
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTopological prediction of palladium coordination cages†
David A.
Poole
III
,
Eduard O.
Bobylev
,
Simon
Mathew
 and
Joost N. H.
Reek
*
and
Joost N. H.
Reek
*
Homogeneous, Supramolecular, and Bio-inspired Catalysis Group, van't Hoff Institute for Molecular Science (HIMS), University of Amsterdam (UvA), Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl
First published on 16th October 2020
Abstract
The preparation of functionalized, heteroleptic PdxL2x coordination cages is desirable for catalytic and optoelectronic applications. Current rational design of these cages uses the angle between metal-binding (∠B) sites of the di(pyridyl)arene linker to predict the topology of homoleptic cages obtained via non-covalent chemistry. However, this model neglects the contributions of steric bulk between the pyridyl residues—a prerequisite for endohedrally functionalized cages, and fails to rationalize heteroleptic cages. We describe a classical mechanics (CM) approach to predict the topological outcomes of PdxL2x coordination cage formation with arbitrary linker combinations, accounting for the electronic effects of coordination and steric effects of linker structure. Initial validation of our CM method with reported homoleptic Pd12LFu24 (LFu = 2,5-bis(pyridyl)furan) assembly suggested the formation of a minor topology Pd15LFu30, identified experimentally by mass spectrometry. Application to heteroleptic cage systems employing mixtures of LFu (∠B = 127°) and its thiophene congener LTh (∠B = 149° ∠Bexp = 152.4°) enabled prediction of Pd12L24 and Pd24L48 coordination cages formation, reliably emulating experimental data. Finally, the topological outcome for exohedrally (LEx) and endohedrally (LEn) functionalized heteroleptic PdxL2x coordination cages were predicted to assess the effect of steric bulk on both topological outcomes and coordination cage yields, with comparisons drawn to experimental data.
Introduction
Spherical coordination cages of type PdxL2x are formed by the self-assembly of palladium ions and organic bipyridine linkers in regular geometric patterns.1 The dynamic nature of Pd–pyridyl coordination has enabled the isolation of PdxL2x assemblies featuring 2–60 metal centers, isolated as the thermodynamic minima of possible structures.1 The internal and external surfaces can be independently functionalized to modulate the sphere environment at the nanoscale for applications.2 Previous reports detail a range of novel properties for guest binding,3 facilitating chemical reactions,4 modifying catalytic processes,5–7 electrochemistry,8 and optoelectronic applications.9 These useful properties are derived from the topology of the assembly,1 creating a significant interest in predicting topological results of self-assembly processes that ultimately lead to production of uniform assemblies of singular topology. Previously, Hay and Young described a model to predict the topology of coordination assemblies based on geometric properties of the metal center and organic linker.10 This model was adapted by Fujita and coworkers, using the bend angle (∠B) of the bipyridine linker to predict the topology of the thermodynamic product, applying it to describe spherical cages containing 3–60 metal centers as a principle for rational design.11 While this approach is sufficient for most homoleptic cages, the model fails to describe assemblies derived from asymmetric, flexible or sterically demanding linkers. This shortcoming is apparent in modern efforts to design heteroleptic cages that feature multiple functional groups to control the assembly outcome in a trial-and-error manner, especially when asymmetric linkers steric bulk or shape-complementarity are employed.12–14 The latter strategy, employed by Clever and colleagues, is especially promising for the design of small M2L4 cages with divergent functionalities.12 A more robust model would provide a better basis of rational design for these new structures, and the application of shape-complementarity to larger designs, motivating the aim of our research.The process of self-assembly leads to formation of supramolecular structures with the most thermodynamically stable topology,11 and a means to identify this product computationally would allow prediction of topologies prior to the experimental identification. While previous efforts have used computational methods to simulate the formation process,15 and determine the guest binding properties of cages,15 there is no available method for in silico screening to identify the preferred topology produced from an arbitrary linker, or set of linkers. Such a method would have practical applications as a tool for the rational design of highly functionalized cage assemblies, and furthermore provide further insight into known assemblies. Distinguishing the true thermodynamic products from trapped states and the identification of minor products are important issues in this regard. A viable method of in silico screening requires accuracy in estimating the free energy coupled with expediency to effectively guide experimental work. Given the size of the assemblies, numerous topologies and permutations of linker combinations, classical mechanics methods are well suited to practically address this task, with modern methodologies showing marked success in describing these assemblies.15–17
Here, we report a strategy that utilizes force field parameters for individual linkers from DFT structure optimization to estimate the relative free energies across 40 topologies containing 3–30 metal centers. The validity of our approach was demonstrated by a detailed study of the topological preferences of homoleptic and heteroleptic assemblies derived from reported linkers. The feasibility of our method is demonstrated by the study of novel heteroleptic assemblies featuring a sterically demanding linker bearing endohedral functionalization. These topological predictions of our CM method are supported by experimental observations.
Methods
Experimental details
Linkers LFu, LTh, LEx, and LEn were synthesized following standard procedures detailed in the ESI (Section D†).2,18 Homoleptic assemblies were formed with a total linker concentration of 10 mM and 0.55 equivalents of Pd(CH3CN)4(BF4)2, at room temperature (LFu and LTh) or 50 °C (LEx and LEn) after 16 hours in CD3CN. The resulting clear solutions were filtered (0.45 μm, PTFE syringe filter), following analysis by NMR and ESI-HRMS. Details of the experimental procedures and subsequent analyses are given in the ESI (Sections E–H†). 1H NMR spectra were measured using either a Bruker DRX 500 (500 MHz) or a Bruker DRX 300 (300 MHz) at 25 °C unless otherwise noted for variable temperature experiments. DOSY NMR spectra were obtained using LED bipolar pulse gradients with a diffusion delay time of 0.1 seconds at 25 °C. Mass spectra were collected on a high-resolution time-of-flight Bruker Impact II ESI-HRMS. Detection was in positive-ion mode with a source voltage between 4 and 6 kV. Samples were prepared in CD3CN with a total linker concentration of 10 mM and a Pd(CH3CN)4(BF4)2 concentration of 5.5 mM, and then analyzed by NMR and ESI–HRMS directly.Computational details
CM calculations were carried out using an Amber-type forcefield19 and structural annealing and optimization were completed with the GPU-enabled Amber16 software suite,20 with run parameters provided in the ESI (Section B†). CM forcefield parameters were developed directly with a model system (Fig. 1).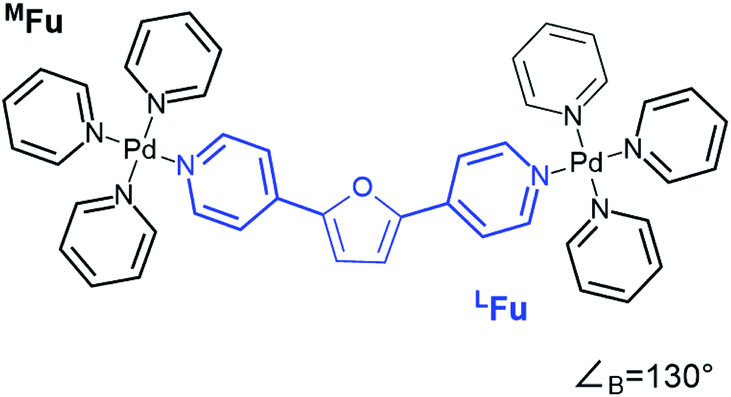 | ||
| Fig. 1 An example of a model system used for DFT dynamics studies (see also Fig. S1†). This model, MFu, features the linker LFu (blue) coordinated to tris-pyridyl Pd2+ centers. The bend angle of the complex is measured from the B3LYP/def2-TZV minimized structure, deviating slightly from the free linker due to electronic effects of metal coordination. Structures, charge assignments (Fig. S2†), dynamics trajectories (Fig. S3†) and other data for model complexes of linkers are provided in the ESI (Section A†). | ||
The optimized structure of the model system was used for charge fitting based on the RESP method employed by antechamber.20 Lennard Jones potentials for organic atom centers were assigned by GAFF atom type,21 palladium centers were parameterized according to previous reports.22 Bond, angle and torsion terms were fit using a genetic algorithm.23 Models of the complete cages were assembled by least-squares fitting placement of the individual linkers in accordance to a template using ProFit.24
The fitting and validation data sets were obtained from trajectories generated using GFN2-xTB,25 supplemented by single point energies of each trajectory frame computed by DFT at a B3LYP/def2-TZV level of theory using Gaussian 16 rev. C.26 A complete discussion of the parameterization (Scheme S1†) and validation (Table S1†) is provided in the ESI (Section A†). While only topologies containing 3–30 metal centers are considered, this approach is readily extended to Pd2L4 based assemblies, as also discussed in the ESI (Section K†).
Results and discussion
Topological survey procedure
In this study we considered the four linkers shown in Fig. 2. LFu (∠B = 127°) and LTh (∠B = 149°) have been previously reported and shown to form both homoleptic and heteroleptic assemblies.24 Furthermore, as the favored topology of the heteroleptic assemblies depends on the composition of linkers in the assembly, this system offers an ideal test for method development. Linkers LEx and LEn are novel linkers featuring exohedral and endohedral functionalization respectively. While these two linkers possess similar bend angles, the latter is sterically strained, which may impact the topological outcome of self-assembly.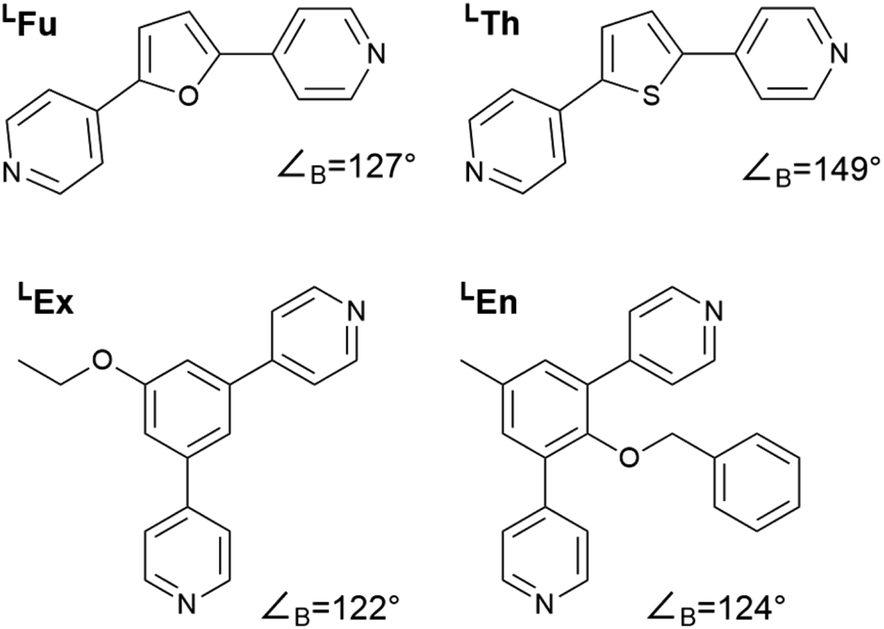 | ||
| Fig. 2 Structures of the bipyridyl linker molecules used in this study. Bend angles, ∠B, are estimated from B3LYP/def2-TZV minimized structures. | ||
Amber type forcefield parameters were developed for the four linkers employed in this study (Fig. 2), which reproduced the relative free energies for the model complex reliable compared to semi-empirical methods (Table S1†). These linker models were then used to construct assembly models in accordance to templated defined for polyhedra containing 3–30 vertices representing many of the known cage topologies. These structures could then be annealed (2 ns) and optimized (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps) expediently to produce a minimum energy structure for each topology, provided in detail in the ESI (Table S4†). The relative energy of these structures was analyzed using a Boltzmann statistical model, with each topology considered a microstate for a given linker weighted by the number of linkers involved as depicted in Scheme 1.
000 steps) expediently to produce a minimum energy structure for each topology, provided in detail in the ESI (Table S4†). The relative energy of these structures was analyzed using a Boltzmann statistical model, with each topology considered a microstate for a given linker weighted by the number of linkers involved as depicted in Scheme 1.
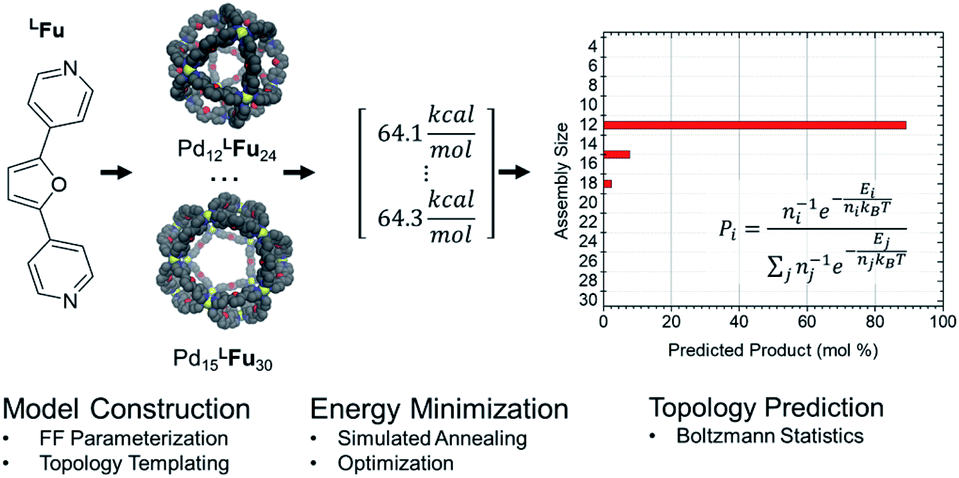 | ||
| Scheme 1 Flow chart for topological prediction of homoleptic assemblies featuring the formation of homoleptic PdxLFu2x assemblies. The linker structure is used to construct a library of possible assembly outcomes, which are subjected to a simulated annealing and structural optimization procedure using implicit solvation. The resulting minimum energies, E, are treated as microstates, and the topological distribution is determined using Boltzmann statistics with a weighting factor ‘n’ corresponding to the number of linker components in the assembly. | ||
A majority of PdxL2x assemblies reported are homoleptic in nature due to the ease of assembly and subsequent analysis. This simple case was used in order to validate our topological survey approach. Specially the method was validated for linkers LFu (∠B = 127°) and LTh (∠B = 149°). The former has been reported to afford a single topology, Pd12LFu24, while the latter is recently known to form a mixture, Pd24LTh48 and Pd30LTh60. These assemblies have been sufficiently characterized by NMR, HRMS, and crystallographic means providing clear and definite knowledge of the assembly outcomes.18,27
Our model predicted a majority (89.1%) presence of M12L24 homoleptic assemblies using linker LFu, which is observed experimentally (Fig. 3). Additionally, our model predicted the presence of a minor (7.8%) Pd15L30 product. We experimentally investigated the minor Pd15L30 species by reproducing assemblies based on reported protocols18 and subsequent characterization of the PdxL2x assembly distribution by DOSY NMR (Fig. S13†) and CSI-HRMS (Fig. S14†). DOSY NMR revealed a single species (log10D = −9.23), consistent with the presence of Pd12LFu24 as previously reported. ESI-HRMS revealed the presence of a Pd15LFu30 assembly (Fig. 3).
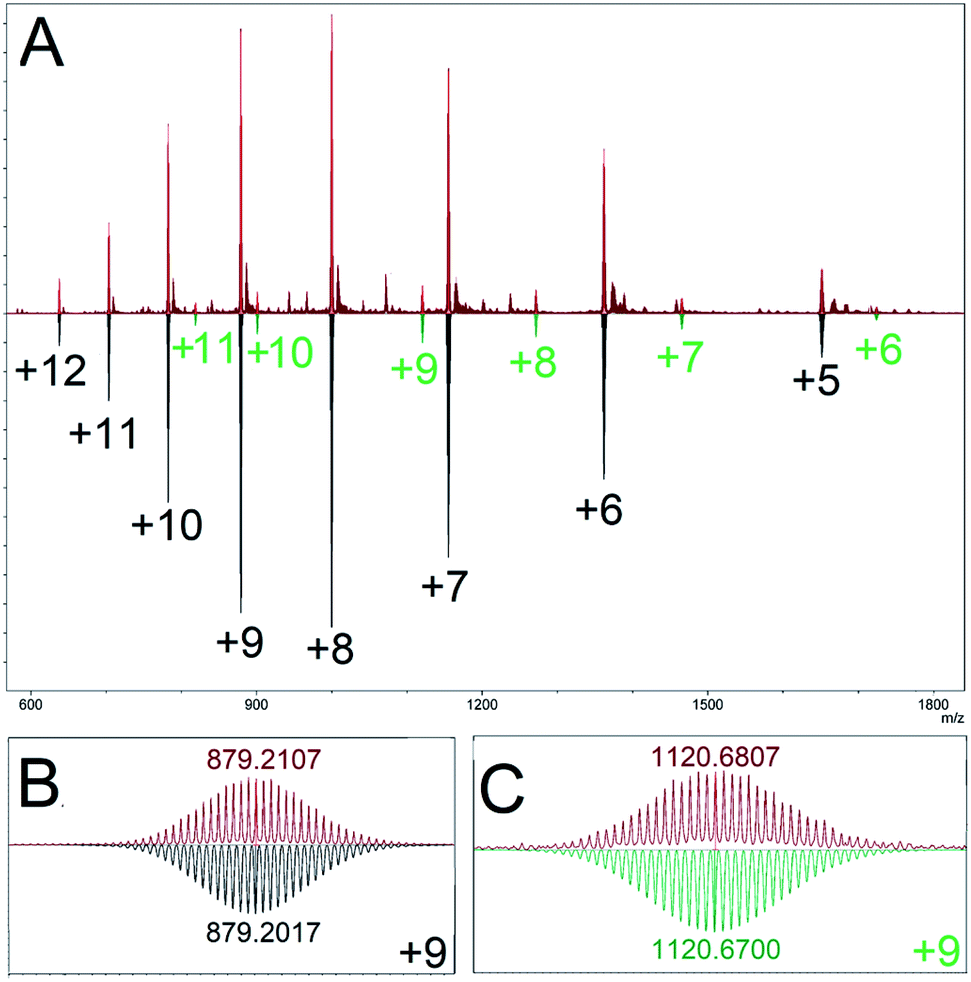 | ||
| Fig. 3 (A) ESI-HRMS characterization of the homoleptic assembly products of linker LFu (red), top. Mass simulations indicate multiple charged species matching simulations for Pd12LFu24, (black), and Pd15LFu30, (green) species. Bottom, an expanded view of the isotope distributions observed for (B) Pd12LFu24(BF4)139+, and (C) Pd15LFu30(BF4)219+, species, annotated with the center peak m/z either observed (red) or calculated (black or green). Data is further detailed in the ESI (Fig. S15†). | ||
Fortuitously, the relative intensity of this minor species (9.4% of Pd15LFu30) correlates well to the prediction of Pd15LFu30 abundance (8.7%) from our computational model, as detailed in the ESI (Table S3†). This highlights the inability of DOSY NMR to distinguish between coexisting Pd12 and Pd15 species originating from significant overlap of the NMR resonances, similar size (and therefore, diffusion coefficient) and the disparity between their relative populations. We applied our method to the PdxL2x assemblies formed with linker LTh (∠B = 149°), reported to generate larger M24L48 and M30L60 assemblies.27 The modelling results of the homoleptic assembly formed with linker LTh predict the formation of assemblies with either M24L48 (67.0%) or M30L60 (32.8%) topologies, further detailed in the ESI (Table S4†).
Initial experimental reports of these assemblies afforded only the M24L48 structure which was characterized by NMR, HRMS, and crystallographic means.24 However, later work afforded a mixture which included both M24L48 and M30L60 topologies, the latter of which was additionally characterized by NMR, HRMS, and crystallographic means.27 Our CM approach was unaffected by the kinetic differences for forming either topologies and the resulting topological distribution contain these two species at relative populations in good agreement with the experimentally reported data.
Topological prediction of heteroleptic PdxL2x assembly distribution
The formation of heteroleptic assemblies is a significant motivation for topological prediction as there is no apparent rule to predict the assembly outcome for the combination of two dissimilar linkers. Therefore, we expanded our survey to consider assemblies composed of two linkers, randomly distributed over the edges of the polyhedral templates. To validate this extension, we considered the previously reported heteroleptic assemblies of LTh and LFu.18A new approach was needed in order to identify the topological preference of mixtures of linkers at arbitrary compositions using a linear of the relative free energy for each topology at various compositions (Fig. 4). This approach is further detailed in the ESI (Section C†).
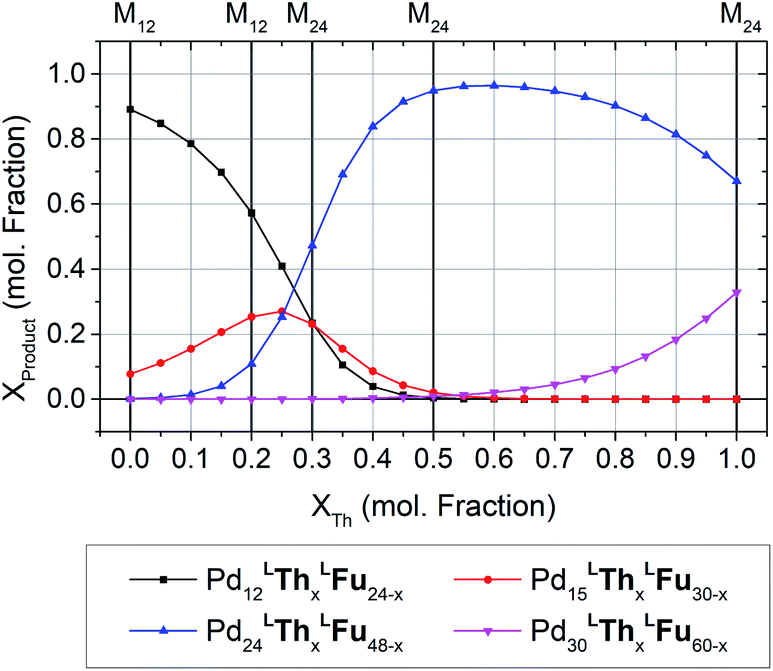 | ||
| Fig. 4 The topological preferences of heteroleptic cages composed of LFu and LTh with the four most major products shown. Referenced experimentally determined topological outcomes shown on upper axis at the reported experimental composition. Preferences at arbitrary compositions were predicted by linear interpolation of the model forcefield energy, and subsequent application of Boltzmann statistical analysis. This procedure is detailed further in the ESI (Section C†), with included analysis and discussion of the fitting results for heteroleptic assemblies of LFu and LTh. | ||
Our model predicts that heteroleptic assemblies with a M12L24 topology are produced from mixtures of LFu and LTh containing less than 27% of LTh. At this critical concentration a transition in topological preference is observed, shifting towards the M24L48 topology. This critical composition is in agreement with the previous report observing the transition occurring at 0.2–0.3 mole fraction LTh.18 The model predicts greater preference for the M15L30 topology at this critical point, suggesting it may be an important intermediate between the two topologies.
The effect of endohedral functionalization on topological distribution
Moving beyond the bend angle, we used our model to study the topological effect of endohedral and exohedral substituents. Linker LEx features external ornamentation that should impart minimal effect on PdxL2x assembly topology, as homoleptic assemblies of similar linkers have been reported to form M12L24 topologies.2 Linker LEn, with internal functionalization demonstrate the same apparent bend angle as LEx suggesting the same topology should form. However, reports of a similar linker featuring a methoxy substituent by Fujita et al., concluded that PdxL2x assembly formation is inhibited assemblies due to the steric encumbrance imparted by substituents in this position.2 To maximize this steric effect, a benzyl group was selected in the design of linker LEn.Shown in Fig. 5, our topological prediction shows the M12L24 topologies are favored over a large range of compositions, as would be suggested by the bend angle. Interestingly, at mixtures containing a larger proportion of LEn, the formation of larger topologies (i.e. M24L48 or M30L60) are favored. As the LEn bend angle (∠B = 124°) favors M12L24 topologies, the preference towards larger assemblies can only be ascribed to the effect of the steric bulk.
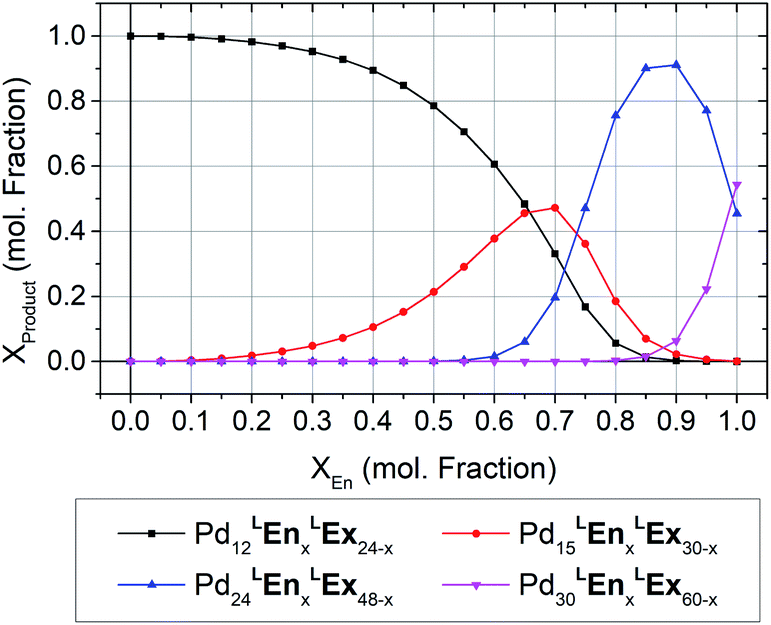 | ||
| Fig. 5 The modeled topologic preference of heteroleptic assemblies of linkers LEx and LEn. The M12L24 is the most preferred topology at lower compositions, with successively larger topologies, M15L30 to M30L60, preferred with increasing LEn. | ||
As expected, homoleptic assemblies were successfully formed with LEx, evidenced by the characteristic shift of α-pyridyl proton resonances in 1H NMR. Subsequent characterization of the assembly by DOSY NMR revealed a hydrodynamic radius of 16.4 Å, identical to the radius found for the Pd12LEx24 model (Table S4†). Finally, isotope pattern analysis within the ESI-HRMS data enabled for unambiguous assignment of Pd12LEx24 cage topologies (Fig. S19†).
Similarly, homoleptic assemblies of LEn were successfully formed on the basis of the characteristic downfield shift of α-pyridyl proton resonances, and the absence of free building block in 1H NMR. However, DOSY NMR indicated that resulting assemblies possess a radius of 27.8 Å (Fig. S22†). While this value is significantly larger than the 17.6 Å predicted by our model of Pd12LEn24 cages, it is a reasonable radius for Pd30LEn60 assemblies. Unfortunately, the difficulty in reliably ionizing this species meant that it's presence could not be confirmed by ESI-HRMS, thus the composition of the assembly (or assemblies) formed experimentally remains ambiguous. These limitations of ESI-HRMS are known for the analysis of larger assemblies, experimentally limiting the pursuit of these structures.27
Heteroleptic assemblies were formed using five different ratios of LEx:LEn linkers, incorporating 4–20 of LEn (mole fractions 0.14–0.86) and 20–4 LEx (Fig. 6). The formation of these assemblies was confirmed by the characteristic downfield shift of the α-pyridyl proton (ca. 9.05 to 9.25 ppm). DOSY NMR indicated that the resulting assemblies were larger than expected for M12L24 topologies (rH 18–21 Å), but within expectations for M24L48 or M30L60 topologies (rH 23–26 Å). These data are provided in the ESI (Section H†). It is well documented that intermediate polymeric species present during assembly formation are NMR silent,28,29 and therefore can be excluded as a major product.
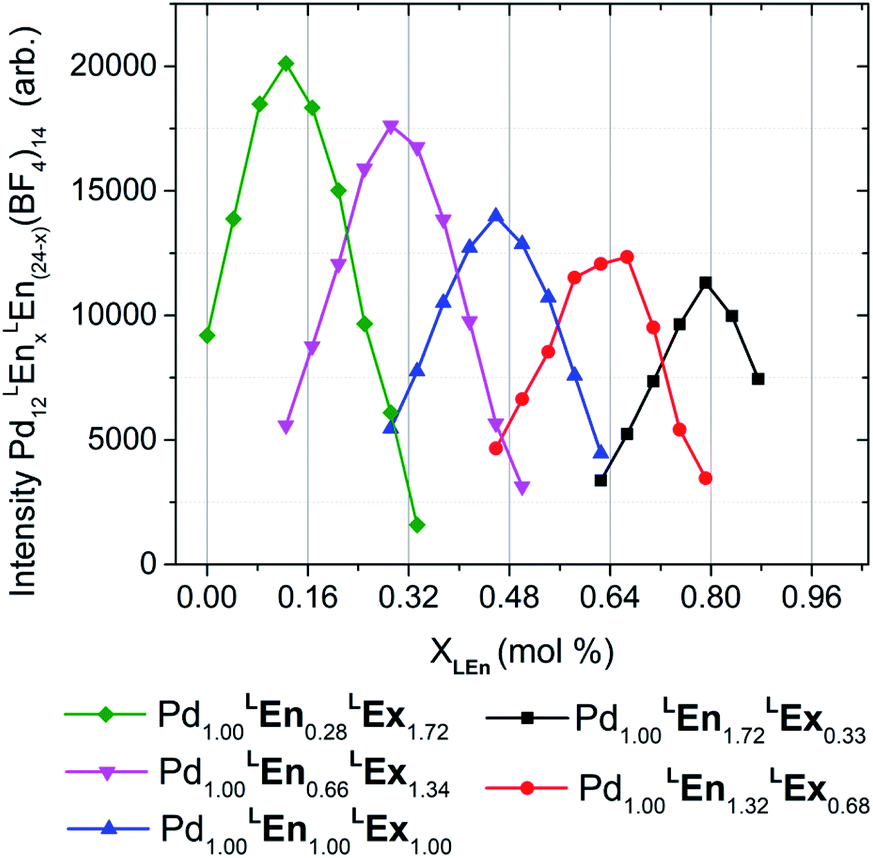 | ||
| Fig. 6 Distributions of heteroleptic assemblies observed by ESI-HRMS. Assembly product mixtures were formed in deuterated acetonitrile from 10.0 mM total concentration of LEn and LEx with 5.0 mM of Pd(BF4)2 and were directly analyzed without dilution by ESI-HRMS. The observed mass spectra of Pd12LEn(x)LEx(24−x)·(BF4)1410+ species suggests a single product topology. These results are further detailed in the ESI (Section H†). | ||
Mass analysis of the heteroleptic assemblies were found to include 0–21 LEn randomly incorporated into the assembly structure with an average ratio corresponding to the composition of the reaction mixture, summarized by Fig. 6. Mass analysis yielded only compositions with a stoichiometry of Pd12LEnxLEx(24−x), corresponding only to the Pd12L24 topologies. This contradicts the DOSY NMR results indicating the formation of larger species containing 24–30 metal centers. Interestingly the overall intensity of the mass signal decreases with the increase in LEn content. If this decrease were due to simple differences in assembly formation, ionizability, or instrument response, a bias should be observed in the distributions of Pd12LEnxLEx(24−x) species formed. Alternatively, this diminished intensity can be attributed to the presence of a secondary species—plausibly Pd24LEnxLEx(48−x) or Pd30LEnxLEx(60−x) cages—which are beyond the current limits of this particular ESI-HRMS analytical technique and therefore cannot be conclusively characterized.
Impaired dihedral rotation and long-range topological effects
From the above data we surmise that heteroleptic assemblies of LEn and LEx form under conditions similar to other palladium–pyridyl coordination cages. Furthermore, these cages possess a larger topology due to the steric effects of the endohedral functionalization of LEn. To understand the mechanism of this steric effect and its impact on topological preferences for heteroleptic assemblies, relaxed potential energy surfaces were computed for the pyridyl–arene dihedral rotation using DFT at a B3LYP/def2-TZV theory level as illustrated in Fig. 7.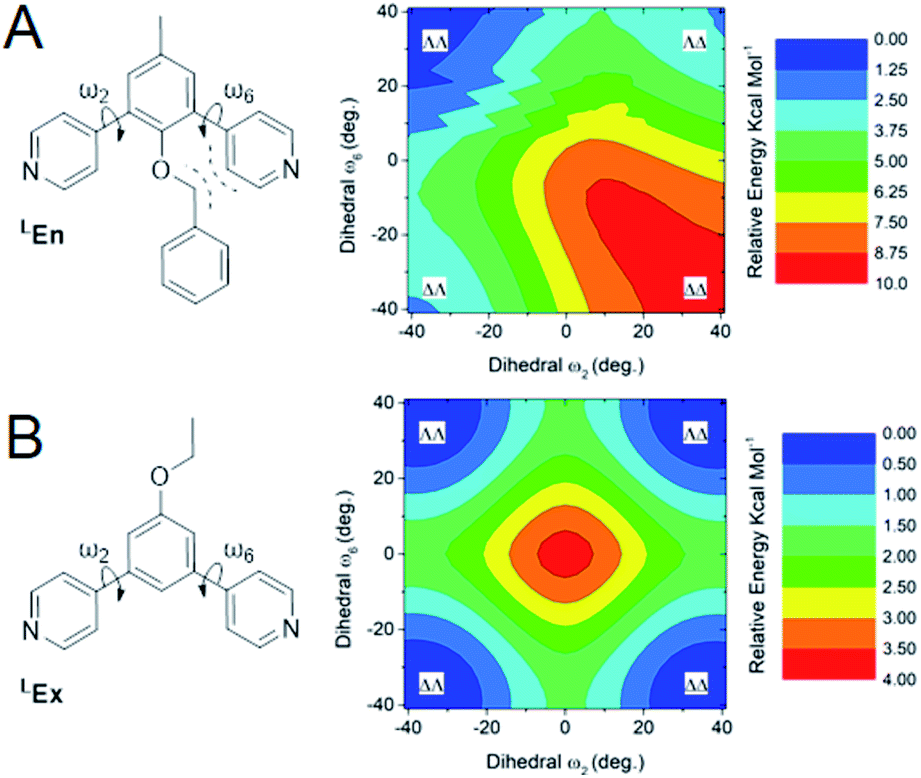 | ||
| Fig. 7 Computed free energy surfaces of dihedral rotations of model systems for LEn (A) and LEx (B). Left, simple drawing of the model system showing the dihedral rotation (arrows), and expected source steric hinderance (dashed lines). Right, potential energy surfaces produced from a relaxed scan of the dihedral angles (ω2, ω6) between ±40° from coplanarity in 28 steps of 3° for each dihedral angle. These DFT calculations were conducted at a B3LYP/def2-TZV level of theory, with the color contour corresponding to the relative free energy of each dihedral pair in kcal mol−1. | ||
While relaxed scans were used, the method converges to local minima resulting in the distinct ΛΛ and ΔΔ atropisomers shown in Fig. 8A. These potential energy surfaces reveal that the pyridyl–arene dihedral rotations for the sterically demanding LEn are asymmetric compared to LEx. The structures generated from this scan were analyzed using our CM parameters, producing similar results as shown in the ESI (Section J†).
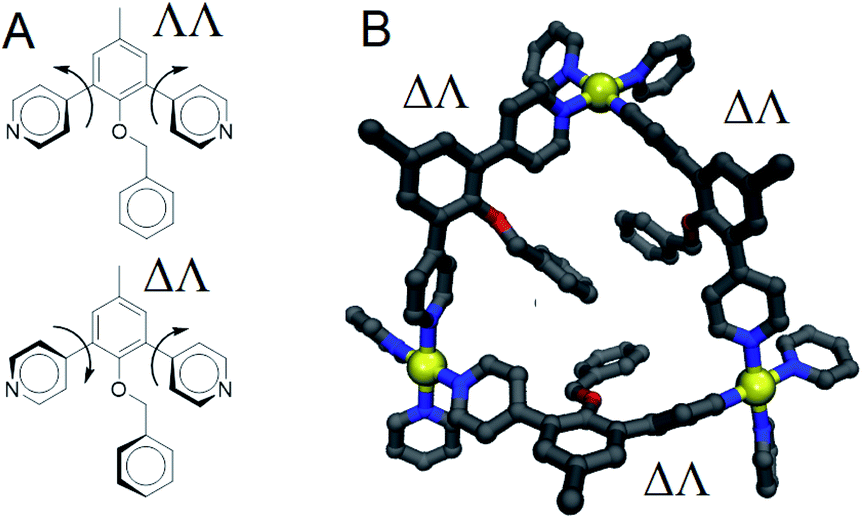 | ||
| Fig. 8 (A) Drawing showing ΛΛ and ΔΛ atropisomers of LEn with out-of-plane rotation shown. (B) Ball and stick model renders of a triangular face from our molecular mechanics model of a cuboctahedral Pd12LEn24 cage including neighboring linker pyridyl groups shown showing requirement for ΔΛ isomers to accommodate the propeller angle about the palladium coordination site. Model features atoms colored by element: carbon as grey; nitrogen, blue; oxygen, red; and palladium as yellow. Hydrogen atoms were omitted from the rendering for clarity. | ||
The DFT potential energy surface shows that ΔΛ or ΛΔ isomers of LEn are less favorable by 2.2 kcal mol−1. The preference for the ΛΛ atropoisomer is easily rationalized by the minimization of steric interactions between the pyridyl groups and the endohedral benzyl functionality. Similarly, the high energy ΔΔ isomer forces the pyridyl groups to rotate into the endohedral benzyl functionality, and therefore such conformation is not physically accessible.
Reproduction of these dihedral scans using our CM method produces similar minima with ΔΛ or ΛΔ isomers being only 1.2 kcal mol−1 higher in energy, as discussed in detail in the ESI (Section J†). Contrary to the computed preference of the ΛΛ isomer, our CM models of assemblies feature predominantly ΔΛ or ΛΔ isomers shown in Fig. 8.
We speculate that this arrangement is a requirement for coordination to satisfy coordination and results in a destabilization of cages proportional to the number of triangular faces involved. This aggregate effect was modelled directly using our CM approach to consider specific arrangements of linkers for Pd12LEn4LEx20 assemblies following the same annealing and optimization methodology used for previously. The relative free energies were computed for four different distributions representative of the intuitive outcomes for different degrees of interaction between LEn linkers.
Based on our model, the non-interacting arrangement (Fig. 9A) is significantly more favorable than co-coordinated or co-facial arrangements (Fig. 9B–D). However, the possibility of forming these favorable arrangements becomes insignificant with increasing LEn content, necessitating the more accessible co-facial arrangements. The relative free energy of co-facial LEn on a triangular face (Fig. 9D) were less favored compared to the co-facial square (Fig. 9C) arrangements by 1.8 kcal mol−1 per linker. In turn this accounts for the observation that assemblies of LEn form larger topologies (i.e. M24L48 and M30L60) as they feature a smaller proportion of triangular faces (8/26 and 8/38 faces, respectively) compared to M12L24 (8/14 faces) topologies. Similarly, the incorporation of the more adaptable LEx alleviates permits the formation of spherical cages with a smaller M12L24 topology that would otherwise be favored by the natural bend angle of both linkers.
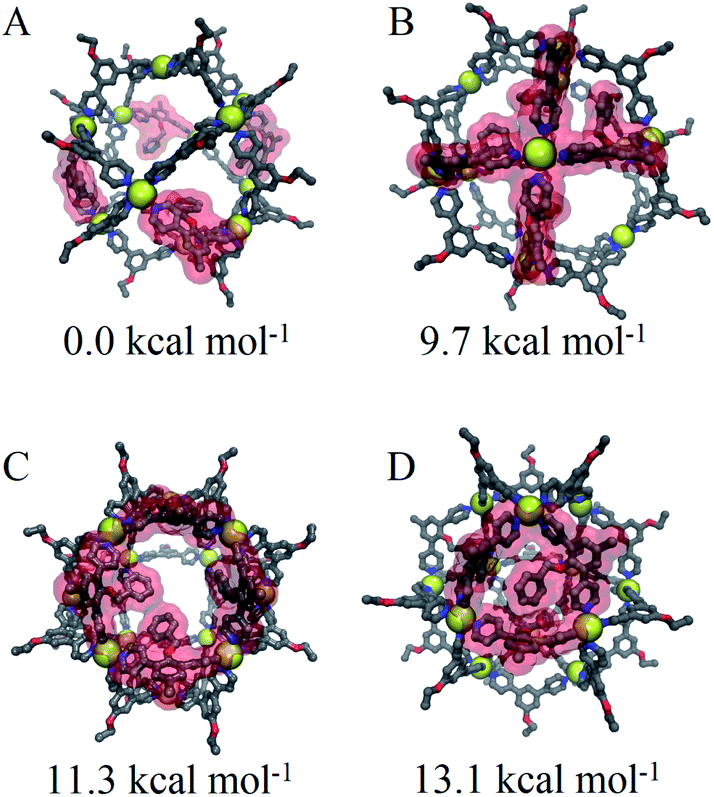 | ||
| Fig. 9 Renders of CM optimized structures of Pd12LEn4LEx20 with forcefield energy per linker relative to the minimum structure. (A) Distributed at distant, non-interacting positions, (B) interaction at a palladium center, (C) interaction as a square face, and (D) interaction as a triangular face, with one distant non-facial linker. Ball-and-stick models of linkers are colored by atom type (carbon (grey), nitrogen (blue) and oxygen (red)) with hydrogen atoms omitted for clarity. Yellow vdW volume spheres (yellow) are shown for palladium centers. A red vdW surface highlights the interacting LEn within the structure. | ||
These computational results account for the experimental observations of heteroleptic assemblies for LEx and LEn, where DOSY NMR shows the formation of a larger complex, and the mass spectra results which present an unexpected loss of intensity. While it is unfortunate that available experimental methods cannot fully characterize and quantitatively assess these larger species, our CM model provides deeper insight into these assemblies and is well supported by both higher-level calculations and available experimental evidence.
Conclusions
In this work we produced a forcefield for pyridyl–palladium complexes as found in a number of coordination assemblies. This forcefield offers significant improvements to accurately estimates of relative free energies of palladium–pyridyl complexes. Using these estimates, we could predict the topological preferences of two previously reported bis-pyridyl linkers, LTh and LFu, reproducing experimentally observed topological preferences for their homoleptic and heteroleptic assemblies. From our results we found a novel minority M15L30 topology which we characterized experimentally. These outcomes demonstrate the viability of a classical mechanics based in silico screening for these topological outcomes.This approach was then used to determine the topological outcome of heteroleptic assemblies formed from two novel linkers, LEx and LEn, respectively featuring exohedral and endohedral functionalization. This study demonstrated the pronounced effect of steric bulk proximal to metal binding sites in topological outcomes and assembly yields, as confirmed by experimental study with high resolution mass spectrometry. These heteroleptic assemblies demonstrate a novel strategy for the inclusion of sterically demanding functionalization within coordination cages, and highlight the effectiveness of our classical mechanics methodology to aid in assembly design.
Future work aims to apply these computational approaches to provide more detailed insight in the formation of these self-assembled nanospheres, and to extend these approaches to include to better suit the analysis of applied supramolecular structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We extend a special thanks to our colleague, Ms. Eline Meijer, for her insightful comments, and providing initial motivation for forming the novel heteroleptic assemblies presented here. This paper is dedicated to Prof. P. H. Dixneuf for his scientific and organisational contributions to field of organometallic chemistry and catalysis. We thank the European Research Council (ERC Adv. Grant 339786-5354 NAT_CAT) and the sustainable chemistry research program from the University of Amsterdam for financial support.References
- For reviews, see: (a) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC; (b) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS; (c) R. A. S. Vasdev, D. Preston and J. D. Crowley, Chem.–Asian J., 2017, 12, 2513–2523 CrossRef CAS; (d) T. K. Ronson, S. Zarra, S. P. Black and J. R. Nitschke, Chem. Commun., 2013, 49, 2476–2490 RSC; (e) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 258, 825–841 CrossRef.
- S. Löffler, J. Lübben, A. Wuttke, R. A. Mata, M. John, B. Dittrich and G. H. Clever, Chem. Sci., 2016, 7, 4676–4684 RSC.
- (a) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (b) F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef; (c) Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS.
- For examples of catalytic cages, see: (a) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS; (b) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS; (c) M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619–1636 RSC; (d) Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS; (e) Y. Fang, J. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC; (f) L. Holloway, P. Bogie, Y. Lyon, C. Ngai, T. Miller, R. Julian and R. Hooley, J. Am. Chem. Soc., 2018, 140, 8078–8081 CrossRef CAS; (g) W. Cullen, M. Misuraca, C. Hunter, M. Williams and M. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS; (h) C. Tan, J. Jiao, Y. Liu and Y. Cui, Angew. Chem., Int. Ed., 2017, 8, 2085–2090 Search PubMed; (i) H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS; (j) See also ref. 16d.
- For reviews, see: (a) V. Mouarrawis, R. Plessi11us, J. I. van der Vlugt and J. N. H. Reek, Front. Chem., 2018, 6, 623 CrossRef CAS; (b) A. C. H. Jans, X. Caumes and J. N. H. Reek, ChemCatChem, 2019, 11, 287–297 CrossRef CAS; (c) S. H. A. M. Leenders, R. Gramage–Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC; (d) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS; (e) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS; (f) M. Raynal, P. Ballester, A. Vidal–Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (g) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS; (h) D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS; (i) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (j) R. J. Hooley and J. Rebek Jr, Chem. Biol., 2009, 16, 255–264 CrossRef CAS; (k) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS; (l) L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS.
- For examples on metal catalyzed processes in cages, see: (a) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2013, 19, 10170–10178 CrossRef CAS; (b) L. J. Jongkind, J. A. A. W. Elemans and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 2696–2699 CrossRef CAS; (c) C. Arroniz, G. Chaubet and E. Anderson, ACS Catal., 2018, 8, 8290–8295 CrossRef CAS; (d) Q. He, X. Li, L. Chen, L. Zhang, W. Wang and C. Y. Su, ACS Catal., 2013, 3, 1–9 CrossRef CAS; (e) V. Welborn, W. Li and T. Head-Gordon, Nat. Commun., 2020, 11, 415 CrossRef CAS; (f) T. A. Bender, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 11806–11810 CrossRef CAS.
- For catalysis in M12L24 spheres, see: (a) Q. Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS; (b) R. Gramage-Doria, J. Hessels, S. H. A. M. Leenders, O. Tröppner, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2014, 53, 13380–13384 CrossRef CAS; (c) F. Yu, D. Poole III, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2018, 57, 11247–11251 CrossRef CAS; (d) S. Gonell, X. Caumes, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Chem. Sci., 2019, 10, 1316–1321 RSC; (e) R. Zaffaroni, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2020, 59, 18485–18489 CrossRef CAS.
- For examples of electrocatalysis within coordination assemblies, see: (a) R. Zaffaroni, E. O. Bobylev, R. Plessius, J. I. van der Vlugt and J. N. H. Reek, J. Am. Chem. Soc., 2020, 142, 8837–8847 CrossRef; (b) C. J. Bruns, D. Fujita, M. Hoshino, S. Sato, J. F. Stoddart and M. Fujita, J. Am. Chem. Soc., 2014, 136, 12027–12034 CrossRef CAS.
- For examples of electro-optical applications of coordination assemblies, see: (a) X. Yan, P. Wei, Y. Liu, M. Wang, C. Chen, J. Zhao, G. Li, M. L. Saha, Z. Zhou, Z. An, X. Li and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 9673–9679 CrossRef CAS; (b) J. Y. Ryu, J. M. Lee, N. Van Nghia, K. M. Lee, S. Lee, M. H. Lee, P. J. Stang and J. Lee, Inorg. Chem., 2018, 57, 11696–11703 CrossRef CAS; (c) D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS.
- N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354–1379 RSC.
- J. Bunzen, J. Iwasa, P. Bonakdarzadeh, E. Numata, K. Rissanen, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 3161–3163 CrossRef CAS.
- For examples of shape-complimentary M2L4 cages, see: (a) W. Bloch, Y. Abe, J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS; (b) S. Saha, B. Holzapfel, Y. T. Chen, K. Terlinden, P. Lill, C. Gatsogiannis, H. Rehag and G. H. Clever, J. Am. Chem. Soc., 2018, 140(50), 17384–17388 CrossRef CAS; (c) R. Zhu, W. Bloch, J. Holstein, S. Mandal, L. V. Schafer and G. H. Clever, Chem.–Eur. J., 2018, 24(49), 12976–12982 CrossRef CAS; (d) R. J. Li, J. Holstein, W. Hiller, J. Andreasson and G. H. Clever, J. Am. Chem. Soc., 2019, 141(5), 2097–2103 CrossRef CAS; (e) B. Chen, J. J. Holstein, S. Horiuchi, W. G. Hiller and G. H. Clever, J. Am. Chem. Soc., 2019, 141, 8907–8913 CrossRef CAS.
- L. R. MacGillivray and J. L. Atwood, Angew. Chem., Int. Ed., 1999, 38, 1018–1033 CrossRef CAS.
- J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–820 CrossRef CAS.
- For relevant studies classical mechanics dynamics of PdxL2x coordination assemblies using non-bonded metal centers, see: (a) M. Yoneya, T. Yamaguchi, S. Sato and M. Fujita, J. Am. Chem. Soc., 2012, 134, 14401–14407 CrossRef CAS; (b) M. Yoneya, S. Tsuzuki, T. Yamaguchi, S. Sato and M. Fujita, ACS Nano, 2014, 8, 1290–1296 CrossRef CAS; (c) Y. Tachi, S. Sato, M. Yoneya, M. Fujita and Y. Okamoto, Chem. Phys. Lett., 2019, 714, 185–189 CrossRef CAS.
- For examples of classical mechanics, semi-empirical and DFT studies to study coordination assembly host-guest chemistry, see: (a) T. A. Young, R. Gheorghe and F. Duarte, J. Chem. Inf. Model., 2020, 60(7), 3546–3557 CrossRef CAS; (b) V. Marti-Centelles, F. Duarte and P. J. Lusby, Isr. J. Chem., 2019, 59, 257–266 CrossRef CAS; (c) R. L. Spicer, A. D. Stergiou, T. A. Young, F. Duarte, M. D. Symes and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 2134–2139 CrossRef CAS; (d) T. A. Young, V. Marti-Centelles, J. Wang, P. J. Lusby and F. Duarte, J. Am. Chem. Soc., 2020, 142(3), 1300–1310 CrossRef.
- In regards to modern methodological development in structural discovery and improved mechanics forcefields, refer to: (a) P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC; (b) S. Spicher and S. Grimme, Angew. Chem., Int. Ed., 2020, 59(36), 15665–15673 CrossRef CAS; (c) See also ref. 25.
- Q. F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- A. W. Götz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- P. Li, B. P. Roberts, D. K. Chakravorty and K. M. Merz, J. Chem. Theory Comput., 2013, 9, 2733–2748 CrossRef CAS.
- R. M. Betz and R. C. Walker, J. Comput. Chem., 2015, 36, 79–87 CrossRef CAS.
- A. D. McLachlan, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1982, 38, 871–873 CrossRef.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. Fujita, Y. Ueda, S. Sato, H. Yokoyama, N. Mizuno, T. Kumasaka and M. Fujita, Chem, 2016, 1, 91–101 CAS.
- S. Kai, T. Shigeta, T. Kojima and S. Hiraoka, Chem.–Asian J., 2017, 12, 3203–3207 CrossRef CAS.
- M. Tominaga, K. Suzuki, M. Kawano, T. Kusukawa, T. Ozeki, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5621–5625 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03992f |
| This journal is © The Royal Society of Chemistry 2020 |