
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInorganic nanocrystal-dynamic porous polymer assemblies with effective energy transfer for sensitive diagnosis of urine copper†
Xujiao
Ma
a,
Yajie
Yang
a,
Rongchen
Ma
a,
Yunfeng
Zhang
a,
Xiaoqin
Zou
a,
Shoujun
Zhu
b,
Xin
Ge
c,
Ye
Yuan
 *a,
Wei
Zhang
*c and
Guangshan
Zhu
*a
*a,
Wei
Zhang
*c and
Guangshan
Zhu
*a
aKey Laboratory of Polyoxometalate Science of Ministry of Education, Northeast Normal University, Changchun 130024, China. E-mail: yuany101@nenu.edu.cn; zhugs@nenu.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China
cKey Laboratory of Automobile Materials MOE, School of Materials Science & Engineering, Electron Microscopy Center, International Center of Future Science, Jilin University, Changchun 130012, China. E-mail: weizhang@jlu.edu.cn
First published on 23rd September 2020
Abstract
Despite their remarkable mechanical, optical, and electrical properties, inorganic particles and dynamic polymer assemblies encounter difficulties in their compatibility with regards to structural order and complexity. Here, covalent organic frameworks (COFs) constructed through reversible coupling reactions were exploited as dynamic porous polymers to prepare inorganic nanocrystal-polymer assemblies. Under an in situ growth process, carbon quantum dots (CDs) were gradually prepared in the COF cavity, with a narrow size distribution (2 ± 0.5 nm). The well-established assemblies achieve effective energy transfer from the inorganic to the organic part (efficiency > 80%), thus rendering a ∼130% increase in quantum yield compared with the pristine COF network. Notably, the hybrid material realizes a simple, selective, and sensitive diagnostic tool for urine copper, surpassing the detection limit of COF solid by 150 times. Beyond the scientific and fundamental interests, such hybrid assemblies are attractive from technological perspectives as well, for example, in energy storage, electronics, catalysis, and optics.
Introduction
Assemblies of biological structures (such as microtubules, viruses, and nanoparticles) and polymers (such as molecular crystals and frameworks) combine their components' strengths and show remarkable mechanical, optical, and electrical properties.1–8 Accordingly, these assemblies have witnessed rapid advancement in the area of selective sorption and separation, sensing, and mechanoactuation.9–12 However, the formation of hybrid materials always involves trade-offs among flexibility, binding pattern, and physical/chemical properties. Along with the increase of atomic or molecular units, composites often face choices between structural order and complexity.1,9 With these parameters in mind, a dynamic polymer with a rationally designed aperture to incorporate the guest object is a pressing need.Covalent organic frameworks (COFs), based on the auto-assembly of aromatic units through the reversible character of polymerization reactions, are a type of dynamic porous polymer.13–16 Due to their designable building blocks and tunable porous structures, COFs have been considered to be functional organic equivalents of inorganic microporous solids for utilization in gas separation,17–19 ion enrichment,20 optoelectronics,21–23 catalysis,24–26 drug transport,27,28 and biosensing.29–32 Based on the reversible formation of covalent bonds, they combine error-correction capability and molecular robustness.14,33 Compared to supramolecular polymers connected by weak noncovalent bonds, the modularity of COF solids endows a well-defined molecular architecture for the integration of guest substances.34,35 Therefore, they have allowed us to explore the exciting applications in host–guest chemistry and nanocomposite fabrication.
Herein, a series of COF samples driven by dynamic imines were utilized as matrices to exploit the novel inorganic nanocrystal-dynamic porous polymer assemblies. Subsequent thermal treatment of sodium citrate precursor afforded the pore-determined CD nanoparticles, denoted as CD@COF samples. Based on the fluorescence resonance energy transfer (FRET) mechanism, the CD donors transfer their emission energy to the energy-absorbing hydrazone groups of the COF skeleton. Accordingly, the CD nanocrystal-doped COF assemblies with high energy transfer efficiency showed a high-fluorescence emission. Thanks to the full host–guest chemistry of the porous structure, Cu2+ ions were concentrated in the fluorescent COF architectures to realize highly sensitive and selective detection.
Results and discussion
To demonstrate the applicability of COF-based inorganic nanocrystal-dynamic porous polymer assemblies, several building blocks were selected to enrich the variation of the COF structures. As illustrated in Fig. 1, BCOF-1, BCOF-2, TCOF-1, TCOF-2, and TCOF-3 were prepared through irreversible Schiff-base copolymerization featuring the energy-absorbing groups (N–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ). Carbon quantum dots with high-profile optical properties as energy donors overcome the limitations of COF's optical performance.36–40 On the strength of their structural traits, CD particles were immobilized on the COF frameworks via hydrogen-bond interaction (Fig. S1†).
). Carbon quantum dots with high-profile optical properties as energy donors overcome the limitations of COF's optical performance.36–40 On the strength of their structural traits, CD particles were immobilized on the COF frameworks via hydrogen-bond interaction (Fig. S1†).
 | ||
| Fig. 1 Scheme for the preparation of COFs and CD-doped COFs (CD@COFs) using different building units (i–v) to prepare five COF skeletons and via in situ introduction of CDs into COF structures: CD@BCOF-1, CD@BCOF-2, CD@TCOF-1, CD@TCOF-2, and CD@TCOF-3. | ||
Experimentally, temperature plays a crucial role in the self-polymerization of the precursor (sodium citrate), which increases the fluorescence performance of CDs with the increase of temperature.41 However, most COF skeletons will decompose at 350 °C, according to thermogravimetric analysis. To establish inorganic particles and dynamic polymer assemblies with effective energy transfer, 200 °C, 250 °C and 300 °C were selected as the temperature to synthesize CD nanoparticles. Loading with the same amount of CD precursors, we found that the CDs synthesized at 300 °C had the strongest fluorescence intensity (Fig. S2†). Then, BCOF-1 constructed with 1,3,5-triformylbenzene and hydrazine hydrate were selected as the subject, through observing fluorescence intensity under different doping amounts to screen for the optimum content of CDs. Sodium citrate, with varied content for each batch, was soaked into BCOF-1 as the precursor. After being heated at 300 °C for 2 hours, CD-doped BCOF-1 samples were obtained and denoted as CD@BCOF-1-X, where X (X = 5/4, 5/8, 5/16, 5/32, and 5/64) represents the initial mass ratio of sodium citrate to COF powder (Table S1†).
Powder X-ray diffraction (PXRD) analysis proved that CD-doped BCOF-1 samples had the same ordered structure as their parent BCOF-1. A sharp diffraction peak at 6.97° corresponds to the (100) crystal plane, and relatively weak signals at 11.99, 13.83, and 18.35° are assigned to the (110), (200), and (120) crystal planes, respectively. The π–π stacking structure between COF layers results in a slightly wider diffraction peak at 26.9° corresponding to the (001) plane. It is indicated that the implanted CD particles do not affect the crystal structure of BCOF-1 (Fig. 2A and S3†). Scanning electron microscopy (SEM) images indicate a consistent cluster-like microscopic morphology of BCOF-1 and CD@BCOF-1-5/4 (Fig. S4†). According to transmission electron microscopy (TEM) images, CD@BCOF-1 usually exhibits clear lattices in a small area, and pores with a pitch of 0.8 nm were detected (Fig. 2B and S5†). Within the COF lattices, the 2–3 nm circled black dots are assigned to the CD particles (Fig. 2B), demonstrating the successful incorporation of carbon dots.42,43 Overall, it is fully proven that the CDs@COF hybrids ensure the orderly structure of the framework while achieving high CD-doped content for structural complexity.
 | ||
| Fig. 2 Characterization of the CD@COF assemblies. (A) PXRD patterns for CD@BCOF-1-X (X = 5/4, 5/8, 5/16, 5/32, 5/64). (B) TEM image of CD@BCOF-1-5/4 showing CD particles embedded in the BCOF-1 matrix. CD nanoparticles are highlighted by the red circle, and the hexagonal pores are highlighted by the blue hexagon. (C) O1s, (D and E) C1s and (F) N1s XPS spectra for BCOF-1 (below) and CD@BCOF-1-5/4 (above). | ||
In the Fourier transform infrared (FT-IR) spectroscopy, the appearance of CN stretching band at 1631 cm−1 demonstrates the formation of imine bonds in COF skeletons (Fig. S6†). With the increased doping amount of CD, there is a red shift for the CN vibration frequency caused by the hydrogen bonding interaction between CD and the COF skeleton. As for CD@BCOF-1-5/4, this change is particularly obvious for the CN band, to 1625 cm−1. The apparent carbonyl stretching vibration at 1695 cm−1 in the IR spectra for CD@BCOF-1-X confirms the successful assembly of CDs (Fig. S6†). The chemical states of the C and O atoms in the CD-doped COF materials were determined by X-ray photoelectron spectroscopy (XPS; Fig. S7†). The high-resolution C1s XPS spectrum for BCOF-1 was deconvoluted into two peaks, located at 284.7 (CC/C–C) and 288.4 eV (CN/CO). As for the C1s spectrum for CD@BCOF-1-5/4, distinct signals were assigned to the C–O (286.2 eV) and CN/CO (288.6 eV) groups in CDs and to CC/C–C (284.7 eV) bonds in BCOF-1 (Fig. 2D). There was a 0.2 eV shift of the CN/CO peak position that could be ascribed to the hydrogen bonding between the O–H groups in the CDs and CN/CO groups in the COF network (Fig. 2E). Meanwhile, the characteristic peak at 532.6 eV in the O1s spectrum is attributed to the CO group at the BCOF-1 skeletal terminal. The high-resolution O1s spectrum for CD@BCOF-1-5/4 is deconvoluted into five peaks. In addition to the CO peak provided by BCOF-1, the signals centered at 531.6, 532.9, 533.4, and 535.2 eV correspond to the O atoms in the CO, C–O, O–CO, and O–H groups of CDs, respectively (Fig. 2C). There is only one convolution peak in the N1s spectrum, assigned to the CN–N group. The hydrogen bonding between the O–H of CDs and CN–N of the COF framework causes the peak position to shift by 0.25 eV (from 400.0 to 399.75 eV) (Fig. 2F).
There is an additional evidence of the successful introduction of CDs via hydrogen bonds in the 13C solid-state nuclear magnetic resonance (NMR) spectra. The distinct signals in the range of 134–124 ppm are assigned to the aromatic carbon atoms in the phenyl ring. The CN characteristic peak shifts from 160 ppm (COF) to the lower field (163 ppm) due to the deshielding effect of the CDs embedded in the COF framework (Fig. S8†). The electron cloud of the CN group in the COF network is pulled by the exposed hydroxyl groups of CDs, leading to the lower binding energy. It is worth mentioning that there is a red shift of the maximum emission wavelength from 635 nm for the original BCOF-1 to 650 nm for CD@BCOF-1-5/4 (Fig. 3A). This increased wavelength (15 nm) is attributed to the hydrogen bonding between CDs and the COF skeleton, which inhibits free rotation within the N–N fragment (Fig. S1†). This restriction of intramolecular rotations (RIR) leads to a reduction of the excited-state non-radiative transitions and induces an increase in the Stokes shift.44
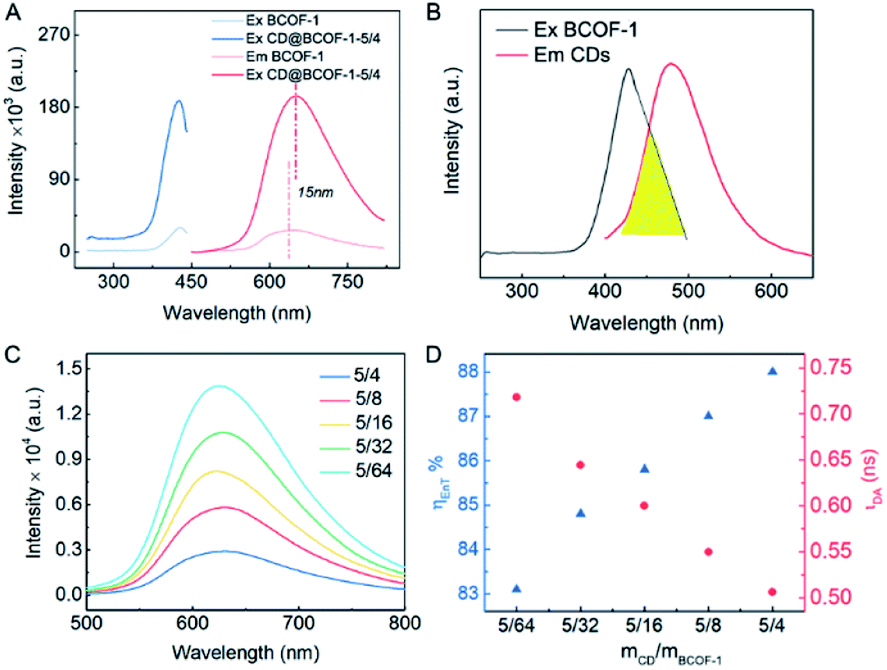 | ||
| Fig. 3 PL performance of CD@BCOFs. (A) PL spectra for COF-1 and CD@COF-1-5/4 (λex = 420 nm, λem = 650 nm). (B) PL emission spectrum for CDs (λex = 380 nm) and PL excitation spectrum for BCOF-1 (λem = 650 nm). (C) Emission spectra for BCOF-1-X (X = 5/4, 5/8, 5/16, 5/32, 5/64; λex = 420 nm). BCOF-1 doped with different amounts of CDs. (D) Dependence of the energy-transfer efficiency and the lifetime of the donor (CDs) excitation at 380 nm as a function of the doping ratio of CDs. | ||
Nitrogen adsorption experiments were performed at 77 K to study the porous nature of the CD-doped COF materials. CD@BCOFs exhibited a type IV isotherm similar to that of BCOF-1. As CD particles increasingly occupied COF channels, the mass per unit increased, and the Brunauer–Emmett–Teller (BET) surface area was reduced from 1222 m2 g−1 for BCOF-1 to 359 m2 g−1 for CD@BCOF-1-5/4 (Fig. S9†). Elemental analysis data showed that the weight of C and N elements in CD@BCOF-1 was significantly reduced compared to BCOF-1. It was calculated that the mass ratio of O element increased from 5.58% to 10.56%, proving the successful incorporation of CDs (Table S2†).
The FRET method refers to transfer of dipole-mediated energy from the donor to the acceptor, as the distance between them is less than 10 nm.45 Its manifestation is the overlapped spectra for the donor molecule in an emissive state (Em1) and the acceptor molecule in an excited state (Ex2) (Fig. 4A). The emission spectrum of CD particles ranged from 405 to 565 nm (centered at 490 nm); the excitation spectrum of BCOF-1 varied within the range of 345–505 nm (centered at 420 nm). There is a spectrum overlap from 405 to 505 nm between the CD-donor emission and the COF-acceptor excitation (Fig. 3B). CDs evidenced a restricted growth in the uniformly distributed binding sites in the framework. The successful coupling between CDs and BCOF-1 resulted in the efficient transfer of fluorescence energy from CDs to BCOF-1. These roles are critical for the successful assembly to endow an efficient FRET behavior.
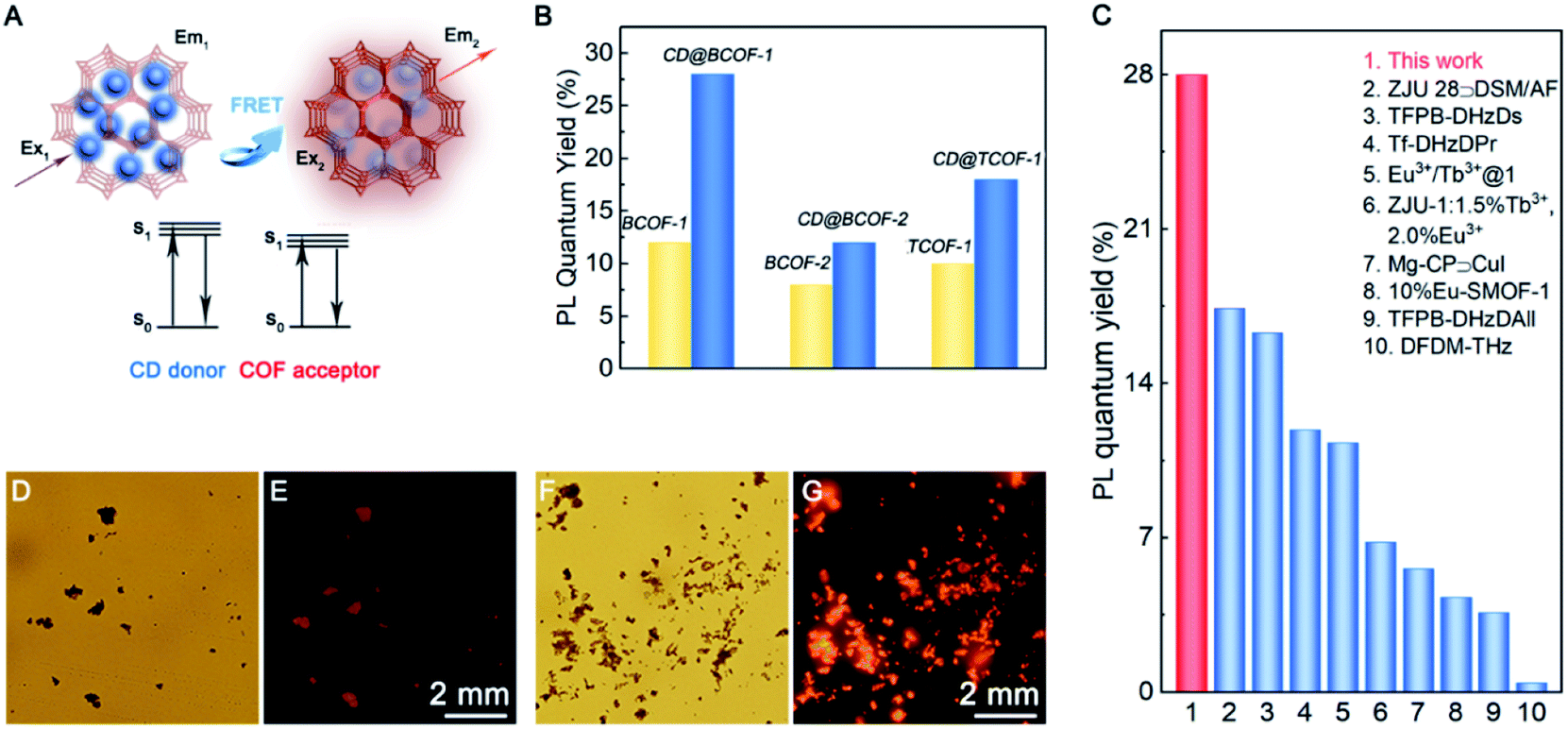 | ||
| Fig. 4 PL mechanism and PL quantum efficiency investigation. (A) Energy transfer mechanism between the CD donor and COF acceptor to obtain fluorescent COFs. (B) PL quantum yield for different COF samples. (C) Histogram of the solid-state fluorescence quantum efficiencies for various samples. LSCM images for BCOF-1 (D and E) and CD@BCOF-1 (F and G) under white light exposure and ultraviolet light (λex = 365 nm). | ||
As shown in Fig. 3C, the fluorescence intensities for the CD@BCOF-1-X materials are significantly increased at 650 nm, corresponding to an orange-red emission (excitation at 420 nm), with the increasing content of loaded CDs. This phenomenon indicates that the energy transfer between CDs and COF structures is gradually enhanced to achieve high fluorescence emission. Significantly, CD@BCOF-1-5/4 gains more than a 10-fold fluorescence intensity increase compared with the original BCOF-1 (Fig. 3A). The fluorescence quantum yield increases from 12% for BCOF-1 to 28% for CD@BCOF-1 (Fig. 4B).
In addition, the time-resolved photoluminescence (TRPL) spectra reveal that CD@BCOF-1 has a prolonged lifetime at the corresponding maximum excitation wavelengths (420 nm) (Fig. S11 and Table S3†). This result further demonstrates the effective energy transfer between CDs and COF structures.35 The time-resolved photoluminescence measurements show that the fluorescence lifetime of CDs is 4.248 ns (excitation at 380 nm). Under the same excitation (380 nm), the fluorescence lifetime for CD@BCOF-1-X decreases with the increase of doping amount of CDs (Fig. S12†).
Correspondingly, the calculated energy transfer (EnT) efficiency, opposite to the fluorescence lifetime, exhibits a linear increase as the doping amount of CDs in the CD@BCOFs increases (Fig. 3D).40,46 This observation provides conclusive evidence for an effective donor–acceptor FRET system. For CD@BCOF-1-5/4 with doping ratio of 5/4, the fluorescence lifetimes decreased to 0.506 ns and EnT efficiency increased to 88.08% (Fig. 3D), which resulted in the KEnT value of 1.74 × 109 S−1. As assessed from laser scanning confocal microscopy (LSCM), all the COF samples were brownish-yellow under sunlight. The solids had orange-red fluorescence under 365 nm of ultraviolet light irradiation, and the fluorescence emission intensities of CD@BCOF-1-5/4 were much stronger than the original BCOF-1 (Fig. 4D–G).
Because CD@BCOF-1-5/4 displayed the highest fluorescence intensity, it was named CD@BCOF-1 to aid in comparisons with the other CD-based COFs. In addition, the optimal content of 5/4 (sodium citrate to COF powder) was utilized to prepare the other CD-doped COF materials, denoted as CD@BCOF-2, CD@TCOF-1, CD@TCOF-2, and CD@TCOF-3 (Fig. 1). The FT-IR and XPS spectra indicate the presence of hydrogen-bonded CD particles in all COF networks. Compared with the FT-IR spectra for the initial monomers, the emerging CN stretching vibration demonstrates the occurrence of Schiff base reaction (Fig. S13†). After doping the CD particles into the COF architectures, red shifts were seen for the CN stretching vibration, from 1631 to 1625 cm−1 for CD@BCOF-1 and from 1656 to 1634 cm−1 for CD@TCOF-1, respectively. These variations suggest the CD particles are fixed in the framework through hydrogen bonding with CN groups (Fig. S6 and S13†). The lower binding energy (from 400.2 to 399.7 eV) belonging to the CN–N convolution peak in the N1s XPS spectra also proves the hydrogen bonding effect between CN groups and CDs in BCOF-1 and TCOF-1 (Fig. S17†). For BCOF-2, TCOF-2, and TCOF-3 with amide groups in the frameworks, there is a blue shift of CO stretching vibration in the FT-IR spectra after doping with CD nanocrystals. This change proves that the CDs are fixed on the carbonyl groups of COF skeletons through hydrogen bonds (Fig. S13†). In addition, CO (COFs) peaks in the O1s XPS spectra shift by 0.3, 0.25, and 0.5 eV in CD@BCOF-2, CD@TCOF-2, and CD@TCOF-3, respectively (Fig. S15†). These augmented binding energies confirm the existence of hydrogen bonds between CDs and CO groups in the COF networks. SEM and TEM images show a random distribution of CDs, and all CD@COFs exhibit excellent thermal stability (Fig. S18 and S19†). All of the characterizations for the CD@COF materials are listed in the ESI.†
Stability is an essential parameter for the practical application of inorganic nanocrystal-dynamic polymer assemblies. Therefore, the fluorescence intensities of CD@BCOF-1 and its supernatant were monitored after dispersion in water for 24 hours. It was found that scarcely any quantum dots overflowed from the COF solid. There was no variation of fluorescence intensity for CD@BCOF-1, confirming its high stability in an aqueous environment (Fig. S20†). In addition, the CD@COF samples could be stored at room temperature for three months without losing their fluorescent quality (Fig. S21†). The extraordinary stability of the CD@COF samples is attributed to the strong affinity (hydrogen bonding interaction) locking the distributed CD particles into the rigid structure, which prevents the collision-induced fluorescence self-quenching phenomenon.47
Similar to CD@BCOF-1, the emission spectrum for the donor CDs overlaps the excitation spectra for BCOF-2 and TCOF-1 (Fig. S22 and S23†). Therefore, a similar FRET phenomenon occurs for CD@BCOF-2 and CD@TCOF-1. They presented enhanced fluorescence intensities, from 8% (BCOF-2) to 12% (CD@BCOF-2) and from 12% (TCOF-1) to 18% (CD@TCOF-1), respectively (Fig. 4B). The time-resolved photoluminescence (TRPL) spectra for CD@BCOF-2 and CD@TCOF-1 indicated that the lifetime was prolonged due to the FRET energy transfer at the maximum excitation wavelength for BCOF-1 (λex = 420 nm) (Fig. S24†). At the maximum excitation wavelength for CDs (λex = 380 nm), the fluorescence lifetimes of CD@BCOF-2 and CD@TCOF-1 were reduced to 0.880 and 0.574 ns, respectively (Fig. S25 and Table S4†). The calculated EnT efficiency reached 79.28% and 86.49% for CD@BCOF-2 and CD@TCOF-1, respectively. Correspondingly, the KEnT values were 0.900 × 109 S−1 for CD@BCOF-2 and 1.506 × 109 S−1 for CD@TCOF-1, respectively. The energy transfer efficiency depends on the distance between the CD and COF materials.45 The different energy transfer efficiencies of CD@BCOF-1, CD@BCOF-2 and CD@TCOF-1 are caused by the different sizes provided by the COFs for CD particles. Hence, we observed significantly stronger fluorescence emission at 365 nm ultraviolet through LSCM photographs of CD@BCOF-2 and CD@TCOF-1 (Fig. S26†). All the conclusions indicated that BCOF-2 and TCOF-1 were able to be used as a platform to accommodate CD nanocrystals, and these assemblies successfully improved the fluorescence performance by means of FRET energy transfer.
In comparison, the excitation spectra for TCOF-2 and TCOF-3 overlapped the absorption spectrum for CD particles. Likewise, the emission spectrum for CDs partially overlapped the excitation spectra for TCOF-2 and TCOF-3. Subsequently, the excitation energy of COFs or CDs was absorbed by CD or COF species, respectively. This internal filter effect indicates a failure of the FRET mechanism for both CD-doped TCOF-2 and TCOF-3 systems, leading to the fluorescence quenching of the CD@COF materials (Fig. S27†).48 Significantly, CD@BCOF-1 exhibits an extraordinary solid-state fluorescence quantum yield that approaches the highest level of fluorescence materials, including TFPB-DHzDs,49 Eu3+/Tb3+@1,50 and Tf-DHzDPr.49 It is much higher than those of traditional porous materials, such as ZJU 28@DSM/AF,51 Mg-CP@CuI,52 and 10%Eu-SMOF-1 (Fig. 4C).53
Copper is a constituent element of human hemocyanin, which has an important influence on the functions of blood, brain, and other central nervous system components. Changes in copper homeostasis will mutate proteins and destroy the liver and nervous system; many diseases are related to the concentration of copper, for instance, Wilson's disease.54 Therefore, the sensitive detection of copper ions is critical for healthcare and environmental monitoring.47 As reported previously, COF architectures offer considerable host–guest chemistry for guest molecules. The N atoms from the adjacent COF layers will concentrate a large number of metal ions through coordination bonds.7,8 Taking advantage of the COF platform, CD@BCOF-1 with a strong emission was selected as a fluorescence detector (Fig. 5).
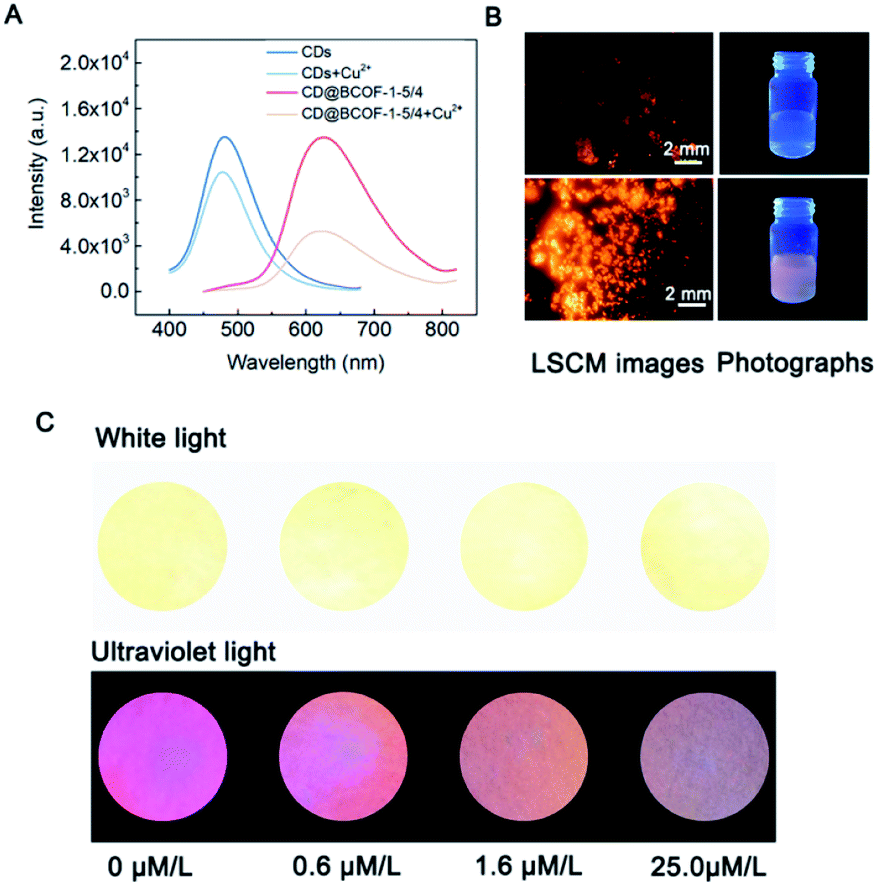 | ||
| Fig. 5 CD@BCOF-1 sensor for Cu2+ ion detection. (A) PL spectra of CDs and CD@BCOF-1 in the presence of 1 mmol L−1 Cu2+ ions. (B) LSCM images and photographs of CD@BCOF-1 under 365 nm UV light in the presence of 1 mmol L−1 Cu2+ ions. (C) Images for Cu2+ ion detection at different stages of Wilson's disease by CD@BCOF-1 fluorescence detector (white light and ultraviolet light with λex = 365 nm). | ||
Solutions of CDs and CD@BCOF-1 with the same PL intensity (3 mL) were added to the same amounts of Cu2+ ions (30 μL, 0.1 mol L−1). CD@BCOF-1 displayed a specific quenching effect for Cu2+ ions (Fig. 5A). The quenched PL intensity (ΔI) of CD@BCOF-1 induced by Cu2+ ions was more obvious than that of CDs. Moreover, the peak position in the emission spectrum was not changed, but the fluorescence lifetime became shorter, indicating the dynamic quenching mode (Fig. S28†). The mechanism was caused by the transition of Cu2+ ions from the excited electronic state to d orbit state, resulting in the non-radiation quenching.
The CD@BCOF-1 sensing system has a comprehensive response range for copper ions. Fitting with eqn (3) in the ESI,† as the concentration changes within the range of 10−7 to 10−8 M, we can obtain the simulated spectral curve F0/F = 1.5003Q + 0.9985 (R2 = 0.998). At a signal-to-noise ratio of 3, the detection limit of the CD@BCOF-1 sensor is 6.60 × 10−9 mol L−1 (Fig. S29†). It is ca. 150 times more sensitive than the BCOF-1 sensor (detection limit of 9.92 × 10−7 mol L−1; Fig. S30†). This higher detection sensitivity is caused by the ordered channels, together with the vast accessible surface, which provides a platform for the full interaction between CD@BCOF-1 and enriched Cu2+ ions. At least 57% reduced fluorescence intensity is detected after CD@BCOF-1 was exposed to Cu2+ ions (10−3 mol L−1). Significantly, the quenching capability maintains its reduced state after 9 minutes of exposure (Fig. S31†).
The selective detection of Cu2+ ions against other interfering ions is necessary for practical application. Various metal ions common in the human body, including Ag+, K+, Ba2+, Ca2+, Co2+, Mg2+, Ni2+, Zn2+, Al3+, and Fe3+, were tested at a concentration of 10−3 mol L−1. As observed in Fig. S32,† there are no apparent changes in PL intensity in the presence of various interfering ions. In the presence of Cu2+ ions, the fluorescence of CD@BCOF-1 is immediately quenched, suggesting the high selectivity of CD@BCOF-1 for Cu2+ ions. Specifically, the fluorescence quenching appearance of the solid powder can be visually observed from LSCM images in the presence of 10−3 mol L−1 Cu2+ ions (Fig. 5B).
As a result of its precious stability, excellent selectivity, and high sensitivity, the CD@BCOF-1 fluorescence sensor was developed for the practical application of trace detection of Cu2+ ions. Wilson's disease can be diagnosed by testing for the presence of Cu2+ ions in urine, as the peak value for a healthy person is about 0.6 μmol L−1; urinary copper usually exceeds 1.6 μmol L−1 in patients with symptoms; children who take copper-discharge drugs will have concentrations above 25.0 μmol L−1.55 Therefore, three concentrations were configured based on actual human urine spiked with copper ions. After CD@BCOF-1 powder was dispersed on a filter paper, we observed that the paper sensor became orange-red under ultraviolet light (λex = 365 nm). However, the fluorescence intensity visibly changed in the 1.6 μmol L−1 copper ion solution; and it was nearly quenched in the presence of 25.0 μmol L−1 copper ion solution (Fig. 5C). This apparent change demonstrates that CD@BCOF-1 exhibits excellent sensing for copper ions at very low levels and is therefore a promising tool for medical diagnosis and treatment.
Conclusion
In summary, we described the design and preparation of COF-based inorganic nanocrystal-dynamic porous polymer assemblies. Due to the existence of pore cavities, the hybrids retain their structural order even when a large number of inorganic nanoparticles are embedded. The construction of an assembled device realizes effective energy transfer, resulting in a selective and sensitive diagnostic tool for copper in human urine. It is possible to provide a good direction for inorganic nanocrystal-polymer assemblies, resulting in devices and sensors with important applications in many fields including healthcare, environmental monitoring, and biodefense.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21975039, 21604008, 21531003 and 91622106), the “111” project (B18012), and the Fundamental Research Funds for the Central Universities (2412020ZD008).Notes and references
- L. Zhang, J. B. Bailey, R. H. Subramanian, A. Groisman and F. A. Tezcan, Nature, 2018, 557, 86 CrossRef CAS.
- Y. Suzuki, G. Cardone, D. Restrepo, P. D. Zavattieri, T. S. Baker and F. A. Tezcan, Nature, 2016, 533, 369 CrossRef CAS.
- A. Worthy, A. Grosjean, M. C. Pfrunder, Y. Xu, C. Yan, G. Edwards, J. K. Clegg and J. C. McMurtrie, Nat. Chem., 2018, 10, 65 CrossRef CAS.
- D. A. Fletcher and R. D. Mullins, Nature, 2010, 463, 485 CrossRef CAS.
- T. Kim, M. K. Al-Muhanna, S. D. Al-Suwaidan, R. O. Al-Kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2013, 52, 6889 CrossRef CAS.
- M. K. Panda, S. Ghosh, N. Yasuda, T. Moriwaki, G. D. Mukherjee, C. M. Reddy and P. Naumov, Nat. Chem., 2014, 7, 65 CrossRef.
- Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193 CrossRef CAS.
- J. Rabone, Y. F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053 CrossRef CAS.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357 CrossRef CAS.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326 CrossRef CAS.
- Q. Chen, Z. Chang, W. C. Song, H. Song, H. B. Song, T. L. Hu and X. H. Bu, Angew. Chem., Int. Ed., 2013, 52, 11550 CrossRef CAS.
- S. Ghosh and C. M. Reddy, Angew. Chem., Int. Ed., 2012, 51, 10319 CrossRef CAS.
- Y. Hu, N. Dunlap, S. Wan, S. Lu, S. Huang, I. Sellinger, M. Ortiz, Y. Jin, S. H. Lee and W. Zhang, J. Am. Chem. Soc., 2019, 141, 7518 CrossRef CAS.
- Q. Wang, C. Yu, H. Long, Y. Du, Y. Jin and W. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7550 CrossRef CAS.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570 CrossRef CAS.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478 CrossRef CAS.
- Y. Yang, M. Faheem, L. Wang, Q. Meng, H. Sha, N. Yang, Y. Yuan and G. Zhu, ACS Cent. Sci., 2018, 4, 748 CrossRef CAS.
- K. Gottschling, L. Stegbauer, G. Savasci, N. A. Prisco, Z. J. Berkson, C. Ochsenfeld, B. F. Chmelka and B. V. Lotsch, Chem. Mater., 2019, 31, 1946 CrossRef CAS.
- P. Das and S. K. Mandal, Chem. Mater., 2019, 31, 1584 CrossRef CAS.
- Q. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Ma, Adv. Mater., 2018, 30, e1705479 CrossRef.
- C. Yang, Z.-D. Yang, H. Dong, N. Sun, Y. Lu, F.-M. Zhang and G. Zhang, ACS Energy Lett., 2019, 4, 2251 CrossRef CAS.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS.
- P. Albacete, J. I. Martinez, X. Li, A. Lopez-Moreno, S. A. Mena-Hernando, A. E. Platero-Prats, C. Montoro, K. P. Loh, E. M. Perez and F. Zamora, J. Am. Chem. Soc., 2018, 140, 12922 CrossRef CAS.
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790 CrossRef CAS.
- B. P. Biswal, H. A. Vignolo-Gonzalez, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082 CrossRef CAS.
- S. Yan, X. Guan, H. Li, D. Li, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 2920 CrossRef CAS.
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Diaz Diaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513 CrossRef CAS.
- V. S. Vyas, M. Vishwakarma, I. Moudrakovski, F. Haase, G. Savasci, C. Ochsenfeld, J. P. Spatz and B. V. Lotsch, Adv. Mater., 2016, 28, 8749 CrossRef CAS.
- X. G. Liu, D. L. Huang, C. Lai, G. M. Zeng, L. Qin, H. Wang, H. Yi, B. S. Li, S. Y. Liu, M. M. Zhang, R. Deng, Y. K. Fu, L. Li, W. J. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266 RSC.
- W. Li, C. X. Yang and X. P. Yan, Chem. Commun., 2017, 53, 11469 RSC.
- Y. W. Peng, Y. Huang, Y. H. Zhu, B. Chen, L. Y. Wang, Z. C. Lai, Z. C. Zhang, M. T. Zhao, C. L. Tan, N. L. Yang, F. W. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698 CrossRef CAS.
- P. Wang, F. Zhou, C. Zhang, S. Y. Yin, L. L. Teng, L. L. Chen, X. X. Hu, H. W. Liu, X. Yin and X. B. Zhang, Chem. Sci., 2018, 9, 8402 RSC.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634 RSC.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575 CrossRef CAS.
- Y. Jin, Y. Hu and W. Zhang, Nat. Rev. Chem., 2017, 1, 0056 CrossRef.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362 RSC.
- J. Li, B. Wang, H. Zhang and J. Yu, Small, 2019, 15, e1805504 CrossRef.
- B. Wang, Y. Mu, H. Zhang, H. Shi, G. Chen, Y. Yu, Z. Yang, J. Li and J. Yu, ACS Cent. Sci., 2019, 5, 349 CrossRef CAS.
- M. J. Sun, Y. W. Zhong and J. Yao, Angew. Chem., Int. Ed., 2018, 57, 7820 CrossRef CAS.
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712 CrossRef CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953 CrossRef CAS.
- Q. Yang, Q. Xu and H. L. Jiang, Chem. Soc. Rev., 2017, 46, 4774 RSC.
- Y. Z. Chen, Z. U. Wang, H. Wang, J. Lu, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035 CrossRef CAS.
- J. Mei, Y. Hong, J. W. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS.
- P. C. Ray, Z. Fan, R. A. Crouch, S. S. Sinha and A. Pramanik, Chem. Soc. Rev., 2014, 43, 6370 RSC.
- Y. Jiang and J. McNeill, Chem. Rev., 2017, 117, 838 CrossRef CAS.
- X. Lin, G. Gao, L. Zheng, Y. Chi and G. Chen, Anal. Chem., 2014, 86, 1223 CrossRef CAS.
- J. Liu, Y. Chen, W. Wang, J. Feng, M. Liang, S. Ma and X. Chen, J. Agric. Food Chem., 2016, 64, 371 CrossRef CAS.
- X. Li, Q. Gao, J. F. Wang, Y. F. Chen, Z. H. Chen, H. S. Xu, W. Tang, K. Leng, G. H. Ning, J. S. Wu, Q. H. Xu, S. Y. Quek, Y. X. Lu and K. P. Loh, Nat. Commun., 2018, 9, 2335 CrossRef.
- C. Y. Sun, X. L. Wang, X. Zhang, C. Qin, P. Li, Z. M. Su, D. X. Zhu, G. G. Shan, K. Z. Shao, H. Wu and J. Li, Nat. Commun., 2013, 4, 2717 CrossRef.
- Y. Cui, T. Song, J. Yu, Y. Yang, Z. Wang and G. Qian, Adv. Funct. Mater., 2015, 25, 4796 CrossRef CAS.
- Z.-F. Wu, B. Tan, Z.-L. Xie, J.-J. Fu and X.-Y. Huang, J. Mater. Chem. C, 2016, 4, 2438 RSC.
- D. F. Sava, L. E. S. Rohwer, M. A. Rodriguez and T. M. Nenoff, J. Am. Chem. Soc., 2012, 134, 3983 CrossRef CAS.
- S. Lee, G. Barin, C. M. Ackerman, A. Muchenditsi, J. Xu, J. A. Reimer, S. Lutsenko, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 7603 CrossRef CAS.
- European Association for Study of Liver, J. Hepatol., 2012, 56, 671 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04359a |
| This journal is © The Royal Society of Chemistry 2020 |