
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light mediated carbonyl trifluoromethylative amination as a practical method for the synthesis of β-trifluoromethyl tertiary alkylamines†
Kavoos
Kolahdouzan
,
Roopender
Kumar
 and
Matthew J.
Gaunt
*
and
Matthew J.
Gaunt
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: mjg32@cam.ac.uk
First published on 9th October 2020
Abstract
We report the development of an operationally straigtforward, visible-light-mediated multicomponent strategy for the construction of β-trifluoromethylated tertiary alkylamines from feedstock aldehydes, secondary amines and a convenient source of trifluoromethyl iodide. The new process does not require a photocatalyst, is metal-free, displays a broad functional group tolerance and offers rapid, one-pot access to trifluoromethylated drug-like compounds that will be of interest in medicinal chemistry.
Introduction
A common strategy by which metabolic stability of alkylamines towards cytochrome P450s can be improved is through the inclusion of an electron-withdrawing functional group in the vicinity of the nitrogen centre.1 By way of example, introduction of the CF3 moiety provides a potentially attractive solution to this problem.2 In addition, the decreased basicity of a nitrogen atom often leads to higher bioavailability, thereby improving the pharmacokinetic profiles of lead compounds.3 In this context, the introduction of a trifluoromethyl group at the β-position of a nitrogen center can result in a large decrease in basicity (≈2 pKa units), making this a prominent tactic for perturbing the physiochemical properties of alkylamine scaffolds towards achieving superior in vivo performance.1a Thus, amines that display the β-trifluoromethyl motif are emerging as important features in pharmaceutical agents, lead compounds and other biologically-active entities (Fig. 1a).4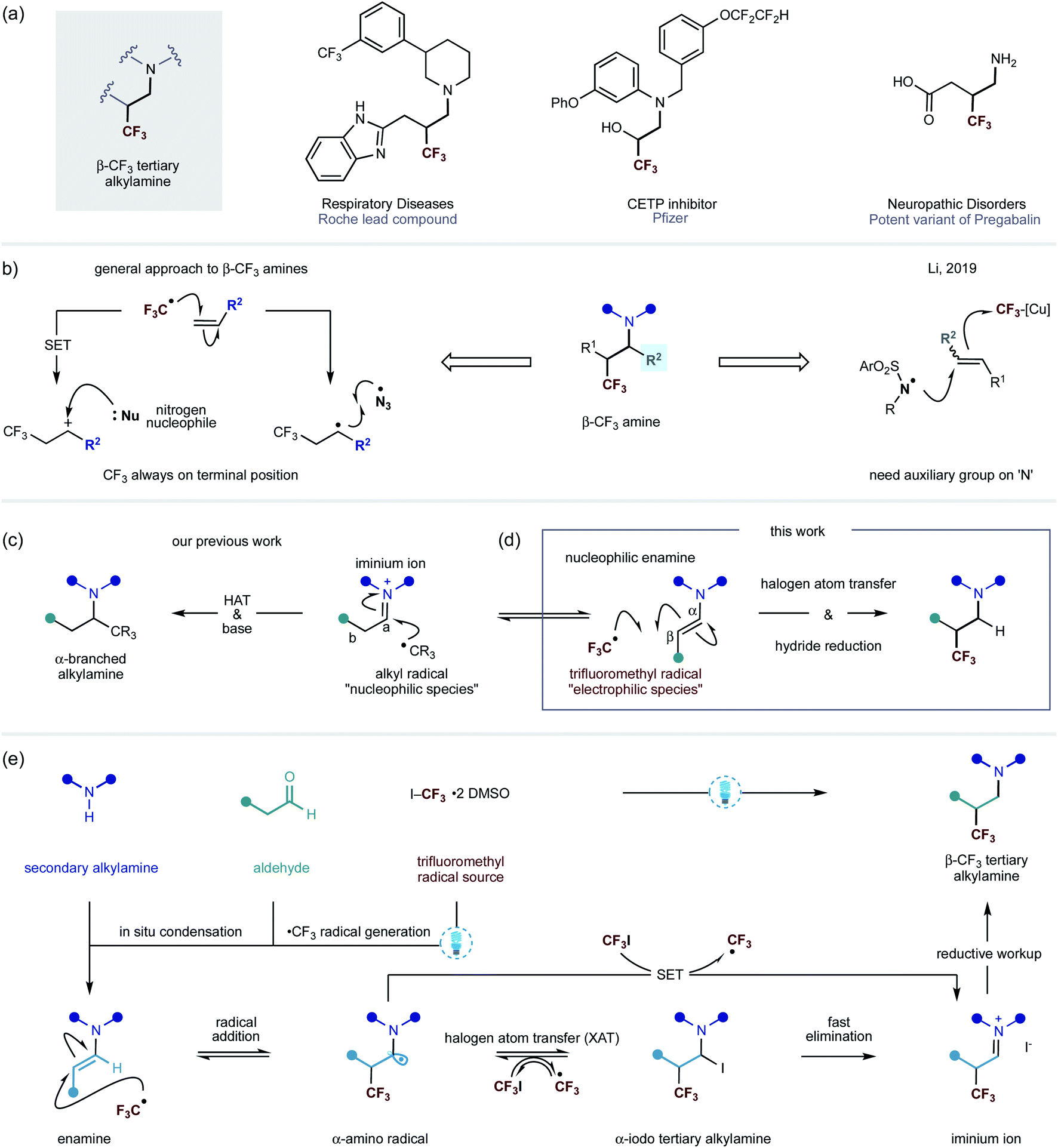 | ||
| Fig. 1 (a) Pharmaceutical compounds containing β-CF3 alkylamines; (b) Reported methods to access β-CF3 amine derivatives; (c) Carbonyl alkylative amination; (d) CF3-radical addition to enamines; (e) This work – carbonyl trifluoromethylative amination: a visible light mediated, metal-free strategy for the addition of CF3-radical to enamines. | ||
Despite the attractive benefits offered by the introduction of a trifluoromethyl group onto an amine scaffold, methods for direct access to this useful class of compounds remain relatively rare. The use of electrophilic trifluoromethyl sources, such as Togni and Umemoto reagents, in combination with Lewis acids as an activator have been the most common strategies for trifluoromethylative amination of substrates containing alkenes. The majority of these synthetic routes involve the trifluoromethyl radical addition onto carbon–carbon double bonds, followed by oxidation of the ensuing open shell species to a carbocation, which can be attacked by an amine nucleophile. Alternatively, the α-trifluoromethyl-alkyl carbon centred radical can recombine with a nitrogen centred radical, such as the azide radical, to give structurally similar products (Fig. 1b).5 However, in all of these protocols, the CF3 group is attached at the least hindered side of the alkene, limiting the potential structures that can be accessed. A rare exception is that of Li's copper-mediated method that leads to the formation of electrophilic nitrogen-centered radicals from N-fluorobis(benzenesulfonyl)-imide (NFSI). The N-centered radical is proposed to add onto alkenes, which is followed by the subsequent trifluoromethylation, through the open shell, with a (bpy)Cu(CF3)2 complex (Fig. 1b).6
Recently, we reported a visible-light-mediated carbonyl alkylative amination method that produces α-branched tertiary alkylamines via the addition of alkyl radicals – generated from alkyl iodides – to in situ formed alkyl iminium ions, followed by hydrogen atom transfer (HAT) with a silane-based reagent to deliver the tertiary alkylamine (Fig. 1c).7 The chemoselectivity observed at the addition of alkyl radicals to electrophilic iminium ions over the corresponding enamine is governed by the polarity match of a nucleophilic alkyl radical and iminium ion; a nucleophilic alkyl radical, by analogy, would expect to be polarity mismatched in a reaction with an enamine.
However, we questioned whether a CF3 radical – an electrophilic species – would, instead, engage the polarity-matched nucleophilic enamine and obviate reaction with the alkyl iminium ion to afford β-trifluoromethyl alkylamine scaffolds (Fig. 1d). On the premise that enamines, upon visible light absorption, can become strong reductants (−2.0 V vs. Ag/Ag+ in CH3CN),8 we further speculated that the excited-state of the enamine could trigger the carbon–iodine bond cleavage in trifluoromethyl iodide to form a CF3 radical through a single-electron pathway (−1.52 V vs. SCE in DMF).9 Such a step could be affected by a direct reduction of carbon–iodine bond by the excited enamine or through visible-light activation of an electron-donor acceptor (EDA) complex between the enamine and trifluoromethyliodide.10 Alternatively, a small amount of the CF3 radical may be formed via homolysis of the weak C–I bond under the action of visible light.11 Either way, the incipient CF3-radical would add to the electron-rich enamine, leading to a transient α-amino radical. This species can undergo a reversible halogen atom transfer (XAT) event with trifluoroiodomethane to produce an α-iodoamine as well as generate another CF3 radical – which then propagates a chain reaction.8,10,12 Due to the instability of the α-iodoamine intermediate, a fast α-iodo-elimination will form a β-trifluoromethylated iminium iodide species. It is also possible that the α-amino radical can engage trifluoroiodomethane through single electron transfer (SET) to form the iminium ion product and generate the CF3 radical. Upon subjection to a reductive work-up, the alkyl iminium will be converted into the β-trifluoromethylated tertiary alkylamine (Fig. 1e). This type of mechanism was reported in 1975 by Cantacuzène et al., wherein they showed that the trifluoromethyl radical – generated using UV-irradiation – could be added to limited range of pyrrolidine derived enamines to form α-trifluoromethyl aldehydes after hydrolytic work-up.13 Interestingly, they later observed that the reaction proceeded under the action of visible-light irradiation.13b In 2009, MacMillan and co-workers reported an enantioselective organocatalytic variant of α-trifluoromethylation, wherein CF3 radicals – generated via visible-light mediated iridium photocatalysis – to chiral imidazolidinone derived enamines.14 As a consequence of the catalysis requirement, the α-CF3 aldehydes are produced in this process alongside liberation of the amine catalyst. Inspired by these studies, we speculated that a straightforward process incorporating a wide range of secondary amines and aldehydes for a direct synthesis of β-CF3 tertiary alkylamines would be of great interest to practitioners of synthetic and medical chemistry. Herein, we describe an operationally simple visible-light mediated process for the synthesis of β-trifluoromethylated tertiary alkylamines. Importantly, this transformation does not require the use of a photocatalyst. The new method effectively combines readily available starting materials – secondary amines and alkyl aldehydes with trifluoroiodomethane – into β-CF3-tertiary alkylamines displaying high levels of functional and structural complexity.
