
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct, stereoselective thioglycosylation enabled by an organophotoredox radical strategy†
Peng
Ji
,
Yueteng
Zhang
,
Feng
Gao
,
Fangchao
Bi
and
Wei
Wang
 *
*
Departments of Pharmacology and Toxicology and Chemistry and Biochemistry, BIO5 Institute, and University of Arizona Cancer Centre, University of Arizona, Tucson, AZ 85721, USA. E-mail: wwang@pharmacy.arizona.edu
First published on 19th October 2020
Abstract
While strategies involving a 2e− transfer pathway have dictated glycosylation development, the direct glycosylation of readily accessible glycosyl donors as radical precursors is particularly appealing because of high radical anomeric selectivity and atom- and step-economy. However, the development of the radical process has been challenging owing to notorious competing reduction, elimination and/or SN side reactions of commonly used, labile glycosyl donors. Here we introduce an organophotocatalytic strategy through which glycosyl bromides can be efficiently converted into corresponding anomeric radicals by photoredox mediated HAT catalysis without a transition metal or a directing group and achieve highly anomeric selectivity. The power of this platform has been demonstrated by the mild reaction conditions enabling the synthesis of challenging α-1,2-cis-thioglycosides, the tolerance of various functional groups and the broad substrate scope for both common pentoses and hexoses. Furthermore, this general approach is compatible with both sp2 and sp3 sulfur electrophiles and late-stage glycodiversification for a total of 50 substrates probed.
Introduction
Despite the fact that O-linked glycosides are a dominant form in biologically important glycoconjugates,1 the replacement of “O” by C-, N- and S-linked glycosides offers the merits of improved hydrolytic stability and/or bioactivity while maintaining similar conformational preferences. 2 In particular, thioglycosides have emerged as a privileged class of structures owing to their broad spectrum of biological activities (see representative examples in Scheme 1).2–5 Moreover, they are widely used as glycosyl donors in glycosylation reactions.6 The broad biological and synthetic utility has triggered significant interest in the development of efficient methods to construct a C–S bond with a defined anomeric configuration, which plays key roles in biological activities.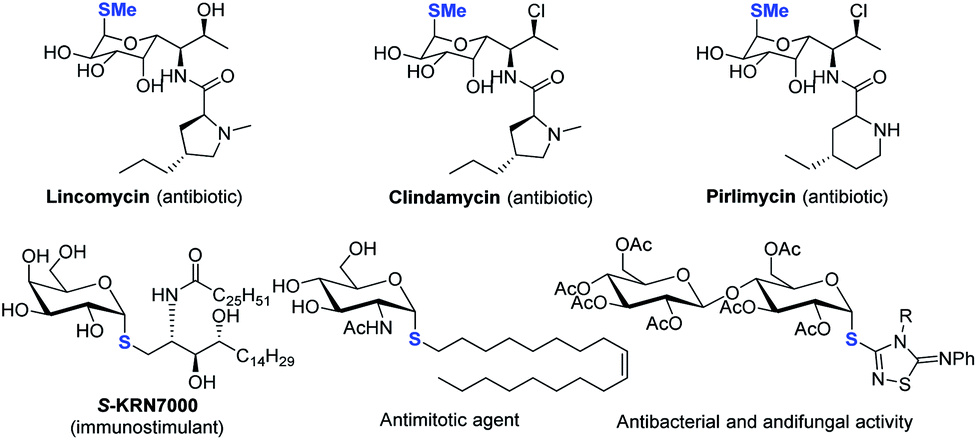 | ||
| Scheme 1 Selected examples of thioglycosides with α-1,2-cis-configuration. | ||
Strategies involving an ionic 2e− transfer pathway have dictated the C–S bond formation development.7–13 Direct replacement by a thiol with a glycosyl donor is an attractive approach in that both starting materials are readily accessible, but gives a mixture of α/β anomers in most cases (Scheme 2a).8 To overcome these limitations, the methods of reversing the polarity at the anomeric carbon have been developed (Scheme 2b).9 These elegant methods enable the stereoselective control formation of both α and β anomers but with limited scope of saccharides.9a Indirect methods using preformed anomeric thiols offer versatile approaches to thioglycosides (Scheme 2c).10–13 Nonetheless, the anomeric stereoselectivity of these processes depends on the nature of the anomeric thiols. In particular, few methods are capable of selectively constructing the challenging α-1,2-cis-thioglycosides,8b featured in a number of natural products and bioactive molecules (Scheme 1).
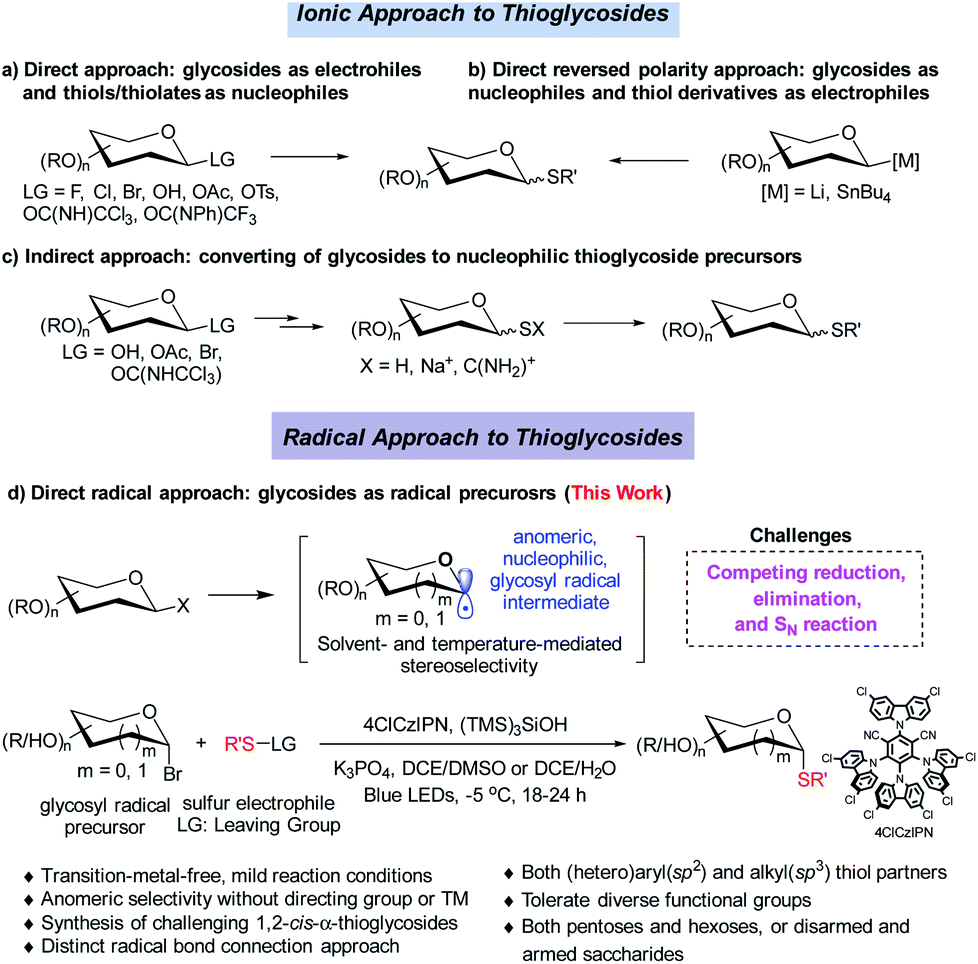 | ||
| Scheme 2 Ionic and radical thioglycosylation. | ||
Radical cross coupling offers a distinct paradigm for stereoselective construction of glycosidic bonds.14 Anomeric radicals have been elegantly explored for highly stereoselective C-glycosidic bond formation with a transition metal (TM).15–17 However, stereoselective C–S bond formation through the glycosyl radical has remained elusive (Scheme 2d).17 This is attributed to: (1) reduction of glycosyl radicals by HAT (hydrogen atom transfer) donors;18 (2) elimination reaction of labile glycosyl donors with a TM catalyst;19 (3) competing SN2 reaction with thiols, which could compromise the anomeric selectivity.2b,7 Therefore, stable radical precursors such as glycosyl stannanes are designed to minimize these issues.17 Given the fact that the glycosyl radical can favour the formation of anomeric C1 conformation, we deliberately push the limit by developing an organophotocatalytic approach without a directing group or a TM for stereoselective S-glycosylation. Herein, we wish to disclose the results of the investigation, which has led to a general organophotocatalyzed thiolation of glycosyl bromides with highly stereoselective control (Scheme 2d).
Results and discussion
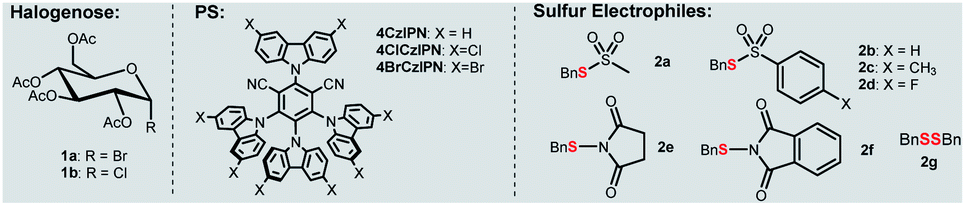
In our own efforts, recently we have developed visible-light-mediated glycosyl radical reactions for the synthesis of C-glycosides.15 In addition, we reported an organophotocatalytic thiolation of acyl radical method with thiosulfonates.20 These chemistries guided us to explore a new thioglycosylation reaction. The reaction of α-glucopyranosyl bromide 1a with thiosulfonate 2a and 4CzIPN21 as a photocatalyst (PS) was probed (Table 1 and Tables S1–S6†). First, we examined several commonly used reductants including iPr2NEt, Hantzsch ester, and ascorbic acid (Table S1,† entries 2, 6 and 7) for the generation of the glycosyl radical. Disappointedly, only the reduced product 4 was obtained. It should be pointed out that this is a general problem in using glycosyl halides as radical progenitors in glycosylation.18 Minimizing the issue requires a radical capable of effective dehalogenation whereas the hydrogenated product should be a weak H-donor. A silyl or a silyloxy radical can induce dehalogenation while the strong Si–H and Si–O–H make them more difficult to abstract.22 Therefore, various silanes were screened and (TMS)3SiOH was the best, giving 3a in 37% yield (Table S1,† entries 3–5 and 8–9). A survey of PSs revealed 4ClCzIPN21b,c as the optimal promoter (65% yield, Table S2† and 1, entries 2–4). The process was also sensitive to bases (entries 4–6 and Table S4†) and K3PO4 gave 3a in high yield. Among the thiosulfonates probed (entries 6–12), methanethiosulfonate (2a) was the best, possibly attributed to the lower hindrance and relatively high redox stability (Ered = −1.65 V vs. SCE, Fig. S3†). Glycosyl chloride (1b) did not undergo the dechlorination presumably due to its strong C–Cl bond (entry 13). To further improve the stereoselectivity (entry 6), we conducted reaction optimization including the solvent and reaction temperature (Table 1, entries 14–15 and Tables S3 and S6†). It was found that the biphasic solvent (DCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 2:1) could not only retain the high anomeric selectivity but also increase the yield (76%, entry 1), and a low temperature (−5 °C) is also required to maintain good yield and anomeric selectivity (entry 14, 15). The control experiments confirmed that base, light, (TMS)3SiOH, and PS were essential for this transformation (entries 16–17).
:H2O = 2:1) could not only retain the high anomeric selectivity but also increase the yield (76%, entry 1), and a low temperature (−5 °C) is also required to maintain good yield and anomeric selectivity (entry 14, 15). The control experiments confirmed that base, light, (TMS)3SiOH, and PS were essential for this transformation (entries 16–17).
|
|
|||
|---|---|---|---|
| Entry | Variation from the “standard conditions”a | Yieldb (3a, %) | α:βc |
|
a Standard conditions: unless specified, a mixture of glycosyl bromide (0.2 mmol), sulfur electrophile (0.1 mmol), 4ClCzIPN (0.005 mmol), K3PO4 (0.4 mmol), and (TMS)3SiOH (0.15 mmol) in DCE/DMSO (1 mL, 1:1, v/v) or DCE/H2O (1.5 mL, 2:1, v/v) was irradiated with 40 W Kessil blue LEDs in a N2 atmosphere at −5 °C for 24 h.
b Yield determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal reference.
c Ratio determined by crude 1H NMR.
d Isolated yield.
|
|||
| 1 | None | 76 (72)d | >20:1 |
| 2 | 4CzIPN (5 mol%), 2c, Na2CO3 (4.0 equiv.), DMSO, rt | 37 | <10:1 |
| 3 | 4BrCzIPN (5 mol%), 2c, Na2CO3 (4.0 equiv.), DMSO, rt | 33 | <10:1 |
| 4 | 4ClCzIPN (5 mol%), 2c, Na2CO3 (4.0 equiv.), DMSO, rt | 65 | <10:1 |
| 5 | Cs2CO3 instead of K3PO4, DCE:DMSO (1:1, v/v), rt |
Trace | — |
| 6 | DCE:DMSO (1:1, v/v), rt |
80 | <10:1 |
| 7 |
2b instead of 2a, DCE:DMSO (1:1, v/v), rt |
72 | <10:1 |
| 8 |
2d instead of 2a, DCE:DMSO (1:1, v/v), rt |
66 | <10:1 |
| 9 |
2d instead of 2a, DCE:DMSO (1:1, v/v), rt |
68 | <10:1 |
| 10 |
2e instead of 2a, DCE:DMSO (1:1, v/v), rt |
Trace | — |
| 11 |
2f instead of 2a, DCE:DMSO (1:1, v/v), rt |
66 | <10:1 |
| 12 |
2g instead of 2a, DCE:DMSO (1:1, v/v), rt |
Trace | — |
| 13 | 1b instead of 1a | Trace | — |
| 14 | DCE instead of DCE:H2O (2:1, v/v), rt |
60 | 17:1 |
| 15 | DCE instead of DCE:H2O (2:1, v/v), −5 °C |
67 | >20:1 |
| 16 | Without 4ClCzIPN, (TMS)3SiOH or K3PO4 | Trace | — |
| 17 | Under dark conditions | Trace | — |
|
|||
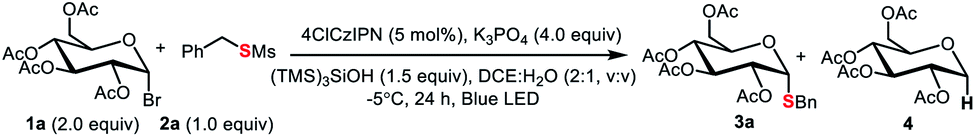
The generality of the new S-glycosylation was examined. We first evaluated the performance using glucosyl bromide (1a) as a radical donor for coupling with various thiosulfonates 2 (Scheme 3). The process serves as a general approach to both aryl and alkyl thioglycosides. Uniformly high axial selectivities are observed regardless of the nature of the sulfur electrophiles. With respect to aryls, electron-neutral (3b), -donating (3c–3d, 3h), and -withdrawing (3e–3f) groups on the phenyl ring and fused aromatic (3g) can be tolerated. Moreover, heteroaromatic thiosulfonates such as thiophenyl (3i) and furanyl (3j) enabled access to medicinally valued thioglycosides. The tetrazole derived disulfide instead of labile thiosulfonate could serve as an alternative and delivered the desired 3k. The reaction performed in DCE:H2O failed for pyridinyl thiosulfonate. Decent results (3l, 79%, α:β > 20:1) were obtained with DCE:DMSO (condition B). The protocol can also be applied in gram scale synthesis. Notably, less reactive sp3 alkyl glycosides 3o–3s could be synthesized with the protocol.17
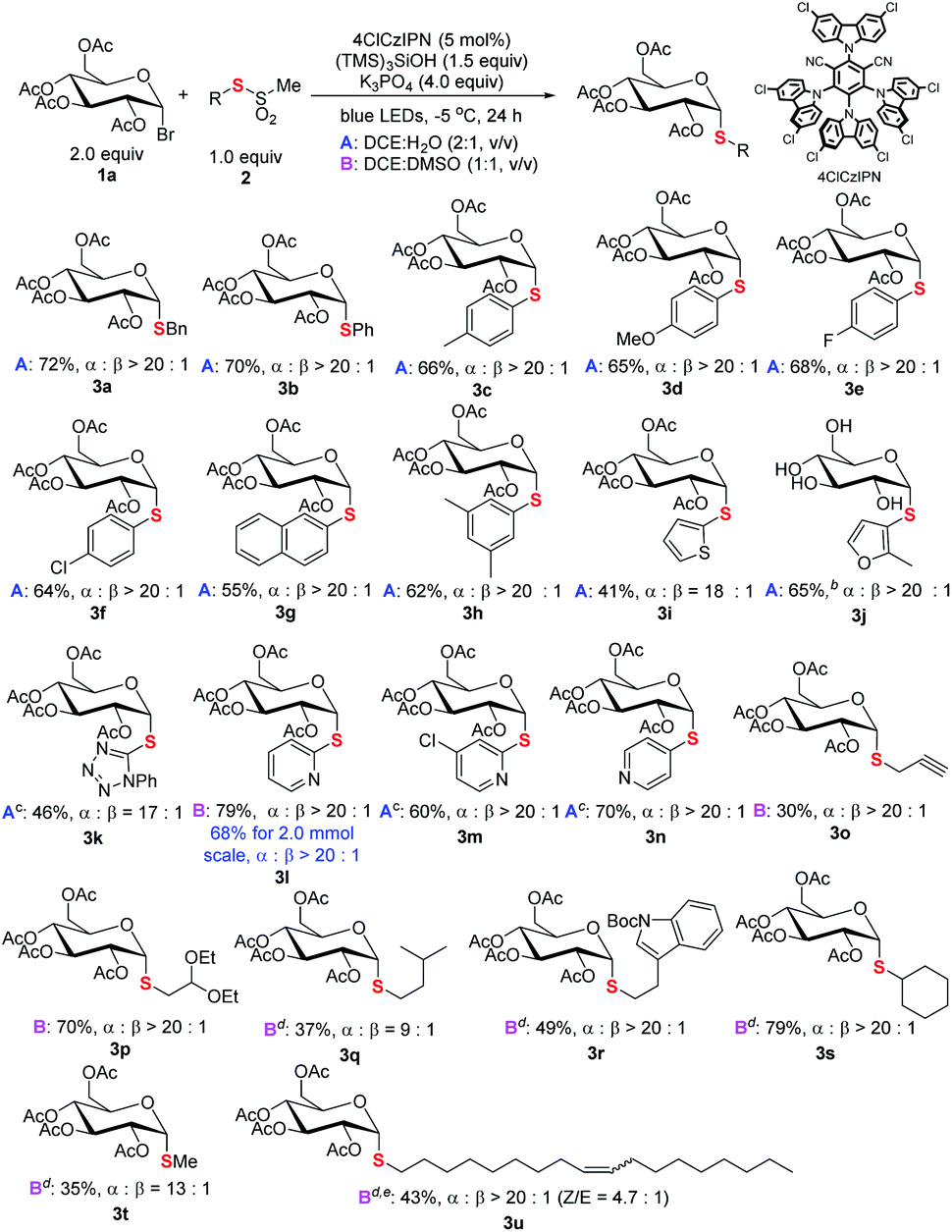 | ||
| Scheme 3 Scope of thiosulfonates. aReaction conditions: unless specified, see footnote a of Table 1 and the ESI;† isolated yield; the ratio of α and β anomers determined by crude 1H NMR. bYield after hydrolysis of the acyl group. cDisulfide used. dToluenethiosulfonate used. eZ/E ratio determined by 1H NMR. | ||
For even less electrophilic substrates, p-tolylthiosulfonates (3q–3u) displayed better performance than methylthiosulfonates. Particularly, a long alkyl chain with a Z-double bond product (3u), which exhibits intriguing antitumor activity (Scheme 1), is efficiently prepared with high diastereoselectivity. The limitation of the method is also realized. C2–N–Ac-saccharides such as D-glucosamine failed to react due to the lability of these reactants (see Fig. S5 in the ESI†).
The alternation of sugars was probed next (Scheme 4). Both common hexoses (glucose 3v–3x, galactose 3y, mannose 3z, fucose 3aa, rhamnopyranose 3ab, and glucuronic acid 3ac) and pentoses (3ad–3af) gave good yields and high stereoselectivity. Among the tested monosaccharides, except ribose (3af) adopting expected β selectivity owning to the steric effect, the others gave expected α-selectivity. Furthermore, disaccharides (3ag and 3ah) could participate in the process smoothly. For xyloses (3ai–3aj), the obtained products adopted β orientation since the anomeric xylosyl radical is β selective.23 Besides pyridyl (Py), other pharmaceutically relevant heteroaromatics such as benzothiazole and oxadiazole (3ai, 3aj) could be efficiently incorporated. This offers a viable strategy for the synthesis of xylose-derived bioactive analogs.4c Finally, the strategy can also be extended for the synthesis of synthetically challenging α-1,2-cis-selenoglycosides (Scheme 4 and Table S7†).24 For example, under the reaction conditions (see footnote a of Table 1, DCE:H2O, v/v, 2:1), four glycosyl bromides could couple with methyl phenylselenyl sulfonate to deliver the corresponding α-seleno-glycosides 3ak–3an with uniformly high stereoselectivity (α:β > 20:1).
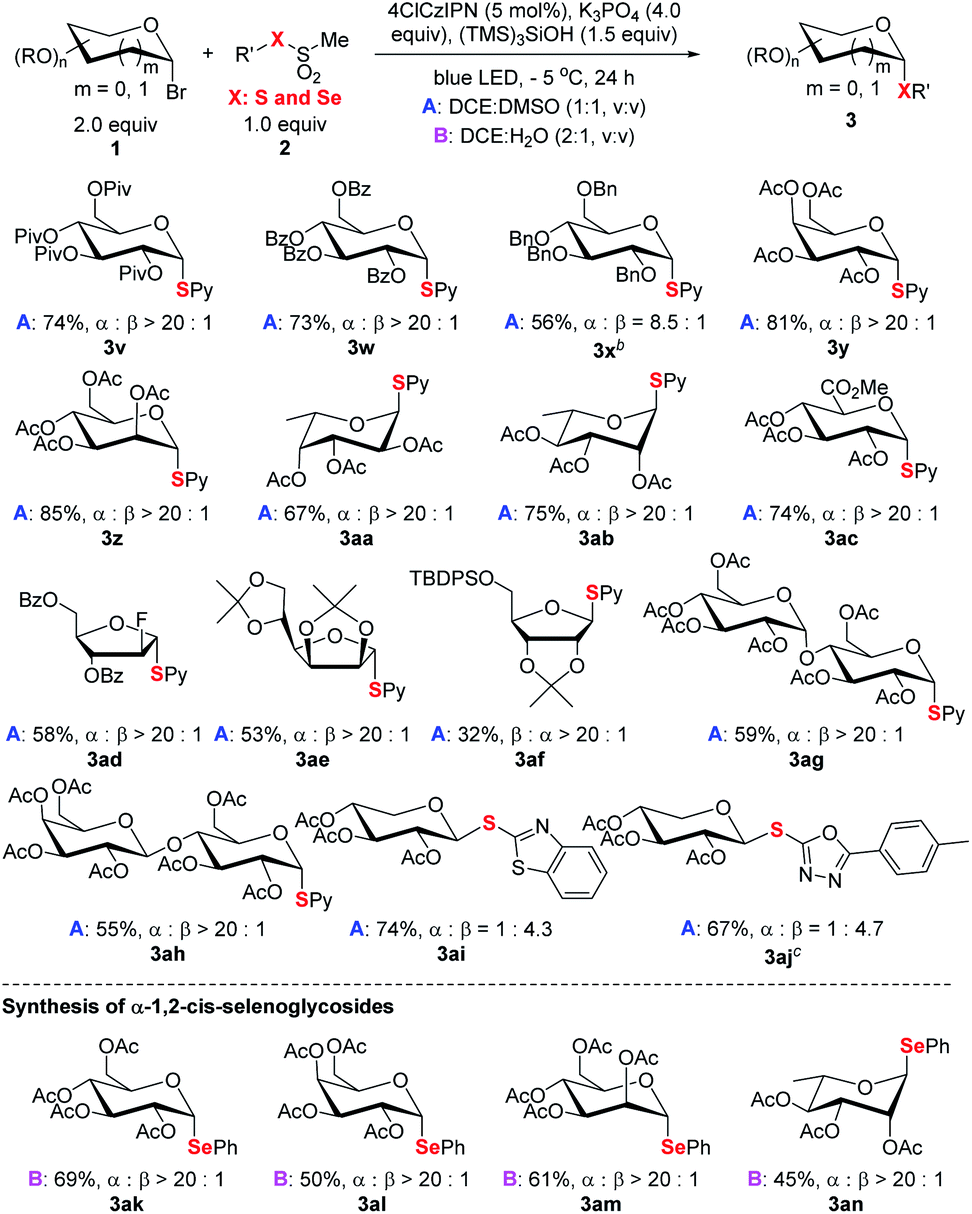 | ||
| Scheme 4 Scope of saccharides and selenoglycosylation. aReaction conditions: unless specified see footnote a of Table 1 and the ESI;† isolated yield; the ratio of α and β anomers determined by crude 1H NMR. b3.0 equiv. of glycosyl bromide used. cDisulfide used. | ||
The capacity of selective functionalization of biologically relevant structures and therapeutics is the testament to the synthetic power of a methodology. As demonstrated (Scheme 5), C1-6′ connected thioglycosides 3ao–3aq were efficiently synthesized. It is noted that a native unprotected saccharide thiosulfonate could be used for efficient cross coupling (3aq). Moreover, it is particularly noteworthy that the protocol is amenable for the synthesis of α-S-linked 1,1′-disaccharides with C1 thiol electrophiles, a synthetic challenge in glycosylation,25 as demonstrated in 1-thiodisaccharides (3ar) and thiotrisaccharide (3as). Furthermore, α-linked thioglycosyl amino acid 3at and peptide 3au could be efficiently constructed. The synthetic manifold was further exemplified by late-stage thioglycosylation of therapeutics. The incorporation of thioglycosyl moieties into estrone (3av), Captopril (3aw), and flavone (3ax) has been realized smoothly.
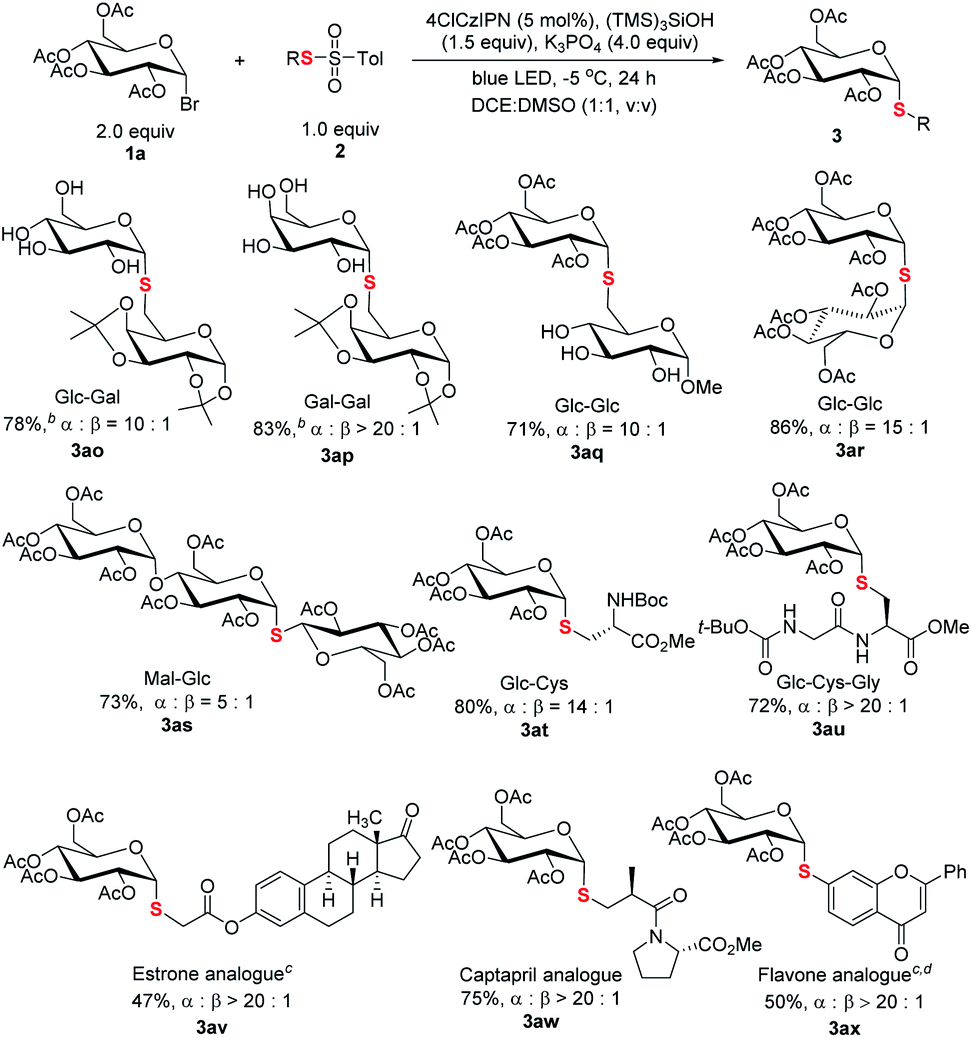 | ||
| Scheme 5 Thiodiversification of pharmaceutically relevant structures. aReaction conditions: unless specified, see footnote a of Table 1 and the ESI;† isolated yield; ratio of α and β anomers determined by crude 1H NMR. bThe product after hydrolysis. cMethylthiosulfonate used. dDCE:H2O (1.5 mL, 2:1, v/v) used as the solvent. | ||
In the new thioglycosylation reaction, critically (TMS)3SiOH was identified as a HAT reagent, which could efficiently suppress the undesired reduction of the radical 8 (Scheme 6a). This may be attributed to the strong O–H bond (calculated BDE = 98 kcal mol−1, see the ESI,† BDE of S–H: 83 kcal mol−1)26,27 and steric hindrance, making the H difficult for 8 to abstract. This strong bond also echoes the use of stronger 4ClCzIPN (E*/E˙− = 1.58 V vs. SCE)21 to oxidize the silyloxide [((TMS)3SiO−/TMS)3SiO˙ = 1.54 V vs. SCE)]. A spontaneous Brook rearrangement of silyloxy radical 6 forms a silicon-centred radical 7,28,21b which acts as an effective debrominator. The anomeric effect makes the radical 8 axially positioned and directs α-selective coupling with thiosulfonate 2. In the reactions, we still observed a notable amount of the reduction product 4. It is believed that it is produced from the reaction of 8 with (TMS)3SiOH, which was confirmed by deuteration experiments with observed deuterated product 4-d (Scheme 6b). This also rationalizes that 2 equiv. of glycosyl bromide 1 is used to ensure high efficiency of the thioglycosylation process. Finally, a radical trapping study with TEMPO and methyl acrylate16d further confirms the radical engaged process (Scheme 6c).
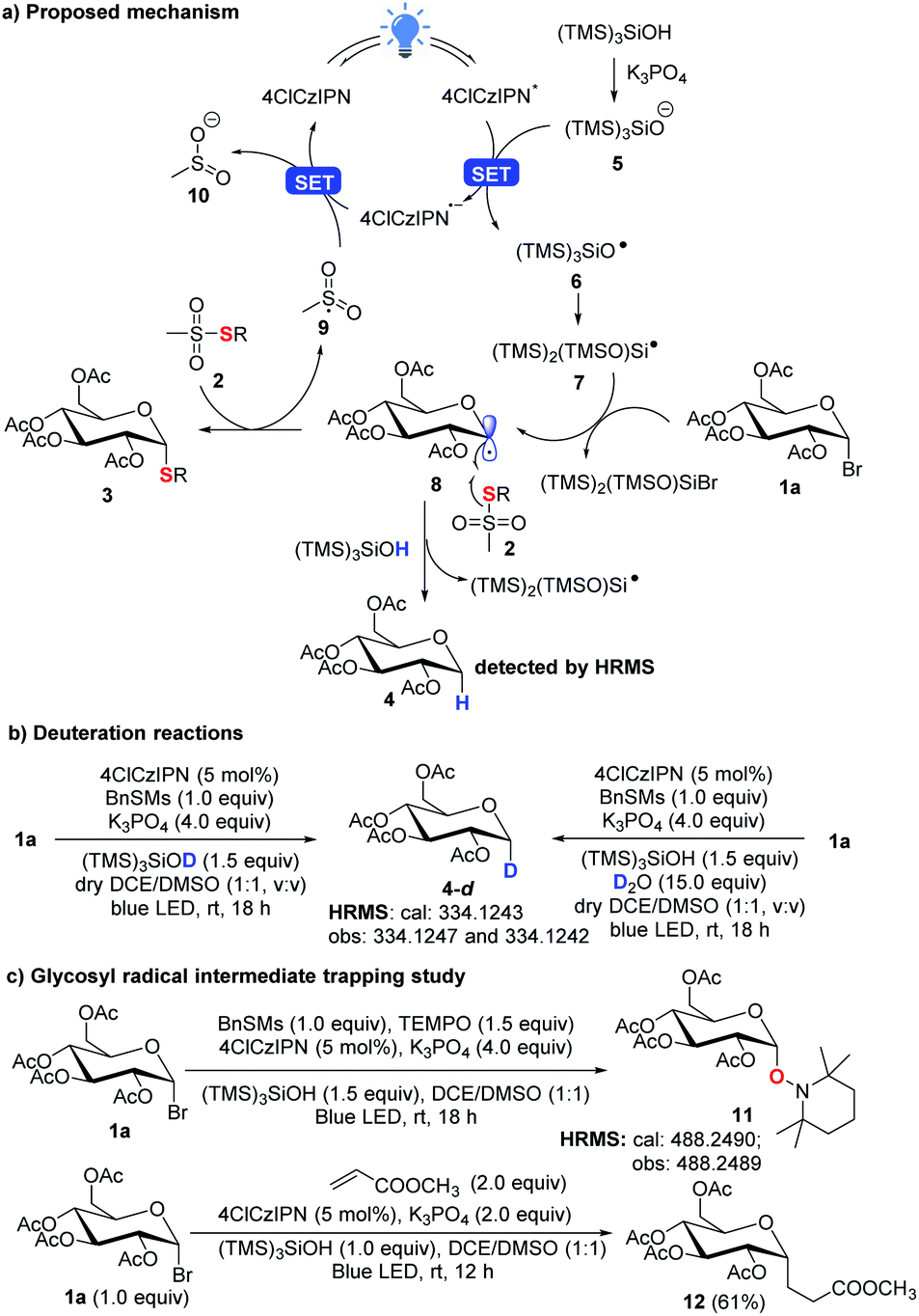 | ||
| Scheme 6 Proposed mechanism and mechanism studies. | ||
Conclusions
In conclusion, we have developed a metal-free, glycosyl radical strategy for the stereoselective synthesis of thioglycosides by employing commonly used glycosyl bromides as radical precursors. The uncovered organophotoredox mediated HAT radical pathway can highly stereoselectively induce the formation of an anomeric C–S bond while minimizing the side reactions. The power of the platform has been underscored by the mild reaction conditions enabling the synthesis of challenging α-1,2-cis-thioglycosides, the tolerance of various functional groups and the broad substrate scope for both common pentoses and hexoses. Furthermore, this general approach is compatible with both sp2 and sp3 sulfur electrophiles and late-stage glycodiversification. It is expected that the strategy enabling the efficient generation of glycosyl radicals from labile glycosyl bromides can offer a reliable alternative for the synthesis of C- and other hetero-glycosides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the NIH (5R01GM125920-04) and the NSF MRI for the acquisition of a 500 MHz NMR spectrometer (1920234).Notes and references
- (a) P. M. Rudd, T. Elliot, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370 CrossRef CAS; (b) B. G. Davis, Chem. Rev., 2002, 102, 579 CrossRef CAS; (c) K. J. Doores, D. P. Gamblin and B. G. Davis, Chem.–Eur. J., 2006, 12, 656 CrossRef CAS; (d) J. B. Lowe, Cell, 2001, 104, 809 CrossRef CAS.
- (a) D. A. Thayer, H. N. Yu, M. C. Galan and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 4596 CrossRef CAS; (b) K. Pachamuthu and R. R. Schmidt, Chem. Rev., 2006, 106, 160 CrossRef CAS; (c) C. S. Rye and S. G. Withers, Carbohydr. Res., 2004, 339, 699 CrossRef CAS; (d) Z. Amso, S. W. Bisset, S.-H. yang, P. W. R. Harris, T. H. Wright, C. D. Navo, M. L. Patchett, G. E. Norris and M. A. Brimble, Chem. Sci., 2018, 9, 1686 RSC.
- (a) S. Schwarz, J. Shen, K. Kadlec, Y. Wang, G. B. Michael, A. T. Feßler and B. Vester, Cold Spring Harb. Perspect. Med., 2016, 6, a027037 CrossRef; (b) T. J. Oman, J. M. Boettcher, H. Wang, X. N. Okalibe and W. A. van der Donk, Nat. Chem. Biol., 2011, 7, 78 CrossRef CAS; (c) Y. S. Y. Hsieh, B. L. Wilkinson, M. R. O'Connell, J. P. Mackay, J. M. Matthews and R. J. Payne, Org. Lett., 2012, 14, 1910 CrossRef CAS; (d) S. Biswas, C. V. Garcia De Gonzalo, L. M. Repka and W. A. van der Donk, ACS Chem. Biol., 2017, 12, 2965 CrossRef CAS.
- (a) Z. J. Witczak, Curr. Med. Chem., 1999, 6, 165 CAS; (b) Z. J. Witczak, P. Kaplon and P. Markus Dey, Carbohydr. Res., 2003, 338, 11 CrossRef CAS; (c) W. A. El-Sayed, N. M. Fathi, W. A. Gad and E. S. H. El-Ashry, J. Carbohyd. Chem., 2008, 27, 357 CrossRef CAS.
- (a) R. N. Comber, J. D. Friedrich, D. A. Dunshee, S. L. Petty and J. A. Secrist, Carbohydr. Res., 1994, 262, 245 CrossRef CAS; (b) D. V. Mangte and S. P. Deshmukh, Heteroat. Chem., 2007, 18, 390 CrossRef CAS; (c) W. A. El-Sayed, N. M. Fathi, W. A. Gad and E. S. H. El-Ashry, J. Carbohyd. Chem., 2008, 27, 357 CrossRef CAS.
- (a) D. Ikuta, Y. Hirata, S. Wakamori, H. Shimada, Y. Tomabechi, Y. Kawasaki, K. Ikeuchi, T. Hagimori, S. Matsumoto and H. Yamada, Science, 2019, 364, 674 CrossRef CAS; (b) M. Lahmann and S. Oscarson, Org. Lett., 2000, 2, 3881 CrossRef CAS; (c) S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687 RSC; (d) M. L. Spell, K. Deveaux, C. G. Bresnahan, B. L. Bernard, W. Sheffield, R. Kumar and J. R. Ragains, Angew. Chem., Int. Ed., 2016, 55, 6515 CrossRef CAS; (e) G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13 CrossRef CAS; (f) B. Dhakal and D. Crich, J. Am. Chem. Soc., 2018, 140, 15008 CrossRef CAS; (g) H.-Y. Wang, S. A. Blaszczyk, G. Xiao and W. Tang, Chem. Soc. Rev., 2018, 47, 681 RSC.
- (a) M. Gerz, H. Matter and H. Kessler, Angew. Chem., Int. Ed. Engl., 1993, 32, 269 CrossRef; (b) J. Ramos-Soriano, U. Niss, J. Angulo, M. Angulo, A. J. Moreno-Vargas, A. T. Carmona, S. Ohlson and I. Robina, Chem.–Eur. J., 2013, 19, 17989 CrossRef CAS; (c) P. J. Pfäffli, S. H. Hixson and L. Anderson, Carbohydr. Res., 1972, 23, 195 CrossRef; (d) T. Fujihira, T. Takido and M. Seno, J. Mol. Catal. A: Chem., 1999, 137, 65 CrossRef CAS.
- (a) S. Ecopy, Y. Singh and A. V. Demchenko, Org. Biomol. Chem., 2019, 17, 8379 RSC; (b) S. Zhu, G. Samala, E. T. Sletten, J. L. Stockdill and H. M. Nguyen, Chem. Sci., 2019, 10, 10475 RSC; (c) D. Hirofumi and N. Yoshihiro, Trends Glycosci. Glycotechnol., 2014, 26, 119 CrossRef.
- (a) K. N. Baryal, D. Zhu, X. Li and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 8012 CrossRef CAS; (b) F. Zhu, E. Miller, S. Zhang, D. Yi, S. O'Neill, X. Hong and M. A. Walczak, J. Am. Chem. Soc., 2018, 140, 18140 CrossRef CAS.
- Alkylation/arylation methods: (a) H. Driguez, Chembiochem, 2001, 2, 311 CrossRef CAS; (b) R. Komor, A. Kasprzycka, G. Pastuch-Gawołek and W. Szeja, Carbohydr. Res., 2014, 396, 37 CrossRef CAS; (c) R. T. Dere, A. Kumar, V. Kumar, X. Zhu and R. R. Scmidt, J. Org. Chem., 2011, 76, 7539 CrossRef CAS; (d) F. M. Ibatullin, K. A. Shabalin, J. V. Jänis and A. G. Shavva, Tetrahedron Lett., 2003, 44, 7961 CrossRef CAS.
- Cross coupling: (a) D. Montoir, M. Amoura, Z. E. A. Ababsa, T. M. Vishwanatha, E. Yen-Pon, E. V. Robert, M. Beltramo, V. Piller, M. Alami, V. Aucagne and S. Messaoudi, Chem. Sci., 2018, 9, 8753 RSC; (b) M. Zhu, G. Dagousset, M. Alami, E. Magnier and S. Messaoudi, Org. Lett., 2019, 21, 5132 CrossRef CAS.
- Thiol-ene reaction and Michael addition: (a) G. Zhao, S. Kaur and T. Wang, Org. Lett., 2017, 19, 3291 CrossRef CAS; (b) M. Fiore, M. L. Conte, S. Pafifico, A. Marra and A. Dondoni, Tetrahedron Lett., 2011, 52, 444 CrossRef CAS; (c) N. Merbouh, F. K. Wallner, O. M. Cociorva and P. H. Seeberger, Org. Lett., 2007, 9, 651 CrossRef CAS; (d) Z. J. Witczak, H. Chen and P. Kaplon, Tetrahedron: Asymmetry, 2000, 11, 519 CrossRef CAS; (e) Y. Zhu and W. A. van der Donk, Org. Lett., 2001, 3, 1189 CrossRef CAS.
- Nucleophilic substitution: (a) D. P. Gamblin, P. Garnier, S. Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828 CrossRef CAS; (b) G. J. L. Bernardes, D. P. Gamblin and B. G. Davis, Angew. Chem., Int. Ed., 2006, 45, 4007 CrossRef CAS; (c) B. G. Davis, M. A. T. Maughan, M. P. Green, A. Ullman and J. B. Jones, Tetrahedron: Asymmetry, 2000, 11, 245 CrossRef CAS; (d) D. P. Galonić, W. A. van der Donk and D. Gin, J. Am. Chem. Soc., 2004, 126, 12712 CrossRef; (e) D. P. Galonić, N. D. Ide, W. A. van der Donk and D. Y. Gin, J. Am. Chem. Soc., 2005, 127, 7359 CrossRef.
- J. M. Smith, S. J. Harwood and P. S. Baran, Acc. Chem. Res., 2018, 51, 1807 CrossRef CAS.
- (a) P. Ji, Y. Zhang, Y. Wei, H. Huang, W. Hu, P. A. Mariano and W. Wang, Org. Lett., 2019, 21, 3086 CrossRef CAS; (b) P. Ji, Y. Zhang, Y. Dong, H. Huang, Y. Wei and W. Wang, Org. Lett., 2020, 22, 1557 CrossRef CAS; (c) Y. Ma, S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang and Y. Zhang, Chem. Commun., 2019, 55, 14657 RSC; (d) N. Kiya, Y. Hidaka, K. Usui and G. Hirai, Org. Lett., 2019, 21, 1588–1592 CrossRef CAS; (e) Y. Hidaka, N. Kiya, M. Yoritate, K. Usui and G. Hirai, Chem. Commun., 2020, 56, 4712–4715 RSC.
- (a) A review, see: Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281 CrossRef CAS; (b) N. Miquel, G. Doisneau and J.-M. Beau, Angew. Chem., Int. Ed., 2000, 39, 4111 CrossRef CAS; (c) H. Abe, S. Shuto and A. Matsuda, J. Am. Chem. Soc., 2001, 123, 11870 CrossRef CAS; (d) R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274 CrossRef CAS; (e) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132 CrossRef CAS; (f) K. Masuda, M. Nagatomo and M. Inoue, Nat. Chem., 2017, 9, 207 CrossRef CAS; (g) A. Dumoulin, J. K. Matsui, A. Gutierrez-Bonet and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6614 CrossRef CAS; (h) S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6610 CrossRef CAS.
- (a) During the preparation of this manuscript, Hong, Walczak and coworkers reported a Cu(I) and blue LED co-catalyzed stereoselective thioglycosylation of glycosyl stannanes with disulfides:F. Zhu, S.-Q. Zhang, Z. Chen, J. Rui, X. Hong and M. A. Walczak, J. Am. Chem. Soc., 2020, 142, 11102 CrossRef CAS; (b) A similar strategy reported by Luo and Nguyen used for impressive stereoselective formation of glycosidic C–O bonds:F. Yu, J. L. Dickson, R. S. Loka, H. Xu, R. N. Schaugaard, H. B. Schlegel, L. Luo and H. M. Nguyen, ACS Catal., 2020, 10, 5990 Search PubMed.
- (a) R. S. Andrews, J. J. Becker and M. R. Gagné, Org. Lett., 2011, 13, 2406 CrossRef CAS; (b) S. Yamago and A. Matsumoto, J. Org. Chem., 2008, 73, 7300 CrossRef CAS; (c) J. Guiard, Y. Rhali and J.-P. Praly, Eur. J. Org. Chem., 2014, 4461 CrossRef CAS.
- (a) N. Miquel, G. Doisneau and J.-M. Beau, Angew. Chem., Int. Ed., 2000, 39, 4111 CrossRef CAS; (b) H. Gong and M. R. Gagné, J. Am. Chem. Soc., 2008, 130, 12177 CrossRef CAS; (c) R. P. Spencer, C. L. Cavallaro and J. Schwartz, J. Org. Chem., 1999, 64, 3987 CrossRef CAS.
- Y. Zhang, P. Ji, W. Hu, Y. Wei, H. Huang and W. Wang, Chem.–Eur. J., 2019, 25, 8225 CrossRef CAS.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS; (b) F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn'ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883 RSC; (c) A. Kretzschmar, C. Patze, S. T. Schwaebel and U. H. F. Bunz, J. Org. Chem., 2015, 80, 9126 CrossRef CAS; (d) a review, see: T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408 RSC.
- (a) C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229 CrossRef CAS; (b) C. Le, T. Q. Chen, C. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010 CrossRef CAS; (c) R. T. Smith, X. Zhang, J. A. Rincon, J. Agejas, C. Mateo, M. Barberis, S. Garcia-Cerrada, O. de Frutos and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 17433 CrossRef CAS; (d) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS.
- (a) B. Giese, J. Dupuis, M. Leising, M. Nix and H. J. Lindner, Carbohydr. Res., 1987, 171, 329–341 CrossRef CAS; (b) H. Abe, S. Shuto and A. Matsuda, J. Am. Chem. Soc., 2001, 123, 11870 CrossRef CAS.
- (a) A. W. McDonagh, M. F. Mahon and P. V. Murphy, Org. Lett., 2016, 18, 552 CrossRef CAS; (b) S. Czernecki and D. Randriamandimby, J. Carbohydr. Chem., 1996, 15, 183 CrossRef CAS; (c) F. Santoyo-Gonzalez, F. G. Calvo-Flores, P. Garcia-Mendoza, F. Hernandez-Mateo, J. Isac-Garcia and R. Robles-Dia, J. Org. Chem., 1993, 58, 6122 CrossRef CAS; (d) V. Di Bussolo, A. Fiasella, F. Balzano, G. Uccello Barretta and P. Crotti, J. Org. Chem., 2010, 75, 4284 CrossRef CAS.
- (a) S. Izumi, Y. Kobayashi and Y. Takemoto, Angew. Chem., Int. Ed., 2020, 59, 14054 CrossRef CAS; (b) J. Defaye and A. Gadelle, Carbohydr. Res., 1991, 217, 51 CrossRef CAS.
- F. Denes, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587 CrossRef CAS.
- http://butane.chem.uiuc.edu/cyerkes/Chem104ACSpring2009/Genchemref/bondenergies.html .
- M. D. Paredes and R. Alonso, J. Org. Chem., 2000, 65, 2292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04136j |
| This journal is © The Royal Society of Chemistry 2020 |