
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA soft molecular 2Fe–2As precursor approach to the synthesis of nanostructured FeAs for efficient electrocatalytic water oxidation†
Rodrigo
Beltrán-Suito‡
a,
Viktoria
Forstner‡
a,
J. Niklas
Hausmann
a,
Stefan
Mebs
b,
Johannes
Schmidt
c,
Ivelina
Zaharieva
b,
Konstantin
Laun
d,
Ingo
Zebger
 d,
Holger
Dau
*b,
Prashanth W.
Menezes
*a and
Matthias
Driess
*a
d,
Holger
Dau
*b,
Prashanth W.
Menezes
*a and
Matthias
Driess
*a
aDepartment of Chemistry: Metalorganics and Inorganic Materials, Technische Universität Berlin, Straße des 17 Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: prashanth.w.menezes@tu-berlin.de; matthias.driess@tu-berlin.de
bFachbereich Physik, Freie Universität Berlin, Arnimallee 14, Berlin 14195, Germany. E-mail: holger.dau@fu-berlin.de
cDepartment of Chemistry: Functional Materials, Technische Universität Berlin, Hardenbergstraße 40, Berlin 10623, Germany
dInstitut für Chemie, Max-Volmar-Laboratorium für Biophysikalische Chemie, Technische Universität Berlin, Straße des 17 Juni 135, Berlin 10623, Germany
First published on 7th October 2020
Abstract
An unprecedented molecular 2Fe–2As precursor complex was synthesized and transformed under soft reaction conditions to produce an active and long-term stable nanocrystalline FeAs material for electrocatalytic water oxidation in alkaline media. The 2Fe2As-centred β-diketiminato complex, having an unusual planar Fe2As2 core structure, results from the salt-metathesis reaction of the corresponding β-diketiminato FeIICl complex and the AsCO− (arsaethynolate) anion as the monoanionic As− source. The as-prepared FeAs phase produced from the precursor has been electrophoretically deposited on conductive electrode substrates and shown to act as a electro(pre)catalyst for the oxygen evolution reaction (OER). The deposited FeAs undergoes corrosion under the severe anodic alkaline conditions which causes extensive dissolution of As into the electrolyte forming finally an active two-line ferrihydrite phase (Fe2O3(H2O)x). Importantly, the dissolved As in the electrolyte can be fully recaptured (electro-deposited) at the counter electrode making the complete process eco-conscious. The results represent a new and facile entry to unexplored nanostructured transition-metal arsenides and their utilization for high-performance OER electrocatalysis, which are also known to be magnificent high-temperature superconductors.
Introduction
The search for alternative energies is one of the most important concerns of the 21st century due to fossil fuel depletion on one side and climate change issues majorly attributed to CO2 emission on the other side.1 Amongst the most frequently proposed ways, the most efficient is the oxidation of hydrogen in fuel cells. Hydrogen is a very promising alternative fuel source because of its high energy density and the generation of only carbon-free products (H2O) during the oxidation process.2 However, current hydrogen production is mainly based on the reforming, i.e. combustion of hydrocarbons, due to low production costs and yet sufficient availability of hydrocarbon sources.3 Another common way to generate hydrogen is the electrolysis of water, where H2O is split into hydrogen (H2) and oxygen (O2) in the hydrogen evolution (HER) and oxygen evolution reactions (OER), respectively.4 The electrocatalytic generation of H2 from water is an effective approach but requires extra energy. This extra energy is expressed in terms of overpotential (η), which is the potential beyond the thermodynamic requirement to break water. It is related to processes that limit the water-splitting reaction such as kinetic hindrances originating from the four sequential proton-coupled electron transfer steps in the OER,5 ohmic voltage loss,6 and other factors related to the device and electrolyte characteristics.7 Therefore, continuous efforts must be made to design and develop suitable and sustainable OER electrocatalysts that entail minimum overpotentials.In this context, non-noble metal-based materials have recently sparked an immense interest in the OER owing to their low cost, abundance, and environment-friendly properties. Additionally, their OER activities are comparable to those of the state-of-the-art electrocatalysts such as RuO2 and IrO2.8 Among them, transition metal (TM) oxides and oxyhydroxides,9 phosphates,10 borophosphates,11 phosphites,12 intermetallics,13 chalcogenides,14,15 and pnictides16–18 have lately been used for the OER, HER and overall water splitting. The latter, notably nitrides and phosphides, have been explored extensively due to their high electroconductivity and greater resistance to harsh electrolyte environments, i.e. under strongly acidic or alkaline conditions, which are used during electrocatalytic water splitting.19 It has been shown that TM pnictides, in most cases, undergo severe transformation under OER conditions forming thermodynamically stable layered oxide/oxyhydroxide species.20–25 The transformation occurs either only at the surface of the electrocatalyst forming a core–shell structure or completely throughout the bulk resulting in defect rich and high surface area TM oxides/oxyhydroxides, whereas arsenates have been reported as stable constituents of a Co-based OER catalyst material.26 TM arsenide electrocatalysts, the heavier congeners, are relatively unknown for the OER except for a recent publication from Schuhmann and co-workers27 where the high-temperature derived NiAs solid phase was used as an electrocatalyst for an alkaline OER with moderate performance. However, insights on the active structure of arsenide materials remain still unexplored and the structural and functional role of arsenic to catalyse the OER is thus meaningful to evaluate.28
As most of the microcrystalline TM arsenides are prepared through high-temperature solid-state approaches, it is highly desirable to discover new synthetic strategies that could produce nanostructured arsenides with a larger surface area and distinct morphology. In this context, an effective pathway to synthesize independently amorphous and crystalline nanostructured materials has emerged in the last few years that utilizes the low-temperature decomposition of molecular single-source precursors (SSPs).22,29–31 Molecular systems enable the adjustment of defined transition-metal to heteroatom ratios of the desired material by manipulation of the synthetic conditions, which is hardly achievable by conventional solid-state, hydrothermal and solvothermal strategies. We have previously isolated several well-defined molecular complexes, containing TM chalcogenide or pnictide cluster cores stabilized by β-diketiminato ligands (L = CH(CRNAr)2 with R = alkyl and Ar = aryl) and some of them have been used as SSPs for catalytic water-splitting applications.22,30–32 Strikingly, molecular TM arsenide structures are less common and have seldom been examined in detail for their suitability in material synthesis, possibly due to their laborious synthetic routes and toxicity. Nevertheless, TM arsenides are useful reference systems for learning about structure–reactivity relationships of TM pnictides towards their utilization in solid-phase energy applications. Among TM arsenides, iron arsenide (FeAs) based materials have attracted much attention and have been studied extensively for their unconventional superconductivity.33–37 Similarly, FeAs has recently been utilized as a promising anode for Li-ion batteries,38 however, its potential application in electrocatalytic water-splitting is currently unexplored due to concerns about toxicity and possible derived contamination. However, pros for the investigation of TM arsenides as OER electro(pre)catalysts have recently been noted in the literature.28
Encouraged by the potential suitability of FeAs in the electrocatalytic OER we focused on the following research questions: (1) could one establish a rational approach to the synthesis of monodisperse and ultra-small FeAs nanostructures from a molecular precursor with an Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :As ratio of 1:1? (2) Is molecularly derived FeAs from (1) suitable for the OER and if so how does its catalytic activity compare to benchmark reference materials? (3) What is the active structure of the FeAs electro(pre)catalyst during OER catalysis? (4) What are the future opportunities of FeAs nanostructures? To answer (1), we designed a molecular β-diketiminato-stabilized 2Fe–2As cluster complex, which turned out to be a suitable precursor for the synthesis of nanostructured FeAs. To our delight, the electrophoretically deposited FeAs on conductive substrates displays superior activity for the OER under alkaline conditions compared to the “Fe-only” reference materials FeOOH, Fe(OH)3 and Fe2O3, respectively, with the same mass loading. State-of-the-art ex situ characterization techniques, as well as quasi in situ XAS, reveal that the FeAs is merely a precatalyst and transforms rapidly under the alkaline OER conditions by complete loss of As into the electrolyte solution to form an catalytically active two-line ferrihydrite phase, a high-surface-area and low-crystalline semiconducting hydrous ferric oxyhydroxide mineral, which has not been yet reported in the literature as an electro(pre)catalyst or as a product of the transformation of Fe-based materials during alkaline OER electrocatalysis.39,40 Furthermore, the dissolved As has been successfully recovered from the electrolyte at the counter electrode (CE) to circumvent any environmental contamination and harmful effects. Our investigation provides a facile access to nanocrystalline FeAs, which opens the door to new applications of the material in magnetism, superconductors, supercapacitors, and photocatalytic hydrogen evolution.41,42 Additionally, the soft molecular precursor approach could be expanded to prepare other nanostructured materials containing TMs and non-metals, and to study their structural and electronic transformation with respect to the OER under alkaline conditions.
:As ratio of 1:1? (2) Is molecularly derived FeAs from (1) suitable for the OER and if so how does its catalytic activity compare to benchmark reference materials? (3) What is the active structure of the FeAs electro(pre)catalyst during OER catalysis? (4) What are the future opportunities of FeAs nanostructures? To answer (1), we designed a molecular β-diketiminato-stabilized 2Fe–2As cluster complex, which turned out to be a suitable precursor for the synthesis of nanostructured FeAs. To our delight, the electrophoretically deposited FeAs on conductive substrates displays superior activity for the OER under alkaline conditions compared to the “Fe-only” reference materials FeOOH, Fe(OH)3 and Fe2O3, respectively, with the same mass loading. State-of-the-art ex situ characterization techniques, as well as quasi in situ XAS, reveal that the FeAs is merely a precatalyst and transforms rapidly under the alkaline OER conditions by complete loss of As into the electrolyte solution to form an catalytically active two-line ferrihydrite phase, a high-surface-area and low-crystalline semiconducting hydrous ferric oxyhydroxide mineral, which has not been yet reported in the literature as an electro(pre)catalyst or as a product of the transformation of Fe-based materials during alkaline OER electrocatalysis.39,40 Furthermore, the dissolved As has been successfully recovered from the electrolyte at the counter electrode (CE) to circumvent any environmental contamination and harmful effects. Our investigation provides a facile access to nanocrystalline FeAs, which opens the door to new applications of the material in magnetism, superconductors, supercapacitors, and photocatalytic hydrogen evolution.41,42 Additionally, the soft molecular precursor approach could be expanded to prepare other nanostructured materials containing TMs and non-metals, and to study their structural and electronic transformation with respect to the OER under alkaline conditions.
Results and discussion
Access to molecular arsenido iron complexes and iron arsenic clusters
Since the first reports on coordinatively stabilized, substituent-free Asn (arsenido) ligands by Dahl in 1969,43 several TM arsenide clusters have been synthesized using yellow arsenic (As4) or AsX3 (X = Cl, H) as sources.44–47 Stable and robust As-carrying molecules could also be employed as suitable As sources. The facile synthesis and isolation of stable and robust arsaethynolate anion (AsCO−) containing compounds were first reported by Goicoechea and co-workers48 and later, we reported the facile one-pot synthesis of the [Na(OCAs)(dioxane)x] salt.49 The AsCO− anion has been shown to react towards electrophiles and to form highly reactive arsinidenes through spontaneous or photocatalytic CO release. Thus, it serves as a building block towards new heterocyclic compounds, diarsenes, and terminal pnictides.46,48,50 The reactivity of this anion towards β-diketiminato stabilized Fe-complexes is yet to be explored. Consequently, the preparation of Fe–As cluster containing molecules is of great interest as they could serve as molecular precursors for materials synthesis.Synthesis and characterization of the 2Fe–2As single-source precursor (SSP)
Treatment of the β-diketiminato ligated chlorido iron(II) LBFeCl 1 (LB = CH(CtBuNDipp)2, Dipp = 2,6-iPr2C6H3) with one molar equivalent of NaOCAs·(dioxane)2.1 at room temperature affords the novel dinuclear arsenido iron complex LBFeAs2FeLB2 in 40% yield according to Scheme 1. Compound 2 was isolated and characterized by elemental analysis, 1H NMR and IR spectroscopy, and a single-crystal X-ray diffraction analysis (see Fig. S1, S2 and Table S1†).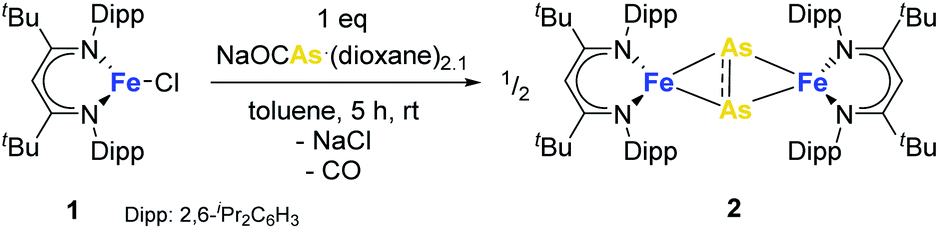 | ||
| Scheme 1 Synthesis of molecular precursor 2. | ||
Complex 2 crystallizes in the monoclinic space group P21/n (Fig. 1) as an FeAs dimer with two co-crystallized toluene molecules in the unit cell. The 2Fe–2As core is symmetrical, with Fe–As distances (2.4023(4) Å and 2.4087(4) Å) in the common range of Fe–As single bonds.51–53 Strikingly, unlike other 2M–2E complexes (M = metal, E = P, As), the iron arsenide cluster core in 2 has not a butterfly-like structure.44–47 The geometry around the iron atoms is tetrahedral instead, generating a planar 2Fe–2As unit. Such a cluster core structure is unprecedented in pnictido transition-metal chemistry. With 2.3447(5) Å, the As–As bond in 2 is significantly longer than expected for an As–As double bond (2.23 Å) in butterfly-like M2As2 complexes.54–56 Contrary to other butterfly-like 2M–2E systems (E = P, As), complex 2 is paramagnetic in solutions as shown by 1H NMR spectroscopy (see Fig. S1†). The experimental shifts are very similar to those from the chlorido iron(II) precursor 1, but the IR spectrum of 2 (see Fig. S2†) shows an irrefutable change in the absorption pattern.
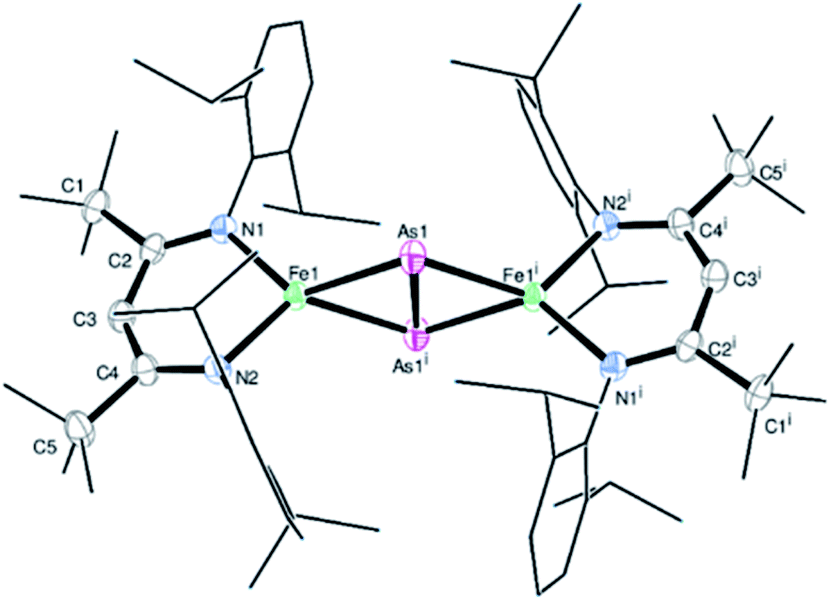 | ||
| Fig. 1 Molecular structure of 2. Ellipsoids are set at 50% probability; hydrogen atoms and co-crystallized toluene have been omitted for clarity. One of the iPr groups in the ligand is disordered over two positions, and hence only one configuration is shown. Selected bond lengths [Å] and angles [°]: As1–As1i 2.3447(5), Fe1–As1 2.4023(4), Fe1–As1i 2.4087(4), Fe1–N1 2.0085(18), Fe1–N2 2.0096(18), Fe1⋯Fe1i 4.201, N1–Fe1–N2 95.95(7), As1–Fe1–As1i 60.9969(13), and Fe1–As1–Fe1i 121.664(12). Symmetry operation for (i): −x + 1, −y + 1, −z + 1 (crystal data and structure refinement are shown in Tables S1–S4†). CCDC 1989143 contains the supplementary crystallographic data for this paper.† | ||
Synthesis and characterization of FeAs
The hot injection of precursor 2 at 250 °C in oleylamine yielded a black powder. Powder X-ray diffraction (PXRD) analysis of the latter revealed reflections corresponding to crystalline FeAs (JCPDS 76-458) (see Fig. S3a†). The solid-state crystal structure of FeAs belongs to the MnP structure type (orthorhombic, Pnma (62) space group, a = 5.442 Å and c = 3.3727 Å) (see Fig. S3b†).57–59 The unit cell of FeAs comprises 4Fe and 4As atoms. Each Fe atom is octahedrally coordinated to six near As atoms, and each As atom is surrounded by six near Fe atoms in a trigonal prismatic configuration. Both coordination polyhedra are far from their regular shapes, leading to a distorted structure.58 Transmission electron microscopy (TEM) showed crystallites of ∼10 nm in size (Fig. 2a and S4†) and high-resolution TEM (HR-TEM) unveils crystalline fringes associated with a lattice spacing of 0.258 ± 0.013 nm corresponding to the (111) plane of the FeAs phase (Fig. 2b). Moreover, in the Fourier transform (FT) of the HR-TEM images, the lattice spacings of 0.255 ± 0.004 and 0.199 ± 0.001 nm were identified and were consistent with the (111) and (211) crystallographic planes of FeAs (see Fig. S5†). In addition, the selected area electron diffraction (SAED) pattern (Fig. 2c) displayed diffraction rings representative of the nanocrystalline FeAs phase and was in agreement with the PXRD. Scanning electron microscopy (SEM) revealed that the solid was composed of agglomerated particles (Fig. 2d and S6†). Elemental mapping by energy-dispersive X-ray spectroscopy (EDX) exhibited a homogenous distribution of Fe and As (Fig. 2e, f, S7 and S8†). The phase identity and composition were also confirmed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) and EDX (see Table S5†), obtaining a ratio of Fe:As of ∼1:1. Elemental analysis revealed the presence of a negligible amount of organic impurities (see Table S6†), derived from the decomposition of the β-diketiminato ligand into unidentified organic species or remaining solvent.21 The Raman active bands obtained within the (resonance) Raman spectroscopic investigations were also consistent with those reported in the literature for FeAs crystals (see Fig. S9†).60 X-ray photoelectron spectroscopy (XPS) was performed to gain information on chemical composition and electronic states. The deconvoluted Fe 2p, As 3d and O 1s XPS spectra of as-prepared FeAs are shown in Fig. S10†. The low binding energy peaks in the Fe 2p are related to Feδ+ in FeAs (2p3/2 705.9 eV and 2p1/2 719.0 eV).61 The remaining higher binding energy peaks are associated with Fe in higher oxidation states (Fe2+/Fe3+), caused by surface passivation (2p3/2 709.8 eV and 2p1/2 723.1 eV for Fe2+; and 2p3/2 711.5 eV and 2p1/2 725.3 eV for Fe3+) (see Fig. S10a†).62 The As 3d spectrum shows a low binding energy peak related to Asδ− in FeAs (40.4 eV), and a smaller peak associated with As3+ in As2O3 (43.2 eV), a consequence of surface passivation (see Fig. S10b†).63 Finally, the O 1s XPS deconvoluted spectrum (see Fig. S10c†) reveals only peaks derived from air exposure: Fe–O (529.4 eV)62 and As–O (530.6 eV).63,64 The peaks at higher binding energy (531.8 and 534.7 eV) are associated with hydroxylation and adsorbed water on the surface.62
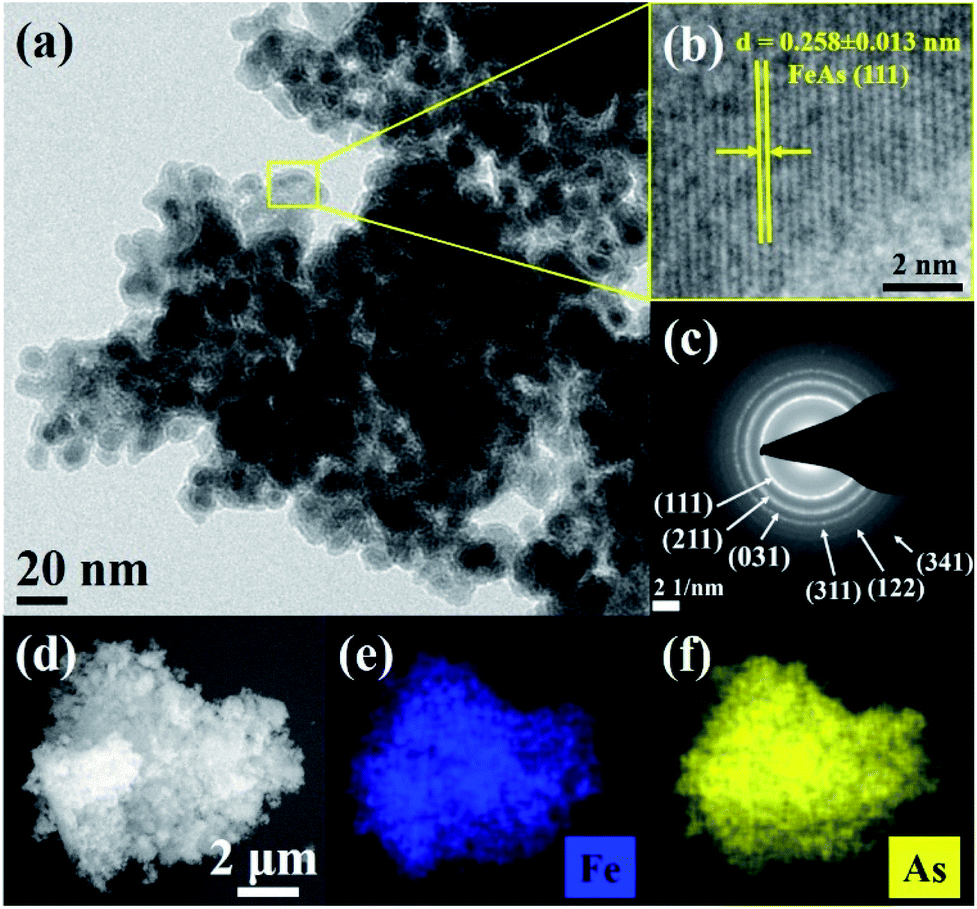 | ||
| Fig. 2 (a) TEM image of agglomerated FeAs nanoparticles of ∼10 nm diameter, (b) HR-TEM image displaying crystalline fringes with an interlayer distance of 0.258 ± 0.013 nm corresponding to the FeAs (111) crystalline plane, (c) SAED pattern with diffraction rings matching the planes of the FeAs phase (JCPDS 76-458) (see Fig. S3a†), and (d) SEM image showing agglomerated particles of FeAs and EDX mapping of homogenously distributed (e) Fe and (f) As. | ||
Electrocatalytic characterization and OER performance
The as-prepared material was electrophoretically deposited on 1 × 1 cm2 nickel foam (NF) with a sample loading of 1 ± 0.1 mg cm−2. The characterization of the films (FeAs/NF) proved that the identity of the material was maintained during the deposition (see Fig. S11–S13†). NF has been considered as an attractive electrode material because of its low cost, high conductivity, large electro-active surface area, mechanical stability, and good corrosion resistance.65 As the oxides, hydroxides, and oxyhydroxides are expected to be the transformation products under the oxidative alkaline conditions,20,66 we synthesized reference Fe(OH)3, FeOOH and Fe2O3 materials (see Fig. S14–S16†), deposited them on NF (1 mg cm−2) and compared their activity to that of FeAs (preparation details in the Experimental section, ESI†). Continuous cyclic voltammetry (CV) experiments in 1 M KOH electrolyte were done first to activate the samples (Fig. S17†). Once the electrodes were activated, the electrocatalytic activity towards the OER of the deposited FeAs films was evaluated by recording linear sweep voltammetry (LSV) curves. An overpotential of η = 252 ± 3 mV (10 mA cm−2) was obtained for crystalline FeAs/NF, which was superior to those of Fe(OH)3/NF (η10 mA = 275 ± 2 mV), FeOOH/NF (η10 mA = 283 ± 2 mV), and Fe2O3/NF (η10 mA = 312 ± 4 mV) (Fig. 3a). The contribution of NF to the activity was proved to be insignificant as the bare NF achieved a high overpotential of η10 mA = 480 ± 5 mV (Fig. 3a) and continuous deactivation and minimum current density (<12 mA cm−2) during 10 h chronoamperometry (see Fig. S18†). The FeAs/NF showed an overpotential of 301 ± 3 mV at 100 mA cm−2 (Fig. 3a).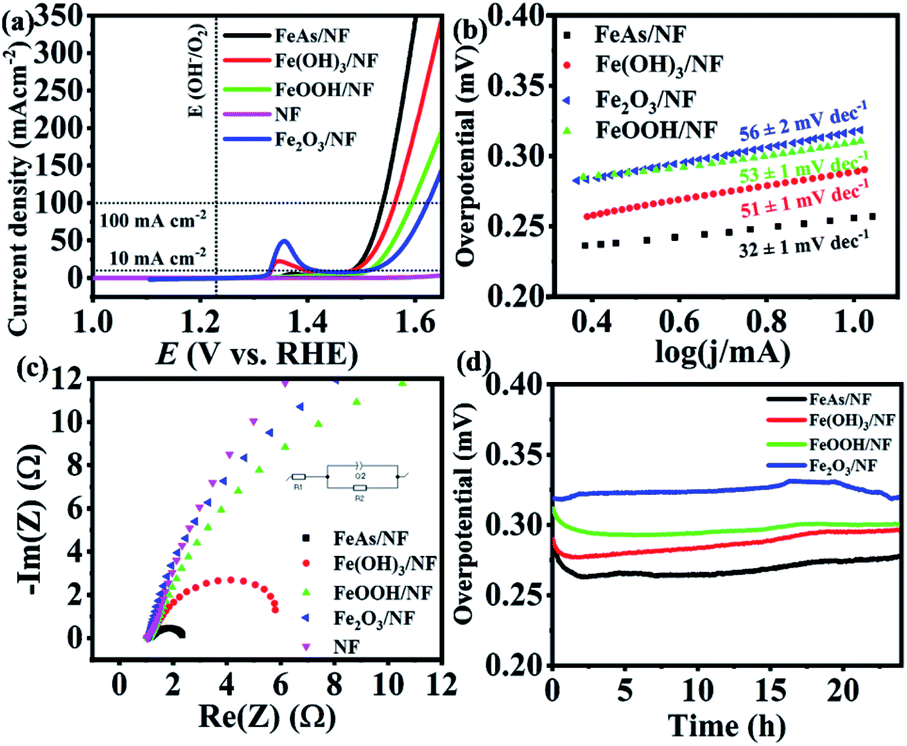 | ||
| Fig. 3 (a) LSV (5 mV s−1) of FeAs and Fe-based reference materials loaded on NF (1 mg cm−2) and bare NF, (b) Tafel slopes obtained from LSV of materials deposited on NF (1 mV s−1), (c) Nyquist plot constructed from the EIS experiment at 1.51 V vs. RHE, and (d) 24 h CP measurement of FeAs/NF and Fe-based reference materials. | ||
Electrocatalytic kinetics were studied by constructing Tafel plots (Fig. 3b) that originated from LSVs with a scan rate of 1 mV s−1. FeAs achieved the lowest Tafel slope of 32 ± 1 mV dec−1 compared to the other prepared materials, indicating more favorable reaction kinetics.67 Electrochemical impedance spectroscopy (EIS) experiments were performed to evaluate the electron transfer efficiency under OER conditions.68 The selected potential for EIS (E = 1.51 V) provides considerable catalytic activity (>10 mA cm−2) for all the materials.69Fig. 3c shows the smallest charge transfer resistance (Rct) across the electrolyte/electrode interface for FeAs compared to the reference Fe materials (for a detailed discussion see Fig. S19 and Table S7†). The lower overpotential observed for FeAs with respect to the examined Fe reference materials is also in accordance with the lower Tafel slope and smaller Rct. A long-term chronopotentiometry (CP) measurement at 10 mA cm−2 was conducted for 24 h to investigate the electrochemical stability, which showed an almost constant overpotential of 265 ± 8 mV, which is lower than that of the Fe-based reference materials (Fig. 3d) and bare NF (see Fig. S18†). CP at higher current density (100 mA cm−2) also revealed a stable overpotential of η100 mA = 330 ± 6 mV for 24 h (Fig. S20†). The contribution of remaining carbon to the activity is minimal, as observed by low activity observed with films of the β-diketiminato ligand (Fig. S21†). A faradaic efficiency of >95% for the OER was calculated by quantifying the evolved O2 by gas chromatography (GC) during electrolysis at a constant current of 50 and 100 mA cm−2 using FeAs/NF as the anode and Pt as the cathode in a closed cell system (see Table S8 and Fig. S22†).
The electrochemical double-layer capacitance (Cdl) was determined by performing continuous CV experiments with different scan rates in a potential range where no apparent faradaic process occurred (see Fig. S23†).70 The difference in cathodic and anodic current was plotted versus the scan rate, from which the value of Cdl was obtained (see Fig. S24†). The electrochemically active surface area (ECSA) is directly proportional to the Cdl. The Cdl was 0.218 ± 0.004 mF cm−2 before the electrocatalytic testing and increased ∼3 times up to 0.629 ± 0.006 mF cm−2 after the CP testing. This change under OER conditions is possibly related to a structural transformation of the FeAs precatalyst to an active structure with more iron sites exposed to the electrolyte by losing most of its As into the electrolyte, and such a phenomenon has already been well demonstrated for non-oxidic based materials.20,71 The overpotential of the prepared FeAs/NF is among the best for iron-based electrocatalysts (see Table S9†) and it is also even 100 mV less than that of the NiAs, the only example of TM arsenides ever reported for the OER (η10 mA = 360 mV).27
The films were deposited on a fluorinated tin oxide (FTO) substrate. The deposited FeAs films on FTO were also characterized, in which the chemical character of FeAs remained (see Fig. S25–S31†). In order to have a fair comparison, Fe(OH)3, FeOOH, and Fe2O3 were also deposited on FTO with the same mass loading. Similarly, the materials were activated by performing continuous CV (see Fig. S32†), followed by measuring the electrocatalytic activity by LSV (see Fig. S33†). The overpotential of the FeAs/FTO was only η = 395 ± 6 mV (10 mA cm−2), which is substantially lower than those of Fe(OH)3/FTO (577 ± 3 mV), FeOOH/FTO (609 ± 5 mV) and Fe2O3/FTO (639 ± 5 mV).
The observed trend in activity is the same as in the NF deposited films, confirming the superior electrochemical performance of FeAs. To verify the effect of mass loading on the OER activity of FeAs, the electrodeposition time was varied, accomplishing different activities (see Table S10, Fig. S34 and S35†). Similar experiments were conducted with Fe(OH)3, the most active Fe-based catalyst among the reference materials (see Table S11, Fig. S36 and S37†). The polarization curves show that the best OER activity was achieved with a loading of 0.4 mg cm−2 and 2.4 mg cm−2 for FeAs/FTO and Fe(OH)3/FTO, respectively.
A similar activity trend in Tafel slopes and Rct results was also observed (see Fig. S38, S39 and Table S12†). The improved electrocatalytic performance of FeAs among the Fe-based catalysts was investigated by ex situ four-point probe resistivity measurements of the films (see Table S13†). The as-deposited FeAs/FTO shows the lowest resistivity, in contrast with the ∼102 to 103 times higher resistivity of the Fe-based reference materials, indicating much slower reaction electron transfer. It should be noted, however, that the ex situ conductivity measurements are not necessarily able to predict the electron transport abilities of materials under OER conditions.72 Long-term CP at 10 mA cm−2 of the FeAs/FTO displayed good stability for 10 h with a slight increase in the overpotential (η = 380 ± 6 mV), in contrast to the Fe-based reference materials (see Fig. S40†).
A redox feature before 1.4 V vs. RHE appeared in the LSVs of the materials deposited on NF (Fig. 3a), but not on FTO (see Fig. S32†). We initially suspected the formation of Ni–FeOOH, which is a benchmark material for the OER.73 However, a comparison of the evolution of the redox peak through the CV of FeAs/NF and bare NF (Fig. S41 and S42†) showed no difference in the peak position and the achieved current. Several investigations have reported that Fe incorporation in Ni–FeOOH generates an anodic shift to higher potentials and a decrease in the current achieved by the peak.73–75 Therefore, we ruled out the formation and contribution of Ni–FeOOH in the electrocatalytic activity. The observed redox peak can only be associated with redox processes occurring on the NF surface (Ni2+/Ni3+). Similar behavior has been observed before for other materials deposited and/or derived from NF.76,77
Post-catalytic characterization
A deep understanding of the structural transformations occurring during catalysis and the origin of the activity was achieved by post-catalytic characterization. The long term CP on FeAs/NF achieved a stable overpotential after ca. 2 h. SEM-EDX performed at this point revealed an almost complete loss of the As, which resulted in a 1:0.06 ± 0.01 ratio of Fe:As (see Fig. S43 and S44†). Similar experiments after the first CV cycle showed an initial loss of As (1:0.91 ± 0.02 ratio of Fe:As, see Fig. S45 and S46†), which confirmed that the complete transformation of the FeAs does not occur with the first CV scan. Consequently, thorough characterization was done after 24 h CP OER (hereafter “after OER”) to ensure the full transformation of the material. PXRD of the film (see Fig. S47†) showed reflections/peaks associated with the crystalline FTO substrate (cassiterite, SnO2, JCPDS 41-1445). No peaks related to remaining FeAs or oxidized arsenic phases were found in the diffraction pattern. SEM showed a possible loss of film material into the electrolyte due to exposure of the FTO substrate (see Fig. S48†). The remaining film was composed of agglomerated particles after OER. Elemental mapping and EDX results revealed an almost complete depletion of As, resulting in a 1:0.05 ± 0.01 ratio of Fe:As (see Fig. S49 and S50†). Similar results were obtained for the films deposited on NF (see Fig. S51–S53†). The loss of the non-metal in solution is a common phenomenon and has been observed before for transition metal chalcogenides and TMP.20,26 Moreover, the high solubility of As under oxidizing and alkaline conditions also contributes to its dissolution into the electrolyte.78 Since a high concentration of As is undesirable due to negative effects on humans and animals,79 and the environment,80 the loss of As was quantitatively determined by ICP-AES (see Table S14†). Although a complete loss of As from FeAs was achieved in 24 h of OER (from EDX), surprisingly, the electrolyte showed only 6.3 ± 0.4% As content of the initial As amount deposited on the film. Puzzled with the results, we closely examined the used Pt CE after OER catalysis that exhibited the deposition of the dark film (see Fig. S54†). As strong arsenic adsorption on Pt electrodes under alkaline electrochemical conditions is already known in the literature,81 we performed SEM and EDX mapping investigations of the black film which confirmed the presence of As at the CE (see Fig. S55†). Consequently, we learned that by replacing the Pt CE with higher surface area CEs (NF, graphite rod), it was possible to recover ∼100% of dissolved As from the electrolyte in its elemental form. The replacement of the Pt CE by a graphitic carbon rod produced no change in the overpotential (see Fig. S56†) because the interactions of As with Pt and carbon electrodes are mechanistically similar under alkaline anodic conditions.82,83
More structural insights on the FeAs electrode gathered by TEM. HR-TEM (Fig. 4a) revealed the formation of crystalline nanodomains with an interlayer distance of 0.25 ± 0.01 nm. The SAED pattern (Fig. 4a, inset) did not show any diffraction rings related to the initial FeAs, instead, two broad diffraction rings at a distance of 0.15 ± 0.01 nm and 0.25 ± 0.01 nm corresponding to the (115) and (110) planes of 2-line ferrihydrite were observed.39,84,85 Fast Fourier transforms (FFT) (see Fig. S57a†) on the nanostructures confirmed a lattice spacing of 0.25 nm (110) which is in agreement with the obtained SAED pattern. TEM-EDX (see Fig. S57b†) showed that only 1.73% of As is left after OER, revealing an almost complete transformation of the FeAs to an oxidized phase. In order to ascertain the identity of the product after OER, FTIR and (resonance) Raman spectroscopies were performed. Both spectroscopic methods confirmed the presence of 2-line ferrihydrite and ruled out other iron (oxy)hydroxide species.40,86,87 The IR spectrum after OER (see Fig. S58†) showed a broad absorption band at 3168 cm−1, attributed to OH stretching vibrations,40 while the bands at 1531 cm−1, 1345 cm−1 and 885 cm−1 were assigned to Fe–O stretching and Fe–OH bending vibrations, respectively.40,88,89 In the (resonance) Raman spectrum taken after OER characteristic bands at 700 cm−1, 510 cm−1 and 360 cm−1 related to 2-line ferrihydrite were detected (see Fig. S59†).87,90–92
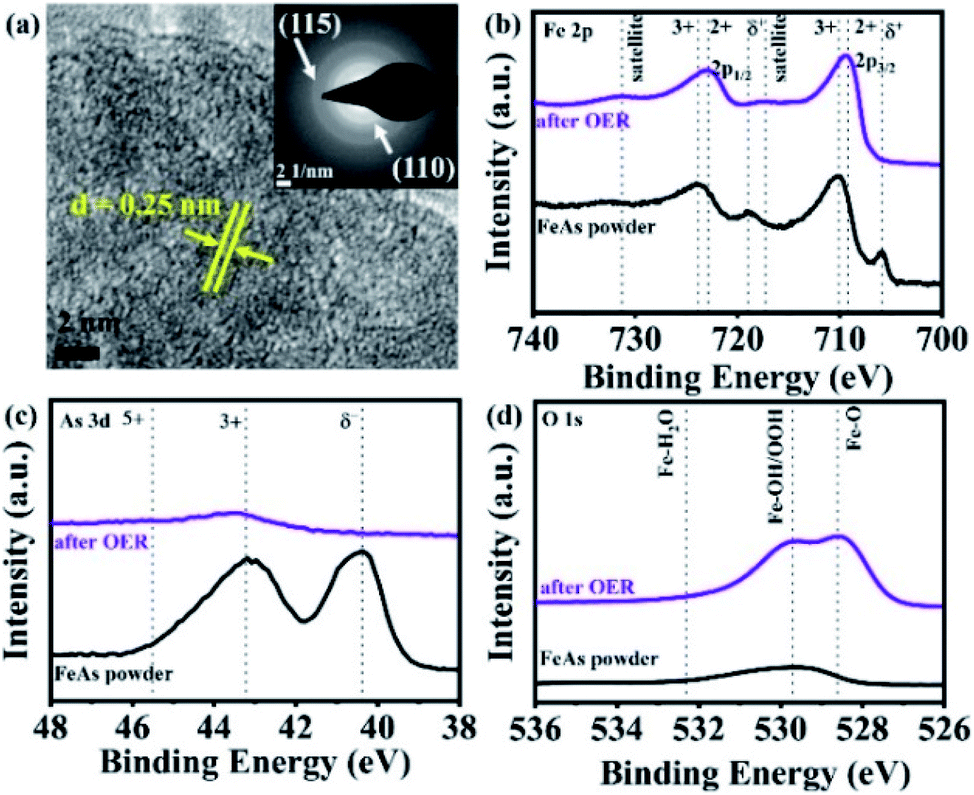 | ||
| Fig. 4 (a) HR-TEM of the FeAs electro(pre)catalyst after OER forming a nanostructure with a lattice spacing of ∼0.25 nm (110). The SAED (a, inset) displayed two broad diffraction rings indicative of 2-line ferrihydrite (see Fig. S57a†). The XPS spectra of the FeAs compared to FeAs after OER for (b) Fe 2p, (c) As 3d and (d) O 1s. | ||
Quasi in situ XAS was carried out to gather further information on the structure and the electronic state of Fe and As during the OER. Post catalytic investigations revealed significant structural and electronic changes suggesting the oxidation and conversion of FeAs to Fe3+OxHy. The Fe X-ray absorption near-edge (XANES) spectra (Fig. 5a) disclosed a change in edge shape and position. The initial half-edge-position for the FeAs powder is close to the one of Fe metal foil, indicating the metallic character of the prepared material. After the OER, the edge position and shape come closer to those observed for α-FeOOH, which reveals complete oxidation of the Fe atoms in the FeAs to Fe3+. This is consistent with the proposed transformation of the FeAs to the 2-line ferrihydrite under alkaline oxidative conditions. Moreover, the edge-energy position of the As-XANES (Fig. 5b) shifts to higher energy during the OER, suggesting an increase in the oxidation state of As, as evidenced by XPS (see Fig. S60†). Besides, the very low raw intensities of the As spectrum confirm that most of the As leaves the structure during the OER (see Fig. S61†), which is in agreement with the ICP-AES, EDX and XPS results.
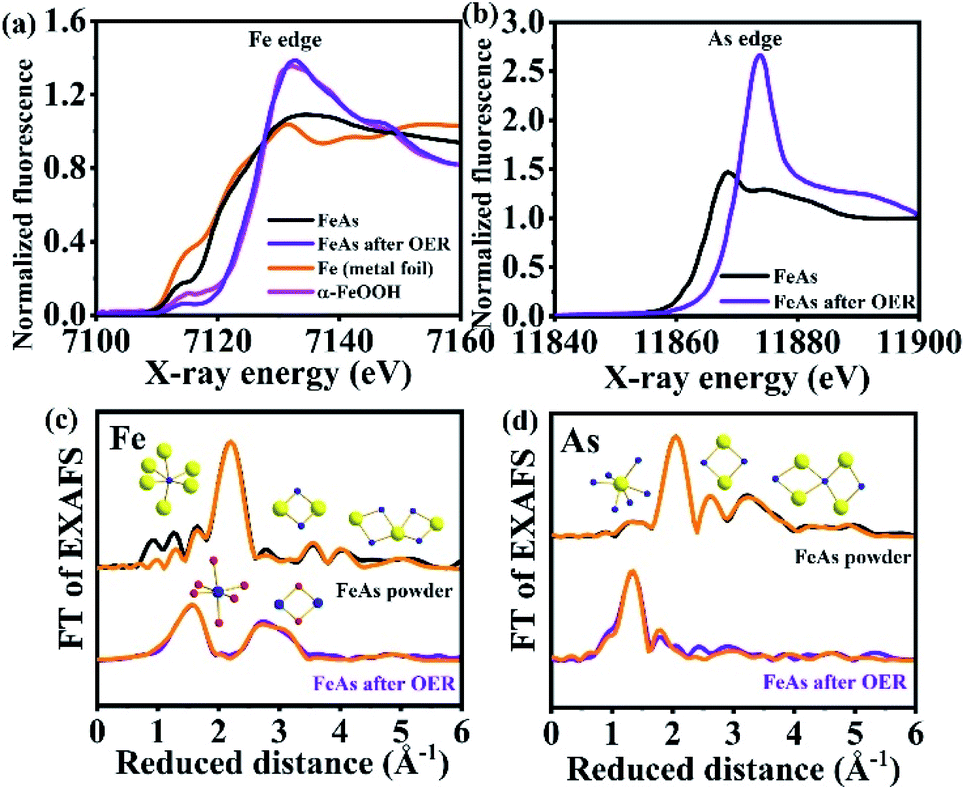 | ||
| Fig. 5 (a) Fe K-edge XANES spectra of metallic Fe and α-Fe3+OOH reference compounds as well as FeAs powder and FeAs after OER, (b) As K-edge XANES spectra of FeAs powder and FeAs after OER, (c) Fe- and (d) As-EXAFS spectra of FeAs powder and FeAs after OER (Fe- and As-EXAFS spectra in k-space are shown in Fig. S62†). Structural motifs of FeAs and 2-line ferrihydrite are included. Blue, yellow and red spheres represent Fe, As and O atoms, respectively. On the top of (c) and (d), FeAs6 octahedron and the trigonal prismatic AsFe6 polyhedron are shown next to the most intense peaks. Low-intensity peaks are related to longer Fe–Fe, As–As and Fe–As distances of two edge-sharing or corner-sharing FeAs6 octahedra. At the bottom of (c), the FeO6 octahedron and the Fe–Fe distance arising from two edge-sharing octahedra in the 2-line ferrihydrite structure are shown. Detailed bond distances and assigns are described in Fig. S63, S64 and Tables S15–S17.† | ||
The Fourier transform (FT) of the Fe-EXAFS spectra (Fig. 5c, for k3-weighted χ spectra of Fe, see Fig. S62a†) of the as-synthesized FeAs showed a low populated Fe–O shell at 2.05 Å, indicating minor contamination with oxidation products due to contact with air. Moreover, the most intense peak at 2.45 Å corresponds to the average Fe–As distance of the distorted octahedrally coordinated Fe, while the minor peaks match to the other Fe–As and Fe–Fe distances on the same structural motif (see Fig. S62†). A similar conclusion can be drawn from the As-EXAFS spectra (k3-weighted χ spectra of As are shown in Fig. S62b†) for the As–As and As–Fe distances on the FeAs powder. A small contribution of As–O at (1.76 Å) was also observed, which could be derived from surface oxidation. A detailed analysis of all shells revealed that the EXAFS distances are in agreement with the crystallographic data of FeAs for the Fe–As, Fe–Fe and As–As bond distances (see Tables S15 and S16,†REXAFSvs. RXRD values and Fig. S63†).
Quasi in situ OER EXAFS taken by freeze quenching at a current density of 10 mA cm−2 (6 h) confirmed the complete transformation of the material. The Fe-EXAFS spectrum could be fitted with the expected 2-line ferrihydrite, revealing an almost perfect agreement in the interatomic distances compared to the crystallographic data (see Table S17,†REXAFSvs. RXRD values). No peaks related to Fe–Fe or Fe–O bond distances from FeAs before catalysis could be observed in the in situ FT-Fe-EXAFS, in accordance with the complete transformation of the FeAs during the OER. The in situ FT Fe-EXAFS was dominated by two intense peaks. The first one corresponds to the nearest oxygen neighbors (O, OH or OH2) (see Fig. S64a†).93 The second peak was broad and a result of two Fe–Fe coordination shells. The first shell with a radius of 3.02 Å can be ascribed to the Fe–Fe distance of edge-sharing FeO6 octahedra (see Fig. S64b†). The second shell of this peak with a radius of 3.45 Å is between two bent corner-sharing FeO6 octahedra (see Fig. S64b and c†).39,93–95 We note here that this distance was also related to tetrahedral FeO4 units in other ferrihydrites. In such cases, a more pronounced pre-edge feature at 7113.5 eV could be observed.93 In the case of the FeAs OER film, the pre-edge feature was even less pronounced than in crystalline α-FeOOH, which contains no tetrahedral units. Therefore, we consider that the FeAs OER film was exclusively comprised of edge and corner-sharing FeO6 octahedra as connective elements. The As-EXAFS spectrum (Fig. 5d) showed the absence of the Fe–As bonds and their oxidation towards the As–O bond (1.70 Å), indicating that the minor amount of remaining As is present as AsOx as a separate phase without Fe bonding.
Therefore, the post-catalysis characterization results can satisfactorily explain the differences in activity between the formed two-line ferrihydrite and the Fe-based reference materials. First, the semiconductor nature of the two-line ferrihydrite96–98 contributes to the high OER electrocatalytic activity, in contrast to the non-conducting FeOxHy phases.99–102 Second, it is also possible that the in situ formation of the new phase may induce some defective edges/sites which could behave as active centers during catalysis, which would explain its higher OER activity.103–106 Additionally, the surface of 2-line ferrihydrite is enriched with tetrahedrally coordinated Fe3+ atoms, that can serve as active sites for the OER.39,81 Finally, the high activity of the 2-line ferrihydrite is related to its extremely high surface area,96–98 as revealed by the increment in the Cdl during the OER.
Conclusions
In summary, the first β-diketiminato 2Fe–2As cluster complex with a planar Fe2As2 core has been synthesized and structurally characterized and utilized as an SSP to access a monodisperse and nanocrystalline FeAs phase which is indeed unachievable by common solid-state approaches. The electrophoretically deposited FeAs films have been examined for the OER in strongly oxidizing alkaline media which exhibited a remarkable catalytic activity, a small Tafel slope, superior long-term stability, and low resistivity compared to the other state-of-the-art Fe-based reference electrodes. Under OER conditions, an electroconversion of the FeAs electro(pre)catalyst to the two-line ferrihydrite occurs as a result of the oxidation mediated dissolution of the As into the electrolyte, as proven by numerous advanced ex situ analytical methods as well as in situ XAS. The complete loss of As and subsequent transformation of the initial FeAs into a two-line ferrihydrite phase enhanced the electrocatalytic surface area tremendously. This transformation was demonstrated to be a crucial factor in accelerating the OER reaction, as well as its semiconducting nature, defective edges/sites, and the presence of surface tetrahedral coordinated Fe3+ atoms, which could behave as active centers for catalysis. Most notably, the dissolved As from the FeAs electrode has been successfully recovered at the cathode/counter electrode making the whole process sustainable and energy-efficient. Our study provides fundamental insights on FeAs as a precatalyst for an efficient OER, where the practical implication of using FeAs for water electrolysis, however, will depend on the surface area of the cathode for enabling arsenic deposition in combination with a suitable type of membrane that allows migration of arsenate anions. Besides, this work presents a facile design and development of unexplored classes of TM-non-metal materials towards several applications beyond the OER, such as magnetism, superconductivity, supercapacitors, light-driven photocatalytic HER, fuel cells, and metal–air batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038 – UniSysCat. The authors are indebted to Mr Christoph Fahrenson (ZELMI, TU Berlin) for SEM and Dr Stefan Berendts, Eva Maria Heppke and Ina Speckmann from the research group of Prof. Martin Lerch (TU Berlin) for PXRD measurements. R. Beltrán-Suito and K. Laun are indebted to the Einstein Foundation Berlin/EC²/BIG-NSE for a PhD fellowship.References
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS.
- G. W. Crabtree and M. S. Dresselhaus, MRS Bull., 2008, 33, 421–428 CrossRef CAS.
- P. Nikolaidis and A. Poullikkas, Renewable Sustainable Energy Rev., 2017, 67, 597–611 CrossRef CAS.
- J. Chi and H. Yu, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. Wiechen, M. M. Najafpour, S. I. Allakhverdiev and L. Spiccia, Energy Environ. Sci., 2014, 7, 2203–2212 RSC.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- P. W. Menezes, C. Panda, C. Walter, M. Schwarze and M. Driess, Adv. Funct. Mater., 2019, 29, 1–12 CrossRef.
- P. W. Menezes, A. Indra, I. Zaharieva, C. Walter, S. Loos, S. Hoffmann, R. Schlögl, H. Dau and M. Driess, Energy Environ. Sci., 2019, 12, 988–999 RSC.
- P. W. Menezes, C. Panda, S. Loos, F. Bunschei-Bruns, C. Walter, M. Schwarze, X. Deng, H. Dau and M. Driess, Energy Environ. Sci., 2018, 11, 1287–1298 RSC.
- P. W. Menezes, C. Walter, J. N. Hausmann, R. Beltrán-Suito, C. Schlesiger, S. Praetz, V. Y. Verchenko, A. V. Shevelkov and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 16569–16574 CrossRef CAS.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- W. Chen, H. Wang, Y. Li, Y. Liu, J. Sun, S. Lee, J. S. Lee and Y. Cui, ACS Cent. Sci., 2015, 1, 244–251 CrossRef CAS.
- X. Peng, C. Pi, X. Zhang, S. Li, K. Huo and P. K. Chu, Sustainable Energy Fuels, 2019, 3, 366–381 RSC.
- H. Du, R. M. Kong, X. Guo, F. Qu and J. Li, Nanoscale, 2018, 10, 21617–21624 RSC.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- R. Beltrán-Suito, P. W. Menezes and M. Driess, J. Mater. Chem. A, 2019, 7, 15749–15756 RSC.
- S. Yao, V. Forstner, P. W. Menezes, C. Panda, S. Mebs, E. M. Zolnhofer, M. E. Miehlich, T. Szilvási, N. Ashok Kumar, M. Haumann, K. Meyer, H. Grützmacher and M. Driess, Chem. Sci., 2018, 9, 8590–8597 RSC.
- P. W. Menezes, A. Indra, C. Das, C. Walter, C. Göbel, V. Gutkin, D. Schmeiβer and M. Driess, ACS Catal., 2017, 7, 103–109 CrossRef CAS.
- C. Panda, P. W. Menezes, M. Zheng, S. Orthmann and M. Driess, ACS Energy Lett., 2019, 4, 747–754 CrossRef CAS.
- J. N. Hausmann, E. M. Heppke, R. Beltrán-Suito, J. Schmidt, M. Mühlbauer, M. Lerch, P. W. Menezes and M. Driess, ChemCatChem, 2020, 12, 1–9 CrossRef.
- J. Villalobos, D. González-Flores, K. Klingan, P. Chernev, P. Kubella, R. Urcuyo, C. Pasquini, M. R. Mohammadi, R. D. L. L. Smith, M. L. Montero and H. Dau, Phys. Chem. Chem. Phys., 2019, 21, 12485–12493 RSC.
- J. Masa, S. Piontek, P. Wilde, H. Antoni, T. Eckhard, Y. Chen, M. Muhler, U. Apfel and W. Schuhmann, Adv. Energy Mater., 2019, 1900796 CrossRef.
- J. Masa and W. Schuhmann, ChemCatChem, 2019, 11, 5842–5854 CrossRef CAS.
- C. Panda, P. W. Menezes and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 11130–11139 CrossRef CAS.
- C. Panda, P. W. Menezes, C. Walter, S. Yao, M. E. Miehlich, V. Gutkin, K. Meyer and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 10506–10510 CrossRef CAS.
- C. Walter, P. W. Menezes, S. Orthmann, J. Schuch, P. Connor, B. Kaiser, M. Lerch and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 698–702 CrossRef CAS.
- B. Chakraborty, S. Kalra, R. Beltrán-Suito, C. Das, T. Hellmann, P. W. Menezes and M. Driess, Chem.–Asian J., 2020, 15, 852–859 CrossRef CAS.
- Y. Kamihara, T. Watanabe, M. Hirano and H. Hosono, J. Am. Chem. Soc., 2008, 130, 3296–3297 CrossRef CAS.
- X. C. Wang, Q. Q. Liu, Y. X. Lv, W. B. Gao, L. X. Yang, R. C. Yu, F. Y. Li and C. Q. Jin, Solid State Commun., 2008, 148, 538–540 CrossRef CAS.
- Z.-A. Ren, J. Yang, W. Lu, W. Yi, X.-L. Shen, Z.-C. Li, G.-C. Che, X.-L. Dong, L.-L. Sun, F. Zhou and Z.-X. Zhao, Europhys Lett., 2008, 82, 57002 CrossRef.
- D. R. Parker, M. J. Pitcher, P. J. Baker, I. Franke, T. Lancaster, S. J. Blundell and S. J. Clarke, Chem. Commun., 2009, 16, 2189–2191 RSC.
- T. Stürzer, C. Stürzer and D. Johrendt, Phys. Status Solidi B, 2017, 254, 1600417 CrossRef.
- J. Chen, H. Zhao, N. Chen, X. Wang, J. Wang, R. Zhang and C. Jin, J. Power Sources, 2011, 200, 98–101 Search PubMed.
- F. M. Michel, L. Ehm, S. M. Antao, P. L. Lee, P. J. Chupas, G. Liu, D. R. Strongin, M. A. A. Schoonen, B. L. Phillips and J. B. Parise, Science, 2007, 316, 1726–1729 CrossRef CAS.
- K. Rout, M. Mohapatra and S. Anand, Dalton Trans., 2012, 41, 3302–3312 RSC.
- M. Manikandan, K. Subramani, M. Sathish and S. Dhanuskodi, ChemistrySelect, 2018, 3, 9034–9040 CrossRef CAS.
- D. Khalafallah, Q. Zou, M. Zhi and Z. Hong, Electrochim. Acta, 2020, 350, 136399 CrossRef CAS.
- A. S. Foust, M. S. Foster and L. F. Dahl, J. Am. Chem. Soc., 1969, 91, 5631–5633 CrossRef CAS.
- O. J. Scherer, M. Ehses and G. Wolmershäuser, Angew. Chem., Int. Ed., 1998, 37, 507–510 CrossRef CAS.
- A. S. Foust, M. S. Foster and L. F. Dahl, J. Am. Chem. Soc., 1969, 91, 5633–5635 CrossRef CAS.
- G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 431–436 CrossRef CAS.
- W. A. Herrmann, B. Koumbouris, T. Zahn and M. L. Ziegler, Angew. Chem., Int. Ed., 1984, 23, 812–814 CrossRef.
- A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 8536–8541 CrossRef CAS.
- S. Yao, Y. Grossheim, A. Kostenko, E. Ballestero-Martínez, S. Schutte, M. Bispinghoff, H. Grützmacher and M. Driess, Angew. Chem., 2017, 129, 7573–7577 CrossRef.
- E. Ballestero-Martínez, T. J. Hadlington, T. Szilvási, S. Yao and M. Driess, Chem. Commun., 2018, 54, 6124–6127 RSC.
- C. Schwarzmaier, S. Heinl, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13116–13121 CrossRef CAS.
- C. von Hänisch and D. Fenske, Z. Anorg. Allg. Chem., 1998, 624, 367–369 CrossRef.
- L. T. J. Delbaere, L. J. Kruczynski and D. W. McBride, J. Chem. Soc., Dalton Trans., 1973, 307–310 RSC.
- O. J. Scherer, Acc. Chem. Res., 1999, 32, 751–762 CrossRef CAS.
- O. J. Scherer, Angew. Chem., Int. Ed., 1990, 29, 1104–1122 CrossRef.
- A. J. Dimaio and A. L. Rheingold, Chem. Rev., 1990, 90, 169–190 CrossRef CAS.
- K. Selte, A. Kjekshus, A. F. Andresen, M. J. Tricker and S. Svensson, Acta Chem. Scand., 1972, 26, 3101–3113 CrossRef CAS.
- K. Selte, A. Kjekshus, J. Sletten, L. Torbjörnsson, P.-E. Werner, U. Junggren, B. Lamm and B. Samuelsson, Acta Chem. Scand., 1969, 23, 2047–2054 CrossRef CAS.
- K. Selte, A. Kjekshus, C. Hyatt, G. D. Stucky, B. Lüning and C.-G. Swahn, Acta Chem. Scand., 1973, 27, 1448–1449 CrossRef CAS.
- P. Desai, K. Song, J. Koza, A. Pariti and M. Nath, Chem. Mater., 2013, 25, 1510–1518 CrossRef CAS.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, Chem. Mater., 1990, 2, 186–191 CrossRef CAS.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- J. M. Epp and J. G. Dillard, Chem. Mater., 1989, 1, 325–330 CrossRef CAS.
- D. E. King, J. E. Fernandez and W. E. Swartz, Appl. Surf. Sci., 1990, 45, 325–339 CrossRef CAS.
- N. K. Chaudhari, H. Jin, B. Kim and K. Lee, Nanoscale, 2017, 9, 12231–12247 RSC.
- G. L. Elizarova, G. M. Zhidomirov and V. N. Parmon, Catal. Today, 2000, 58, 71–88 CrossRef CAS.
- F. Zhou, A. Izgorodin, R. K. Hocking, L. Spiccia and D. R. MacFarlane, Adv. Energy Mater., 2012, 2, 1013–1021 CrossRef CAS.
- R. L. Doyle and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 5224–5237 RSC.
- S. Anantharaj and S. Noda, ChemElectroChem, 2020, 7, 2297–2308 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS.
- M. Görlin, P. Chernev, J. F. De Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef.
- M. Gorlin, J. F. De Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef.
- H. Sun, Z. Ma, Y. Qiu, H. Liu and G. g. Gao, Small, 2018, 14, 1–12 Search PubMed.
- M. S. Wu, C. Y. Jao, F. Y. Chuang and F. Y. Chen, Electrochim. Acta, 2017, 227, 210–216 CrossRef CAS.
- P. H. Masscheleyn, R. D. Delaune and W. H. Patrick, Environ. Sci. Technol., 1991, 25, 1414–1419 CrossRef CAS.
- A. Vahidnia, G. B. Van Der Voet and F. A. De Wolff, Hum. Exp. Toxicol., 2007, 26, 823–832 CrossRef CAS.
- J. C. Ng, J. Wang and A. Shraim, Chemosphere, 2003, 52, 1353–1359 CrossRef CAS.
- L. J. Gao and B. E. Conway, J. Electroanal. Chem., 1995, 395, 261–271 CrossRef.
- R. Santoyo-Cisneros, J. R. Rangel-Mendez, J. L. Nava, E. R. Larios-Durán and L. F. Chazaro-Ruiz, J. Hazard. Mater., 2020, 392, 122349 CrossRef CAS.
- E. Morallón, J. Arias-Pardilla, J. M. Calo and D. Cazorla-Amorós, Electrochim. Acta, 2009, 54, 3996–4004 CrossRef.
- D. E. Janney, J. M. Cowley and P. R. Buseck, Clays Clay Miner., 2000, 48, 111–119 CrossRef CAS.
- J. L. Jambor and J. E. Dutrizac, Chem. Rev., 1998, 98, 2549–2586 CrossRef CAS.
- L. Mei, L. Liao, Z. Wang and C. Xu, Adv. Mater. Sci. Eng., 2015, 2015, 1–10 Search PubMed.
- L. Mazzetti and P. J. Thistlethwaite, J. Raman Spectrosc., 2002, 33, 104–111 CrossRef CAS.
- M. Seehra, P. Roy, A. Raman and A. Manivannan, Solid State Commun., 2004, 130, 597–601 CrossRef CAS.
- H. Tüysüz, E. L. Salabaş, C. Weidenthaler and F. Schüth, J. Am. Chem. Soc., 2008, 130, 280–287 CrossRef.
- S. Das, M. Jim Hendry and J. Essilfie-Dughan, Appl. Geochem., 2013, 28, 185–193 CrossRef CAS.
- M. Hanesch, Geophys. J. Int., 2009, 177, 941–948 CrossRef CAS.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- X. Wang, M. Zhu, L. K. Koopal, W. Li, W. Xu, F. Liu, J. Zhang, Q. Liu, X. Feng and D. L. Sparks, Environ. Sci. Nano, 2016, 3, 190–202 RSC.
- A. Manceau and V. A. Drits, Clay Miner., 1993, 28, 165–184 CrossRef CAS.
- F. Maillot, G. Morin, Y. Wang, D. Bonnin, P. Ildefonse, C. Chaneac and G. Calas, Geochim. Cosmochim. Acta, 2011, 75, 2708–2720 CrossRef CAS.
- L. Yang, C. I. Steefel, M. A. Marcus and J. R. Bargar, Environ. Sci. Technol., 2010, 44, 5469–5475 CrossRef CAS.
- S. Lan, X. Wang, Q. Xiang, H. Yin, W. Tan, G. Qiu, F. Liu, J. Zhang and X. Feng, Geochim. Cosmochim. Acta, 2017, 211, 79–96 CrossRef CAS.
- S. Kato, K. Hashimoto and K. Watanabe, Environ. Microbiol., 2012, 14, 1646–1654 CrossRef CAS.
- L. Lei, D. Huang, C. Zhou, S. Chen, X. Yan, Z. Li and W. Wang, Coord. Chem. Rev., 2020, 408, 213177 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS.
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, ACS Catal., 2020, 10, 4019–4047 CrossRef CAS.
- J. Hu, S. Li, J. Chu, S. Niu, J. Wang, Y. Du, Z. Li, X. Han and P. Xu, ACS Catal., 2019, 10705–10711 CrossRef CAS.
- M. Lee, H. S. Oh, M. K. Cho, J. P. Ahn, Y. J. Hwang and B. K. Min, Appl. Catal., B, 2018, 233, 130–135 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518–9523 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1989143 for compound 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04384b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |