
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDigitising chemical synthesis in automated and robotic flow
Tomas
Hardwick
abc and
Nisar
Ahmed
 *a
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: AhmedN14@cardiff.ac.uk
bNational Graphene Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cDepartment of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 7th October 2020
Abstract
Continuous flow chemical synthesis is already known to have many attributes that give it superiority over batch processes in several respects. To expand these advantages with those from automation will only drive such enabling technologies further into the faster producing, more efficient 21st century chemical world. In this report we present several examples of algorithmic chemical search, along with flow platforms that link hardware and digital chemical operations on software. This enables organic syntheses to be automatically carried out and optimised with as little human intervention as possible. By applying such enabling technologies to the production of small organic molecules and pharmaceutical compounds in end-to-end multistep processes, a range of reaction types can be accessed and, thus, the flexibility of these single, compact flow designs may be revealed. Automated systems can allow several reactions to take place on the same setup, enabling direct comparison of reactions under different conditions. Moreover, the production of new and known target compounds can be made faster and more efficient, the recipes of which can then be stored as digital files. Some of the automating software has employed machine-powered learning to assist the chemist in developing intelligent algorithms and artificial intelligence (AI) driven synthetic route planning. This ultimately produces a continuous flow platform that can design its own viable pathway to a particular molecule and then carry it out on its own, allowing the chemists, at the same time, to apply their expertise to other pressing challenges in their fields.
 Tomas Hardwick | Tomas Hardwick obtained his master's degree in chemistry at Cardiff University, performing organic chemistry research focused on electrochemical organic synthesis in batch and flow. During this time he spent a year working on heterogeneous catalysis at the University of Florida. He is currently a PhD student in the University of Manchester working with the Advanced Nanomaterials Group, with research interests involving two-dimensional materials for photovoltaics and photoelectrochemistry and flow technology. |
 Nisar Ahmed | Nisar Ahmed obtained his PhD in organic chemistry (2012) under Brain Korea BK21 fellowship working in the group of Prof. Kwang S. Kim (POSTECH, Korea). Then, he moved to the University of Zurich, Switzerland (2013) for a postdoctoral stay with a Novartis Fellowship. Subsequently (2015), he joined the University of Bristol as a research associate. He started his research career in late 2016 at the School of Chemistry, Cardiff University, United Kingdom with early career EU-COFUND grant as Sêr Cymru Fellow. However, his recent research support includes an EPSRC grant (UKRI). He has also been an Adjunct Faculty at HEJ Research Institute of Chemistry, ICCBS since 2018. His research interests are the development of green & sustainable technology in organic synthesis using batch & microflow chemistry and molecular recognition of biomolecules. |
Introduction
Compared to batch processing, chemical synthesis performed in continuous flow offers the user a versatile and a more advantageous approach to product manufacturing, as evidenced by the significant attention this enabling technology has received in recent years.1–6 Such a methodology offers an enhanced safety perspective by flowing material within channels from stock containers to reactors, thus minimising human contact. It also opens the door to scalability by intercepting the flow path with a range of highly adaptive process modules (reactors, separators, filters and analytics). Thus, sequential, non-simultaneous reactions can be combined to perform multistep processes.7–14 Continuous flow gives better reproducibility, smaller time scales due to efficient mixing and heat and mass transfer, variable flow rates that can accelerate reaction rates through process intensification,7,8,15 and reconfiguration and reuse of reactors under different conditions. Other attributes include real-time reaction monitoring, storage of intermediates, a possible lowering of production costs and an increase in the quality of the products.16,17In addition to the benefits of flow chemistry, automating synthesis platforms can assist the human experimenter and overcome other outstanding issues. For example, typical published works in journals and reaction databases are plagued by human bias that leads the chemist away from performing certain experiments, while incomplete and ambiguous recordings of specific details hinder reproducibility.10 Furthermore, the huge knowledge banks of chemistry are growing so fast that there are now millions of reaction combinations and details that cannot be considered by people. Modern computers, while heavily underused in chemistry, can overcome such issues by digitising chemistry in coded formats that can be easily accessed and operated at the push of a button. The creation and rapid search of complete molecular chemical spaces from reaction databases can be streamlined by a machine, which can then design an optimal synthesis pathway to existing compounds, and even discover new compounds and novel chemistries. This will allow the more efficient creation of new materials, drugs and on-demand synthesis of several chemical types, which is ideal for pharmaceuticals during a shortage or for those with short-lifetime intermediates. Standard chemical processes are designed so that the pathway is separated into individual and independent steps. Each step involves laborious processes including workup, purification and characterisation. These are preferably avoided in automated flow, and reaction conditions compatible with all steps are carefully chosen.18
On the other hand, most continuous flow setups focus on core motifs rather than on a specific compound. Extending their capabilities to automatic and reconfigurable systems will require multistep syntheses of complex molecules, involving several unit operations that do not occur in the same order. These may demand different residence times, making them challenging and laborious to design and operate. It is, therefore, desirable to develop a flexible means of running flow syntheses that enables a comparable or enhanced performance compared to batch processes, with a reduction in reaction times and manual labour. A system that could predict its own reaction pathways without the necessity to be physically reconfigured by a human operator for each different synthesis is an ideal candidate. The absence of detailed information in reaction databases means that in order to digitise chemistry in automated flow, open-source data that specifies conditions and agreed-upon data standardisation metrics are necessary for future progress.19 The collaboration and advancement of machine-learning-assisted chemistry will require publications and datasets to be written in machine-readable formats that are also contextualised, transparent and traceable.20 Moreover, information concerning side-products is often left out and so an improved prediction of the product distribution is needed, which can be reviewed as a prediction of the major recorded (>50% yield) product.19 There is also a bias in the literature towards reporting only successful reactions. This is a shame because data about negative and failed reactions can guide the machine model to understand reactivity trends, mimic patterns and provide complementary knowledge, even from poor yielding, a typical reactions. Failed reactions have the potential to present future research opportunities and have already been of use in materials chemistry.21 Of course, digitising all the necessary reaction rules and other required elements is impractical to do by hand. It is not scalable due to the massive amount of data out there, not to mention the full substrate scope and reaction incompatibilities, and that it is currently dependant on a small number of chemists with computational experience (something that most chemists have never been taught).22 Machine learning and AI are, therefore, desirable for this task.
To that end, several recent efforts by chemists and chemical engineers have tried to employ digital chemistry to automatically generate and discover new (and known) products with machine learning in a way that enhances both reproducibility and productivity, with as little human intervention as possible.10–14,23,24 In addition, removing the physical barriers to the organic synthesis will enable processes to be accelerated and experimental setups to be practically simplified. Accelerated production of lab-scale and commercial-scale quantities of small molecules to larger compounds such as biopolymers (e.g. peptides) can be attained by automated processes in safer, faster and reproducible ways.25 In some cases, different synthetic routes can be directly compared on the same system under different conditions.14 Although many automated multistep syntheses that have been reported have shown advances in technology and reduced human intervention, they still rely on time-consuming manually performed tasks. These include the design of synthetic routes, system reconfiguration13 for specific chemistries and some only perform some types of chemical reactions.10,26,27 Note that for those who are familiar with continuous flow but less familiar with the digital side, we have written this work in a manner that can provide an overview of some recent developments and breakthroughs in the combination of chemistry with computer science. We appreciate that greater learning about some of the concepts may be desirable after reading; however, in order not to overcomplicate things or to take anything away from the original papers, we remind you that there are many citations throughout this review for you to access for a deeper understanding of the key citations.
To date, there has been no universal method of automating chemical synthesis and, therefore, this should be a goal that we as chemists should strive towards. The combination of flow with automation will allow the evolution of chemical laboratories into a faster producing and more efficient future. Step one will be designing a flow system that is both reconfigurable and robust in its pursuit of different multistep chemical synthesis outcomes. Step two would then be linking the hardware to user-friendly software that would not only remotely control the setup, but also digitally store known optimised recipes that can be rerun, even by unskilled chemical operators. Finally, the third step in the pursuit of the lab of the future will be to upgrade the simple automated platforms with machine-powered learning and artificial intelligence (AI) that can design its own synthetic routes and carry them out on its own.
Digitising continuous flow chemistry
An emerging virtual tool to assist synthetic chemists in finding better pathways is to combine chemically relevant hardware with software that executes computer-aided synthesis planning (CASP): digitised chemical knowledge in the form of an executable program.19 Originally used to predict routes before they were carried out in practice, CASP is envisioned to accelerate processes and to be combined with robotic platforms for faster experimental testing and de novo synthesis.28 A decent program would be one that inputs a chemical structure and outputs a detailed list of plausible reaction pathways to the target from commercially available materials. Notwithstanding nearly 60 years of research efforts, however, CASP has not yet been widely accepted.28 This may be because of pessimism regarding limitations when it is applied to complex molecules (i.e. natural products) and intricacies of process and medicinal chemistry.19 There is also criticism of digitally extracting chemical information from databases (published journals) due to high noise and lack of raw data/“chemical intelligence”, the cost of computationally expensive templates, or that it does not scale to the ever-growing knowledge banks.28–31 Nevertheless, a study by Adamo et al. combines the formulation of the final product with multiple complex syntheses, purifications and in-system reaction monitoring in a digitally controlled, reconfigurable continuous flow platform.11 Attention was focused on aqueous or alcohol-based concentrated formulations that could be stored and remain stable for one month. The use of solid formulations (i.e. tablets) was beyond the scope of their work since modules to perform drying, powder transport, blending and tableting operations would require much more additional space. Their reconfigurable system, the size of a refrigerator, consisted of an upstream unit containing stocks, pumps, pressure regulators, reactors and separators, and a downstream unit for precipitation, crystallisation and formulation (Fig. 1). Real-time monitoring could occur through a FlowIR. Hardware was then expanded to include flow rate, pressure and temperature sensors so that LabVIEW programs and the modular X Series data acquisition (DAQ) device could be employed to implement syntheses and for automation. Four pharmaceutical products with different molecular structures were produced in hundreds to thousands of oral or topical liquid doses per day. These were diphenhydramine hydrochloride, also known as Nytol (UK) or Benadryl (USA), lidocaine hydrochloride, diazepam (Valium) and fluoxetine hydrochloride (Prozac or Sarefem). They were obtained in good yields of 82, 90, 94 and 43%, respectively, in production times ranging from 12.2 to 44.7 h (the bulk of which was dominated by downstream precipitation steps). As a comparison of time between this flow approach and a conventional batch process, diphenhydramine hydrochloride was complete within 15 min (a batch process in contrast would require over 5 h),32 lidocaine hydrochloride took 36 min (a batch process takes 60 min of refluxing in toluene33 to 4–5 h in benzene)34 and diazepam in 13 min (compared to 24 h for a batch process).35 The advantage of this system is contingent on the production of pharmaceutical compounds still being heavily reliant on batch synthesis, usually taking about 12 months to complete, with multiple fragments being made at different locations to construct the active pharmaceutical ingredient (API), which is finalised at a different plant.11 This may result in long production times, possible supply chain disruptions, variations in quality control and drug shortages due to a limited number of venders (particularly when there is an increase in demand, e.g. during an epidemic or pandemic). It would be much more desirable to formulate high-quality APIs in a more flexible and robust manner, such as through the use of continuous flow.17,36 | ||
| Fig. 1 Refrigerator-sized, reconfigurable flow system for fabricating APIs with upstream synthesis modules and downstream purification and formulation modules shown. Reproduced from ref. 11. | ||
The next logical step to accompany the inherent advantages of continuous flow (efficient mixing, heat and mass transfer, etc.) is the development and incorporation of in-line analytical techniques, such as IR, NMR and MS.37 Spectral responses will, therefore, enable real-time data acquisition in synergy with continuous flow attributes: e.g., high-speed monitoring of large amounts of reaction progression data, precise control over experimental parameters and hence the final outcome. In addition, novel discoveries and new advances can be attained in a more timely manner. The Jensen and Jamison group have built an automated and reconfigurable continuous flow platform, with the intention of optimising above-the-arrow conditions for a broad scope of reaction types.12 This can help enhance yields and selectivities and reduce labour times by constantly receiving feedback from online analytics (IR, MS, Raman and HPLC). Optimal conditions can then be transferred to another lab and repeated with high fidelity. Control of reagents, hardware modules and analytics can be performed in user-friendly MATLAB and LabVIEW software and, thus, will allow remote progress monitoring. Their system is also compatible with optimisation algorithms, e.g. flexibility and generality can be provided by the stable noisy optimisation by a branch and fit (SNOBFIT)38 algorithm. Once an optimisation is discovered, it can be stored and reused by others in downloadable electronic files. As shown in Fig. 2, the setup contains several types of plug-in reactors, a liquid–liquid separation membrane, sensors and analytics. With thousands of possible reactor module configurations, the system provides good sorting after generality and ease of use that can realise optimisations and evaluations within hours or days. This is also ideal for easy reaction comparisons. Its competency was then examined by generating over 50 compounds from seven chemical transformation types, with good to excellent yields and practical flow rates. These were the Buchwald–Hartwig amination (72–99% yield), Horner–Wadsworth–Emmons olefination (67–97%), reductive amination (67–97%), Suzuki–Miyaura cross-coupling (88–99%), a nucleophilic aromatic substitution (88–99%), photoredox catalysis (73–93%), and a ketene generation (47–90%).
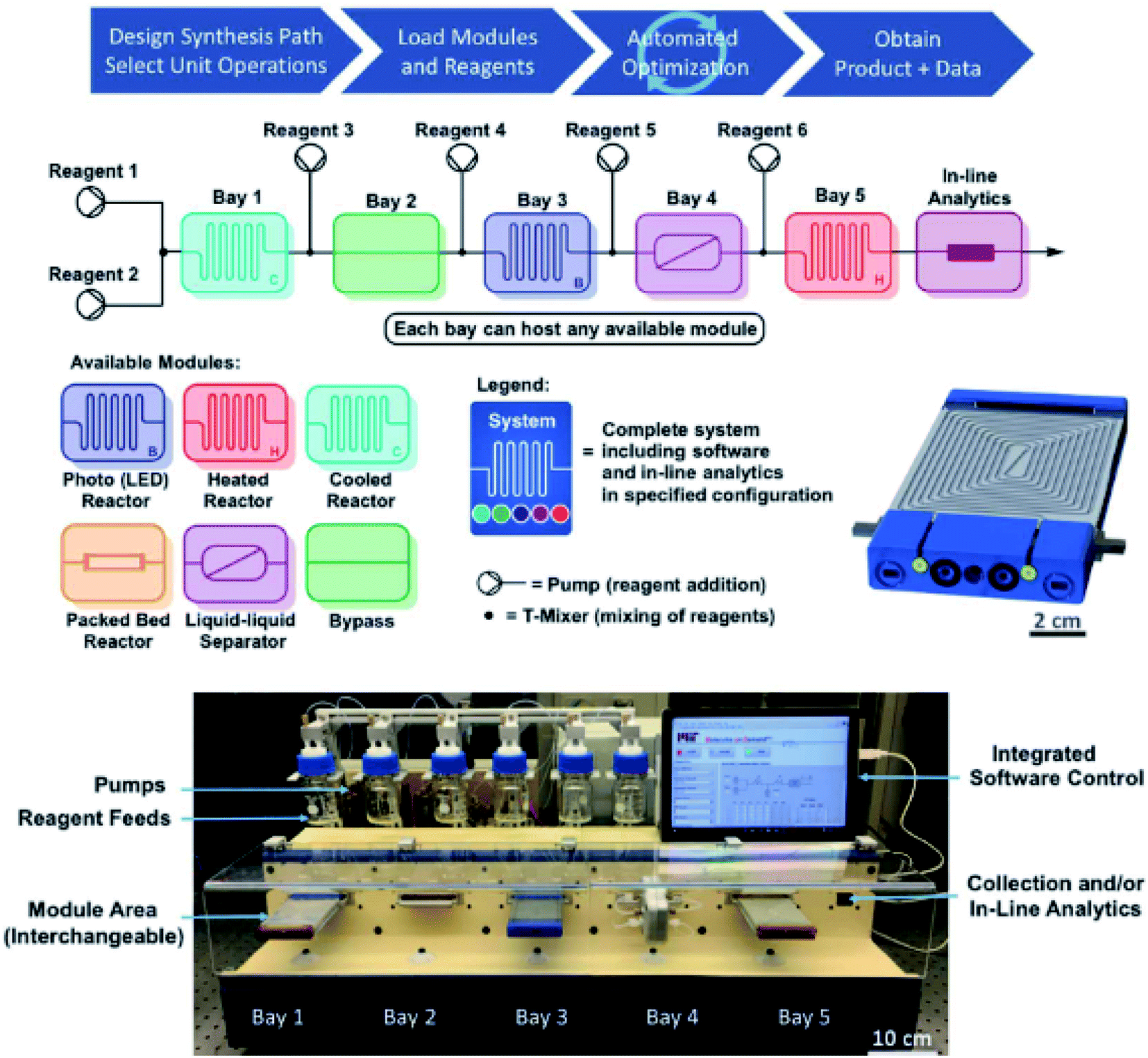 | ||
| Fig. 2 Plug-and-play continuous-flow setup illustrating its operation via a general four-step protocol, schematic diagrams of the overall arrangement with interchangeable process modules, and a CAD (computer-aided design) LED reactor. Reproduced from ref. 12. | ||
These two flow platforms above are both reconfigurable, but in different ways, i.e. the first is reconfigured in silico, allowing the user to change the flow route within the system, while the plug-and-play approach involves the physical removal and insertion of reactor modules. The latter has the added merit of incorporating analytics in the flow path, enabling faster analysis and, hence, optimisation compared to the former which spends a lot of time on precipitation and crystallisation before product identification. Both of them also operate in a linear fashion in the same way that humans perform batch reactions, one after the other. But is this the optimal way to perform flow chemistry? Chatterjee et al. have argued against this typical method by designing a radial synthesiser that can run single or multiple reactions in both linear (conventional) and convergent strategies (Fig. 3), with automatic reconfiguration.14 This is motivated by constraints in mass flow that dictate that the flow of the input reagents must equal the output flow. This can be affected by temperature, reactor volume and type, and so a specific sequence of modules and conditions is required for each synthesis. For a flow system to be competitive, it must therefore be reconfigurable and have different reactors that can comply with a range of optimal syntheses. The system is comprised of a central hub that uses a 16-way valve to direct reagent flow to surrounding storage and stock containers, reactors and in-line analytics; intermediates can be stored so that they may join together at a later phase. This allows reactions to be performed under their individual optimum conditions and in silico use and reuse of reaction modules. Using LabVIEW, software is remotely controlled by inputting the necessary reaction details into the graphical interface containing a series of virtual instruments that link and control the hardware and software (timed to allow for full automation).
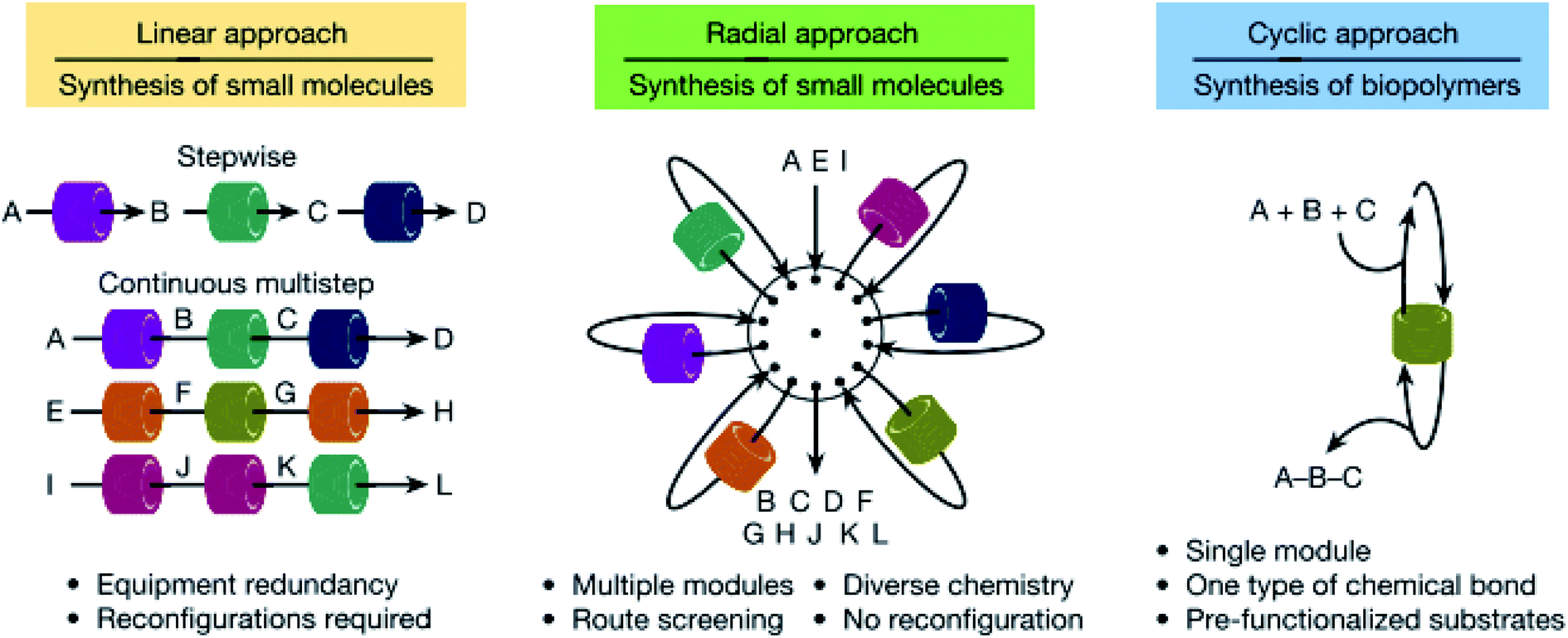 | ||
| Fig. 3 Pictorial examples of three different approaches to organic synthesis in flow. The advantages of the linear and cyclic approaches are combined to give the radial approach which has a central switching station surrounded by process modules for maximum versatility with minimal equipment. Reproduced from ref. 14. | ||
To compare this radial approach with a typical linear method, the authors applied the system to the optimisation of the multistep synthesis of the anticonvulsant drug, rufinamide (Scheme 1). Monitored by FlowIR, the azide and amide intermediates were independently synthesised before the concluding copper-catalysed cycloaddition was optimised, affording the target molecule in 70% yield (88% NMR yield).14 In conjunction, the linear method afforded rufinamide in 45% yield (83% NMR yield) using near identical conditions. The exception was the concentrations of the two (blue) starting materials which were diluted from 1.5 to 1.0 M because the triazole intermediate (green) was found to be insoluble at the original concentration.14 Thus, less concentrated solutions (which may also explain the lower yield due to the lower final concentration) and due to the addition of water, resulted in a more complex purification. This study is noteworthy because it shows the decoupling of reaction steps by automatically directing effluent lines to the appropriate reactor or storage facility; therefore, reactions are independent of one another, thus removing mass flow constraints. Furthermore, this one system can be used for different target molecules and its ability to reconfigure itself can reduce development times.
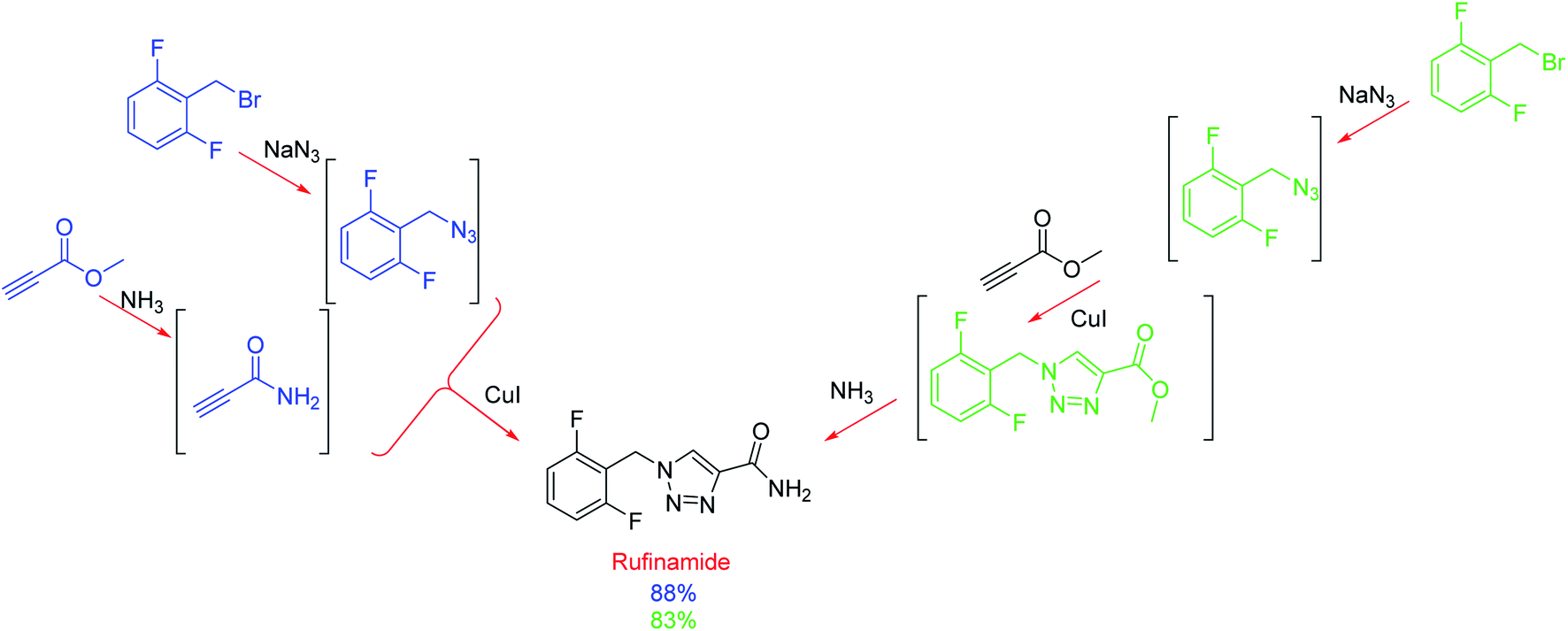 | ||
| Scheme 1 Radial (blue) and linear (green) synthetic routes of rufinamide. | ||
On a slight tangent and in addition to machine learning and analytics, reactor equipment can be exchanged to suit a given synthesis or even printed for integration into a flow platform. Cronin et al. sealed 3D print polypropylene reactionware that could be used as part of a digital synthesis platform.39 This was constructed via a chemical to computer-automated design (ChemCAD) approach, and, henceforth, allows the user to load chemicals in a simple manner for benchtop-scale reactions. Multiple components were printed to conduct small-scale productions of three drug molecules: namely baclofen, zolimidine and lamotrigine. The objective here is to digitise the chemical manufacturing of fine molecules and pharmaceuticals in a cheap and reproducible (standardised) way. The authors describe the process as a “platform-independent digital code” which would be useful for non-chemists (medical professionals and biologists) to print and recreate the synthesis, simply by following a given set of instructions. Compounds that are needed then and there or that have short shelf lives are ideal candidates for this. Their particular design is for existing compounds and each different compound requires a different 3D printed module. For example, the baclofen reactor, which consists of two liquid–liquid separators, a set of evaporations and filtrations for three reactions, would not be suitable for lamotrigine. It should be noted that the efficiencies of the polypropylene reactors were slightly lower than those of the glass reactors, attributed to lower product recovery owing to the higher degree of surface roughness of the polypropylene surface. Moreover, the efficiency of the zolimidine (copper-catalysed iodination) reaction was significantly lower than those of the other two drugs, likely due to unwanted side reactions of iodine with the polypropylene.
While the aforementioned strategies combine hardware with simple software, computers are used mainly to perform and analyse the chemicals produced from a small number of reactions. On the other hand, machine learning and AI have been accepted to manage and analyse big data, while flow chemistry, despite its advances, is yet to fully accept high-throughput reaction screening with multiple continuous (temperature, pressure, residence time, etc.) and discrete (catalyst, ligand base, etc.) variables.40 Therefore, leading on from reconfigurable flow systems and in-line analytics, the data can then be digitally analysed by intelligent algorithms and autonomously used to create a chemical space that will allow fast optimisation and give the opportunity for a dial-a-molecule request. Techniques that study the effect of multiple parameters such as “Design of Experiments” (DoE) are also useful to place in tandem with flow and automation. Digitising chemical objectives in this way will minimise human intervention, as the synthesis platform will be controlled by an algorithm making decisions based on the user's desires (yield, time, selectivity etc.)
The use of high-throughput experimentation (HTE) is becoming increasingly more desirable since it can significantly accelerate the exploration of chemical space and be performed by increasingly available off-the-shelf robotics.41 HTE is particularly renowned for being able to practically perform thousands of different nanomole-scale reactions on a daily basis, using an appropriate search algorithm that has defined the scheduled chemical space experiments by certain merits: i.e. time, cost and resources.41 Moreover, HTE is ideal to provide AI and other models with large amounts of information and has already proven itself useful in scale-up optimisations of known compounds as well as the discovery of new reactions.42–47 HTE can be coupled with advanced analysis techniques to decrease the analysis time, e.g. matrix-assisted laser desorption ionisation-time-of-flight spectrometry (MALDI-TOF), which has can handled thousands of experiments in minutes.48 Scientists at Merck have developed a nanomole-scale synthesis platform to successfully optimise Pd-catalysed Buchwald–Hartwig C–O, C–N and C–C cross-couplings in dimethyl sulfoxide (DMSO) at room temperature.47 Automated reactions were optimised using robotics from biotechnology and mass spectrometry (MS)-based high-throughput analysis, producing drug-like fragments by iterative reaction screening in 1.0 ml volumes. 1536 reactions could be evaluated in 2.5 h using only 0.02 mg of starting material per reaction. Later, this was extended to include the affinity of a compound to a target protein (called NanoSAR) that allows for in situ analysis of structure–activity relationships.49 Off-the-shelf robots are becoming more and more commonplace and so Merck has provided a good example of how chemistry can be devised for the robot's capabilities. This work does, however, suffer from limitations, such as the need for non-volatile solvents (e.g. DMSO), low-resolution MS and no heating to prevent solvent evaporation. In contrast, Perera et al. have used in-line high resolution liquid chromatography-MS for real-time analysis of a flow system that deals with nanomole to micromole solutions.40 This makes it ideal for automated biological testing. The team demonstrated the capabilities with high-throughput reaction screening of Suzuki–Miyaura coupling reactions under a range of variables (volatile and non-volatile solvents, temperature, pressure, residence time, catalyst, ligand and base) that totalled 5760 reactions. Only ∼0.05 mg of substrate per reaction was required, enabling >1500 reactions to be screened per 24 h. Scaling up the HTE capacity for more useful material quantities was demonstrated by injecting 100 consecutive segments to produce 10–100 mg of a specific compound per hour whilst preventing cross-contamination between segments.
Doyle et al. have also predicted the yield of Pd-catalysed Buchwald–Hartwig C–N cross-coupling products using a random forest algorithm that calculated the multidimensional chemical space via HTE from 4608 reactions.50 Components of the Buchwald–Hartwig amination, i.e. atomic, molecular and vibrational descriptors, were generated by the random forest model which increased the yield prediction efficiency to a degree that outperformed other linear regression analysis. The reaction descriptors and yields were then used as inputs and outputs, respectively. The overall prediction is generated by a random forest (a nonlinear approach) constructing decision trees from random data samples. As the number of data points increases, the model is updated and chemical space can be navigated better. Similarly, Doyle and co-workers also used the random forest algorithm to accelerate the yield prediction and identification of optimal conditions for the deoxyfluorination of a range or alcohols using sulfonyl chlorides.51
Nowadays, however, large reaction databases (USPTO, Reaxys, SciFinder) are available to train CASP approaches for integration with machine learning. This can streamline the search for known molecule syntheses and help to create and corroborate new synthesis planning methods. Two retrosynthetic examples that have emerged are Chematica,52,53 a program which uses rule-based code, and the Monte Carlo tree search algorithm employed by Segler et al.28
Creating a library of retrosynthetic templates by hand-coding reaction rules has been attempted in the past, but has been plagued by a user-unfriendly syntax and incomplete databases.19 However, with adequate investment in time and labour, beginning in 2001 a program called Chematica (commercialised as Synthia) has been able to gather a network of ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hand-coded rules which, for validation, were sufficient to experimentally improve the yields, time and cost of several medically relevant compounds.53 It works via an algorithm tree search (Fig. 4) coupled to undisclosed AI heuristics, allowing Chematica to discover new pathways, have lists of functional groups that are incompatible for each template and possess a user-friendly graphical interface. As a guide, tree-based heuristics are advantageous as they allow design flexibility mimicking chemical intuition, favour synthetic routes that can be allocated with the knowledge of other alternatives and increase the efficiency of navigating through chemical space. The tree is attuned to terminate its braches at commercial reactants, can penalise pathways that involve strained intermediates or practically infeasible structures and non-selective reactions, and can store and reuse routes as part of another strategy if one of its products is required. This saves the need for further search expansion. Due to its reliance on digitising the rules by hand, the growth of this program is hampered by the increasing volume of literature.
000 hand-coded rules which, for validation, were sufficient to experimentally improve the yields, time and cost of several medically relevant compounds.53 It works via an algorithm tree search (Fig. 4) coupled to undisclosed AI heuristics, allowing Chematica to discover new pathways, have lists of functional groups that are incompatible for each template and possess a user-friendly graphical interface. As a guide, tree-based heuristics are advantageous as they allow design flexibility mimicking chemical intuition, favour synthetic routes that can be allocated with the knowledge of other alternatives and increase the efficiency of navigating through chemical space. The tree is attuned to terminate its braches at commercial reactants, can penalise pathways that involve strained intermediates or practically infeasible structures and non-selective reactions, and can store and reuse routes as part of another strategy if one of its products is required. This saves the need for further search expansion. Due to its reliance on digitising the rules by hand, the growth of this program is hampered by the increasing volume of literature.
 | ||
| Fig. 4 Complex tree-search network in Chematica. Nodes can be displayed as 2D or 3D molecular structures (for basic modelling calculations) and can be expanded down the retrosynthetic path. Blue nodes denote products; green are minor/side-products, red are commercially available substances, and yellow halos denote regulated substances. Reproduced from ref. 53. | ||
In contrast to this hand-coding approach for Chematica, Segler et al. described a neural network, guided by a heuristic best first search (BFS), for retrosynthesis prediction that used reactant fingerprints to rank 8720 extracted reaction templates whilst avoiding reactivity conflicts.54 These came from 3.5 million reactions from the Reaxys database with up to 78% accuracy. This was then extended to a Monte Carlo tree search and symbolic AI approach, trained on every published organic reaction (12.4 million), to predict full retrosynthetic routes.28 Their algorithm used reactant fingerprints from the reactions to create reaction templates; rules that occurred more than 50 times in the database were considered, totalling 17134 and 301671 for 52% and 79% of all single-step reactions, respectively. It also acted as an “in-scope filter” to remove poor-quality suggestions, e.g. not practically feasible or too long and complex, with excellent speed (30 times faster while solving for twice as many molecules compared to the traditional computer-aided search method based on extracted rules and hand-designed heuristics).28 Moreover, for testing, 100 million negative reactions were generated. For verification, expert chemists underwent double-blind A/B testing which revealed that the quality of the digitised AI routes was equivalent to those of a human. This work is commendable because of the high level of efficiency, accuracy and sophistication that is both faster and comparable to a human, has the ability to minimise the number of reaction steps and has employed such a large number of published experimental examples which previous works have not done due to the complexity and nature of only publishing successful reactions. These retrosynthetic strategies, however, do not determine the feasibility of the forward reaction, directly provide reaction conditions, or have their code open-source/model available for comparison (negatively impacting reproducibility and progress), and their reliance on manual labour hinders scalability and standardisation.
From another angle, a similarity-based approach to automated retrosynthesis has been suggested by Coley and Jensen et al. whereby the similarity between product and reactant is used to identify the forward strategy.55 This system works by mimicking how a chemist would think about synthesising a compound. It is based on previously reported syntheses (40000 reactions from Reaxys or SciFinder) from similar motifs of other compounds and then determines whether the suggested routes are appropriate. The similarity between the target and reactants is calculated and used to quantify and rank the proposed reaction. Here, a highly generalised template is produced, but like most other template-based works, e.g. that of Segler et al., the templates only consist of the bare minimum of chemically relevant information (only the atoms involved in the immediate reaction).28 This eliminates the need to define heuristics for their extraction, to code conflicts in reactivity by hand, and since the template is not as extensive as usual, the computational speed is not as impeded. Out of 5000 test reactions, proposed reactions within a specific reaction class were successful 52.9% of the time. When 5 and 10 disconnections were suggested, the success rate increased to 81.2% and 88.1%, respectively. While this study cannot be directly compared to Segler et al.'s because their open-source code is not available, it can be compared to the template-free seq2seq model reported by Liu et al.56 The similarity approach exceeds the seq2seq model (81.2% compared to 57% for top-5 accuracy) and can be applied to more complex pathways (i.e. to drug compounds).55 This method is, however, simplistic in the sense that it does not include information about conditions such as temperature, catalysts, reagents or solvents. Also, the routes do not consider other experimental merits like yield, cost, safety, workup etc. nor do they suggest any major benefits over a human chemist's knowledge of the reaction types in question. Moreover, this similarity search is not AI and does not account for stereochemistry, unlike the Chematica tree search or Segler et al.'s Monte Carlo tree search algorithms.57 Compared to Chematica, the speed and amount of data used are similar; however, Chematica has much more experimental validation. Segler et al.'s Monte Carlo tree search uses a lot more data and is much faster than the other two, but has much less experimental validation than Chematica (and is more similar to the similarity search in this respect). Furthermore, Chematica and the Monte Carlo tree search are very competitive with humans, whereas this aspect is not really known for the similarity search.57
Moving on from this and to gain a better insight into true chemical intuition and to overcome some of the flaws of previous strategies, Coley at al. compiled a reaction database of 15000 US patented reactions with plausible but negative reaction examples.22 Given a certain set of reactants, a neural network model is trained to predict the major product of the reaction and in doing so learns the probability of producing a compound based on certain modes of reactivity. The pool of potential products is overgeneralised to increase product coverage rather than specificity. The machine learning operates in a two-step manner (Fig. 5a); the first step is to produce a library of forward reaction templates to define chemically plausible products. In addition, since a large number of potential products could be formed by a template, a filter is put on that excludes any reaction that proceeds at a rate that is insignificant compared to others. This overcomes the constraint of only considering high-yielding data and allows more information to be extracted from the other plausible reactions.22 Secondly, a 5-fold cross-validation of each reaction candidate selects and assigns the major (recorded) product as “true” and ranks it as rank 1 in 71.8%, rank ≤3 in 86.7%, and rank ≤5 in 90.8% of cases. The “false” products are the generated plausible alternative products. Product likelihood scores could then be mapped and compared as a distribution of probabilities in a softmax network layer. The most abundant products can then be predicted in a way that focuses on the fundamental reactant-to-product transformation instead of molecular fingerprints. Therefore, when implemented, an accuracy of 72% exhibited for the top-ranked products suggests that the method has realistic practicality, because one product can be determined out of a variety of competing reactions in the glassware.
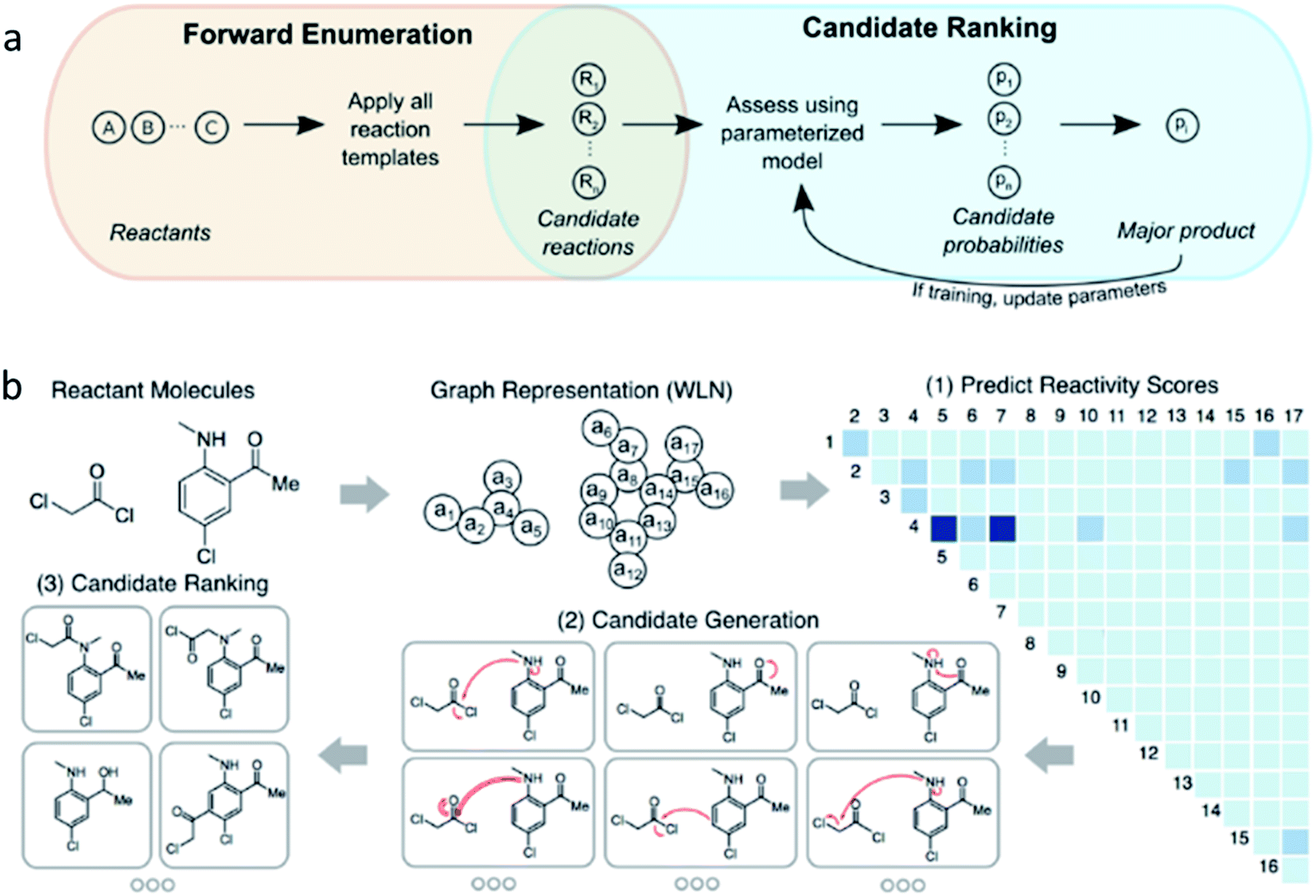 | ||
| Fig. 5 Two approaches to predicting outcomes of organic reactions. (a) Model framework combining template-based enumeration and neural network-based candidate ranking. (b) A graph convolutional neural network, where reactants are represented as a graph, learns to calculate likelihood reactivity scores of each bond change between each atom pair for focused enumeration of possible products, which are then filtered by certain rules and ranked. Reproduced from ref. 19 and 22. | ||
Later, this work was extended to include solvent information and atom-mapped molecular graphs were constructed from changes in bond order from the reactant pool.58 This model saw an improvement in interpretability and performance, and provided a way to understand reactivity by viewing molecular structures in terms of bonds being formed/broken. Likely reaction sites are identified, then products are enumerated (rather than digitising the molecule's functional groups to code) and ranked. The graphs (Fig. 5b) have edges corresponding to bonds and nodes as atoms. From here, structural information (e.g. aromaticity, atomic number, degree of connectivity) and geometrical and electronic features (e.g. surface area contribution, charge) are observed. This time the major product was correctly predicted from reactivity in over 85% of cases, notably higher than previous machine learning attempts, with only 100 ms of calculations required per example. The model is designed to mimic the rationale of a human chemist and was found to be competitive with a group of human chemists. Drawbacks of this approach include the sample size limiting the statistical power and templates limiting scalability (outside of which predictions cannot be made). Nevertheless, a broad range of reaction types can be studied using a knowledge bank much larger than that of a person, and a mixture of products (not just the major ones) can be catalogued, which is ideal for impurity identification and quantification.
Coley et al. have also developed an open source software framework (they call ASKCOS) for CASP to integrate a robotically reconfigurable flow apparatus with an AI-driven retrosynthesis prediction algorithm.13 Using protocols they have previously developed,22,58 mentioned above, a library of 163723 rules were algorithmically extracted from 12.5 million published single-step reactions from Reaxys. To reduce the probability of proposals that would be unfeasible in practice, the program incorporated RDKit and RDChiral to perform reactions and to make sure they occurred in a consistent manner when dealing with stereochemistry.59 Forward reaction templates were then assessed by examples of around 15 million published positive reactions and 115 million artificial negative reactions, via a binary classifier based on Segler et al.'s “in-scope filter” which removes low-quality suggestions.13,28 After this, a forward predictor model36,60 would predict the generated product and side products. A neutral network model would be used to provide reaction conditions to reach a specific target, i.e. solvents, temperature, catalysts and reagents.61 Finally, plausible templates were combined as chemical recipe files (CRFs) that act as the intermediate between the robotic flow platform and the software.13 The CRFs contain practical information, such as the location of stock solutions, their paths and flow rates throughout the system and the sequence of modular process units that are to be moved to and from the platform and the storage stack for a particular synthesis. The physical system, shown in Fig. 6, consists of a robotic arm which connects plug-in flow reactors, membrane separators and reagent lines on a fluidic switchboard with computer-controlled pumps. Selector values can choose up to 24 stock solutions, including a cleaning solvent which flushes the system. As a means of demonstrating the utility of this system, 15 medically relevant small drug-like molecules were synthesised in good yields.13
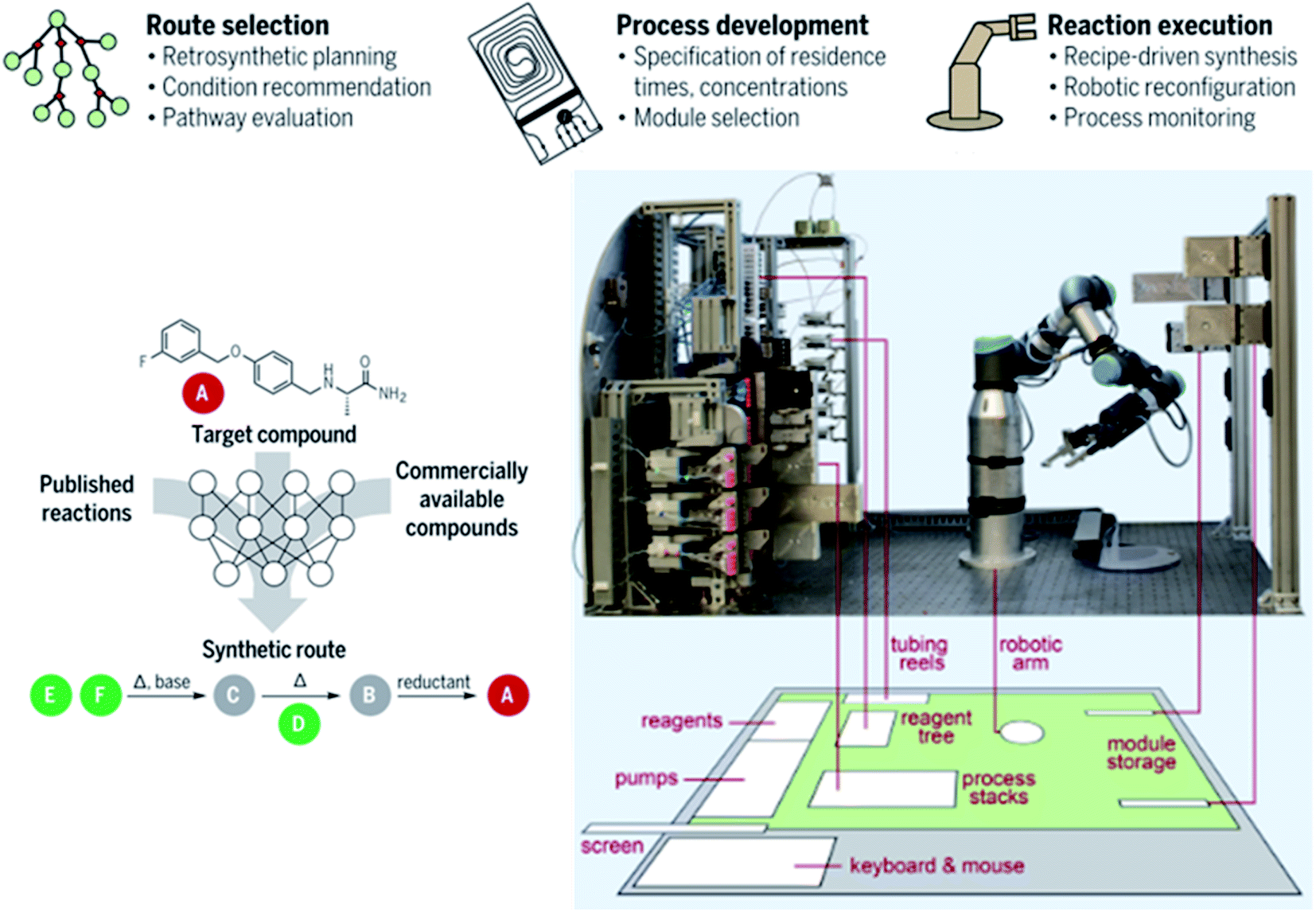 | ||
| Fig. 6 Photograph of a robotically controlled, reconfigurable continuous flow system showing a 6 × 4 ft working table floorplan (grey), ventilated enclosure (green) and multistep synthesis route planning thought process assisted by AI. Reproduced from ref. 13. | ||
The robotic arm is an interesting approach, different from the aforementioned in silico reconfigurable flow setups (Fig. 1 and 3), and driven by a more advanced algorithm that allows the robot to design and carry out its own reactions once an operator has told it what the desired product is while it shares some similarities with the plug-and-play system where modules are physically moved in and out of the flow path, the robotic arm acts as a sort of mini-chemist operating its own little chemical platform. What if, however, this idea could be taken one step further to a real laboratory-sized robot arm that could work ‘at the same time as’ humans? This question could also be extended to ‘instead of’ humans. Normally this would seem like an odd question; however, at the time of writing (during the global COVID-19 pandemic) many scientists have been stopped from physically entering the lab. A unique study from the University of Liverpool has employed a mobile robotic chemist (Fig. 7), driven by a Bayesian optimisation algorithm, to assist and mimic the researcher.62 Its human-like dimensions and compliance with safety standards for collaborative robots make it suitable to work alongside humans in a typical laboratory. Movement is guided by touch feedback and laser scanning, imparting the ability to both work in the dark (good for light-sensitive reactions) and giving it a high positioning and orientational precision to perform dexterous human manipulations (e.g. instrument operation, handling sample vials, etc.) Excluding the time the robot needs to charge, with optimal scheduling it can operate for up to 21.6 h a day performing experiments 1000 times faster than a human. Cooper et al.'s research objective was to use the robot to search for bio-derived hole scavengers to accompany the conjugate polymer photocatalyst P10, during the water splitting reaction to produce hydrogen under light.62 The experiment begins with the robot loading solid components onto a solid-dispensing station to be weighed into vials, which are then transported to a liquid-dispensing station, 16 vials at a time. It loads the vials onto a capping station to be capped under nitrogen or sonication, then delivers them to a photolysis station to be irradiated, and subsequently transferred to a gas chromatography station for H2 analysis and finally stores the finished vials. Automatic operation went on for 8 days in this case, searching a ten-variable experimental space in 688 experiments.
 | ||
| Fig. 7 Photographs of the mobile robotic chemist performing a six-point calibration with respect to the black location cube attached to the bench, the unmodified lab space (and its map) used for the autonomous experiments, and the multipurpose gripper shown with and without it griping a capped sample vial. Reproduced from ref. 62. | ||
L-Cysteine displayed good performance as a hole scavenger with P10; however, it was still lower than its petrochemical TEAO competitor. Next, the authors came up with five hypotheses to increase the performance, which the robot tested simultaneously. These amounted to a search space of over 98 million points and so, guided by the Bayesian algorithm, the robot started with random conditions and was eventually able to make its own decisions on what was and what was not important. In doing so the machine identified a number of scientific conclusions, such as increasing the pH and ionic strength are favourable for H2 generation, with the former being more profound (and also increases ionic strength). Moreover, mixtures that were 6 times more active than the original catalyst were found by choosing or rejecting components. These five hypotheses would have taken several months for a human to study. For 1000 such experiments, with 1/2 a day spent on researcher time per experiment, it would take a human 500 days. This is in contrast to the robot chemist which could perform 1000 experiments in 10 days, five of which are dedicated researcher time. After initial setup which took 1/2 a day, the machine ran autonomously over multiple days; therefore, 1/2 a day of researcher time for 1000 experiments, making this method 1000× faster than manual and >10× faster than other non-autonomous robotic follow systems.62 It took around two years to build this system at first, but once working with a low error rate it can be used as a useful tool (particularly during a lockdown scenario) and can be introduced into a new lab much more quickly and be extended to use other instruments (e.g. NMR). It is also advantageous when dealing with dangerous material, or for intricate pharmaceutical processes and can be extended to territories other than chemistry, such as material science. It should also be made clear that this is an enabling technology, not a replacement for the scientist: for example, the robot did not generate its own hypotheses and there is currently no computational brain.
Going back to the linear flow platform design and addressing the incomplete way in which chemical syntheses are reported, Cronin and co-workers developed what they call a “Chemputer” – a universal chemical programing language that operates an automated synthesis.10,63 The physical operations that control an automated batch synthesis platform are bridged with an organic synthesis abstraction that embodies reaction, workup, isolation and purification; the four stages of synthetic protocols (Fig. 8). While the layout of this physical setup is inferior to the aforementioned linear and radial approaches, the predominant area of interest for this group was in the software design and control of retrosynthetic strategies. The design began with the development of a digital flow system that relied on sensor feedback from changes in IR spectra to navigate a closed-loop chemical space search, based on reactivity rather than on conventional synthetic rules (i.e. not just following yield).64 In short, their algorithm was attuned automatically to select the most reactive pathways (based on previous experiments) within a network of 64 possible reaction combinations, without having to do every experiment. This was done in real time, many reactions could be linked together, and prior chemical knowledge or work-up and purification steps were not required. A simple metric then ranked the reactivity of all reactant combinations, resulting in the discovery of new molecules with only a fraction of the reactions needing to be performed. In this case, only 19% of all possible reactions needed to be explored. Furthermore, the screening time and material could be significantly reduced in each subsequent reaction step. This work was extended to include the use of more in-line analytics (NMR, MS and IR) and to predict the reactivity of 1000 reaction combinations, achieving over 80% accuracy with only ∼10% of the dataset being conducted in practice (the rest being predicted).65 It should be noted that this chemically unbiased methodology led to the discovery of four new reactions. To begin with, reaction mixtures were deemed either reactive or non-reactive and then digitised by a binary number (0 or 1) using a support vector machine. Secondly, a reaction descriptor was created with the number of starting materials defining its width and with a bit string representing the presence (1) or absence (0) of a chemical. This allowed the machine to make decisions that were assessed in real time via a comparison of theoretical and experimental spectra. This database was then coupled to a linear description analysis model which could assign a probability of reactivity and construct a chemical space architecture. The highest probability reactions are then conduced and analysed autonomously, enabling non-reactive mixtures to be avoided and the processing time to be decreased. This data would then update the machine learning model and close the loop. For exploration, the Suzuki–Miyaura reaction was investigated by the algorithm randomly choosing 10% of the possible reactions to train the neural network. The highest predicted yield reactions were then carried out and the rest were rated by the machine model in batches of 100. The initial random guess produced a mean yield of 39% with a standard deviation (SD) of 27%; the first batch then had a mean yield of 85% with an SD of 14%, and then subsequent batches rely on progressively fewer starting materials until the non-reactive parts of the chemical space are reached.
 | ||
| Fig. 8 Schematic representation of the Chemputer with four unit operations attached to the fluidic backbone. Reproduced from ref. 10. | ||
For clarity, these efforts allowed an autonomous system combining a robotic platform with AI, to make its own decisions based on previous experiments. This could then give way to the beating heart of the Chemputer, a program that produces the low-level, specific code instructions that command the hardware to operate a written synthesis, namely the “Chempiler”.10,63 In correlation with the above works of Coley and Jenson (Fig. 5) the platform and abstraction are represented as a graph,19,58 making it possible to digitise and run published syntheses, assuming the required process modules are present within the setup, without manual configuration. The workflow, shown in Fig. 9, automatically generates and optimises a valid synthesis that can be executed by a continuous flow platform. As a proof-of-concept, three pharmaceuticals were automatically produced in yields and purities comparable to manual synthesis. These were: diphenhydramine hydrochloride, 58% yield over four steps in 77 h vs. 68% yield manually in 4 days, sildenafil (Viagra), 44% yield in 102 h, and rufinamide, 46% yield in 38 h vs. 38% manually. In this case however, for simplicity and reproducibility, the flow setup consisted of batch glassware (since this is most commonly found in today's laboratories) rather than flow reactors. Naturally, synthetic routes will have to be digitised and validated one by one, but eventually databases will be created that will allow automated platforms to directly convert a reaction from the database to a code and/or run known operations from an electronic file.
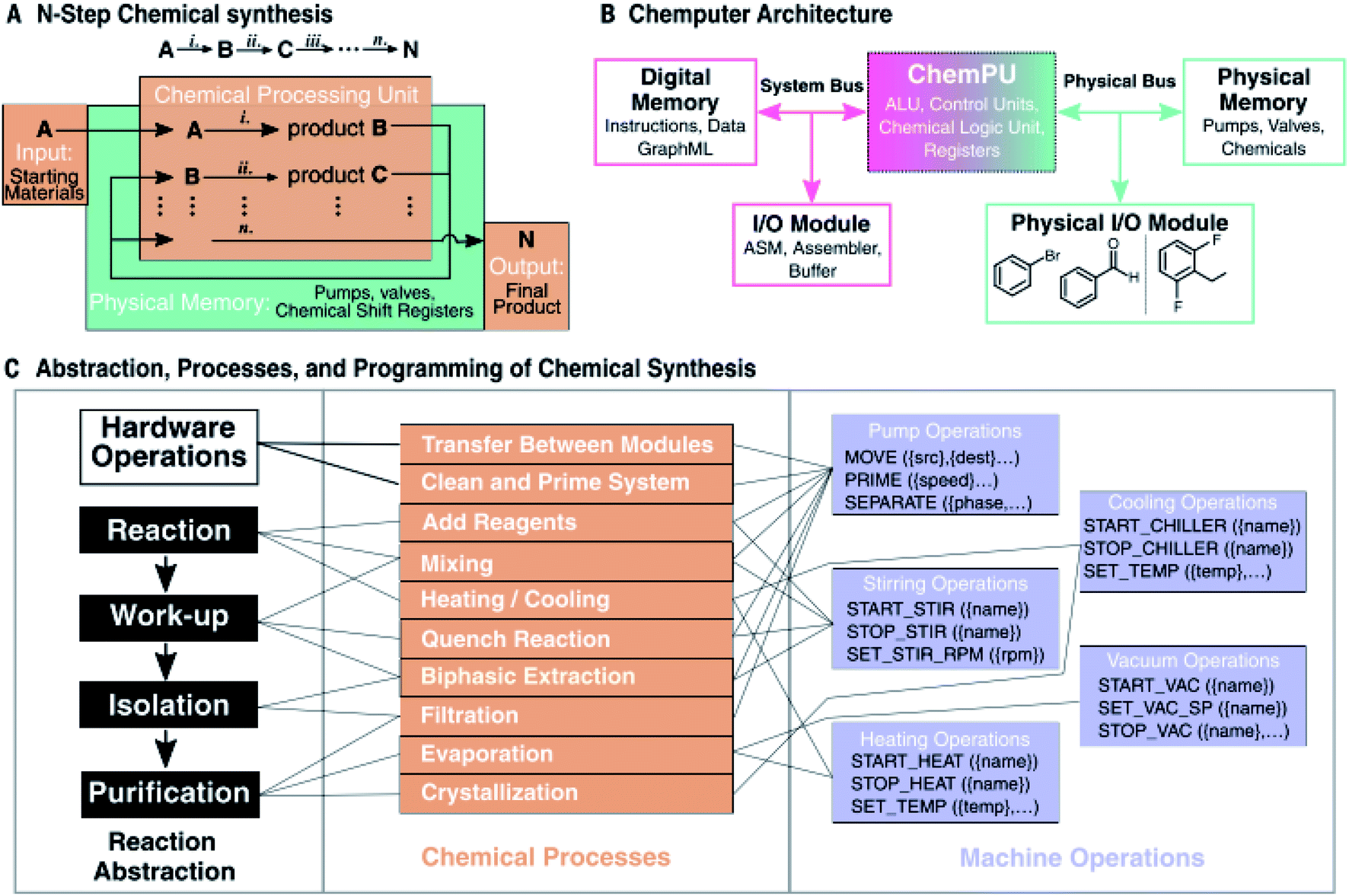 | ||
| Fig. 9 Operating codes for the Chemputer. (A) Schematic illustration of an organic synthesis where reagents are treated as inputs and the final product as an output. (B) Chemputer architecture outline (ALU = arithmetic logic unit; I/O = input/output; ASM = assembly language). (C) Abstraction of chemical synthesis able to be universally programmed using a machine. Reagents and products are represented by a memory bus that splits a complex pathway into steps or cycles that can be accessed by the hardware. Reproduced from ref. 10. | ||
With all these advances in mind, an automated synthesis platform, “AutoSyn CityScape”, which emulates a miniaturised chemical plant, has been designed to produce milligram-to-gram quantities of small organic molecules.18 The configuration, shown in Fig. 10, resembles a city's high-rise landscape, with a subway map of flow components that can be operated with minimal intervention by a single-user. This is similar to the flow pattern of Chatterjee et al.'s radial synthesiser14 that is guided through fluid circuits and can choose when and which modules to go to. Digitised chemical processes then guide the platform to select suitable flow paths between various reaction modules and in-line analytics. In this work there are 3887 possible routes that can be taken. Multistep syntheses can, therefore, be combined to generate a wide variety of target compounds. These digital procedures enable reproducibility and the ability to transfer to and from different labs. The main merit of the system was demonstrated by its versatility, producing 10 known drugs in a range of yields (6–100%), purities (17–91%) and lengths of time (0.75–3.25 h). The synthesis planning is conducted in a three-level process. Level 1 has to do with the manual or algorithmic design of a synthesis route, including relevant solvents, reagents and conditions. Level 2 automatically converts level 1 into a flow process with operating parameters that correspond to the available hardware. Here, reagent and solvent compatibility with the machinery are taken into account as well as possible side products. Level 3 then converts the level-2 process map into the hardware “subway map” that runs the digital synthesis as computer scripts. The flexibility of AutoSyn was chosen as the predominant attribute (rather than yield or full optimisation), demonstrating a step closer towards a “universal synthesiser”.18 Further optimisations of the synthesis procedures will in the future help increase yields and efficiency. For example, levels 1 and 2 are similar to the processes of Chematica and Cronin, respectively;10,52,53,63 however, benefit would be revealed by introducing machine learning techniques incorporating the huge number of reactions stored in databases as in the works of Segler et al.28 and Coley and Jenson et al.22,55,58 Moreover, AutoSyn is designed to produce products in usable quantities (mg to g) which is a necessity for large, complicated (but well-known) multistep syntheses, i.e. this will be hugely beneficial for making useful quantities of peptides and DNA sequences.
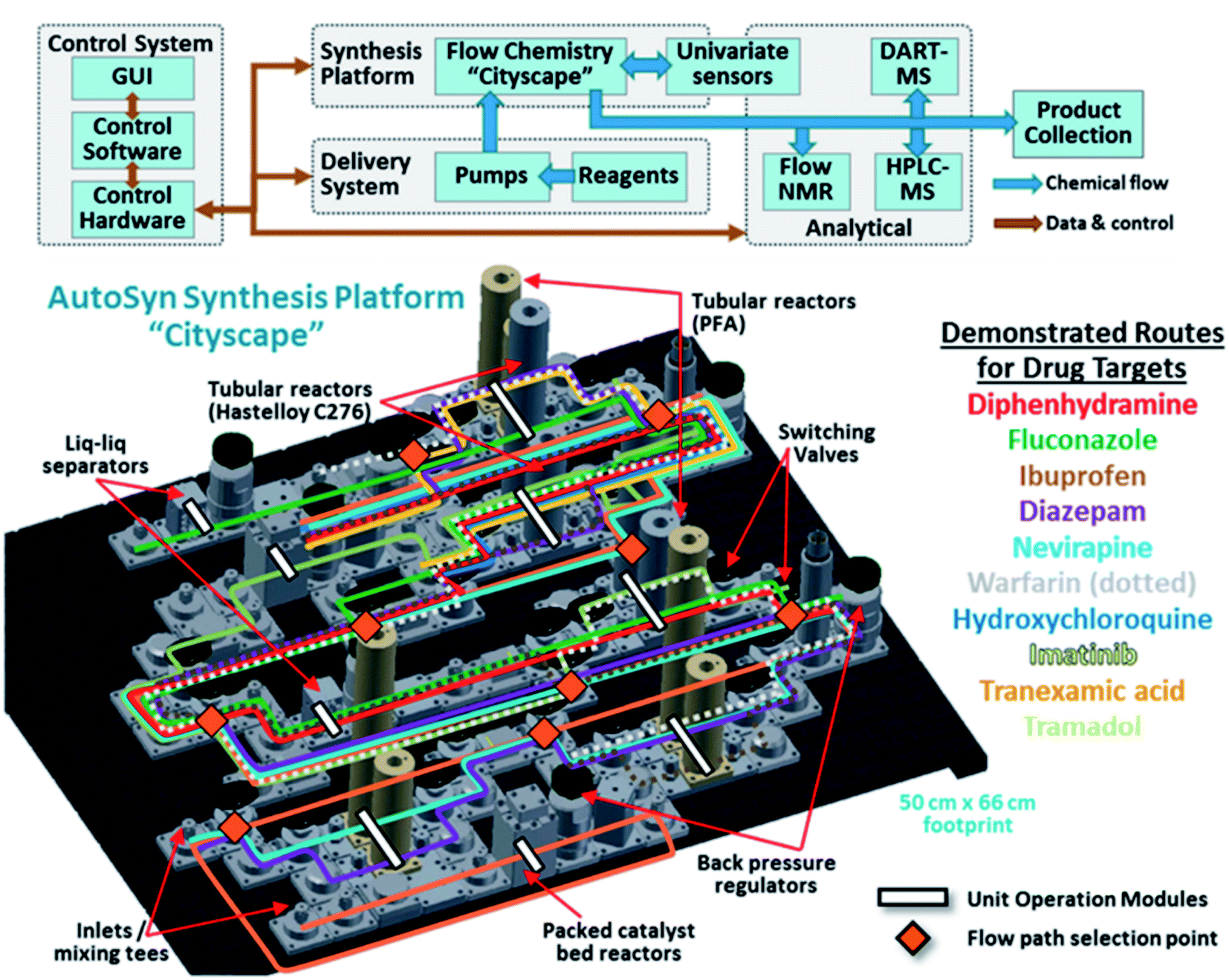 | ||
| Fig. 10 AutoSyn CityScape comprising a flow chemistry platform, a reagent delivery system, and process analytical technologies controlled by integrated software. A number of chemical syntheses can be carried out, such as the pharmaceutical targets listed with their corresponding highlighted “subway map” of the routes through the platform and their unit operation modules. Reproduced from ref. 18. | ||
There are, however, challenges that are standing in the way of automated synthesis progression and its widescale adoption. While some methods can address stereochemistry, not all of them can and, thus, reliable predictions are going to depend on this becoming commonplace.28 Concurrently, quantitative evaluation of enantiomeric or diastereomeric ratios remains a problem, and relies on expensive quantum mechanical calculations.28,53,66 It may be possible to get around this with stereochemistry-aware descriptors.28 Complex natural products, with their elaborate pathways and unpredictability, are troublesome even for expert chemists and are still beyond the capabilities of most digitised flow platforms. Also, many prediction algorithms exclude or struggle to complete feasible reaction conditions. When creating a forward predictor model, there is the question of what defines a good reaction pathway. This will differ for different areas of science: organic chemists might want yield or stereospecificity, medical professionals could be time conscious (e.g. on-demand pharmaceuticals during a shortage or for convenience), industrialists may value the cost of the chemicals and equipment, or safety may be the main concern. User-friendly software with a “filter” option would be a good idea. Retrosynthetic algorithms work by simplifying a target molecule into smaller components, but do not consider the judicious approach of employing protecting or directing groups, since this would mean increasing the size of the intermediate compounds. In addition, the algorithms should account for the availability of reagents since suitable precursors can streamline routes to complex molecules.28,53 Finally, there is the physiological aspect of removing the scepticism chemists may have of this new technology. While well established in other disciplines, practical chemists may doubt current machine learning's ability to grasp the “art” of organic chemistry.67 Others may be under the belief that robots will replace them and so will need to be shown that automation is actually an enabling tool to assist them in a very effective manner. The things machine learning can and cannot do, so as to avoid misconceptions, will need to be communicated. Moreover, many chemists do not have sufficient knowledge of coding, machine learning and AI. It would be beneficial for large chemical organisations such as the Royal Society of Chemistry and ACS to make available webinars and courses that address this deficiency. Some universities teach their undergrad students courses such as “maths of chemists” (when maths was not their strong point prior joining the course). This could similarly be done for chemists of all disciplines and experience (not just for organic chemistry or for undergrads), i.e. “computer science” or “coding for chemists”.
Conclusion
To summarise, we have shown that the synthesis of small molecules and pharmaceutical compounds can proceed via automated continuous flow platforms, guided by machine learning and AI with little human interaction. Some retrosynthetic algorithms can work relatively independently while others rely a bit more on chemists digitising reactions. At first, the latter will be tedious and time consuming but will eventually lead to results that could be used to generate databases for known processes. There are still some challenges and further advances to be overcome, including convincing experimental scientists that automated flow is not designed to be their replacement but rather to assist them and to make experiments and discovery faster, more efficient and reproducible. The reality is that modern computers and advanced algorithms can use the massive amounts of information in the literature and reaction databases to build the chemical space of a molecule in a way that is far superior to that of a human. From here, known targets can be synthesised in practice, optimised by merits chosen by the operator, and new compounds and novel chemistries can be discovered. Constructing models and carrying out optimised forward reactions are ideal tasks for automation. As we have outlined herein, the quality of reactions performed via digitised chemistry operating in automated and robotic flow can not only compete with expert chemists, but also be a hundred to a thousand times faster. In order to further the goal of automated synthesis in a flow system, not only would reactions have to occur as independently from humans as possible, but it would also be able to design and predict its own viable routes to particular chemical compounds so that it may then carry them out and subsequently enable scale-up. An ideal system would be fully automated, in the sense that it would be able to create the chemical space of a molecule, search through it to design and predict the best possible (feasible) routes to a target compound and then automatically implement them in practice. Moreover, a universal standard for a coding language and online, available chemical spaces or digital, forward reaction recipe files do not yet exist. By taking the context of this paper into consideration, however, they could one day become an additional part of the chemist's ever-expanding arsenal, with machine-powered learning and humans working side-by-side to produce lab- and industrial-scale material more efficiently and safely. Multistep syntheses to target molecules could then be performed in a matter of hours to days rather than weeks to months.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Support from the EU-COFUND Horizon 2020 project (Grant No 663830) and the School of Chemistry, Cardiff University, UK, is gratefully acknowledged.References
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 13–20 CrossRef CAS.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- J. Zhang, C. Gong, X. Zeng and J. Xie, Coord. Chem. Rev., 2016, 324, 39–53 CrossRef CAS.
- J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- T. Hardwick and N. Ahmed, RSC Adv., 2018, 8, 22233–22249 RSC.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- R. Gérardy, N. Emmanuel, T. Toupy, V. Kassin, N. N. Tshibalonza, M. Schmitz and J. M. Monbaliu, Eur. J. Org. Chem., 2018, 20–21, 2301–2351 CrossRef.
- J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, M. Granda, G. Keenan, T. Hinkley, G. Aragon-camarasa, P. J. Kitson, D. Angelone and L. Cronin, Science, 2019, 363, eaav2211 CrossRef CAS.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor and D. R. Snead, Science, 2016, 352, 61–67 CrossRef CAS.
- A. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef.
- C. W. Coley, D. A. T. Iii, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 557, eaax1566 CrossRef.
- S. Chatterjee, M. Guidi, P. H. Seeberger and K. Gilmore, Nature, 2020, 579, 379–384 CrossRef CAS.
- R. D. Padmaja and K. Chanda, Org. Process Res. Dev., 2018, 22, 457–466 CrossRef CAS.
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V Moore, X. Y. Lawrence and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- C. Badman and B. L. Trout, J. Pharm. Sci., 2015, 104, 779–780 CrossRef CAS.
- N. Collins, D. Stout, J. Lim, J. P. Malerich, J. D. White, P. B. Madrid, M. Latendresse, D. Krieger, J. Szeto, V. Vu, K. Rucker, M. Deleo, Y. Gorfu, M. Krummenacker, L. A. Hokama, P. Karp and S. Mallya, Org. Process Res. Dev., 2020, 24(10), 2064–2077 CrossRef CAS.
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS.
- W. Ian, Nature, 2019, 570, 175–181 CrossRef.
- P. Raccuglia, K. C. Elbert, P. D. F. Adler, C. Falk, M. B. Wenny, A. Mollo, M. Zeller, S. A. Friedler, J. Schrier and A. J. Norquist, Nature, 2016, 533, 73–76 CrossRef CAS.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2017, 3, 434–443 CAS.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS.
- M. Peplow, Nature, 2014, 512, 20 CrossRef CAS.
- R. B. Merrifield, Science, 1965, 150, 178–185 CrossRef CAS.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef.
- A. G. Godfrey, T. Masquelin and H. Hemmerle, Drug Discovery Today, 2013, 18, 795–802 CrossRef CAS.
- M. H. S. Segler, M. Preuss and P. Mark, Nature, 2018, 555, 604–610 CrossRef CAS.
- A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz and A. Simon, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 79–107 CAS.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef.
- W. Ihlenfeldt and J. Gasteiger, Angew. Chem., Int. Ed. Engl., 1996, 34, 2613–2633 CrossRef.
- J. G. Rieveschl, US Pat., 2,421,714A, 1947.
- T. J. Reilly, J. Chem. Educ., 1999, 76, 1557 CrossRef CAS.
- N. M. Löfgren and B. J. Lundqvist, US Pat., 2,441,498, 1948.
- T. Sugasawa, M. Adachi, T. Toyoda and K. Sasakura, J. Heterocycl. Chem., 1979, 16, 445–448 CrossRef CAS.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC.
- V. Sans and L. Cronin, Chem. Soc. Rev., 2016, 45, 2032–2043 RSC.
- W. Huyer and A. Neumaier, ACM Trans. Math. Softw., 2008, 35, 1–25 CrossRef.
- P. J. Kitson, G. Marie, J. Francoia, S. S. Zalesskiy, R. C. Sigerson, J. S. Mathieson and L. Cronin, Science, 2018, 359, 314–319 CrossRef CAS.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, Science, 2018, 434, 429–434 CrossRef.
- P. S. Gromski, A. B. Henson, J. M. Granda and L. Cronin, Nat. Rev. Chem., 2019, 3, 119–128 CrossRef.
- D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS.
- K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859 CrossRef CAS.
- J. R. Schmink, A. Bellomo and S. Berritt, Aldrichimica Acta, 2013, 46, 71–80 Search PubMed.
- A. Bellomo, N. Celebi-Olcum, X. Bu, N. Rivera, R. T. Ruck, C. J. Welch, K. N. Houk and S. D. Dreher, Angew. Chem., 2012, 124, 7018–7021 CrossRef.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr and M. R. Smith III, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS.
- A. B. Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L. Campeau, J. Schneeweis, S. Berritt, Z. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 443–448 Search PubMed.
- S. Lin, S. Dikler, W. D. Blincoe, R. D. Ferguson, R. P. Sheridan, Z. Peng, D. V. Conway, K. Zawatzky, H. Wang, T. Cernak, I. W. Davies, D. A. Dirocco, H. Sheng, C. J. Welch and S. D. Dreher, Science, 2018, 361, eaar6236 CrossRef.
- N. J. Gesmundo, B. Sauvagnat, P. J. Curran, M. P. Richards, C. L. Andrews, P. J. Dandliker and T. Cernak, Nature, 2018, 557, 228–232 CrossRef CAS.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS.
- M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef CAS.
- B. A. Grzybowski, S. Szymku, E. P. Gajewska, K. Molga, P. Dittwald and A. Wołos, Chem, 2018, 4, 390–398 CAS.
- E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef.
- M. H. S. Segler and M. P. Waller, Chem.–Eur. J., 2017, 23, 5966–5971 CrossRef CAS.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2017, 3, 1237–1245 CAS.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, Acc. Chem. Res., 2017, 3, 1103–1113 CAS.
- A. F. de Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem., 2019, 3, 589–604 CrossRef.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jenson, Chem. Sci., 2019, 10, 379–377 RSC.
- C. W. Coley, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2019, 59, 2529–2537 CrossRef CAS.
- W. Jin, C. Coley, R. Barzilay and T. Jaakkola, in Advances in Neural Information Processing Systems, 2017, pp. 2607–2616 Search PubMed.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 4, 1465–1476 CAS.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS.
- P. S. Gromski, J. M. Granda and L. Cronin, Trends Chem., 2020, 2, 4–12 CrossRef CAS.
- V. Dragone, V. Sans, A. B. Henson, J. M. Granda and L. Cronin, Nat. Commun., 2017, 8, 15733 CrossRef.
- J. M. Granda, L. Donina, V. Dragone, D. Long and L. Cronin, Nature, 2018, 559, 377–381 CrossRef CAS.
- Q. Peng, F. Duarte and R. S. Paton, Chem. Soc. Rev., 2016, 45, 6093–6107 RSC.
- G. Skoraczyński, P. Dittwald, B. Miasojedow, S. Szymkuć, E. P. Gajewska, B. A. Grzybowski and A. Gambin, Sci. Rep., 2017, 7, 1–9 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |