
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnesium-mediated sp3 C–H activation in cascade cyclization of 1-arylethynyl-2-alkyl-o-carboranes: efficient synthesis of carborane-fused cyclopentanes†
Jie
Zhang‡
,
Cen
Tang‡
and
Zuowei
Xie
 *
*
Department of Chemistry, State Key Laboratory of Synthetic Chemistry, The Chinese University of Hong Kong, Shatin, N. T., Hong Kong, China. E-mail: zxie@cuhk.edu.hk
First published on 2nd September 2020
Abstract
This work reports an unprecedented cascade cyclization of 1-arylethynyl-2-alkyl-o-carboranes promoted by magnesium-mediated sp3 C–H activation. Treatment of 1-arylethynyl-2-alkyl-o-carboranes with MeMgBr gives a series of carborane-fused cyclopentanes in very good yields. Deuterium labelling and control experiments suggest that HMgBr, resulting in situ from the nucleophilic substitution of cage B–H bonds with Grignard reagent, initiates the reaction, in which magnesium-promoted intramolecular sp3 C–H activation serves as a key step. This work not only offers a new route for the synthesis of carborane-fused cyclopentanes, but also sheds some light on Mg-mediated C–H activation and functionalization.
Introduction
Grignard reagents are a well-known class of C-nucleophiles and have been intensively used in organic synthesis for the formation of C–C bonds.1 They have also been employed as coupling partners in the functionalization of carboranes, for example, in Pd-catalyzed cage B–C cross-coupling,2 and Ni-catalyzed cage C–C cross-coupling3 (Scheme 1). Very recently, we disclosed that Grignard reagents can activate the cage B–H bond in o-carboranes via nucleophilic cage BH substitution reaction for regioselective functionalization of o-carboranes.4 | ||
| Scheme 1 Grignard reagents in the functionalization of o-carboranes. | ||
Carboranes, a class of carbon–boron molecular clusters, possess unique properties such as remarkable thermal and chemical stability, spherical geometry, and highly electron-delocalized hydrophobic surface.5 These features have made carboranes useful functional blocks in materials,6 organometallic/coordination chemistry,7 boron neutron capture therapy agents,8 pharmacophores,9 and more.10 To this end, functionalization of carboranes have attracted enormous research interest. Considerable progress has recently been made in selective functionalization of carboranes at either BH5,11 or CH5,12 vertices.
As an ongoing project in our laboratory, we studied the nucleophilic substitution reaction of 1-phenylethynyl-2-methyl-o-carborane (1a) with MeMgBr. No expected B(3,6)-dimethylated o-carborane was isolated, rather, an unprecedented compound 2a was obtained from a mixture of polymethylated o-carboranes. Its 1H coupled 11B NMR spectrum indicated that 2a is not a cage B-substituted species. The 13C NMR spectrum showed the absence of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C unit. Its 1H NMR spectrum displayed a pentet at 4.05 ppm (1H), two double doublets at 2.89 ppm (2H) and 2.62 ppm (2H), respectively, in addition to phenyl protons. This compound was later unambiguously confirmed by single-crystal X-ray analysis to be 1,2-[CH2CH(Ph)CH2]-o-carborane (2a; Fig. 1). This result indicates clearly that the alkyne unit –CC– in 1a has been completely reduced to the alkyl by addition of three H atoms, forming a C–C single bond. To address the reaction pathway of this unprecedented Mg-mediated cyclization, we studied this reaction in detail and the results are presented in this article.
C unit. Its 1H NMR spectrum displayed a pentet at 4.05 ppm (1H), two double doublets at 2.89 ppm (2H) and 2.62 ppm (2H), respectively, in addition to phenyl protons. This compound was later unambiguously confirmed by single-crystal X-ray analysis to be 1,2-[CH2CH(Ph)CH2]-o-carborane (2a; Fig. 1). This result indicates clearly that the alkyne unit –CC– in 1a has been completely reduced to the alkyl by addition of three H atoms, forming a C–C single bond. To address the reaction pathway of this unprecedented Mg-mediated cyclization, we studied this reaction in detail and the results are presented in this article.
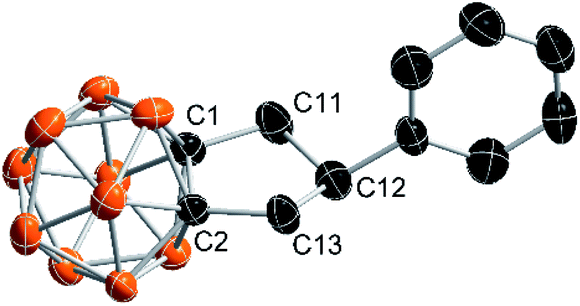 | ||
| Fig. 1 Molecular structure of 2a with the thermal ellipsoids shown at 50% probability level (all H atoms are omitted for clarity). Selected bond distance (Å) and angels (°): C(1)–C(2) 1.641(2), C(2)–C(13) 1.519(3), C(13)–C(12) 1.569(3), C(12)–C(11) 1.566(3), C(11)–C(1) 1.512(3); C(1)–C(2)–C(13) 106.9(2), C(2)–C(13)–C(12) 106.3(2), C(13)–C(12)–C(11) 107.1(2), C(1)–C(11)–C(12) 106.9(2), C(11)–C(1)–C(2) 106.8(2). | ||
Results and discussion
We first optimized the reaction conditions for the formation of 2a. Treatment of 1a with 2 equiv. of MeMgBr in a mixed solvent of THF/toluene (1/10 in v/v) at 80 °C for 36 h gave 2a in 80% isolated yield (entry 1, Table S1†). Replacement of MeMgBr with other Grignard reagents or organic lithium reagents led to a dramatically decreased yield of 2a (entries 2–4, Table S1†). Screening of the reaction time, solvents and amount of MeMgBr did not offer better results (entries 5–8, Table S1†).With the optimal reaction conditions (entry 1, Table S1†) in hand, we examined the substrate scope, and the results were summarized in Table 1. Both electron-donating and electron-withdrawing groups at para position of the phenyl ring except for p-F and p-CF3 gave the corresponding carborane-fused cyclopentanes in 72–80% yields (2a–2i). For 1g with p-F or 1i with p-CF3 on phenyl ring, the desired product 2g or 2i was isolated in 35% or 7% yield along with 1g/1i being recovered in 47%/80% yield, whereas m-F substituted 1k afforded 2k in 72% yield. On the other hand, the cyclization of ortho-substituted substrates gave 2m and 2n in 43–45% yields because of steric reasons. Replacement of the methyl group at cage-C with n-butyl, the cyclization reaction occurred smoothly to generate 2p in 77% yield. In contrast, the benzyl group at the cage-C lowered the cyclization efficiency, leading to 2q in 27% yield along with 1-(PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)-2-Bn-o-C2B10H10 as the major product. No reaction was observed if Ph and –CC– was separated by a –CH2CH2– unit (2r), suggesting an important role of phenylacetylene moiety in the reaction. These results offer some hints into the reaction mechanism (vide infra).
CH)-2-Bn-o-C2B10H10 as the major product. No reaction was observed if Ph and –CC– was separated by a –CH2CH2– unit (2r), suggesting an important role of phenylacetylene moiety in the reaction. These results offer some hints into the reaction mechanism (vide infra).
|
a Reactions were conducted on 0.1 mmol scale of 1 in a mixed solvent of THF/toluene (0.8 mL, 1/10 in v/v) in a closed flask at 80 °C for 36 h.
b Isolated yields.
c 47% starting material recovered.
d 80% starting material recovered.
e Reaction was conducted at 120 °C for 7 d. 1-(PhCHCH)-2-Bn-o-C2B10H10 was isolated in 60% yield.
f 100% starting material recovered.
|
|---|

|
Compounds 2 were characterized by 1H, 13C, and 11B NMR spectroscopy as well as high-resolution mass spectrometry. Their 11B NMR spectra exhibited a similar pattern of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:1:2 spanning the range from −14 to −7 ppm. The molecular structures of 2a and 2p have been further confirmed by single-crystal X-ray analyses.13
:1:2:1:2 spanning the range from −14 to −7 ppm. The molecular structures of 2a and 2p have been further confirmed by single-crystal X-ray analyses.13
The most interesting part of this reaction is the reaction mechanism by which compounds 2 are formed. To figure out the sources of the three added H atoms in 2a, several deuterium labelling experiments were carried out (Scheme 2). When the reaction was conducted in a mixed deuterated solvent (THF-d8/toluene-d8), 2a was obtained without any D incorporation in 80% yield (Scheme 2a). Treatment of 1a with fully deuterated ethyl magnesium bromide (C2D5MgBr)14 gave 2a in 50% yield without any incorporation of deuterium. These results clearly indicated that (1) the three added H atoms come from neither solvent nor Grignard reagent, and (2) ethyl magnesium bromide is less reactive than methyl magnesium bromide. If the reaction was quenched with D2O, 2a-d1 was isolated in 78% yield with 100% D incorporation (Scheme 2c). On the other hand, reaction of 1-(PhCC)-2-CD3-o-carborane (1a-d3) with MeMgBr under the standard conditions, followed by quenching with H2O, afforded 2a-d3 in 76% yield. Surprisingly, one of the methyl deuterium atoms migrated to C(5) with 100% D incorporation (Scheme 2d).
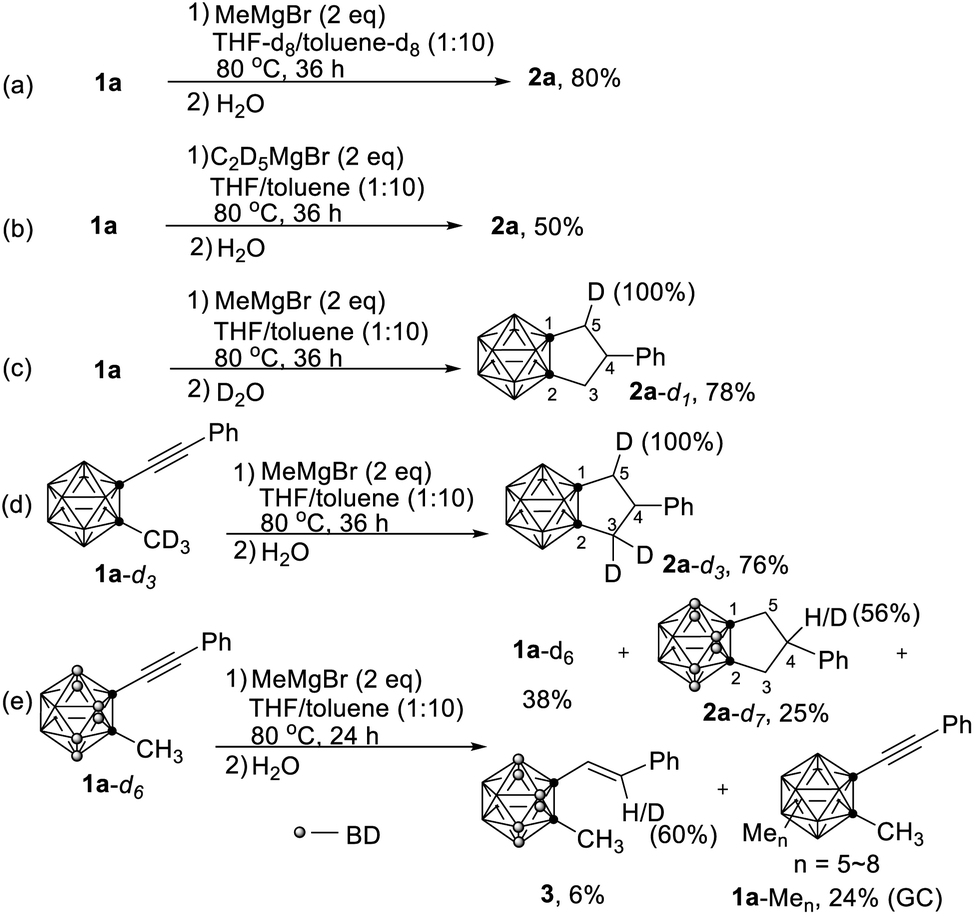 | ||
| Scheme 2 Deuterium labelling experiments: (a) reaction run in deuterated solvents (THF-d8/toluene-d8). (b) Reaction of 1a with C2D5MgBr. (c) Quenching of reaction by D2O. (d) Reaction of 1-(PhC≡C)-2-CD3-o-carborane (1a-d3) with MeMgBr. (e) Reaction of 1-(PhC≡C)-2-CH3-3,4,5,6,7,11-D6-o-carborane (1a-d6) with MeMgBr. | ||
Having identified the sources of two out of three added hydrogen atoms in 2a, the question then arises as to what is the origin of the third added hydrogen in 2a. We learnt from our previous work that HMgBr was generated as a by-product in the nucleophilic cage BH substitution of o-carboranes with RMgBr.4 It was then assumed that the cage B–H may serve as H source for the third H atom. To verify this hypothesis, compound 1-(PhCC)-2-CH3-3,4,5,6,7,11-D6-o-carborane (1a-d6) was synthesized. Treatment of 1a-d6 with MeMgBr under the standard conditions gave the corresponding product 2a-d7 in 25% yield with 56% D incorporation at C(4)-position (Scheme 2e). This result suggested that cage BH indeed served as H source via the formation of HMgBr, which was further supported by the observation of a mixture of multi-B-methylated o-carboranes 1a-Men on GC-MS.
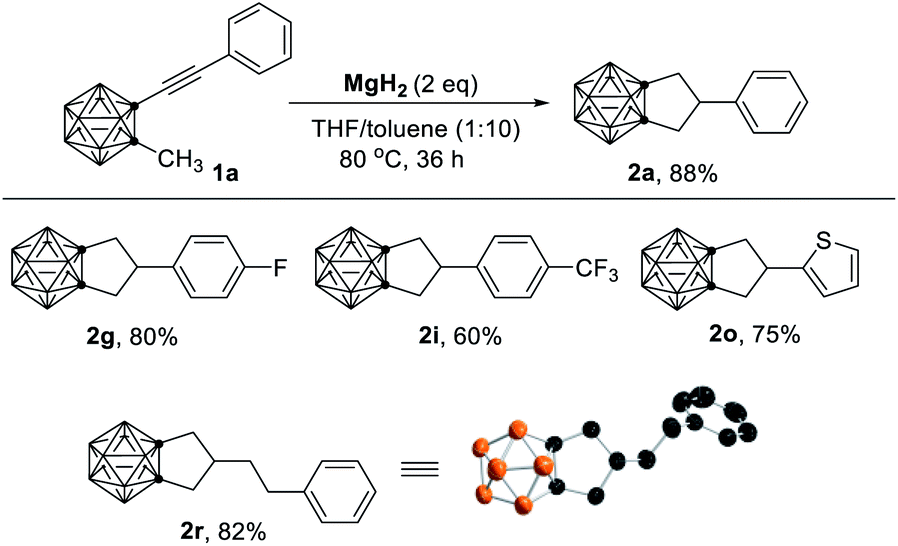
To further understand the role of HMgBr, the easily accessible MgH2 (ref. 15) was selected as the reagent. Treatment of 1a with 2 equiv. of MgH2 in THF/toluene at 80 °C for 36 h afforded 2a in 88% isolated yield (Table 2). Substrates that showed no or poor activity using MeMgBr as a reagent (1g, 1i, 1o, 1r in Table 1) worked well in the reaction with MgH2 to give the corresponding cyclization products (2g, 2i, 2o, 2r) in 60–82% yields (Table 2). These results strongly indicated that HMgBr was essential for this cyclization reaction.
To gain more insight into the reaction pathway, additional control experiments were carried out (Scheme 3). Treatment of 1a with MeMgBr in the presence of 2 equiv. of 1,1-diphenylethylene16 under standard reaction conditions gave 2a in 80% yield (Scheme 3a). No obvious change was observed when the reaction was run in the dark. These results suggested that the above cascade cyclization reaction may not involve a radical pathway. Quenching the reaction with TMSCl after 18 h generated a compound 4 in 35% isolated yield in addition to 2a (38% yield) (Scheme 3b). On the other hand, compound 5 was isolated in 65% yield if the reaction was quenched by I2 after 36 h (Scheme 3c). The isolation of 4 and 3 (Scheme 2e) shed some light on the reaction intermediates. Moreover, quenching the reaction of 1i with MeMgBr after 36 h with D2O led to the 80% recovery of 1i without any D incorporation, suggesting that the cage C–CH3 proton was not deprotonated by MeMgBr. This result was in agreement with the D-labelling experiments shown in Scheme 2d. If the cage C-CD3 was deprotonated by MeMgBr, the D would not migrate to C(5) with 100% D incorporation in 2a-d3.
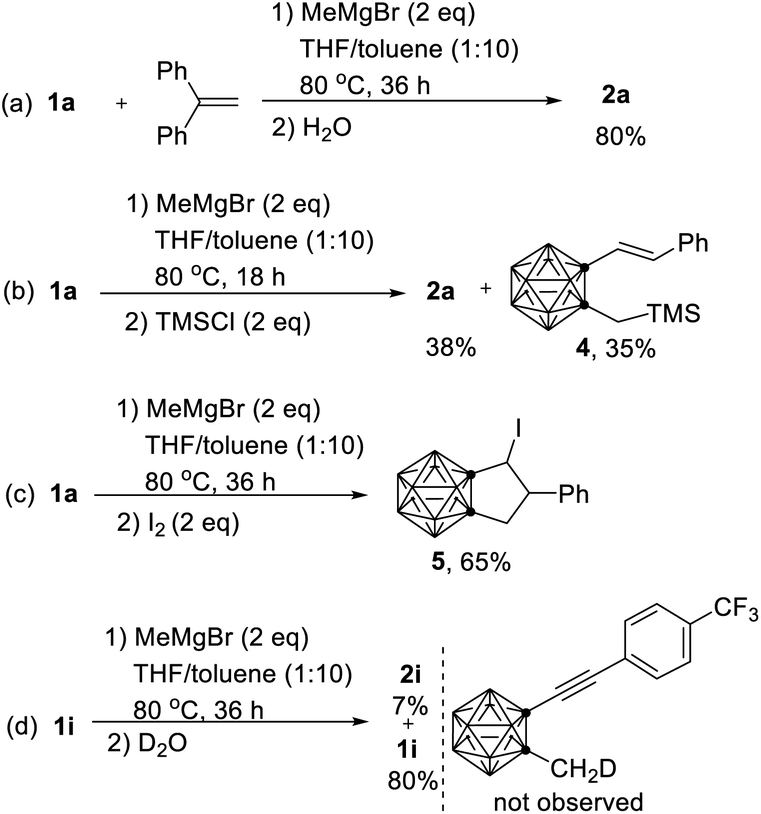 | ||
| Scheme 3 Control experiments: (a) reaction in the presence of 1,1-diphenylethylene. (b) Quenching reaction of 1a by TMSCl. (c) Quenching reaction of 1a by I2. (d) Quenching reaction of 1i by D2O. | ||
On the basis of the aforementioned results, a plausible mechanism was proposed in Scheme 4 in an example of 2a. Multiple nucleophilic substitution reaction of cage B–H bonds in 1a with MeMgBr leads to the formation of HMgBr and multi B-methylated carboranes 1a-Men (Scheme 2e).4trans-Hydromagnesiation of the triple bond in 1a furnishes an intermediate A (3 in Scheme 2e).17 The regioselectivity is controlled by the polarity of CC triple bond as carboranyl is a stronger electron-withdrawing group than phenyl. Intramolecular methyl proton–MgBr exchange in A proceeds to form B (Schemes 2d and 3b).18,19 Intramolecular ring closure affords C (Schemes 2c and 3c). Quenching of C with water generates 2a.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
We report an unprecedented cascade cyclization of 1-arylethynyl-2-alkyl-o-carboranes with Grignard reagent for highly efficient synthesis of various carborane-fused cyclopentanes. On the basis of D-labelling and control experiments, a HMgBr-initiated reaction mechanism is proposed, in which the magnesium-mediated intramolecular sp3 C–H activation serves as a key step. These results not only shed some light on magnesium-promoted C–H activation and functionalization, but also offer references for the design of cascade reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Research Grants Council of HKSAR (Project No. 14305918) and NSFC/RGC Joint Research Scheme (Project No. N_CUHK402/18), as well as the Impact Postdoctoral Fellowship Scheme (IPDFS), CUHK, to Z. J.Notes and references
- (a) R. E. Mulvey and S. D. Robertson, Top. Organomet. Chem., 2013, 45, 103–140 CrossRef CAS; (b) H. Shinokubo and K. Oshima, Eur. J. Org. Chem., 2004, 2004, 2081–2091 CrossRef; (c) J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545–1554 CrossRef CAS; (d) M. R. Luderer, W. F. Bailey, M. R. Luderer, J. D. Fair, R. J. Dancer and M. B. Sommer, Tetrahedron: Asymmetry, 2009, 20, 981–998 CrossRef CAS; (e) Y. Muramatsu, S. Kanehira, M. Tanigawa, Y. Miyawaki and T. Harada, Bull. Chem. Soc. Jpn., 2010, 83, 19–32 CrossRef CAS; (f) M. B. Smith, Organic Synthesis, 4th edn, 2017, pp. 547–603 Search PubMed.
- (a) F. Teixidor, G. Barberà, C. Viñas, R. Sillanpää and R. Kivekäs, Inorg. Chem., 2006, 45, 3496–3498 CrossRef CAS; (b) W. Jiang, C. B. Knobler, C. E. Curtis, M. D. Mortimer and M. F. Hawthorne, Inorg. Chem., 1995, 34, 3491–3498 CrossRef CAS; (c) C. Viñas, G. Barberà, J. M. Oliva, F. Teixidor, A. J. Welch and G. M. Rosair, Inorg. Chem., 2001, 40, 6555–6562 CrossRef; (d) R. M. Dziedzic, L. M. A. Saleh, J. C. Axtell, J. L. Martin, S. L. Stevens, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 9081–9084 CrossRef CAS; (e) K. P. Anderson, H. A. Mills, C. Mao, K. O. Kirlikovali, J. C. Axtell, A. L. Rheingold and A. M. Spokoyny, Tetrahedron, 2019, 75, 187–191 CrossRef CAS; (f) R. M. Dziedzic and A. M. Spokoyny, Chem. Commun., 2019, 55, 430–442 RSC; (g) Y. Ge, J. Zhang, Z. Qiu and Z. Xie, Angew. Chem., Int. Ed., 2020, 59, 4851–4855 CrossRef CAS.
- C. Tang and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 7662–7665 CrossRef CAS.
- (a) C. Tang, J. Zhang and Z. Xie, Angew. Chem., Int. Ed., 2017, 56, 8642–8646 CrossRef CAS; (b) C. Tang, J. Zhang, J. Zhang and Z. Xie, J. Am. Chem. Soc., 2018, 140, 16423–16427 CrossRef CAS; (c) Y. Quan, C. Tang and Z. Xie, Dalton Trans., 2019, 48, 7494–7498 RSC.
- (a) R. N. Grimes, Carboranes, Academic Press, Amsterdam, 3rd edn, 2016 Search PubMed; (b) N. S. Hosmane, Boron Science: New Technologies and Application, CRC Press, Boca Raton, FL, 2011 Search PubMed; (c) Z. Xie and G. X. Jin, Carborane Themed Issue, Dalton Trans., 2014, 43, 4911–5232 RSC; (d) V. I. Bregadze and Z. Xie, Boron Chemistry Themed Issue, Eur. J. Inorg. Chem., 2017, 4344–4692 Search PubMed.
- Selected reviews for applications in materials: (a) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef; (b) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC; (c) X. Li, H. Yan and Q. Zhao, Chem.–Eur. J., 2016, 22, 1888–1898 CrossRef CAS.
- Selected reviews for applications in organometallic/coordination chemistry: (a) N. S. Hosmane and J. A. Maguire, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, ch. 5, vol. 3 Search PubMed; (b) Z. Xie, Acc. Chem. Res., 2003, 36, 66–77 CrossRef; (c) W.-B. Yu, P.-F. Cui, W.-X. Gao and G.-X. Jin, Coord. Chem. Rev., 2017, 350, 300–319 CrossRef CAS; (d) Z. Qiu, S. Ren and Z. Xie, Acc. Chem. Res., 2011, 44, 299–309 CrossRef CAS; (e) S. P. Fisher, A. W. Tomich, S. O. Lovera, J. F. Kleinsasser, J. Guo, M. J. Asay, H. M. Nelson and V. Lavallo, Chem. Rev., 2019, 119, 8262–8290 CrossRef CAS; (f) J. Zhang and Z. Xie, Acc. Chem. Res., 2014, 47, 1623–1633 CrossRef CAS.
- Selected reviews for applications in medicine: (a) M. F. Hawthorne, Angew. Chem., Int. Ed. Engl., 1993, 32, 950–984 CrossRef; (b) M. F. Hawthorne and A. Maderna, Chem. Rev., 1999, 99, 3421–3434 CrossRef CAS; (c) A. F. Armstrong and J. F. Valliant, Dalton Trans., 2007, 4240–4251 RSC.
- (a) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS; (b) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS.
- (a) H. Jude, H. Disteldorf, S. Fischer, T. Wedge, A. M. Hawkridge, A. M. Arif, M. F. Hawthorne, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 12131–12139 CrossRef CAS; (b) B. H. Northrop, H.-B. Yang and P. J. Stang, Chem. Commun., 2008, 5896–5908 RSC; (c) J. M. Ludlow III, M. Tominaga, Y. Chujo, A. Schultz, X. Lu, T. Xie, K. Guo, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, Dalton Trans., 2014, 43, 9604–9611 RSC; (d) D. Jung, L. M. A. Saleh, Z. J. Berkson, M. F. El-Kady, J. Y. Hwang, N. Mohamed, A. I. Wixtrom, E. Titarenko, Y. Shao, K. McCarthy, J. Guo, I. B. Martini, S. Kraemer, E. C. Wegener, P. Saint-Cricq, B. Ruehle, R. R. Langeslay, M. Delferro, J. L. Brosmer, C. H. Hendon, M. Gallagher-Jones, J. Rodriguez, K. W. Chapman, J. T. Miller, X. Duan, R. B. Kaner, J. I. Zink, B. F. Chmelka and A. M. Spokoyny, Nat. Mater., 2018, 17, 341–348 CrossRef CAS; (e) J. Guo, D. Liu, J. Zhang, J. Zhang, Q. Miao and Z. Xie, Chem. Commun., 2015, 51, 12004–12007 RSC; (f) R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC; (g) N. S. Hosmane and R. Eagling, Handbook of Boron Science with applications in organometallics, catalysis, materials and medicine, World Scientific, Singapore, 2019 Search PubMed.
- (a) Z. Qiu, Tetrahedron Lett., 2015, 56, 963–971 CrossRef CAS; (b) D. Olid, R. Núñez, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC; (c) W.-B. Yu, P.-F. Cui, W.-X. Gao and G.-X. Jin, Coord. Chem. Rev., 2017, 350, 300–319 CrossRef CAS; (d) Y. Quan, Z. Qiu and Z. Xie, Chem.–Eur. J., 2018, 24, 2795–2805 CrossRef CAS; (e) R. M. Dziedzic and A. M. Spokoyny, Chem. Commun., 2019, 55, 430–442 RSC; (f) Y. Quan and Z. Xie, Chem. Soc. Rev., 2019, 48, 3660–3673 RSC; (g) X. Zhang and H. Yan, Coord. Chem. Rev., 2019, 378, 466–482 CrossRef CAS.
- (a) Z. Xie, Sci. China: Chem., 2014, 57, 1061–1063 CrossRef CAS; (b) Z. Qiu and Z. Xie, Dalton Trans., 2014, 43, 4925–4934 RSC; (c) D. Zhao and Z. Xie, Coord. Chem. Rev., 2016, 314, 14–33 CrossRef CAS.
- CCDC 1998059 (2a), CCDC 1998060 (2p) and CCDC 1998061 (2r) contain the ESI crystallographic data for this paper.†.
- E. Johansson, P. T. Hurley, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2009, 113, 15239–15245 CrossRef CAS.
- M. J. Michalczyk, Organometallics, 1992, 11, 2307–2309 CrossRef CAS.
- For review using Ph2CHCH2 as radical scavenger, see: S. Tang, K. Liu, C. Liu and A. Lei, Chem. Soc. Rev., 2015, 44, 1070–1082 RSC.
- (a) B. Wang, D. Y. Ong, Y. Li, J. H. Pang, K. Watanabe, R. Takita and S. Chiba, Chem. Sci., 2020, 11, 5267–5272 RSC; (b) L. Garcia, C. Dinoi, M. F. Mahon, L. Maron and M. S. Hill, Chem. Sci., 2019, 10, 8108–8118 RSC.
- Y. Suzuki, M. Kanai and S. Matsunaga, Chem.–Eur. J., 2012, 18, 7654–7657 CrossRef CAS.
- R. Rochat, K. Yamamoto, M. J. Lopez, H. Nagae, H. Tsurugi and K. Mashima, Chem.–Eur. J., 2015, 21, 8112–8120 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1998059–1998061. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04465b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |