
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Et3SiH + KOtBu provide multiple reactive intermediates that compete in the reactions and rearrangements of benzylnitriles and indolenines†
Andrew J.
Smith‡
a,
Daniela
Dimitrova‡
a,
Jude N.
Arokianathar
a,
Kenneth F.
Clark
a,
Darren L.
Poole
 b,
Stuart G.
Leach
b and
John A.
Murphy
*a
b,
Stuart G.
Leach
b and
John A.
Murphy
*a
aDepartment of Pure and Applied Chemistry, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: john.murphy@strath.ac.uk
bGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
First published on 21st October 2020
Abstract
The combination of potassium tert-butoxide and triethylsilane is unusual because it generates multiple different types of reactive intermediates simultaneously that provide access to (i) silyl radical reactions, (ii) hydrogen atom transfer reactions to closed shell molecules and to radicals, (iii) electron transfer reductions and (iv) hydride ion chemistry, giving scope for unprecedented outcomes. Until now, reactions with this reagent pair have generally been explained by reference to one of the intermediates, but we now highlight the interplay and competition between them.
Introduction
A novel reducing system, consisting of the reagent-pair, triethylsilane and potassium tert-butoxide was reported by Stoltz, Grubbs et al. in 2013.1 The combination of the two reagents has since been investigated by a number of research groups2–15 and provides a range of distinctive reaction types, arising through an unprecedented menu of reactive intermediates formed in the reaction, including triethylsilyl radicals 1, silanates 2 as hydrogen atom donors to both closed shell molecules and to radicals, and as potential hydride ion donors, and tert-butoxytriethylsilyl radical anions 3 as a very powerful electron donor. Exposing substrates simultaneously to multiple reactive intermediates is not routinely encountered in organic chemistry, other than in modelling of prebiotic conditions,16 and so the variety of reactive intermediates produced by this reagent pair provides opportunities to witness unusual outcomes.Thus, triethysilyl radicals 1 are candidates for the conversions of substrates 4–7 (ref. 2–6, 8 and 13) to their products 11–14. (Note that silylation reactions, as in formation of 13 usually occur at lower temperatures, here 45 °C). On the other hand, Jeon has established9 that silanate complex 2′ (and less efficiently 2) conducts a potassium ion-dependent H-atom transfer to afford hydrosilylation products 15 from styrenes such as 8 at 80 °C. Tuttle, Murphy et al. have reported that N-benzylindoles 9 are deprotected by electron transfer reactions with 3 acting as electron donor.7 In each of the above cases, the products can be attributed to one of the reactive intermediates. Most recently, a more complex rearrangement of N-aryl indoles (e.g.10) to dihydroacridines (in this case, 17) features sequential electron transfer from 3 and H-atom transfer from 2.14 In addition to these transformations, the reagent pair Et3SiH/KOtBu has found wider applications in silylation of alcohols10 and amines,11 as well as the silylation of terminal alkynes.15 The broad range of possible pathways featuring different reactive intermediates is what makes this reagent-pair so fascinating (Scheme 1).12
 | ||
| Scheme 1 | ||
During a recent study, we showed that Et3SiH/KOtBu carries out reductive decyanation of benzylic nitriles (e.g.18 → 19, Scheme 2)7,17 and our starting point for this current study was to find out more about the reactivity of the substrates and intermediates.
 | ||
| Scheme 2 | ||
In 2017, Chiba et al. uncovered18 a probe for hydride-based reduction of nitriles, where substrates e.g.20 reacted with a composite of NaH and LiI to form an iminyl anion 21 that displaced the o-MeO group in a concerted cSNAr reaction19 to form indolenine 22 [R = (CH2)4]. In his elegant paper, aminyl anions also underwent efficient cyclisation. We wondered whether nitriles that are subjected to the Et3SiH + KOtBu reagent would behave similarly, giving evidence for formation of iminyl anion intermediates through hydride ion delivery from 2.20
Results and discussion
Substrates 23, 25 and 27 (Scheme 2) were prepared (see ESI†) and reacted with the Et3SiH/KOtBu mixture. In each case, cyclisation with displacement of the methoxy group was observed. Our initial conversion of 23 → 24 occurred in 32% yield (Scheme 2), but upon optimisation, the yield of 24 was increased to 72% by lowering the temperature to 70 °C (Scheme 3 and ESI†). The detection of imine 30 during the optimisation studies suggests that an iminyl anion 21 (R = nPr) was a key intermediate in the reactions. These reactions are therefore proposed to occur by hydride ion delivery to the nitrile by intermediate 2. The mechanism of conversion of imines e.g.30 to amines (in this case, 24) comes up for discussion later in this paper.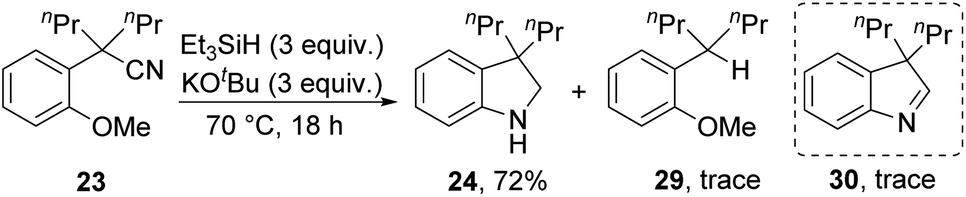 | ||
| Scheme 3 | ||
The effect of the identity of the base and silane present was then investigated. On changing the counter-ion on butoxide from potassium to sodium or lithium, no reaction was observed (see ESI†). Other potassium bases such as KHMDS, KOH, and KOEt were also unsuccessful. KH was somewhat successful with 24 being isolated in 12% yield, whereas NaH gave no reaction. These results underline the special reactivity of potassium tert-butoxide in this reagent system, which cannot be replicated by sodium tert-butoxide or lithium tert-butoxide. The effect of solvent on the reaction was also investigated, and solvent-free conditions were found to be optimal for the cyclisation (see ESI†).
The optimised conditions were then used to further study the scope of the reaction with substrates related to 23. Firstly, the ethoxy derivative 31 was also successful, with cyclised product 24 (R = Et) isolated in 65% yield. Halide leaving groups were then tested (Table 1, entries 3–6). Interestingly, methoxide out-performed halide leaving groups for the formation of 24, with the halides following the general trend of SNAr reactivity (F > Cl > Br = I). Bromo- and iodo-substituted substrates 34 and 35 did not afford any cyclised products and instead, dehalogenated compound 37 was isolated, suggesting that dehalogenation of iodides and bromides was more rapid than activation of the nitrile. Although many mechanisms can be considered, dehalogenation is a hallmark of reactions of silyl radicals 1 or can result from electron transfer chemistry of 3.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | 24/% | 29/% | 30/% | 37/% |
| 1 | 23 | 72 | 11 | — | — |
| 2 | 31 | 65 | 8 | — | — |
| 3 | 32 | 44 | — | Trace | — |
| 4 | 33 | 19 | — | — | — |
| 5 | 34 | — | — | — | 57 |
| 6 | 35 | — | — | — | 78 |

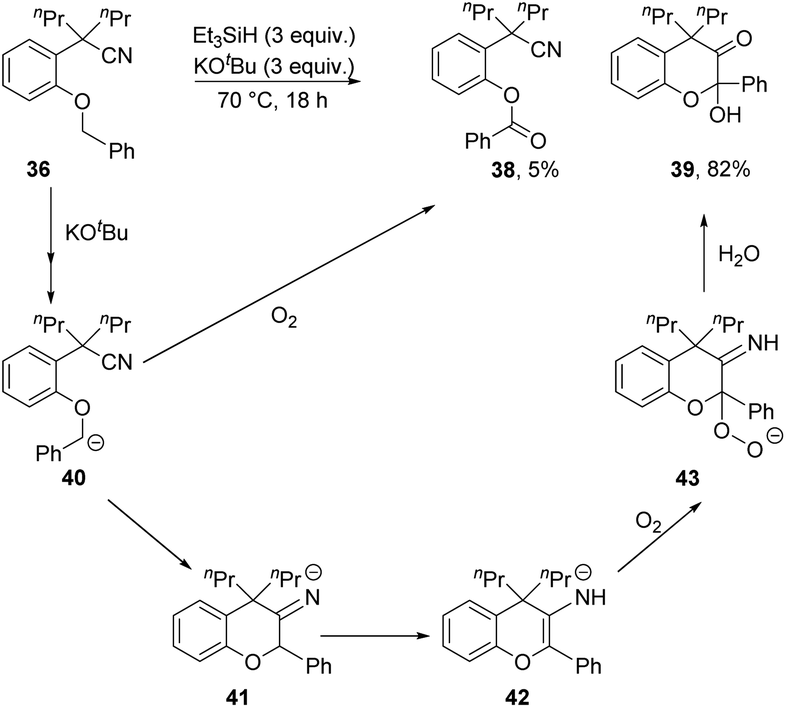
Changing the methoxy group to benzyloxy in 36 brought about different chemistry. No displacement of the benzyloxy group was detected and, instead, compounds 38 (5%) and 39 (82%) were isolated (Scheme 4). Both products suggested an initial activation at the benzyl position, most likely via anion 40. Cyclisation onto the nitrile would afford 41 which would be converted to 42 through a proton shift. The electron-rich alkene in 42 will readily undergo electron transfer and coupling to molecular oxygen to afford 43.21–23 If this can convert into a hydroperoxide, then reductive cleavage of the O–O bond can occur during the reaction. Otherwise, 43 could protonate on workup to a hydroperoxyketal, which can lose hydroperoxide anion in a hydrolysis that then leads to 39. Alternatively, any residual anion 40 would also react with air on work up, ultimately leading to ester 38.
 | ||
| Scheme 4 | ||
Our next steps were to establish the necessary components for the cyclisation of 23 to 24. Control reactions were now performed (Table 2). The parent reaction is shown as entry 1.

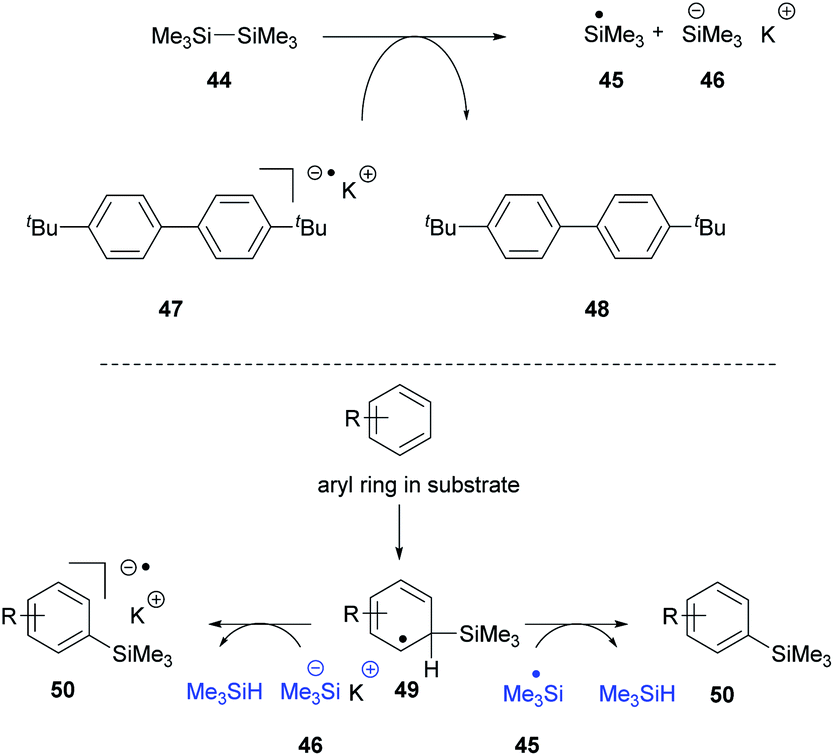
Recently, we proposed14 that the reactivity of the Et3SiH/KOtBu couple could be reproduced in the absence of the silane, provided that an alternative source of silyl radicals was present. To this end, entry 2 shows that when the silane was replaced by the disilane 44 (Scheme 5) in the presence of the electron donor 47,24 the radical anion of di-p-tert-butylbiphenyl, the cyclisation reaction was still observed, affording 24 (19%).
 | ||
| Scheme 5 | ||
Entries 3 and 4 show that in the absence of a silyl source or an electron donor source, the reaction is not observed, while entry 5 shows that KOtBu alone cannot bring about the reaction.
Our explanation for entry 2 is that the electron donor 47 can cleave the disilane 44 to a silyl radical 45 and a silyl anion 46 as shown in Scheme 5.25 The silyl radical can react with many species in solution. Notably, it can add to arene rings in the substrate to generate intermediates 49 that feature a labile H atom.2–6,13 This can react with a silyl radical 45 to form trimethylsilane or with silyl anion 46 to form trimethylsilane as shown in Scheme 5. This would mean that the missing trialkylsilane reagent (Me3SiH in this experiment) would be generated in situ, starting from the disilane.
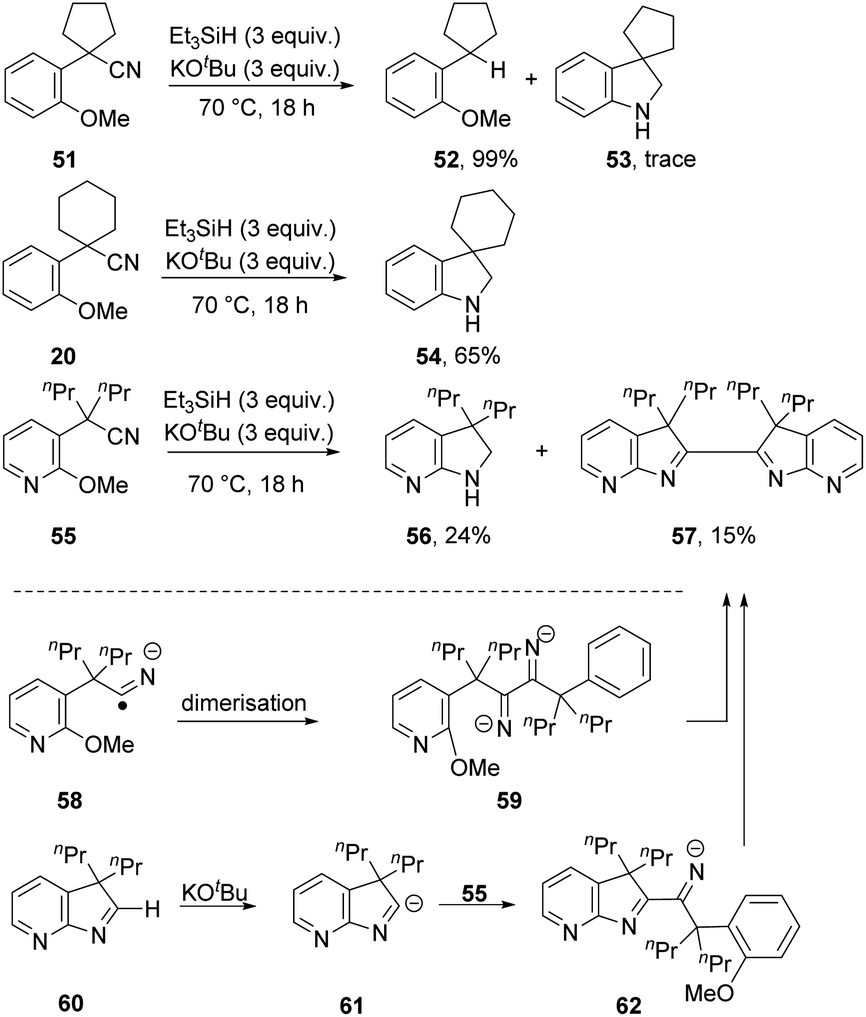
A series of substrates, 51, 20 and 55 (Scheme 6), was now prepared and tested under the optimised conditions, with surprising results. From substrate 51, reductive decyanation to 52 was observed in 99% yield, with only a trace amount of cyclised product 53 detected. However, from the analogous substrate 20, cyclisation to 54 was observed in 65% yield. Pyridine-containing substrate 55 afforded product 56 (24%), along with dimer 57 (15%). This compound 57 might arise by dimerisation of radical anion 58, e.g. if electron transfer occurred from radical anion 3, followed by double cSNAr cyclisation. Alternatively, and more probably, cyclic imine 60 could be deprotonated under the basic conditions to anion 61,26 which could then attack another molecule of 55 to give anion 62, which affords bis-indolenine 57 by cSNAr cyclisation. Observation of this dimerisation solely for this substrate could then be attributed to enhancement of the acidity of the iminyl proton in 60 by the pyridine ring.
 | ||
| Scheme 6 | ||
The results to date are consistent with bicyclic imines such as 60 as key intermediates in the formation of the final indolines, and so we were curious to probe the behaviour of related imines in the presence of the reductive silane–butoxide reagent pair.
To access imines related to 30 (Scheme 3), we considered that an iminyl anion could form by addition of a Grignard reagent to a nitrile and then undergo cyclisation, in the manner of Gademann et al.27 To test this, substrate 23 was treated with MeMgBr at 70 °C, however no reaction occurred. Upon warming to 130 °C, however, products 63–65 were isolated with optimum yields arising from 4 equiv. of Grignard reagent (Scheme 7 and ESI†). Compounds 63 and 64 are indicative of the proposed mechanism for indolenine formation. Compound 65 could arise by deprotonation of the iminyl-CH3 group of 64 by MeMgBr, before attack onto the nitrile group of another molecule of 23. The resulting imine anion can then undergo cSNAr and tautomerism to yield 65.
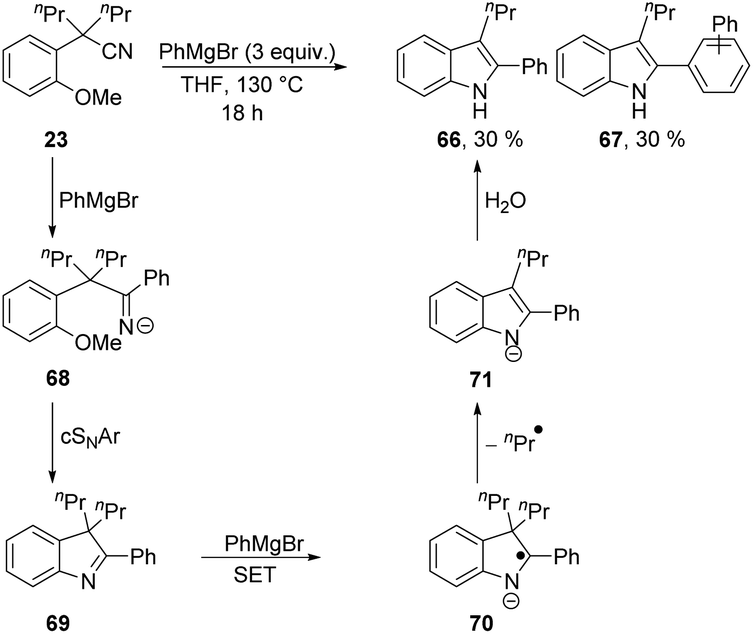
The complications in Scheme 7 leading to a low yield of 64 arose from the ease of deprotonation of the methyl group in 64. To prevent such complications, a Grignard reagent was used that cannot be deprotonated in the α-position, i.e. PhMgBr. Interestingly, cyclisation to an inseparable mixture of indoles 66 and 67 (in 30% yield each, calculated by NMR internal standard) was observed (Scheme 8). This transformation shows loss of a propyl substituent, and aromatisation of the ring system to give indole products undoubtedly provides the driving force for this.
 | ||
| Scheme 7 | ||
 | ||
| Scheme 8 | ||
Grignard reagents have been previously reported in the literature to facilitate SET reactions to reducible substrates.28 Therefore, we propose that electron transfer from PhMgBr must occur to the conjugated indolenine 69. First, PhMgBr adds to the nitrile of 23, forming 68, which can undergo cSNAr to form 69. Electron transfer to the conjugated indolenine 69 then occurs to form 70, which can aromatise with loss of an alkyl group to form 71, which ultimately protonates to 66 upon work-up. The formation of compound 67 is rationalised by the presence of phenyl radicals, generated upon single electron oxidation of PhMgBr, initiating a BHAS mechanism as previously reported in the literature.29
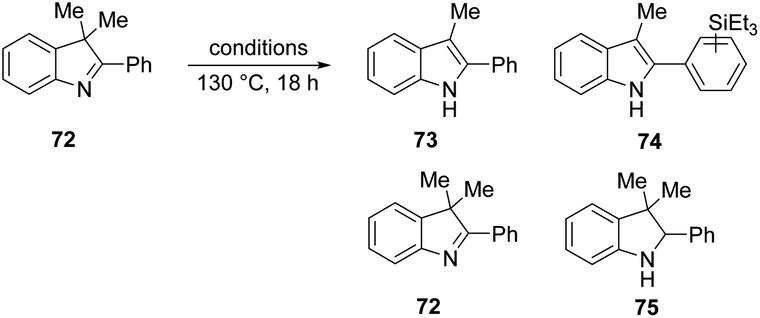
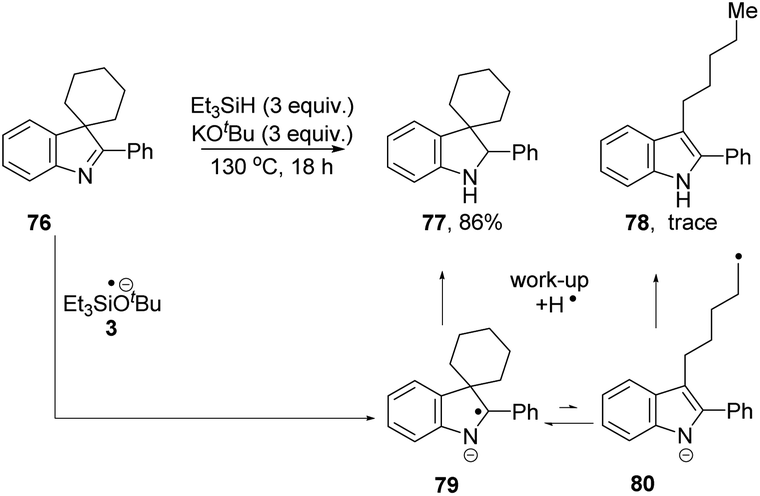
We then investigated if the reducing mixture resulting from the combination of Et3SiH and KOtBu, could perform the same transformation of indolenine to indoline and thereby give evidence of electron transfer from intermediate 3. Compound 72 was treated with Et3SiH and KOtBu, and compounds 73 and 74 were isolated in 45% and 24% respectively (Table 3, entry 1). The elimination of a methyl group from 72 clearly mirrors the electron transfer reactions seen with PhMgBr. Moreover, the formation of silylated derivative 74 (an inseparable mixture of 2 regioisomers was isolated) results from the presence of triethylsilyl radicals, analogous to the phenyl radicals above. The reaction was repeated in the presence of TEMPO. The outcome was to improve the yield of indole 73 to 84% (entry 2). This outcome likely arises, at least in part, from trapping of triethylsilyl radicals by TEMPO, thereby inhibiting the formation of 74. Two further pieces of evidence support the electron transfer proposal: (i) exposure of substrate 72 to potassium metal and KOtBu also afforded 73 (36%), 72 (28%) and 75 (trace amounts) (entry 3); (ii) analogue 76 (Scheme 9) underwent reaction with Et3SiH + KOtBu to afford principally indoline 77 (86%) together with indole 78 (trace amounts). Product 78 arises from an analogous cleavage in radical anion 79 to that seen for radical anion 70. The pentyl side-chain of 78 shows the fate of the cleaved radical, which simply abstracts a hydrogen atom from silane or hydrogen atom donor 2. The difference in outcome for substrates 72 and 76 relates to the fragmentation of their radical anions – when the radical anion of 72 fragments, a methyl group is lost and diffuses away from the substrate. In contrast, fragmentation of radical anion 79 sees the fragmented radical tethered to the indole structure in 80. Radical re-addition to the indole anion reforms radical anion 79, which then abstracts an H-atom (e.g. from triethylsilane or from species 2) to give indoline 77.
 | ||
| Scheme 9 | ||
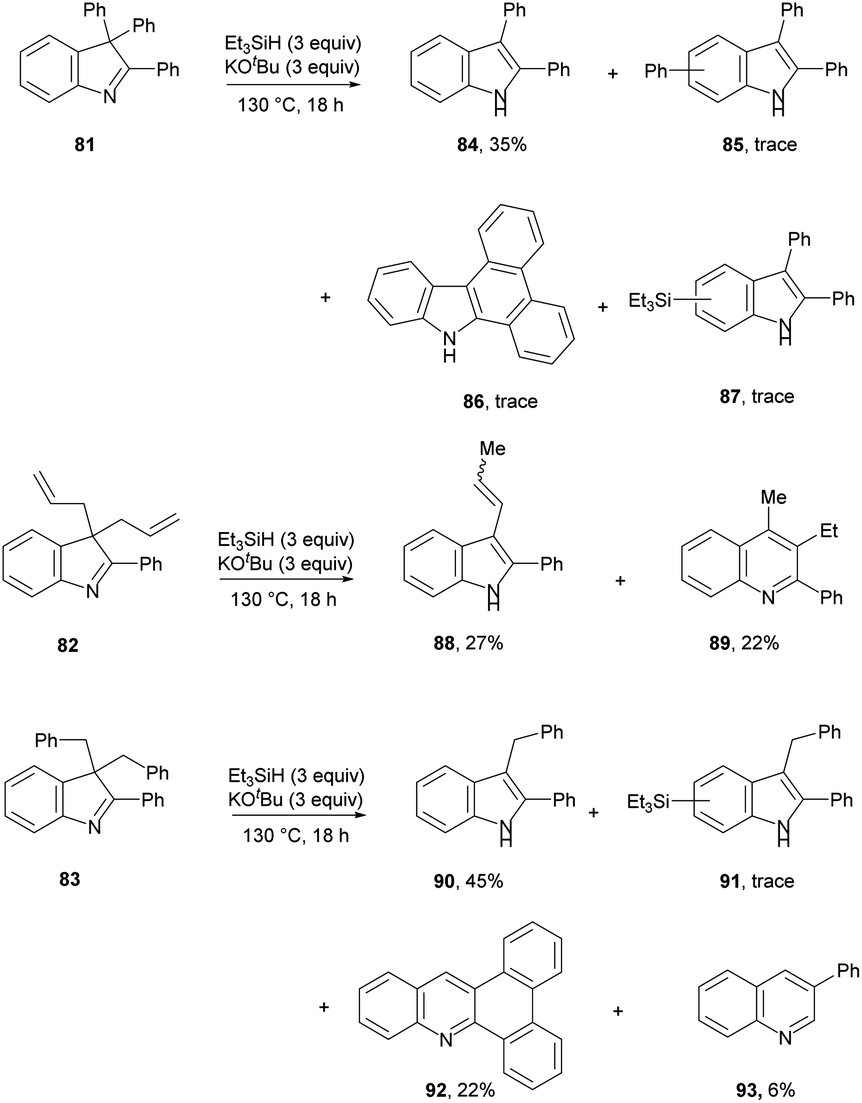
The scope of the groups that can be expelled upon aromatisation was also investigated (Scheme 10). These results show that phenyl, allyl and benzyl are feasible leaving groups.
 | ||
| Scheme 10 | ||
Firstly taking substrate 81, this mirrors the reactions of imines 69, 72 and 76. Loss of a phenyl radical is more difficult than loss of an alkyl radical, but indole 84 is still formed in 35% yield. Also detected were products 85 and 87, resulting from attack on 84 by phenyl or triethylsilyl radicals and subsequent rearomatisation. Again, this mimics the addition of phenyl radicals and triethylsilyl radicals seen respectively in 67 and 74. In addition, compound 86 was detected in an inseparable mixture with compound 85 with 1H NMR data and GC-MS data consistent with those previously reported in the literature. The mechanism envisaged for the formation of 86 is somewhat analogous to that for compound 92 (see below).
For substrate 82, the products 88 (27%) and 89 (22%) can be explained by invoking KOtBu-induced isomerisation of a terminal allyl group to internal alkene 96 (Scheme 11). We have recently reported that allyl groups undergo base-induced isomerisation under the Et3SiH/KOtBu conditions.14 Subjecting this compound, 96, to electron transfer from donor 3 gives radical anion 94. Expulsion of an allyl radical accounts for the formation of indole 88.
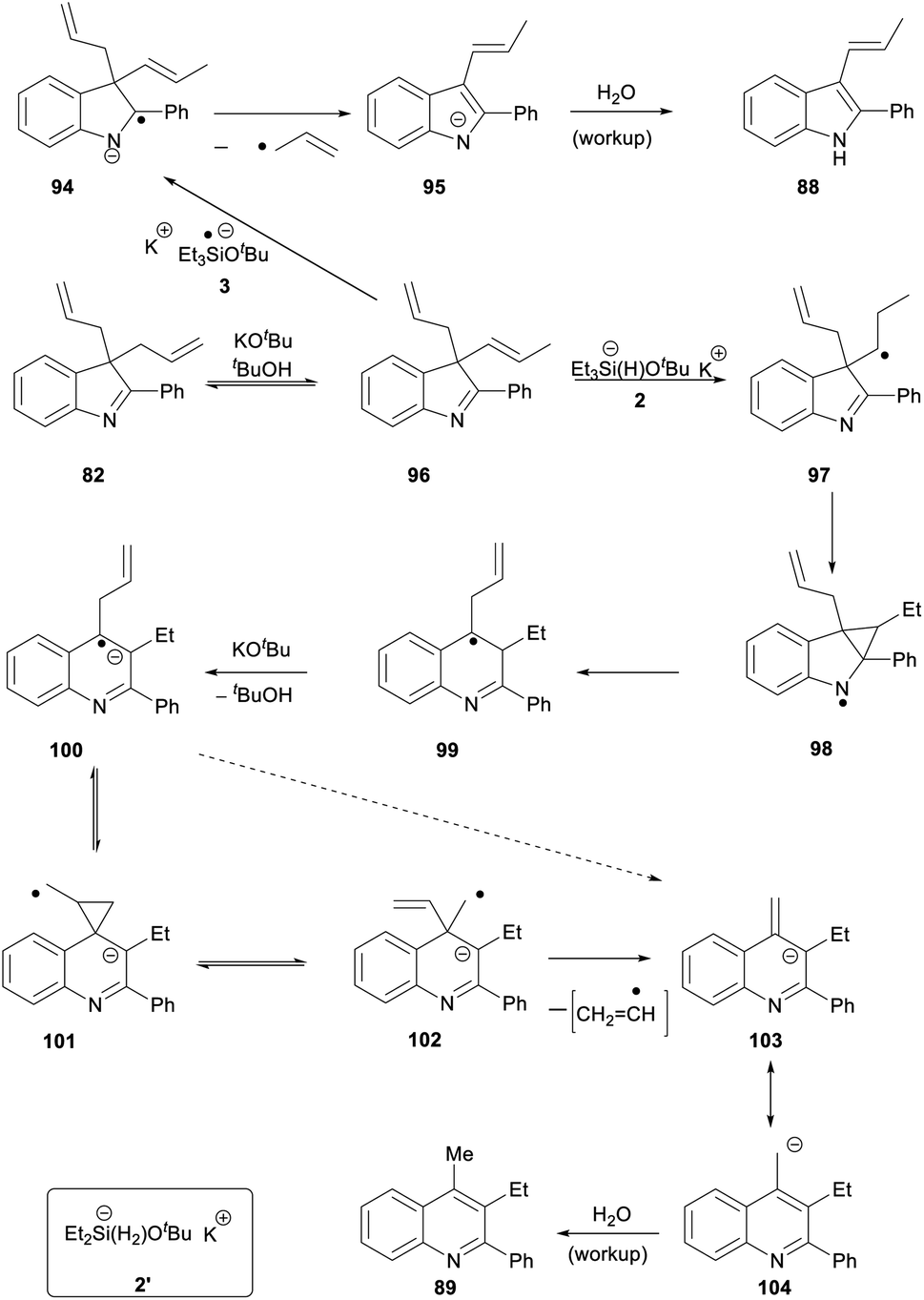 | ||
| Scheme 11 | ||
The second product formed from substrate 82 is the quinoline 89 (22%). This is a really interesting product. Focusing on the 6 carbons of the allyl substituents in 82, it appears that one carbon has been incorporated into the ring system during a ring-expansion, two carbons have been lost during the rearrangement, and the remaining three carbons end up as the methyl and ethyl substituents on neighbouring ring carbons in 89 – a rearrangement of serious complexity. Our working hypothesis is that the product 89 arises also from intermediate diene 96. Jeon recently demonstrated H-atom addition to styrenes by reactive intermediate 2′ (inset, Scheme 11) formed from diethylsilane. A K+ ion, complexed by the aromatic ring in the styrene, held the silanate anion in 2′ (and analogues) close to the aromatic ring, and this complexation was essential for H-atom addition. Here, the feasibility of H-atom addition from 2, rather than 2′, to a side-chain alkene should also increase when the alkene is nearer to the aromatic ring, thereby directing H-atom addition to 96 to form radical 97. An aza-version of a cyclopropylcarbinyl rearrangement governs the ring-expansion to radical 99. This radical has an adjacent H-atom that is easily acidic enough to be removed by KOtBu, affording the quinoline radical anion 100. This undergoes reversible cyclisation to cyclopropylcarbinyl radical 101, which must very occasionally fragment to distal radical anion 102; expulsion of a vinyl radical (or a vinyl anion) then affords benzylic anion 103 (or its benzylic radical counterpart) which affords 89 on workup.30
Having proposed a route to the quinoline 89 from substrate 82, we note that two further quinolines, 92 and 93, which arise from substrate 83, require explanation. We have recently shown that benzylic C–H bonds can undergo abstraction of an H-atom under the conditions of these reactions, by triethylsilyl radicals 1.13 In this case, this would lead to radical 105 (Scheme 12). Cyclopropylcarbinyl radical rearrangement would lead to ring-expansion to radical 107, which, following deprotonation, would expel a benzyl radical to yield quinoline anion 109. Protonation from tBuOH, followed by electron transfer from 3 would give radical anion 110. Expulsion of a phenyl radical affords anion 111 that abstracts a proton (from tBuOH or on workup) to give 93.
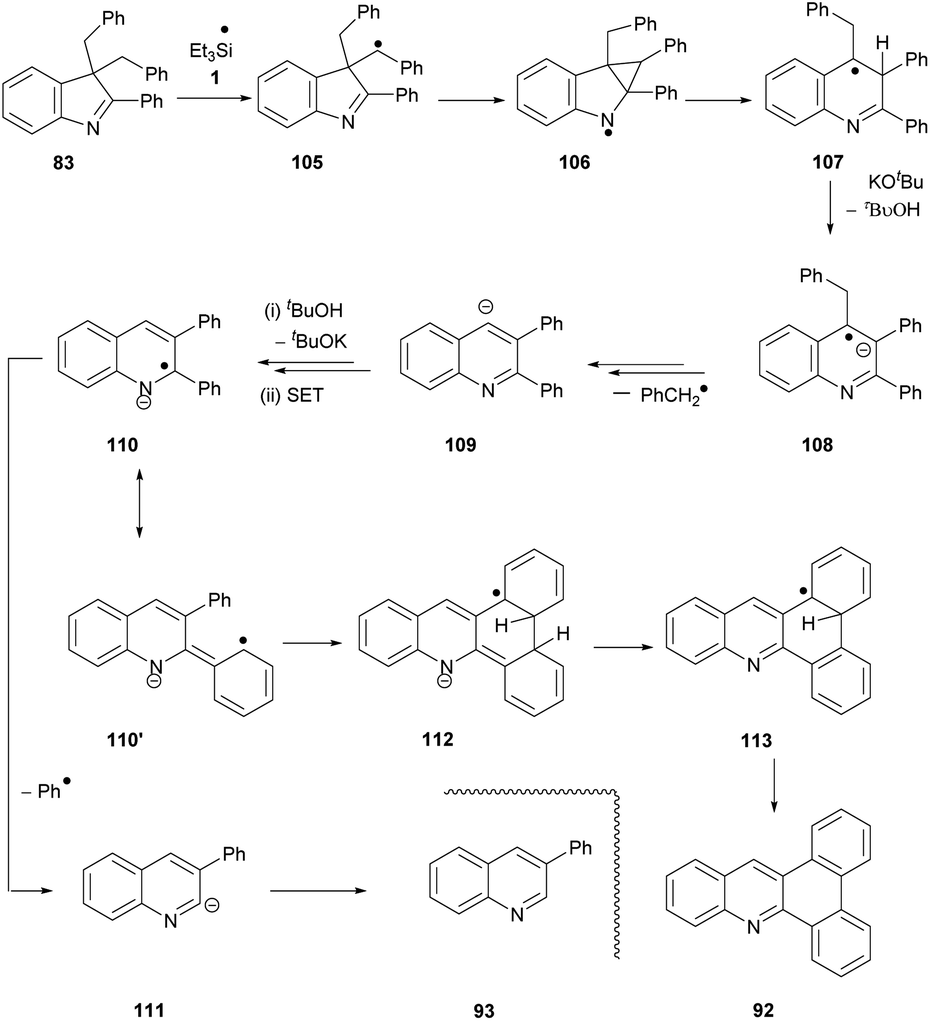 | ||
| Scheme 12 | ||
We again propose 110′ as the source of the other product, 92. Cyclisation to the neighbouring phenyl ring gives radical anion 112. The drive to aromaticity can then oversee the expulsion of an H atom (or a proton followed by an electron) and a hydride ion to give product 92.
2-Phenyl-substituted indolenines 81–83 are likely to be more receptive towards electron transfer than analogues with H or alkyl groups substituted in the 2-position, but these substrates illustrate well here the array of reactive intermediates in the KOtBu/Et3SiH reagent pair.
Conclusions
The reagent pair KOtBu + Et3SiH provides a unique interplay of reactive intermediates to react with substrates. This study of benzyl nitriles and indolenines features products arising from (i) hydride addition from silanate complex 2, (ii) electron transfer from 3 (iii) hydrogen atom transfer from anion 2, and (iv) hydrogen atom abstraction by silyl radicals 1. The range of product types observed illustrates a unique diversity of outcomes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank GSK and EPSRC for i-CASE awards to AJS and DD. We thank (i) the National Mass Spectrometry Service, Swansea and (ii) the Cronin group and Dr Emma Carrick (University of Glasgow) for high resolution mass spectra.References
- A. Fedorov, A. A. Toutov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640–1645 RSC.
- A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS.
- A. A. Toutov, W.-B. Liu, K. N. Betz, B. M. Stoltz and R. H. Grubbs, Nat. Protoc., 2016, 10, 1897–1903 CrossRef.
- W.-B. Liu, D. P. Schuman, Y.-F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS.
- S. Banerjee, Y.-F. Yang, I. D. Jenkins, Y. Liang, A. A. Toutov, W.-B. Liu, D. P. Schuman, R. H. Grubbs, B. M. Stoltz, E. H. Krenske, K. N. Houk and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6880–6887 CrossRef CAS.
- A. A. Toutov, M. Salata, A. Fedorov, Y.-F. Yang, Y. Liang, R. Cariou, K. N. Betz, E. P. A. Couzijn, J. W. Shabaker, K. N. Houk and R. H. Grubbs, Nat. Energy, 2017, 17008, DOI:10.1038/nenergy.2017.
- A. J. Smith, A. Young, S. Rohrbach, E. F. O'Connor, M. Allison, H.-S. Wang, D. L. Poole, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2017, 56, 13747–13751 CrossRef CAS.
- (a) A. A. Toutov, K. N. Betz, A. Fedorov, B. M. Stoltz, W.-B. Liu and R. H. Grubbs, US2019/0048030A1, 2019; (b) A. A. Toutov, K. N. Betz, A. Fedorov, B. M. Stoltz, W.-B. Liu and R. H. Grubbs, US2020/0115396A1, 2020.
- P. Asgari, Y. Hua, C. Thiamsiri, W. Prasitwatcharakorn, A. Karedath, X. Chen, S. Sardar, K. Yum, G. Leem, B. S. Pierce, K. Nam, J. Gao and J. Jeon, Nat. Catal., 2019, 2, 164–173 CrossRef CAS.
- A. A. Toutov, K. N. Betz, A. M. Romine and R. H. Grubbs, US2019/0241587A1, 2019.
- (a) A. A. Toutov, K. N. Betz, A. M. Romine and R. H. Grubbs, US 2019/0218232A1, 2019; (b) F. Palumbo, S. Rohrbach, T. Tuttle and J. A. Murphy, Helv. Chim. Acta, 2019, e1900235 CAS.
- I. D. Jenkins and E. H. Krenske, ACS Omega, 2020, 5, 7053–7058 CrossRef CAS.
- J. N. Arokianathar, K. Kolodziejczak, F. Bugden, K. Clark, T. Tuttle and J. A. Murphy, Adv. Synth. Catal., 2020, 362, 2260–2267 CrossRef CAS.
- A. J. Smith, D. Dimitrova, J. N. Arokianathar, K. Kolodziejczak, A. Young, M. Alison, D. L. Pole, S. G. Leach, J. A. Parkinson, T. Tuttle and J. A. Murphy, Chem. Sci., 2020, 11, 3719–3726 RSC.
- A. A. Toutov, K. N. Betz, D. P. Schuman, W. B. Liu, A. Fedorov, B. M. Stoltz and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 1668–1674 CrossRef CAS.
- S. L. Miller, Science, 1953, 117, 528–529 CrossRef CAS.
- For some previous papers on decyanation, see (a) J. M. R. Mattalia, Beilstein J. Org. Chem., 2017, 13, 267–284 CrossRef CAS; (b) P. C. Too, G. H. Chan, Y. L. Tnay, H. Hirao and S. Chiba, Angew. Chem., Int. Ed., 2016, 55, 3719–3723 CrossRef CAS; (c) E. Doni and J. A. Murphy, Org. Chem. Front., 2014, 1, 1072–1076 RSC.
- A. Kaga, H. Hayashi, H. Hakamata, M. Oi, M. Uchiyama, R. Takita and S. Chiba, Angew. Chem., Int. Ed., 2017, 56, 11807–11811 CrossRef CAS.
- (a) E. E. Kwan, Y. Zeng, H. A. Besser and E. N. Jacobsen, Nat. Chem., 2018, 10, 917–923 CrossRef CAS; (b) A. J. Lennox, Angew. Chem., Int. Ed., 2018, 57, 14898–14900 CrossRef; (c) S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 16368–16388 CrossRef CAS.
- We verified that such benzylic nitrile substrates can be cyclised also with LiAlH4 at 130 °C, although not at 70 °C, again indicative of cyclisation of an iminyl anion – See ESI†.
- J. A. Murphy, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS.
- S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS.
- R. A. Aitken, A. D. Harper and A. M. Z. Slawin, Synlett, 2017, 28, 1738–1742 CrossRef CAS.
- A. Guijarro and M. Yus, Tetrahedron Lett., 1993, 34, 3487–3490 CrossRef CAS.
- C. Däschlein and C. Strohmann, Dalton Trans., 2010, 39, 2062–2069 RSC.
- N-Methylindoles have also been proposed to be deprotonated in the 2-position by Et3SiH + KOtBu5.
- F. Huber, J. Roesslein and K. Gademann, Org. Lett., 2019, 21, 2560–2564 CrossRef CAS.
- (a) T. Holm, H. Baumann, H. Lönn, J. Lönngren, H. Nyman and H. Ottosson, Acta Chem. Scand., Ser. B, 1983, 37, 567–584 CrossRef; (b) T. Matsuyama, H. Yamataka and T. Hanafusa, Chem. Lett., 1988, 1367–1370 CrossRef CAS; (c) J. McKinley, A. Aponick, J. C. Raber, C. Fritz, D. Montgomery and C. T. Wigal, J. Org. Chem., 1997, 62, 4874–4876 CrossRef CAS; (d) T. Ramnial, S. A. Taylor, J. A. C. Clyburne and C. J. Walsby, Chem. Commun., 2007, 2066–2068 RSC; (e) N. Uchiyama, E. Shirakawa and T. Hayashi, Chem. Commun., 2013, 49, 364–366 RSC; (f) E. C. Ashby and T. L. Wiesemann, J. Am. Chem. Soc., 1974, 96, 7117–7119 CrossRef CAS; (g) E. C. Ashby, Acc. Chem. Res., 1988, 21, 414–421 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS.
- An alternative direct fragmentation from 100 → 103 is also considered. Vinyl radical fragmentations often have high energy transition states, but additional driving force is expected in converting 102 → 103 as the nitrogen-containing ring in 102 is not aromatic but becomes aromatic in 103.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04244g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |