
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA case of chain propagation: α-aminoalkyl radicals as initiators for aryl radical chemistry†
Timothée
Constantin
 a,
Fabio
Juliá
a,
Nadeem S.
Sheikh
b and
Daniele
Leonori
*a
a,
Fabio
Juliá
a,
Nadeem S.
Sheikh
b and
Daniele
Leonori
*a
aDepartment of Chemistry, University of Manchester, Manchester M13 9PL, UK. E-mail: daniele.leonori@manchester.ac.uk; Web: https://leonoriresearchgroup.com
bDepartment of Chemistry, College of Science, King Faisal University, P. O. Box 400, Al-Ahsa 31982, Saudi Arabia
First published on 20th October 2020
Abstract
The generation of aryl radicals from the corresponding halides by redox chemistry is generally considered a difficult task due to their highly negative reduction potentials. Here we demonstrate that α-aminoalkyl radicals can be used as both initiators and chain-carriers for the radical coupling of aryl halides with pyrrole derivatives, a transformation often employed to evaluate new highly reducing photocatalysts. This mode of reactivity obviates for the use of strong reducing species and was also competent in the formation of sp2 C–P bonds. Mechanistic studies have delineated some of the key features operating that trigger aryl radical generation and also propagate the chain process.
Introduction
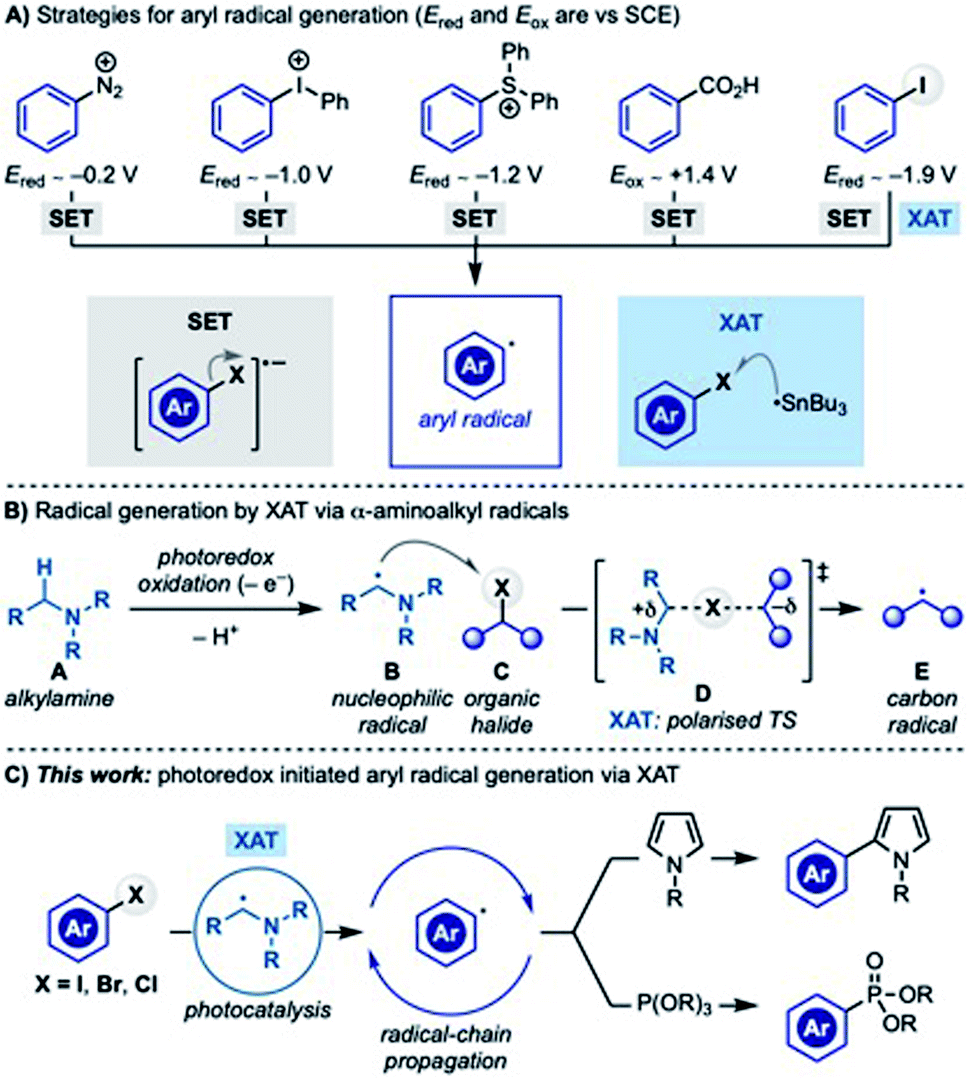
Aryl radicals are versatile synthetic intermediates for the assembly of sp2 C–C and C–Y (Y = heteroatom) bonds.1 Reactions like the Meerwein cyclization and arylation,2 the Pschorr cyclization3 and the Gomber–Bachmann biphenyl synthesis4 are text-book examples of aryl radical reactivity and are still used in the assembly of high-value materials.5Historically, aryl radicals have been generated by SET (single-electron transfer) reduction of aryl diazonium,6 iodonium7 and sulfonium salts8 or oxidation of benzoates9 (Scheme 1A). Despite their synthetic versatility, these substrates can sometimes be difficult to prepare or unstable or they might require forcing reaction conditions which has somewhat limited their application especially on large-scale settings.10
 | ||
| Scheme 1 Most common approaches for aryl radical generation and this work. | ||
Aryl halides are a large class of stable and commercial building blocks, which are routinely used in synthetic chemistry through transition-metal-catalysed cross-coupling reactions. Aryl radical generation by SET reduction followed by heterolytic sp2 C–X (X = halogen) bond fragmentation is feasible for substrates containing electron withdrawing groups (e.g. CN, NO2…) but challenging for electron neutral/rich ones owing to their highly negative reduction potentials (Ered < −2 V vs. SCE). Nevertheless, recent work based on electrochemistry,11 sulfoxylate radical anion chemistry,12 organic super electron donors13 and photo-electrochemistry14 has successfully addressed some of these issues. Within the field of visible-light photoredox catalysis,15 targeting such negative reduction potentials has been tackled using homoleptic Ir(III)-photocatalysts16 and organic dyes able of sequentially absorbing two photons,17 both in combination with excess of amines as sacrificial electron donors.
An alternative approach for aryl radical generation involves the homolytic sp2 C–X bond cleavage via halogen-atom transfer (XAT) which has historically been achieved using tin radicals.18 Strategies based on silicon radicals19 have circumvented the use of toxic tin hydrides and more recently have been successfully exploited in metallaphotoredox manifolds, retrieving an increasingly renewed interest.20
We have recently demonstrated that generating α-aminoalkyl radicals from alkylamines (A → B) under photoredox catalysis represents an effective gateway to access alkyl and aryl radicals from the corresponding halides (Scheme 1B).21 In this reactivity, the highly nucleophilic α-aminoalkyl radical B homolytically activates the sp3 or sp2 C–X bond by XAT (B + C → E) through a polarised transition state (D) stabilised by extensive charge-transfer character.
As part of our ongoing interest in exploring this activation mode, we recently questioned if α-aminoalkyl radical-mediated XAT could be leveraged to enable divergent arene functionalization under photoredox conditions. Our interest was mainly focused on the radical coupling of aryl halides with pyrrole derivatives, a benchmark transformation often used to assess the reductive ability of new photoredox catalysts.22 In these examples, the photocatalyst (either in the excited or the reduced state) triggers the SET reduction of the aryl halide, while stoichiometric amounts of amines are employed as sacrificial reductants. In general, the absence of the amine completely switches off the reactivity and other types of electron donors are either not discussed or not feasible. We were therefore intrigued by the possibility of establishing a mechanistically alternative approach where α-aminoalkyl radicals and XAT deliver the key aryl radicals thus bypassing the need for strongly reducing photocatalysts. Herein we report the successful implementation of this proposal which demonstrates how photoredox catalysis and α-aminoalkyl radical chemistry can be used to generate and explore the reactivity of aryl radicals in chain propagating manifolds (Scheme 1C).
Results and discussion
Mechanistic considerations
We initially questioned whether photoredox catalysis could be used to generate α-aminoalkyl radicals to enable C–H arylation of electron rich heteroaromatics with aryl halides. Our interest in exploring this reactivity stem from preliminary results demonstrating that coupling between 4-acetyl-iodobenzene with N-Me-pyrrole (1 + 2 → 3) was efficiently achieved using 4CzIPN as the photocatalyst, n-Bu3N as the amine and KH2PO4 as the base in DMSO–H2O under blue light irradiation (Scheme 2A). We were initially puzzled by this experimental outcome as we recognized an inherent chemical reactivity issue that should have thwarted product formation. As shown in Scheme 2B, the reductive-quenching photoredox cycle required for α-aminoalkyl radical generation (n-Bu3N → B1) followed by XAT (B1 + 1 → E1 + F1) and radical addition to pyrrole (E1 + 2 → E2) generates a redox imbalance as it requires the simultaneous oxidation of 4CzIPN˙− and the intermediate E2. This should disfavour a mechanism based on a closed photoredox system and, as a result, hamper catalysis. | ||
| Scheme 2 (A) Initial results on the radical addition of iodide 1 to pyrrole 2. (B) Mechanistic analysis of a photoredox manifold reveals a redox imbalance. (C) Mechanism based on a radical-chain propagation. (D) Control experiments supporting initiation by α-aminoalkyl radicals. | ||
We therefore considered a different mechanistic scenario where photoredox catalysis would serve, through the generation of B1, as initiation step for a radical-chain propagating system (Scheme 2C).23 This proposed reactivity hinged however on the ability of the electron rich 5π-radical E2 to regenerate the aryl radical E1 by reaction with 1. While this mechanistic possibility has not been considered in the analysis of previous radical arylation processes, we were intrigued by the fact that E2 is electron rich and, crucially, also an extended aminoalkyl radical. Hence, we postulated that it might be able to participate in XAT (or SET) events with 1. Two major mechanistic aspects became a focal point: (1) which is the species involved in the key C–X activation step and (2) whether a radical propagation pathway could be operative.
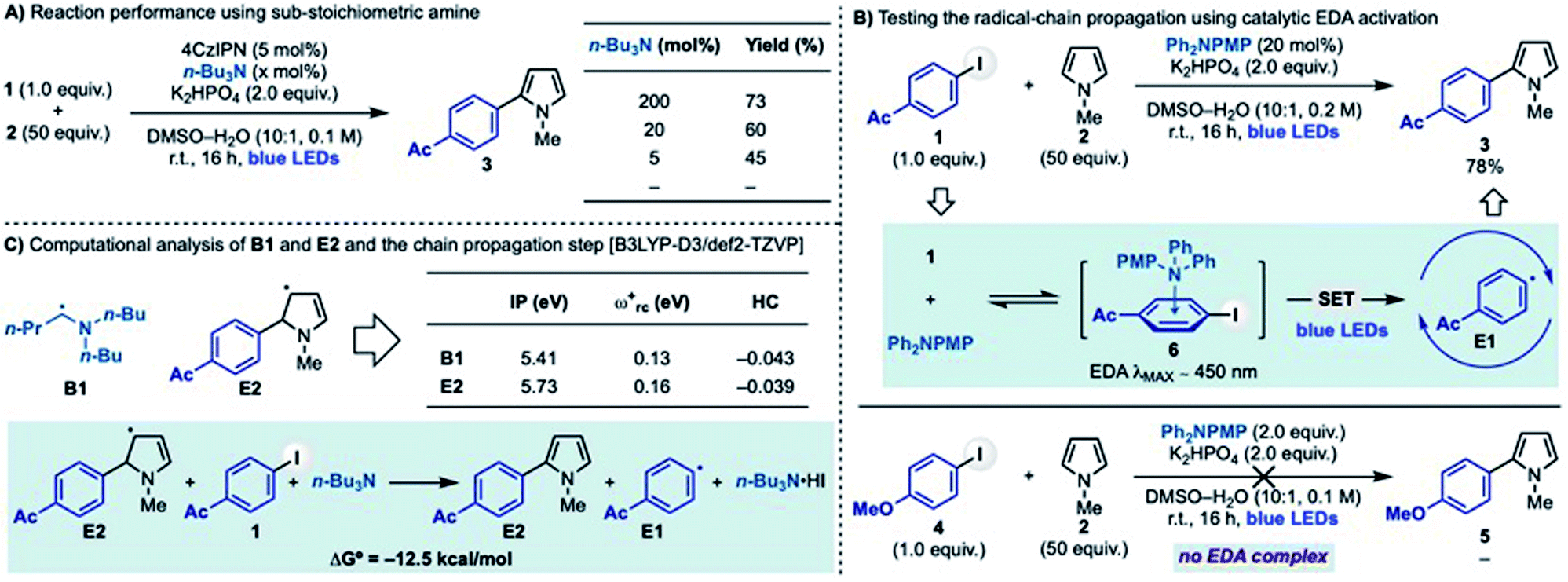 | ||
| Scheme 3 (A) Impact of the n-Bu3N stoichiometry on the radical coupling between 1 and 2. (B) Radical couplings via EDA. (C) Computational studies on key properties of α-aminoalkyl radical (B1) and extended aminoalkyl radical (E2) and the thermodynamic feasibility of the chain propagation step. PMP = p-OMe-C6H4. | ||
The stabilised 5π-radical E2 is the key intermediate enabling aryl radical regeneration in the proposed chain-propagating process. As mentioned earlier, the reaction between E2 and an aryl iodide can be considered to go via XAT (or SET). To further support this hypothesis, we have conducted preliminary computational studies (Scheme 3C). Our results support the hypothesis that α-aminoalkyl radical B1 and 5π-radical E2 should display similar reactivity by virtue of their very similar electronic features. In particular, the close values obtained for their ionization potential (IP), global electrophilicity (ωrc+) and Hirshfeld charge (HC) demonstrate E2 is a highly nucleophilic radical that could therefore participate in XAT processes and benefit from related polar effects in the transition state. It is however important to note that due to the high-degree of charge transfer in XAT processes, a SET between E2 and the aryl iodide might be operative in the case of highly electron poor systems (e.g. 4-NO2-iodobenzene). Moreover, the overall reaction leading to the aryl radical regeneration (E2 + 1 + n-Bu3N → 3 + E1 + n-Bu3N·HI) was found to be feasible according to the exothermic ΔG° energy of this step.24
Taken together, these experimental and computational results support the feasibility of a radical chain propagation as the operating mechanism in these radical coupling reactions. Furthermore, we believe the ability of this α-aminoalkyl radicals-mediated strategy to activate both electron rich and electron poor aryl iodides exemplifies its versatility as initiation mode for aryl radical-based chain propagations.
In order to have more insights on the extent of the chain propagation we determined the quantum yield for the reaction between 1 and 2 to give 3 under standard conditions. This analysis provided Φ = 0.37. Such a moderate value can be rationalised with the process being supported by either short-lived radical chain propagations or, as demonstrated by Scaiano,27 an inefficient initiation process.28
Substrate scope
After identifying optimal reaction conditions, we explored the scope of the transformation by evaluating different aryl halides (Scheme 4A). Aryl iodides substituted with electron withdrawing groups at the para-position reacted in good to high yields (3, 7–11). More challenging electron-neutral substrates as well as the ones equipped with electron-donating groups, which are elusive in some redox-based approaches, reacted well delivering the desired 2-aryl-pyrroles in good yields (11, 13 and 5). Pleasingly, the reaction was also tolerant of polar functional groups such as free anilines (14) thus showcasing high chemoselectivity. The effect of the substituent position was also evaluated using meta- or ortho-substituted rings. Little difference in yields were observed for both meta- (15–18) and ortho-substituted (18, 19, 29 and 32) aryl halides in comparison with their para-functionalised counterparts. Other aryl iodides like 1- and 2-iodo-napthalenes, 2-iodo-pyrazine, 4-iodo-pyridine and 2-iodothiophenes were also suitable substrates for the transformation giving 21–26 in moderate to good yields. Furthermore, the weaker nature of sp2 C–I bonds compared to sp2 C–Br/Cl bonds meant that aryl iodides could be selectively functionalised in the presence of sp2 C–Br and C–Cl bonds (28 and 29).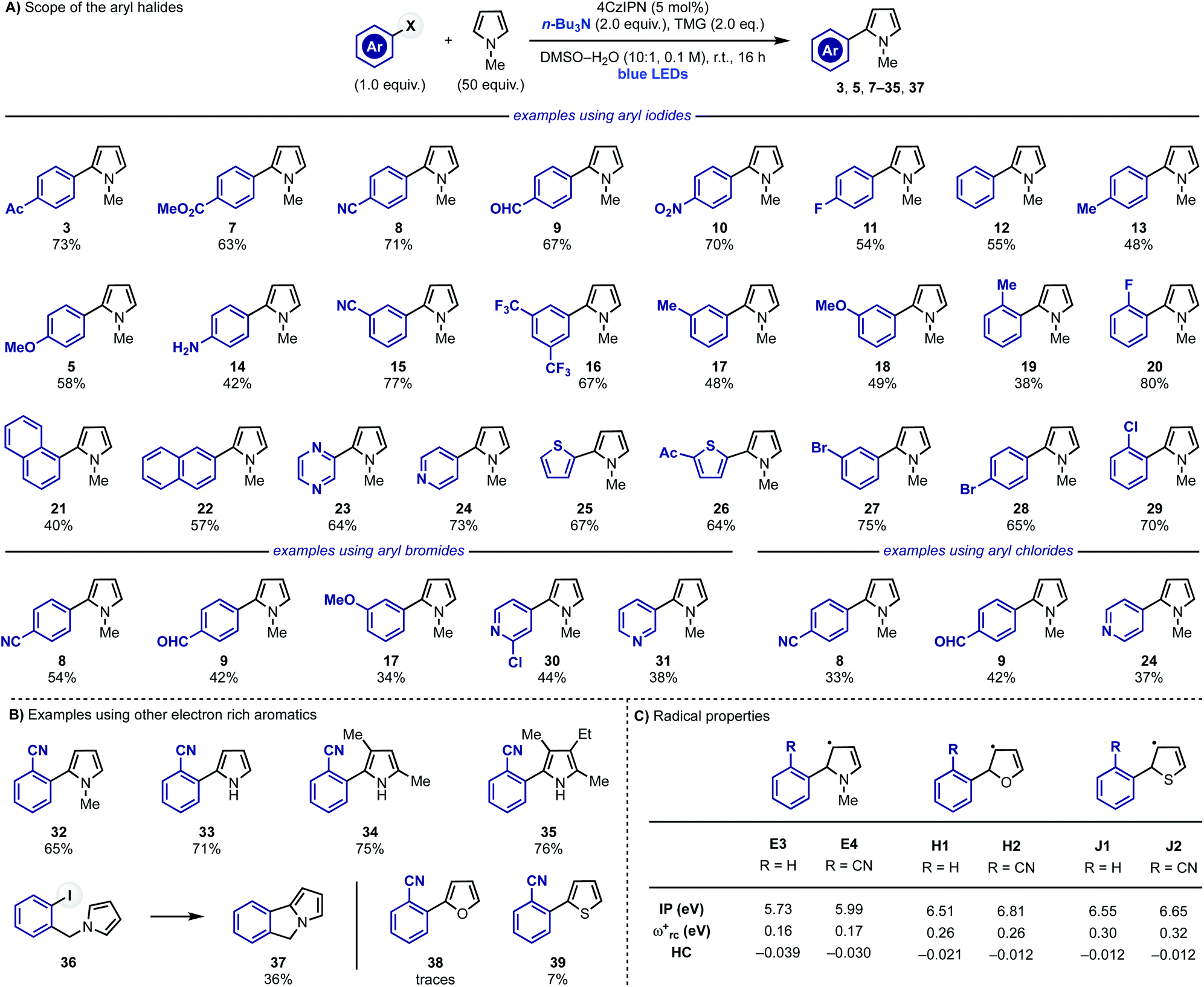 | ||
| Scheme 4 (A) Scope of the radical coupling between pyrrole 2 and aryl iodides, bromides and chlorides. (B) Scope of the heteroaromatics. (C) Computational studies of different 5π-radicals. | ||
This strategy was also extended to (hetero-)aryl bromides (8, 9, 30 and 31) and even weakly electron-rich aryl bromides (17) which are often challenging substrates. Consistently with the trend observed in the aryl iodide scope, electron-poor systems gave higher yields than the electron-rich ones. Electron-deficient aryl chlorides were also tested and they could be engaged in this reactivity albeit in moderate conversions (8, 9 and 24).
Having evaluated the scope around the aryl halide partner we decided to investigate other electron rich aromatics (Scheme 4B). o-CN-iodobenzene displayed good reactivity with 2 (to give 32) and could also be used for the C-2 arylation of unprotected pyrrole (33) as well as poly-substituted derivatives (34 and 35) including the highly-hindered cryptopyrrole. Furthermore, we also evaluated the possibility to render these reactions intramolecular, which was successfully achieved by preparing 36 that provided the tricyclic heterocycle 37.
While this approach enables the efficient C-2 arylation of pyrrole and its derivatives, we did not succeed in extending it to other electron rich heteroaromatics like furan (38) and thiophene (39). We were initially surprised by this lack of reactivity especially considering (1) the lower degree of aromaticity of these systems29 and (2) their successful engagement in radical arylations based on diazonium precursors.30 We propose that these failed reactions are a clear manifestation of the intrinsic reactivity difference between α-N vs. α-O/S-5π-radical intermediates Evs.H–J. The latter species should be less nucleophilic and therefore radical-chain propagation might now be hampered by their lower ability to regenerate the key aryl radical by XAT/SET. Indeed, computational analysis of H1,2 and J1,2 demonstrated their attenuated nucleophilic character which is in line with our working hypothesis (Scheme 4C).24,31
Expanding α-aminoalkyl radicals initiation to phosphonylation
Our current mechanistic understanding of these radical arylations suggests that α-aminoalkyl radical-mediated XAT can serve as initiation step to access chain propagations where “in cycle” aryl radical re-generation is possible. To test the validity of this hypothesis, we decided to extend this concept to other arene-functionalization processes. We were particularly interested by recent reports demonstrating the ability of the ethyl radical to act as chain carrier species.32 We therefore decided to evaluate the ability of α-aminoalkyl radicals to initiate the sp2 phosphonylation of aryl halides with trialkyl phosphites.17b,22a,33 We were hopeful that upon XAT-initiated aryl radical generation and addition to the phosphite, the resulting P-radical intermediate would undergo a thermodynamically driven β-scission resulting in the formation of a strong P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and an ethyl radical that could continue the chain propagation by reacting with the aryl halide.
O bond and an ethyl radical that could continue the chain propagation by reacting with the aryl halide.
Pleasingly, exposure of aryl iodides and several phosphites [P(OEt)3, P(OMe)3 and P(Oi-Pr)3] to conditions identical to the ones developed for pyrrole arylation provided the desired phosphonates 40–45 in good to moderate yield (Scheme 5). It is worth mentioning that this reactivity enabled engagement of both electron poor and electron rich substrates and could be extended, albeit with lower chemical yields, to aryl bromides and, in the case of substrates with electron withdrawing groups, also to aryl chlorides.
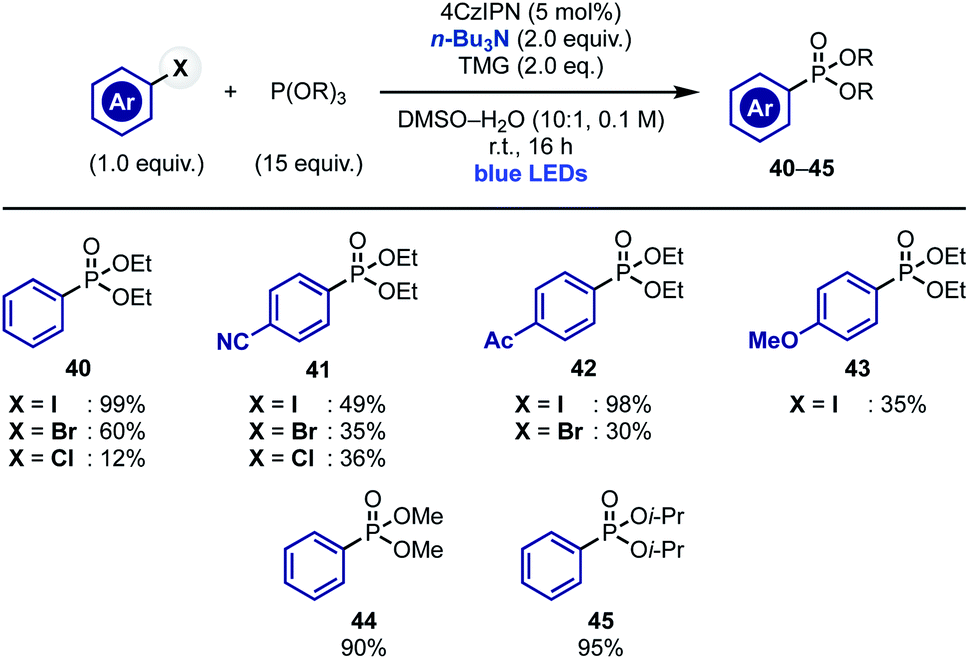 | ||
| Scheme 5 XAT-initiated radical chain phosphonylation of aryl halides. | ||
Conclusions
We have reported here that the photoredox generation of α-aminoalkyl radicals can be used as initiation step for the development of aryl-radical-based chain propagations. This approach by-passes the requirement for strongly reducing photocatalysts if an appropriate chain-carrier radical species is generated. In the case of the coupling with pyrroles, these results point for a unique ability of aminoalkyl radicals to participate as both initiators and then chain carriers owing to their high nucleophilicity. In light of these results, we suggest that this alternative mechanism should be taken in consideration when benchmarking new photoredox catalysts with the radical arylation of pyrroles in the presence of alkylamines.We believe the results presented here demonstrate that XAT-based aryl radical generation using simple alkylamines and photocatalysis can be used as an entry point for radical chains and we expect that might be extended to other transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. L. thanks EPSRC for a Fellowship (EP/P004997/1), and the European Research Council for a research grant (758427). We thank Dr Anne-Laure Barthelemy for help with the UV/Vis studies.Notes and references
- G. Pratsch and M. R. Heinrich, Top. Curr. Chem., 2012, 320, 33–59 CrossRef CAS.
- (a) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef CAS; (b) A. L. J. Beckwith and G. F. Meijs, J. Org. Chem., 1987, 52, 1922–1930 CrossRef CAS.
- D. F. DeTar, in Organic Reactions, 2011, pp. 409–462, DOI:10.1002/0471264180.or009.07.
- J. J. Li, Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, Springer, Berlin Heidelberg, 2006, pp. 267–268, DOI:10.1007/3-540-30031-7_118.
- F.-X. Felpin and S. Sengupta, Chem. Soc. Rev., 2019, 48, 1150–1193 RSC.
- (a) C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS; (b) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS.
- (a) M. Hartmann, Y. Li, C. Mück-Lichtenfeld and A. Studer, Chem. – Eur. J., 2016, 22, 3485–3490 CrossRef CAS; (b) D. Wang, F. Szillat, J. P. Fouassier and J. Lalevée, Macromolecules, 2019, 52, 5638–5645 CrossRef CAS.
- Á. Péter, G. J. P. Perry and D. J. Procter, Adv. Synth. Catal., 2020, 362, 2135–2142 CrossRef.
- X.-Q. Hu, Z.-K. Liu, Y.-X. Hou and Y. Gao, iScience, 2020, 23, 101266 CrossRef CAS.
- (a) A. A. Bondarev, E. V. Naumov, A. Z. Kassanova, E. A. Krasnokutskaya, K. S. Stankevich and V. D. Filimonov, Org. Process Res. Dev., 2019, 23, 2405–2415 CrossRef CAS; (b) C. Schotten, S. K. Leprevost, L. M. Yong, C. E. Hughes, K. D. M. Harris and D. L. Browne, Org. Process Res. Dev., 2020, 24, 2336–2341 CrossRef CAS.
- G. Sun, S. Ren, X. Zhu, M. Huang and Y. Wan, Org. Lett., 2016, 18, 544–547 CrossRef CAS.
- F. Yu, R. Mao, M. Yu, X. Gu and Y. Wang, J. Org. Chem., 2019, 84, 9946–9956 CrossRef CAS.
- (a) J. A. Murphy, S.-z. Zhou, D. W. Thomson, F. Schoenebeck, M. Mahesh, S. R. Park, T. Tuttle and L. E. A. Berlouis, Angew. Chem., Int. Ed., 2007, 46, 5178–5183 CrossRef CAS; (b) D. S. Lee, C. S. Kim, N. Iqbal, G. S. Park, K.-s. Son and E. J. Cho, Org. Lett., 2019, 21, 9950–9953 CrossRef CAS.
- (a) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS; (b) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS.
- D. P. Hari, T. Hering and B. Konig, in Visible Light Photocatalysis in Organic Chemistry, pp. 253–281, DOI:10.1002/9783527674145.ch8.
- (a) J. M. R. Narayannam and C. R. J. Stephenson, Chem. Soc. Rev., 2008, 40, 102 RSC; (b) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS.
- (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS; (b) R. S. Shaikh, S. J. S. Düsel and B. König, ACS Catal., 2016, 6, 8410–8414 CrossRef CAS; (c) C. J. Zeman, S. Kim, F. Zhang and K. S. Schanze, J. Am. Chem. Soc., 2020, 142, 2204–2207 CrossRef CAS.
- C. Chatgilialoglu, in Radicals in Organic Synthesis, 2001, pp. 28–49, DOI:10.1002/9783527618293.ch3.
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS.
- (a) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS; (b) J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CrossRef CAS; (c) D. J. P. Kornfilt and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 6853–6858 CrossRef CAS.
- (a) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS; (b) R. K. Neff, Y.-L. Su, S. Liu, M. Rosado, X. Zhang and M. P. Doyle, J. Am. Chem. Soc., 2019, 141, 16643–16650 CrossRef CAS; (c) J. Lalevée, D. Gigmes, D. Bertin, B. Graff, X. Allonas and J. P. Fouassier, Chem. Phys. Lett., 2007, 438, 346–350 CrossRef.
- (a) I. Ghosh, R. S. Shaikh and B. König, Angew. Chem., Int. Ed., 2017, 56, 8544–8549 CrossRef CAS; (b) M. Neumeier, D. Sampedro, M. Májek, V. A. de la Peña O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem. – Eur. J., 2018, 24, 105–108 CrossRef CAS; (c) S. O. Poelma, G. L. Burnett, E. H. Discekici, K. M. Mattson, N. J. Treat, Y. Luo, Z. M. Hudson, S. L. Shankel, P. G. Clark, J. W. Kramer, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2016, 81, 7155–7160 CrossRef CAS; (d) Z. Tang, J. Li, F. Lin, W. Bao, S. Zhang, B. Guo, S. Huang, Y. Zhang and Y. Rao, J. Catal., 2019, 380, 1–8 CrossRef CAS; (e) R. Matsubara, T. Yabuta, U. Md Idros, M. Hayashi, F. Ema, Y. Kobori and K. Sakata, J. Org. Chem., 2018, 83, 9381–9390 CrossRef CAS; (f) Z.-J. Li, S. Li, E. Hofman, A. Hunter Davis, G. Leem and W. Zheng, Green Chem., 2020, 22, 1911–1918 RSC.
- An additional chain-carrying step would involve XAT between E2 and F1. However, this process was determined to be highly endothermic24.
- See ESI† for more information.
- (a) J. Haimerl, I. Ghosh, B. König, J. Vogelsang and J. M. Lupton, Chem. Sci., 2019, 10, 681–687 RSC; (b) X. Guo and O. S. Wenger, Angew. Chem., Int. Ed., 2018, 57, 2469–2473 CrossRef CAS.
- (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS; (b) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS; (c) J. Davies, S. G. Booth, S. Essafi, R. W. A. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017 CrossRef CAS.
- S. P. Pitre, C. D. McTiernan, W. Vine, R. DiPucchio, M. Grenier and J. C. Scaiano, Sci. Rep., 2015, 5, 16397 CrossRef CAS.
- We have performed intermittent irradiation experiments to support an inefficient initiation step.24.
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777–2812 CrossRef CAS.
- D. P. Hari, P. Schroll and B. Konig, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS.
- According to our calculations, the XAT reactions between Ph–I and H1/J1 are significanlty more endothermic than the one between Ph–I and E3.24.
- (a) D. Dolenc and B. Plesničar, J. Org. Chem., 2006, 71, 8028–8036 CrossRef CAS; (b) T. Nakamura, H. Yorimitsu, H. Shinokubo and K. Oshima, Synlett, 1999, 1415–1416 CrossRef CAS.
- (a) A. Inial, F. Morlet-Savary, J. Lalevée, A. C. Gaumont and S. Lakhdar, Org. Lett., 2020, 22, 4404–4407 CrossRef CAS; (b) A. F. Garrido-Castro, N. Salaverri, M. C. Maestro and J. Alemán, Org. Lett., 2019, 21, 5295–5300 CrossRef CAS; (c) W. G. Bentrude, J.-J. L. Fu and C. E. Griffin, Tetrahedron Lett., 1968, 9, 6033–6036 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04387g |
| This journal is © The Royal Society of Chemistry 2020 |