
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to P-chiral sec- and tert-phosphine oxides enabled by Le-Phos-catalyzed asymmetric kinetic resolution†
Haile
Qiu
a,
Qiang
Dai
a,
Jiafeng
He
a,
Wenbo
Li
a and
Junliang
Zhang
 *ab
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai, P. R. China. E-mail: jlzhang@chem.ecnu.edu.cn
bDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai 200438, P. R. China. E-mail: junliangzhang@fudan.edu.cn
First published on 2nd September 2020
Abstract
The synthesis of P-stereogenic building blocks is extremely difficult. Herein we report an efficient kinetic resolution of secondary phosphine oxides via a Le-Phos-catalyzed asymmetric allylation reaction with Morita–Baylis–Hillman carbonates. This method provides facile access to enantioenriched secondary and tertiary P-chiral phosphine oxides with broad substrate scope, both of which could serve as P-stereogenic synthons, and can be rapidly incorporated into a given scaffold bearing a P-stereocenter. The highly desirable late stage modifications demonstrate the practicability of our method and can be a critical contribution to obtaining optimal P-chiral catalysts and ligands.
Introduction
P-Stereogenic phosphines represent a class of highly efficient ligands or catalysts as their stereocenters and active centers converge at one point and have been used in countless catalytic processes.1 However, the less availability and synthetic challenges are the hurdles for exploration of these privileged compounds and thus restrict their further applications.2 In this context, during the past two decades, catalytic asymmetric synthesis of P-stereogenic phosphines, as a more efficient alternative, has been an intensive field of research.3–5 The direct cross-coupling of secondary phosphines3 or secondary phosphine oxides (SPOs)4 with a variety of specific partners provided a more straightforward approach to access P-chiral compounds with diverse functional groups (Scheme 1a). The toxicity and liability of the former, however, largely deterred them from being used in many laboratories.6 As a promising alternative, SPOs were bench stable, less toxic, and odorless. Apart from the widespread use of their racemates in asymmetric catalysis to construct P-stereogenic compounds, their chiral version also served as a key intermediate synthon that could be rapidly incorporated into a given ligand scaffold (Scheme 1b).7 | ||
| Scheme 1 Application of the SPOs. (a) Catalytic asymmetric synthesis of P-chiral TPOs. (b) Construction and application of P-chiral SPOs. (c) Kinetic resolution of H-phosphinates. (d) This work: access to P-Chiral SPOs and TPOs. | ||
On the other hand, specific asymmetric catalytic systems usually achieved single transformation and were hardly used as general systems to construct multiple types of P-chiral compounds. Moreover, they often required an excess amount of SPOs to secure better enantioselectivities and yields due to the difficult interconversion of both enantiomers in the reaction system.4a–c Although some studies have attempted to use this feature to achieve a standard kinetic resolution (KR) of SPOs in order to obtain P-chiral tertiary phosphine oxides (TPOs) as well as enantioenriched SPOs, the elusive degree of racemization of SPOs made the process very challenging.4b–d In 2009, the Tan group achieved the kinetic separation of H-phosphinates in the phospha-Mannich reaction, obtaining enantiomerically pure H-phosphinates in 87% ee as only one example (Scheme 1c).8 To date, P-chiral SPOs were typically prepared using enantiopure starting materials, and chiral auxiliaries, or by recrystallization from the racemates.9 Very little attention has been paid to the acquisition of P-chiral SPOs by kinetic resolution, which still remains in its infancy. Herein we report a highly efficient kinetic resolution of SPOs via a Le-Phos-catalyzed asymmetric allylation reaction with Morita–Baylis–Hillman (MBH) carbonates (Scheme 1d). The developed process holds some promise as a potentially straightforward route to enantioenriched P-chiral SPOs as well as TPOs, both of which could serve as P-stereogenic synthons, and would undoubtedly spur on more detailed studies of their chemistry and synthetic utility.
Results and discussion
Our initial investigation was carried out by using Boc-protected MBH adduct 2a (0.5 equiv.) and racemic mesityl(phenyl)phosphine oxide 1a in the presence of multifunctional chiral phosphine catalyst P1 (Wei-Phos, 10 mol%) in toluene (Table 1, entry 1).10 The reaction gave the desired allylation product 3aa in 34% yield with 7% ee, along with recovered 1a in 30% yield with 17% ee, correlating with an s-factor of 1.3. Then, Xiao-Phos P2 was tested but a little better result was obtained (Table 1, entry 2).11 Gratifyingly, the use of (RP,S,S,RS)-Le-Phos P3 and (SP,R,S,RS)-Le-Phos P4 as the catalysts, especially the latter one increased the ees of both 3aa and recovered (R)-1a (Table 1, entries 3 and 4). This result indicated that the configuration of the catalyst backbone has a remarkable impact on the enantioselectivities of the products.|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Recovery of 1a | 3 | s factor |
| a All reactions were performed using 1a (0.2 mmol), 2 (0.1 mmol) and an organocatalyst (10 mol%) in 1.5 mL of solvent at r.t. All yields were determined by 1H NMR analysis of the crude mixture and CH2Br2 as the internal standard. Enantiomeric excesses were determined by HPLC. C (calculated conversion) = eeSM/(eeSM + eePR). s (selectivity) = ln[(1 − C)(1 − eeSM)]/In[(1 − C) (1 + eeSM)]. b 2b instead of 2a. c PhCO2H (50 mol%) used as an additive. d PhOH (20 mol%) used as an additive. e Mesitylene instead of toluene. f tBuPh instead of toluene. g At 10 °C. h At 0 °C. i rac-1a (0.205 mol) was used. | ||||
| 1 | P1 | 30%, 17% ee | 34%, 7% ee | 1.3 |
| 2 | P2 | 38%, −26% ee | 36%, −21% ee | 1.9 |
| 3 | P3 | 42%, 10% ee | 44%,10% ee | 1.3 |
| 4 | P4 | 30%, 86% ee | 32%, 48% ee | 7.4 |
| 5 | P5 | 40%, 78% ee | 42%, 63% ee | 10.4 |
| 6 | P6 | 41%, 64% ee | 43%, 55% ee | 6.5 |
| 7 | P7 | 73%, −6% ee | 18%, −35% ee | 2.2 |
| 8b | P5 | 40%, 82% ee | 42%, 81% ee | 24.1 |
| 9b,c | P5 | 42%, 79% ee | 40%, 79% ee | 20.3 |
| 10b,d | P5 | 40%, 75% ee | 40%, 78% ee | 18.2 |
| 11b,e | P5 | 35%, 88% ee | 38%, 78% ee | 23.4 |
| 12b,f | P5 | 36%, 89% ee | 44%, 75% ee | 20.5 |
| 13b,e,g | P5 | — | Trace | — |
| 14b,f,g | P5 | 37%, 93% ee | 44%, 86% ee | 44.4 |
| 15b,f,h,i | P5 | 45%, 89% ee | 43%, 90% ee | 57 |
| 16b,f,h,i | P8 | 65%, 17% ee | 20%, 66% ee | 5.7 |

Encouraged by this finding, several modified chiral phosphine catalysts (SP,R,S,RS)-Le-Phos P5–P7 with the variation of the substituent R at the center of the chiral sulfinamide were then tested.12 The enantioselectivity of 3aa could be improved to 63% along with increased yields of 3aa and recovered (R)-1a when the phosphine catalyst was switched to (SP,R,S,RS)-Le-Phos P5 (R = cyclopentyl) (Table 1, entry 5). However, isopropyl- and phenyl-substituted P6 and P7 resulted in lower enantioselectivities (Table 1, entries 6 and 7). More importantly, when the benzoyl protected MBH adduct 2b instead of 2a was used under the catalysis of P5 in toluene at room temperature, the product 3ab was obtained in 42% yield with 81% ee and (R)-1a was recovered in 40% yield with 82% ee, further improving the s-factor to 24.1 (Table 1, entry 8). The additives such as PhCO2H and PhOH have no obvious effect on both yield and enantioselectivity (Table 1, entries 9 and 10). Further solvent screening revealed that the kinetic resolution of secondary phosphine oxide rac-1a afforded the allylation product 3ab in better yields and recovered (R)-1a in higher ee by using tert-butylbenzene as solvent in a shorter reaction time (Table 1, entry 12). Lowering the reaction temperature to 10 °C improved the ees of 3ab and (R)-1a to 86% and 93% for 8 h, respectively (Table 1, entry 14). It is noteworthy that only a trace amount of the corresponding product was detected with the use of mesitylene as the solvent at 10 °C (Table 1, entry 13). Fortunately, the enantioselectivity of the desired product 3ab could be further improved to 90% by lowering the reaction temperature to 0 °C and increasing the equivalent of rac-1a to 2.05 (Table 1, entry 15). Lowering the temperature also largely improved the s-value to 57. We also tried out the classical chiral phosphine catalyst (R,R)-Me-DuPhos (P8), but unfortunately the ee value and s-value were lower compared with those of P5 (Table 1, entry 16).
Under the optimized reaction conditions, the scope of the kinetic resolution reaction of SPOs was examined by performing asymmetric allylic alkylation in the presence of (SP,R,S,RS)-Le-Phos P5 (Scheme 2). The steric hindrance of the mesityl group of SPOs has a significant impact on the reaction reactivity and enantioselectivity.13 Substrates SPOs with iPr or Me at the meta-position of another aromatic ring afforded the corresponding tertiary phosphine oxides 3bb and 3cb in 39–40% yields with 87–91% ees, and secondary phosphine oxides 1b and 1c were recovered in 38–39% yields with 93–98% ees. A series of SPOs with a para-substituted aryl group were well tolerated in this reaction, producing the corresponding allylation products 3db–3gb in good yields (30–40%) with high enantioselectivities (89–93% ees). The chiral diarylphosphine oxides (R)-1d–1g could be recovered in 32–43% yields with 84–90% ees, and s factors were in the range of 51–76. Multi-substituted aromatic groups such as 3,5-di-tert-butylphenyl, 3,5-dimethylphenyl and 3,4,5-trimethoxylphenyl were also compatible, and both the alkylation products and the recovered SPOs were formed in good to excellent enantioselectivities (s factors = 92–167).14 Fortunately, by the variation of the mesityl group to the 2,5-dimethylphenyl group, the allylic alkylation kinetic resolution reactions gave comparable yields and enantioselectivities ((R)-1k and (R)-1l: 37–38% yields, 85–98% ees; 3kb and 3lb: 38–39% yields, 90–91% ees; s factors = 53–76). The absolute configuration of recovered SPO 1l was unambiguously confirmed as R by X-ray crystallographic analysis.15
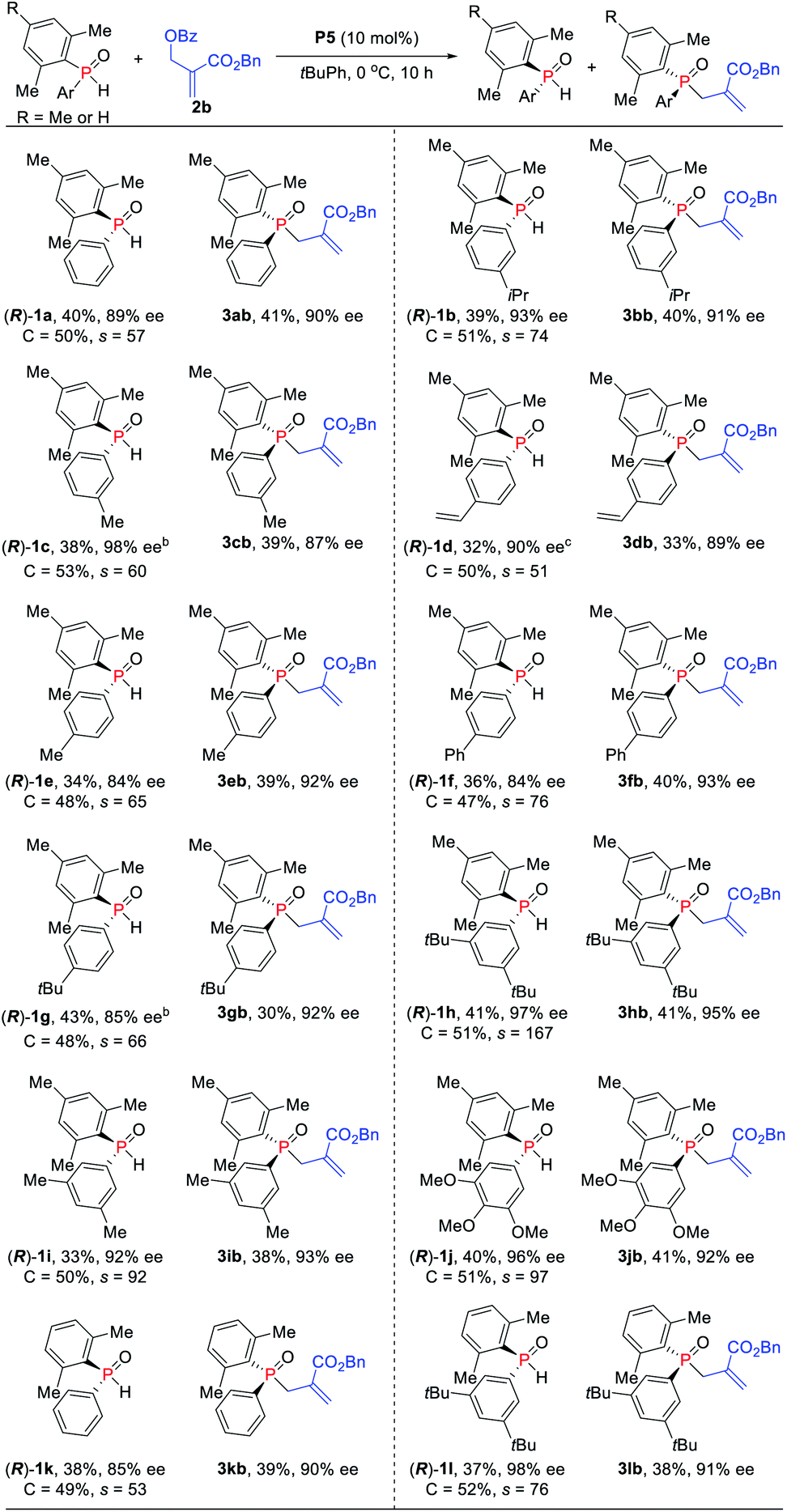 | ||
| Scheme 2 Asymmetric allylic alkylation of SPOs and MBH carbonates. arac-SPO (0.205 mmol), 2b (0.1 mmol), and P5 (10 mol%) in 1.5 mL of tBuPh for 10 h at 0 °C. Isolated yields. brac-SPO (0.21 mol) was used. crac-SPO (0.22 mol) was used. | ||
To further increase the enantioselectivity of SPOs, the reaction conditions were modified by reducing the amount of SPOs (Scheme 3). Indeed, the kinetic resolution reaction of SPOs 1g and 1k provided higher ees with relatively lower s factors (27–33). Moreover, electron-withdrawing and electron-donating aryl groups on the phosphine atom were all tolerated and up to 99% ee was obtained (s factors = 17–64). The naphthyl-substituted phosphine oxide 1p was also kinetically well resolved in 30% yield with 90% ee. The heteroaryl-substituted substrate was compatible with the standard reaction conditions, producing P-stereogenic diaryl phosphine oxide in 35% yield and 96% ee, and achieving the corresponding TPO 3rb in 33% yield and 89% ee (s factors = 67).
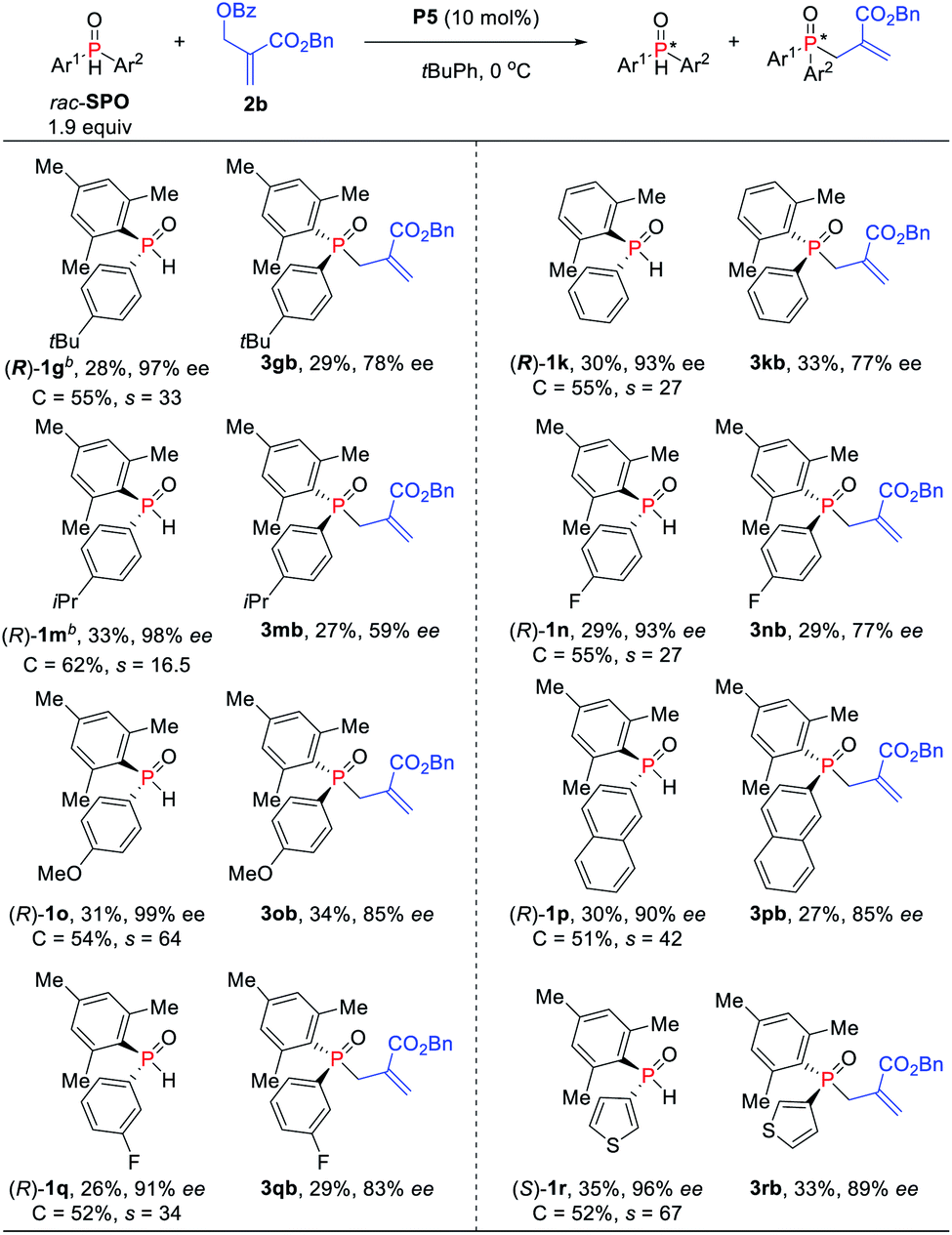 | ||
| Scheme 3 Asymmetric allylic alkylation of SPOs and MBH carbonates. aIsolated yields based on the amount of SPOs. brac-SPO (0.20 mol) was used. | ||
Alternatively, in order to obtain tertiary phosphine oxides with good ee, the asymmetric kinetic resolution of SPOs was further investigated with slightly adjusting the ratio of SPOs to MBH carbonates (2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Scheme 4). Substrates bearing an ortho-, meta- or para-substituted aryl on the phosphorus center could be accommodated for the reaction, furnishing the corresponding TPOs in 63–83% yields with 86–94% ees (s-factors = 23–38). Electron-withdrawing and -donating substituents on the aromatic ring of SPOs have little influence on the reaction yield and enantioselectivity. Naphthyl and thienyl on the SPOs were also compatible with the allylic alkylation process, and good yields with high enantioselectivities were obtained for 3pb (71%, 91% ee), 3ub (70%, 90% ee) and 3rb (79%, 90% ee), respectively. More hindered SPO was found to be less efficient compared to the mesityl-substituted SPO under these conditions (3obvs.3wb).
:1) (Scheme 4). Substrates bearing an ortho-, meta- or para-substituted aryl on the phosphorus center could be accommodated for the reaction, furnishing the corresponding TPOs in 63–83% yields with 86–94% ees (s-factors = 23–38). Electron-withdrawing and -donating substituents on the aromatic ring of SPOs have little influence on the reaction yield and enantioselectivity. Naphthyl and thienyl on the SPOs were also compatible with the allylic alkylation process, and good yields with high enantioselectivities were obtained for 3pb (71%, 91% ee), 3ub (70%, 90% ee) and 3rb (79%, 90% ee), respectively. More hindered SPO was found to be less efficient compared to the mesityl-substituted SPO under these conditions (3obvs.3wb).
 | ||
| Scheme 4 Phosphine-catalyzed asymmetric allylic alkylation of secondary phosphine oxides. aIsolated yields of 3 based on the amount of 2b. | ||
The reactions of more challenging alkyl-substituted phosphine oxides and MBH carbonate 2c were carried out with 20 mol% catalyst P5, which proceeded well to afford the desired TPOs 3xc–3yc in moderate yields with 82–87% ees (Scheme 5). To probe the efficiency of the currently studied kinetic resolution strategy in the synthesis of chiral SPOs and TPOs, gram scale reactions were investigated. To our delight, the desired products TPOs (3ab and 3lb) and recovered SPOs ((R)-1a and (R)-1l) were obtained without any loss of enantioselectivity even with only 5 mol% catalyst P5.
 | ||
| Scheme 5 Phosphine-catalyzed asymmetric allylic alkylation of secondary phosphine oxides. aIsolated yields based on the amount of 2c. bIsolated yields based on the amount of SPOs. | ||
Further investigation of the substrate scope was focused on MBH carbonate substrates 2 (Scheme 6). The Ac-protected MBH carbonates 2d afforded the target products 3ab in 35% yield with 88% ee, and unreacted 1a was recovered in 37% yield with 83% ee. The benzyl substituted MBH carbonates obtained the corresponding TPOs 3ae–3af in moderate to excellent yields (32–39%) with excellent enantioselectivities (89–92% ees). The chiral diarylphosphine oxides were also compatible. It is also worth mentioning that we achieved 3ae in 16% yield with 24% ee via N-methylated chiral phosphine catalyst N-Me-P5, thus indicating that the hydrogen-bonding interaction might indeed play an important role in the catalytic activity and enantioselectivity control.
 | ||
| Scheme 6 Substrate scope of MBH carbonate substrates. | ||
Given their potential of synthetic versatility, various transformations of chiral SPOs were conducted. The chiral SPO 1l with an R configuration at the phosphorus atom underwent an allylic alkylation reaction with MBH carbonate 2b to provide the enantiomer of TPO 3lb, that is, ent-3lb in 86% yield and 96% ee (Scheme 7A). Further cross-coupling of (R)-1l with 2-iodothiophene under the catalysis of palladium and xantphos afforded the enantioenriched triaryl P-stereogenic oxide 4 in 84% yield with 96% ee.16 The direct alkylation of (R)-1l with benzyl chloride in the presence of KOH, furnished the novel TPO 6 in 82% yield with slight erosion of ee to 87% ee.17 When (R)-1 was used for this transformation, the corresponding products were obtained without loss ofenantioselectivity.18 Then the demethylation of TPO 6 in the presence of BBr3 furnished the catalyst 7, which has potential application in the asymmetric catalytic Mitsunobu reaction.19 We also utilized (R)-1a to synthesize chiral pincer-type ligand 8 in 47% yield with 81% ee. Moreover, we demonstrated the utility of these enantioenriched TPOs through further transformations of the P-chiral scaffold. Cyclopropanantion of this enantiopure TPO 3ab was carried out and the cyclopropane product 9 was converted into P-stereogenic phosphine 10 without loss of chirality (Scheme 7B).16,20
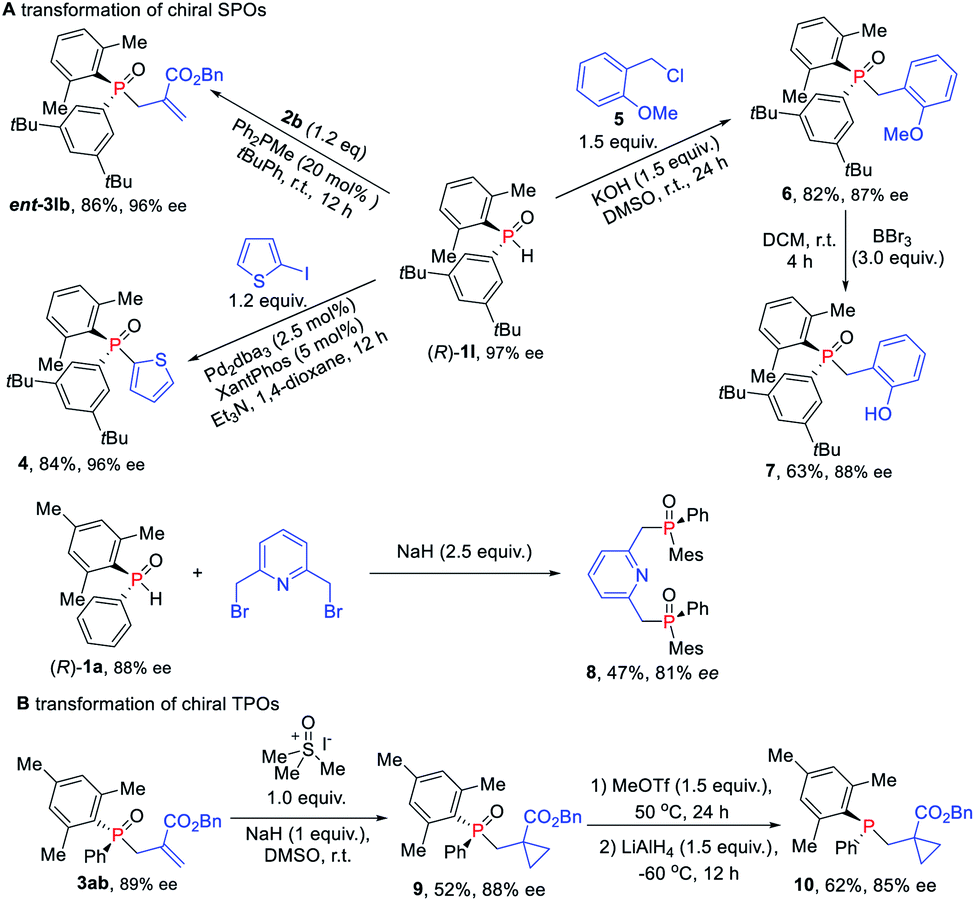 | ||
| Scheme 7 (A) Transformations of chiral SPOs. (B) Transformations of TPOs. | ||
Conclusions
In summary, we have developed a novel type of bifunctional chiral sulfinamide cyclic phosphine catalyst Le-Phos, which can be easily prepared in gram scale from inexpensive commercially available starting materials in short steps. (SP,R,S,RS)-Le-Phos has shown excellent performance in the enantioselective γ-addition reactions of various N-centered nucleophiles to γ-substituted allenoates, acquiring a series of γ-addition adducts in high yields with up to 98% ees and excellent regioselectivity and diastereoselectivity under mild conditions. Its prominent characteristics are general substrate scope, mild reaction conditions, good yields, high enantioselectivities, ease of scale-up to gram scale, and further synthetic transformations of the products. Further explorations of Le-Phos as an organocatalyst and chiral ligand of transition metals in asymmetric catalysis are currently underway in our group and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the funding support of the National Natural Science Foundation of China (21672067, 21702063 and 21921003) and the Program of Eastern Scholar at Shanghai Institution of Higher Learning.References
- (a) A. Grabulosa, P-Stereogenic Ligands in Asymmetric Catalysis, RSC Publishing, Cambridge, 2011 Search PubMed; (b) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS; (c) I. O. Kolodiazhnyi, Top. Curr. Chem., 2014, 360, 161 CrossRef; (d) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771 RSC; (e) T. Imamoto, Yuki Gosei Kagaku Kyokaishi, 2007, 65, 1060 CrossRef CAS; (f) M. Benaglia and S. Rossi, Org. Biomol. Chem., 2010, 8, 3824 RSC; (g) M. Dutartre, J. Bayardon and S. Juge, Chem. Soc. Rev., 2016, 45, 5771 RSC; (h) T. Imamoto, Chem. Rec., 2016, 16, 2659 CrossRef; (i) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049 CrossRef CAS; (j) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344 CrossRef CAS; (k) T. Ayad, A. Gernet, J.-L. Pirat and D. Virieux, Tetrahedron, 2019, 75, 4385 CrossRef CAS.
- (a) O. Korpiun, R. A. Lewis, J. Chickos and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4842 CrossRef CAS; (b) E. Bergin, C. T. O'Connor, S. B. Robinson, E. M. McGarrigle, C. P. O'Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566 CrossRef CAS; (c) D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728 CrossRef CAS; (d) O. Berger and J.-L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377 CrossRef CAS; (e) K. Nikitin, K. V. Rajendran, H. Müller-Bunz and D. G. Gilheany, Angew. Chem., Int. Ed., 2014, 53, 1906 CrossRef CAS; (f) S. Jugé and J. P. Genet, Tetrahedron Lett., 1989, 30, 2783 CrossRef; (g) T. León, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2011, 133, 5740 CrossRef; (h) Z. S. Han, L. Zhang, Y. Xu, J. D. Sieber, M. A. Marsini, Z. Li, J. T. Reeves, K. R. Fandrick, N. D. Patel, J.-N. Desrosiers, B. Qu, A. Chen, D. M. Rudzinski, L. P. Samankumara, S. Ma, N. Grinberg, F. Roschangar, N. K. Yee, G. Wang, J. J. Song and C. H. Senanayake, Angew. Chem., Int. Ed., 2015, 54, 5474 CrossRef CAS.
- (a) J. R. Moncarz, N. F. Laritcheva and D. S. Glueck, J. Am. Chem. Soc., 2002, 124, 13356 CrossRef CAS; (b) V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786 CrossRef CAS; (c) D. S. Glueck, Synlett, 2007, 2627 CrossRef CAS; (d) V. S. Chan, M. Chiu, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021 CrossRef CAS; (e) Y. Huang, Y. Li, P.-H. Leung and T. Hayashi, J. Am. Chem. Soc., 2014, 136, 4865 CrossRef CAS; (f) C. Li, Q.-L. Bian, S. Xu and W.-L. Duan, Org. Chem. Front., 2014, 1, 541 RSC; (g) Y.-C. Song, G.-F. Dai, F. Xiao and W.-L. Duan, Tetrahedron Lett., 2016, 57, 2990 CrossRef CAS; (h) B. J. Anderson, S. C. Reynolds, M. A. Guino-o, Z. Xu and D. S. Glueck, ACS Catal., 2016, 6, 8106 CrossRef CAS.
- (a) Y. Zhang, H. He, Q. Wang and Q. Cai, Tetrahedron Lett., 2016, 57, 5308 CrossRef CAS; (b) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183 CrossRef CAS; (c) Q. Dai, W. Li, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 20556 CrossRef CAS; (d) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, J. Am. Chem. Soc., 2019, 141, 16584 CrossRef CAS.
- For examples of desymmetrization of prochiral phosphorus molecules, see: (a) J. S. Harvey, S. J. Malcolmson, K. S. Dunne, S. J. Meek, A. L. Thompson, R. R. Schrock, A. H. Hoveyda and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 762 CrossRef CAS; (b) Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734 CrossRef CAS; (c) Z. J. Du, J. Guan, G. J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS; (d) Z. Huang, X. Huang, B. Li, C. Mou, S. Yang, B.-A. Song and Y. R. Chi, J. Am. Chem. Soc., 2016, 138, 7524 CrossRef CAS; (e) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364 CrossRef CAS; (f) K. M. Lim and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 8122 CrossRef CAS; (g) Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981 RSC; (h) B. M. Trost, S. M. Spohr, A. B. Rolka and C. A. Kalnmals, J. Am. Chem. Soc., 2019, 141, 14098 CrossRef CAS; (i) G.-H. Yang, Y. Li, X. Li and J.-P. Cheng, Chem. Sci., 2019, 10, 4322 RSC; (j) R.-Y. Zhu, L. Chen, X.-S. Hu, F. Zhou and J. Zhou, Chem. Sci., 2020, 11, 97 RSC. For selected examples of other strategies, see: (k) K. V. L. Crepy and T. Imamoto, Top. Curr. Chem., 2003, 229, 1 CrossRef CAS; (l) J. J. Gammon, V. H. Gessner, G. R. Barker, J. Granander, A. C. Whitwood, C. Strohmann, P. O'Brien and B. Kelly, J. Am. Chem. Soc., 2010, 132, 13922 CrossRef CAS; (m) K. V. Rajendran, L. Kennedy and D. G. Gilheany, Eur. J. Org. Chem., 2010, 2010, 5642 CrossRef; (n) W. Tang, B. Qu, A. G. Capacci, S. Rodriguez, X. Wei, N. Haddad, B. Narayanan, S. Ma, N. Grinberg, N. K. Yee, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2010, 12, 176 CrossRef CAS; (o) T. Imamoto, K. Tamura, T. Ogura, Y. Ikematsu, D. Mayama and M. Sugiya, Tetrahedron: Asymmetry, 2010, 21, 1522 CrossRef CAS.
- Phosphine–boranes were usually used as an alternative: B. Join, D. Mimeau, O. Delacroix and A. C. Gaumont, Chem. Commun., 2006, 3249 RSC.
- (a) M. Stankeviča, G. Andrijewskib and K. M. Pietrusiewicz, Synlett, 2004, 2, 311 Search PubMed; (b) D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728 CrossRef CAS.
- X. Fu, W.-T. Loh, Y. Zhang, T. Chen, T. Ma, H. Liu, J. Wang and C.-H. Tan, Angew. Chem., Int. Ed., 2009, 48, 7387 CrossRef CAS.
- (a) W.-M. Dai, K. K. Y. Yeung, W. H. Leung and R. K. Haynes, Tetrahedron: Asymmetry, 2003, 14, 2821 CrossRef CAS; (b) J. Drabowicz, P. Łyzwa, J. Omelanczuk, K. M. Pietrusiewicz and M. Mikołajczyk, Tetrahedron: Asymmetry, 1999, 10, 2757 CrossRef CAS; (c) A. Leyris, J. Bigeault, D. Nuel, L. Giordano and G. Buono, Tetrahedron Lett., 2007, 48, 5247 CrossRef CAS.
- W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853 CrossRef CAS.
- X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6874 CrossRef CAS.
- H. Qiu, X. Chen and J. Zhang, Chem. Sci., 2019, 10, 10510 RSC.
- Experimental comparison of several SPOs: see the ESI (Table S3) for details.†.
- (a) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS; (b) M. D. Greenhalgh, J. E. Taylor and A. D. Smith, Tetrahedron, 2018, 74, 5554 CrossRef CAS; (c) L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 16260 CrossRef CAS.
- CCDC: 1985770 ((R)-1l) contains the supplementary crystallographic data for this paper.†.
- A. J. Bloomfield and S. B. Herzon, Org. Lett., 2012, 14, 4370 CrossRef CAS.
- S.-Z. Nie, Z.-Y. Zhou, J.-P. Wang, H. Yan, J.-H. Wen, J.-J. Ye, Y.-Y. Cui and C.-Q. Zhao, J. Org. Chem., 2017, 82, 9425 CrossRef CAS.
- Base-promoted alkylation of (R)-1: see ESI 5.7 for details.†.
- H. R. Beddoe, G. K. Andrews, V. Magné, D. J. Cuthbertson, J. Saska, L. A. Shannon-Little, E. S. Shanahan, F. H. Sneddon and M. R. Denton, Science, 2019, 365, 910 CrossRef.
- (a) T. N. Tzvetkov, T. Arndtb and J. Mattay, Tetrahedron, 2007, 63, 10497 CrossRef; (b) K. V. Rajendran and D. G. Gilheany, Chem. Commun., 2012, 48, 817 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1985770 ((R)-1l). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04041j |
| This journal is © The Royal Society of Chemistry 2020 |