deeper local minimum on S1 with increasing size. This is consistent with an increasingly distorted excited-state geometry in the capsule that inhibits the initial in-plane motion along the inversion-assisted rotational reaction coordinate. More distortion due to increased crowding results in a larger barrier to reach the region of the conical intersection, and therefore the longer excited-state lifetimes compared with solution. The minimum-energy pathway in solution starts from a planar structure on the S1 surface, for which the barrier height is smallest.20 At the same time that the S1 lifetime increases, we also observe a significant distortion of the 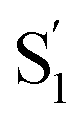 spectrum with increasing chain length. The splitting of the
spectrum with increasing chain length. The splitting of the 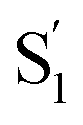 SAS into two bands suggests that the electronic structure of the
SAS into two bands suggests that the electronic structure of the 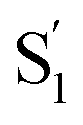 state changes across the series of encapsulated molecules. The progressive change in the
state changes across the series of encapsulated molecules. The progressive change in the 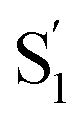 SAS for the mono-substituted compounds probably represents increasing distortion as the chain length increases, a trend that is also evident in the ground-state where the ππ* absorption bands indicate a distorted, nonplanar geometry of the longest mono-substituted compounds, t-EtAz@OA2 and t-PrAz@OA2.
SAS for the mono-substituted compounds probably represents increasing distortion as the chain length increases, a trend that is also evident in the ground-state where the ππ* absorption bands indicate a distorted, nonplanar geometry of the longest mono-substituted compounds, t-EtAz@OA2 and t-PrAz@OA2.
The dynamics are even more sensitive to encapsulation for the di-substituted compounds, which prefer to remain in the trans structure due to stable packing of the alkyl tails in the ends of the capsule. For example, the absence of a fast 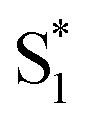 →
→ 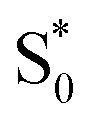 relaxation channel for the disubstituted compounds indicates that encapsulation prevents these molecules from easily accessing the region of the conical seam, even after excitation to the higher-energy ππ* state. Favorable packing of the alkyl tails into the two ends of the OA2 capsule favors the extended trans geometry, and possibly stabilizes an excited-state structure that then inhibits the isomerization. The favorable packing of the trans isomer of the disubstituted compounds is also reflected in the very low isomerization quantum yields for the two encapsulated molecules compared with solution and with the other encapsulated compounds (Table 1).
relaxation channel for the disubstituted compounds indicates that encapsulation prevents these molecules from easily accessing the region of the conical seam, even after excitation to the higher-energy ππ* state. Favorable packing of the alkyl tails into the two ends of the OA2 capsule favors the extended trans geometry, and possibly stabilizes an excited-state structure that then inhibits the isomerization. The favorable packing of the trans isomer of the disubstituted compounds is also reflected in the very low isomerization quantum yields for the two encapsulated molecules compared with solution and with the other encapsulated compounds (Table 1).
The interaction of the alkyl tails with the capsule provides an explanation for the variation of the 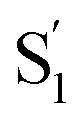 SAS across the series of encapsulated compounds. An unsubstituted phenyl ring does not fit into the ends of the capsule as well as the smaller alkyl tails, therefore crowding favors the more compact structure of the cis isomer to compensate the lengthening of the molecule with increasing alkyl chain length. The unsubstituted and mono-substituted compounds therefore increasingly favor the cis structure as the length of the tail increases, resulting in the higher quantum yields and stronger perturbation of the SAS as crowding becomes more severe.35 In contrast, the di-substituted compounds always favor the trans structure, and even the
SAS across the series of encapsulated compounds. An unsubstituted phenyl ring does not fit into the ends of the capsule as well as the smaller alkyl tails, therefore crowding favors the more compact structure of the cis isomer to compensate the lengthening of the molecule with increasing alkyl chain length. The unsubstituted and mono-substituted compounds therefore increasingly favor the cis structure as the length of the tail increases, resulting in the higher quantum yields and stronger perturbation of the SAS as crowding becomes more severe.35 In contrast, the di-substituted compounds always favor the trans structure, and even the 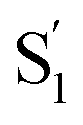 state probably retains a more distorted trans-like structure for these molecules in the capsule. Excited-state simulations for the encapsulated compounds would be helpful to elucidate the structures, but the very low quantum yield for disubstituted compounds clearly shows the impact of alkyl tails in changing the reaction path inside the OA2 capsule.
state probably retains a more distorted trans-like structure for these molecules in the capsule. Excited-state simulations for the encapsulated compounds would be helpful to elucidate the structures, but the very low quantum yield for disubstituted compounds clearly shows the impact of alkyl tails in changing the reaction path inside the OA2 capsule.
Finally, we point out that confinement probably also affects the topology at the conical seam directly, leading to a lower quantum yield for ππ* excitation in the capsule (5%) compared with solution (11%) even for unsubstituted t-Az. The impact becomes greater with increased crowding, and it is interesting to note that the isomerization quantum yield actually increases in the capsule compared with solution for the longest mono-substituted compound. This result is not entirely surprising considering the change in relative stability of the trans and cis isomers with increasing chain length,35 but nevertheless highlights the sensitivity of the excited-state dynamics and reaction pathways to confinement.
5 Conclusion
The ultrafast spectroscopy for a series of encapsulated alkyl-substituted azobenzene derivatives provides insight into the fundamental photoisomerization reaction following nπ* and ππ* excitation. In comparison with the azobenzene derivatives in solution we observe the encapsulated azobenzene molecules to have increasing excited-state lifetimes with increasing molecular length when relaxing from the S1 state due to the increasing sterical hindrance for phenyl rotational motion within the capsule. The confined environment opens up an additional relaxation pathway leading to a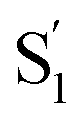 species, which for the mono-substituted derivatives resembles the formation of the cis excited-state isomer. Restricting the excited-state dynamics with di-substitution at the 4 and 4′ positions of the azobenzene molecule greatly impedes the inversion mechanism, which results in significantly lower trans → cis quantum yields and an
species, which for the mono-substituted derivatives resembles the formation of the cis excited-state isomer. Restricting the excited-state dynamics with di-substitution at the 4 and 4′ positions of the azobenzene molecule greatly impedes the inversion mechanism, which results in significantly lower trans → cis quantum yields and an 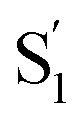 species that has a highly out-of-plane distorted geometry. The fundamentally different behavior observed in the confined environment demonstrates the importance of understanding the unique mechanisms for the isomerization of azobenzene, including the non-Kasha behavior that results in different quantum yields for nπ* and ππ* excitation.
species that has a highly out-of-plane distorted geometry. The fundamentally different behavior observed in the confined environment demonstrates the importance of understanding the unique mechanisms for the isomerization of azobenzene, including the non-Kasha behavior that results in different quantum yields for nπ* and ππ* excitation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under grants CHE-1151555 (C. G. E.) and CHE-1807729 (V. R.). C. J. O. also acknowledges support by the US Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through Argonne National Laboratory under Contract No. DE-AC02-06CH11357.Notes and references
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- M. Vengris, M. A. van der Horst, G. Zgrablić, I. H. M. van Stokkum, S. Haacke, M. Chergui, K. J. Hellingwerf, R. van Grondelle and D. S. Larsen, Biophys. J., 2004, 87, 1848–1857 CrossRef CAS PubMed.
- P. Kukura, D. W. McCamant, S. Yoon, D. B. Wandschneider and R. A. Mathies, Science, 2005, 310, 1006 CrossRef CAS PubMed.
- D. Polli, P. Altoè, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies, M. Garavelli and G. Cerullo, Nature, 2010, 467, 440 CrossRef CAS PubMed.
- S. Gozem, H. L. Luk, I. Schapiro and M. Olivucci, Chem. Rev., 2017, 117, 13502–13565 CrossRef CAS PubMed.
- Y. Norikane and N. Tamaoki, Org. Lett., 2004, 6, 2595–2598 CrossRef CAS PubMed.
- A. Sobolewska, S. Bartkiewicz, A. Miniewicz and E. Schab-Balcerzak, J. Phys. Chem. B, 2010, 114, 9751–9760 CrossRef CAS PubMed.
- E. A. Dolgopolova, T. M. Moore, O. A. Ejegbavwo, P. J. Pellechia, M. D. Smith and N. B. Shustova, Chem. Commun., 2017, 53, 7361–7364 RSC.
- H. Rau, Angew. Chem., Int. Ed., 1973, 12, 224–235 CrossRef.
- H. Rau, in Photochromism: Molecules and Systems, ed. H. Durr and H. Bouas-Laurent, Elsevier, 1990, ch. 4, pp. 165–192 Search PubMed.
- G. Kumar and D. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- Y. Kim, J. A. Phillips, H. Liu, H. Kang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6489–6494 CrossRef CAS PubMed.
- J. Wang, H.-B. Liu and C.-S. Ha, Tetrahedron, 2009, 65, 9686–9689 CrossRef CAS.
- E. Wei-Guang Diau, J. Phys. Chem. A, 2004, 108, 950–956 CrossRef.
- I. K. Lednev, T.-Q. Ye, R. E. Hester and J. N. Moore, J. Phys. Chem., 1996, 100, 13338–13341 CrossRef CAS.
- C. M. Stuart, R. R. Frontiera and R. A. Mathies, J. Phys. Chem. A, 2007, 111, 12072–12080 CrossRef CAS PubMed.
- T. Fujino and T. Tahara, J. Phys. Chem. A, 2000, 104, 4203–4210 CrossRef CAS.
- H. Satzger, C. Root and M. Braun, J. Phys. Chem. A, 2004, 108, 6265–6271 CrossRef CAS.
- T. Fujino, S. Y. Arzhantsev and T. Tahara, J. Phys. Chem. A, 2001, 105, 8123–8129 CrossRef CAS.
- C.-W. Chang, Y.-C. Lu, T.-T. Wang and E. W.-G. Diau, J. Am. Chem. Soc., 2004, 126, 10109–10118 CrossRef CAS PubMed.
- H. Rau and E. Luddecke, J. Am. Chem. Soc., 1982, 104, 1616–1620 CrossRef CAS.
- H. Rau, J. Photochem., 1984, 26, 221–225 CrossRef CAS.
- L. Blancafort, ChemPhysChem, 2014, 15, 3166–3181 CrossRef CAS PubMed.
- Y. Hirose, H. Yui and T. Sawada, J. Phys. Chem. A, 2002, 106, 3067–3071 CrossRef CAS.
- A. Nenov, R. Borrego-Varillas, A. Oriana, L. Ganzer, F. Segatta, I. Conti, J. Segarra-Marti, J. Omachi, M. Dapor, S. Taioli, C. Manzoni, S. Mukamel, G. Cerullo and M. Garavelli, J. Phys. Chem. Lett., 2018, 1534–1541 CrossRef CAS PubMed.
- N. Turro, V. Ramamurthy, W. Cherry and W. Farneth, Chem. Rev., 1978, 78, 125–145 CrossRef CAS.
- T. Itoh, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS PubMed.
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- S. R. Samanta, A. Parthasarathy and V. Ramamurthy, Photochem. Photobiol. Sci., 2012, 11, 1652–1660 RSC.
- A. Parthasarathy and V. Ramamurthy, Photochem. Photobiol. Sci., 2011, 10, 1455–1462 RSC.
- C. J. Otolski, A. M. Raj, V. Ramamurthy and C. G. Elles, J. Phys. Chem. Lett., 2019, 10, 121–127 CrossRef CAS PubMed.
- C. J. Otolski, A. M. Raj, G. Sharma, R. Prabhakar, V. Ramamurthy and C. G. Elles, J. Phys. Chem. A, 2019, 123, 5061–5071 CrossRef CAS PubMed.
- A. Parthasarathy, L. S. Kaanumalle and V. Ramamurthy, Org. Lett., 2007, 9, 5059–5062 CrossRef CAS PubMed.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS PubMed.
- V. Ramamurthy and A. Parthasarathy, Isr. J. Chem., 2011, 51, 817–829 CrossRef CAS.
- V. Ramamurthy, Acc. Chem. Res., 2015, 48, 2904–2917 CrossRef CAS PubMed.
- A. Mohan Raj and V. Ramamurthy, Org. Lett., 2017, 19, 6116–6119 CrossRef CAS PubMed.
- I. L. Zheldakov, J. M. Wasylenko and C. G. Elles, Phys. Chem. Chem. Phys., 2012, 14, 6211–6218 RSC.
- A. L. Houk, I. L. Zheldakov, T. A. Tommey and C. G. Elles, J. Phys. Chem. B, 2015, 119, 9335–9344 CrossRef CAS PubMed.
- C. L. Ward and C. G. Elles, J. Phys. Chem. A, 2014, 118, 10011–10019 CrossRef CAS PubMed.
- L. Landt, M. Staiger, D. Wolter, K. Klünder, P. Zimmermann, T. M. Willey, T. v. Buuren, D. Brehmer, P. R. Schreiner, B. A. Tkachenko, A. A. Fokin, T. Möller and C. Bostedt, J. Chem. Phys., 2010, 132, 024710 CrossRef PubMed.
- M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756–8771 CrossRef CAS PubMed.
- P. Bortolus and S. Monti, J. Phys. Chem., 1979, 83, 648–652 CrossRef CAS.
- M. Moreno, R. Gelabert and J. M. Lluch, ChemPhysChem, 2016, 17, 2824–2838 CrossRef CAS PubMed.
- I. Lednev, T.-Q. Ye, P. Matousek, M. Towrie, P. Foggi, F. Neuwahl, S. Umapathy, R. Hester and J. Moore, Chem. Phys. Lett., 1998, 290, 68–74 CrossRef CAS.
- R. W. Hendler and R. I. Shrager, J. Biochem. Biophys. Methods, 1994, 28, 1–33 CrossRef CAS PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- T. Nagele, R. Hoche, W. Zinth and J. Wachtveitl, Chem. Phys. Lett., 1997, 272, 489–495 CrossRef.
- J. Azuma, N. Tamai, A. Shishido and T. Ikeda, Chem. Phys. Lett., 1998, 288, 77–82 CrossRef CAS.
- F. Aleotti, L. Soprani, A. Nenov, R. Berardi, A. Arcioni, C. Zannoni and M. Garavelli, J. Chem. Theory Comput., 2019, 15, 6813–6823 CrossRef CAS PubMed.
- I. Conti, M. Garavelli and G. Orlandi, J. Am. Chem. Soc., 2008, 130, 5216–5230 CrossRef CAS PubMed.
- Y. Lu, E. Diau and H. Rau, J. Phys. Chem. A, 2005, 109, 2090–2099 CrossRef CAS PubMed.
- S. Yuan, Y. Dou, W. Wu, Y. Hu and J. Zhao, J. Phys. Chem. A, 2008, 112, 13326–13334 CrossRef CAS PubMed.
- J. Bahrenburg, F. Renth, F. Temps, F. Plamper and W. Richtering, Phys. Chem. Chem. Phys., 2014, 16, 11549–11554 RSC.
- M. Boggio-Pasqua, M. Ravaglia, M. J. Bearpark, M. Garavelli and M. A. Robb, J. Phys. Chem. A, 2003, 107, 11139–11152 CrossRef CAS.
- R. S. H. Liu and G. S. Hammond, Photochem. Photobiol. Sci., 2003, 2, 835–844 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional ground-state spectra, transient absorption spectra, and species-associated spectra from kinetic fits to the data (PDF). See DOI: 10.1039/d0sc03955a |
| ‡ Present address: Chemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois, USA. |
| This journal is © The Royal Society of Chemistry 2020 |