Results and discussion
We began our investigations with a reaction between morpholine and hydrocinnamaldehyde, as representative coupling partners, and Ritter's trifluoroiodomethane-DMSO complex (as the source of trifluoromethyl radical) under visible-light irradiation; the process was terminated using the simple reductant, sodium triacetoxylborohydride [NaBH(OAc)3]. After assessment of the reaction parameters, namely solvent and a base (Table 1), we found that a robust protocol combined near equimolar quantities of secondary alkylamine, alkyl aldehyde with 1.5 equivalents of CF3I·2 DMSO and 1.5 equivalents of triethylamine in a 0.5 M solution of DMF containing 4 Å molecular sieves.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Variation | Yielda |
| a Yields determined by 19F NMR analysis using trifluorotoluene as internal standard. Reactions performed with a 30 W CFL lamp, equimolar amounts of aldehyde and amine, 1.5 equiv. of CF3I, 1.5 equiv. of base in a 0.5 M solution and followed by a reductive workup. b 2.5 equiv. of TEMPO added. | ||||
| 1 | DCM | Cs2CO3 | — | 60 |
| 2 | DCM | Et3N | — | 52 |
| 3 | DMF | Et3N | — | 83 |
| 4 | DMF | Et3N | In dark | 0 |
| 5 | DMF | Et3N | TEMPOb | 0 |
| 6 | DMF | — | — | Trace |
| 7 | DMF | Et3N | 40 W blue LED | 75 |
| 8 | DMF | Et3N | 455 nm filter | 48 |
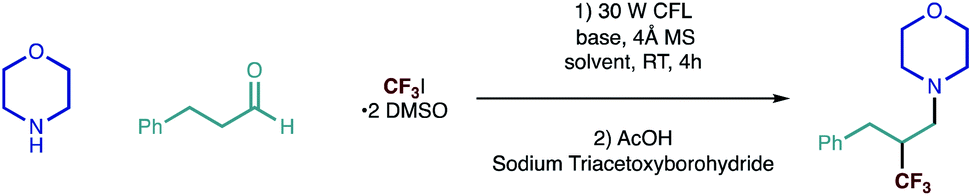
The reaction was stirred at room temperature for 4 hours, before in situ reduction of the transient β-CF3 iminium ion was affected by treatment with sodium triacetoxyborohydride (STAB) forming the desired amine in good yield (Fig. 2a). Reaction in the absence of the base led to dramatically reduced product formation, presumably due to the build of the hydroiodic acid byproduct that is generated under the reaction conditions. Exclusion of light completely suppressed the reaction and the carbonyl trifluoromethylative amination reaction was also inhibited by the addition of TEMPO (2.5 equiv.), supporting our hypothesis for a radical-based process. The efficiency was maintained when the reaction mixture was subjected to irradiation with a 40 W blue LED (entry 7, Table 1) as opposed to CFL lamp. Interestingly, the carbonyl trifluoromethylative amination process still worked, giving a 45% yield when a 455 nm long-pass filter (which removes the minor UV and near-UV components of the blue LED) was employed in the reaction with the blue LED. This suggests that C–I bond homolysis is unlikely to be the pathway through which the trifluoromethyl radical is generated or the chain is initiated. We surmise that reduction of CF3–I is most likely affected by interaction with the excited state enamine via an electron-donor acceptor complexation. Despite our best efforts, however, we have not yet been able to observe any red-shift band in UV-vis spectrum from any combination of alkylamine, aldehyde, enamine and CF3I·2 DMSO, which suggests that trifluoromethyl radical formation could proceed by an as yet unknown pathway.
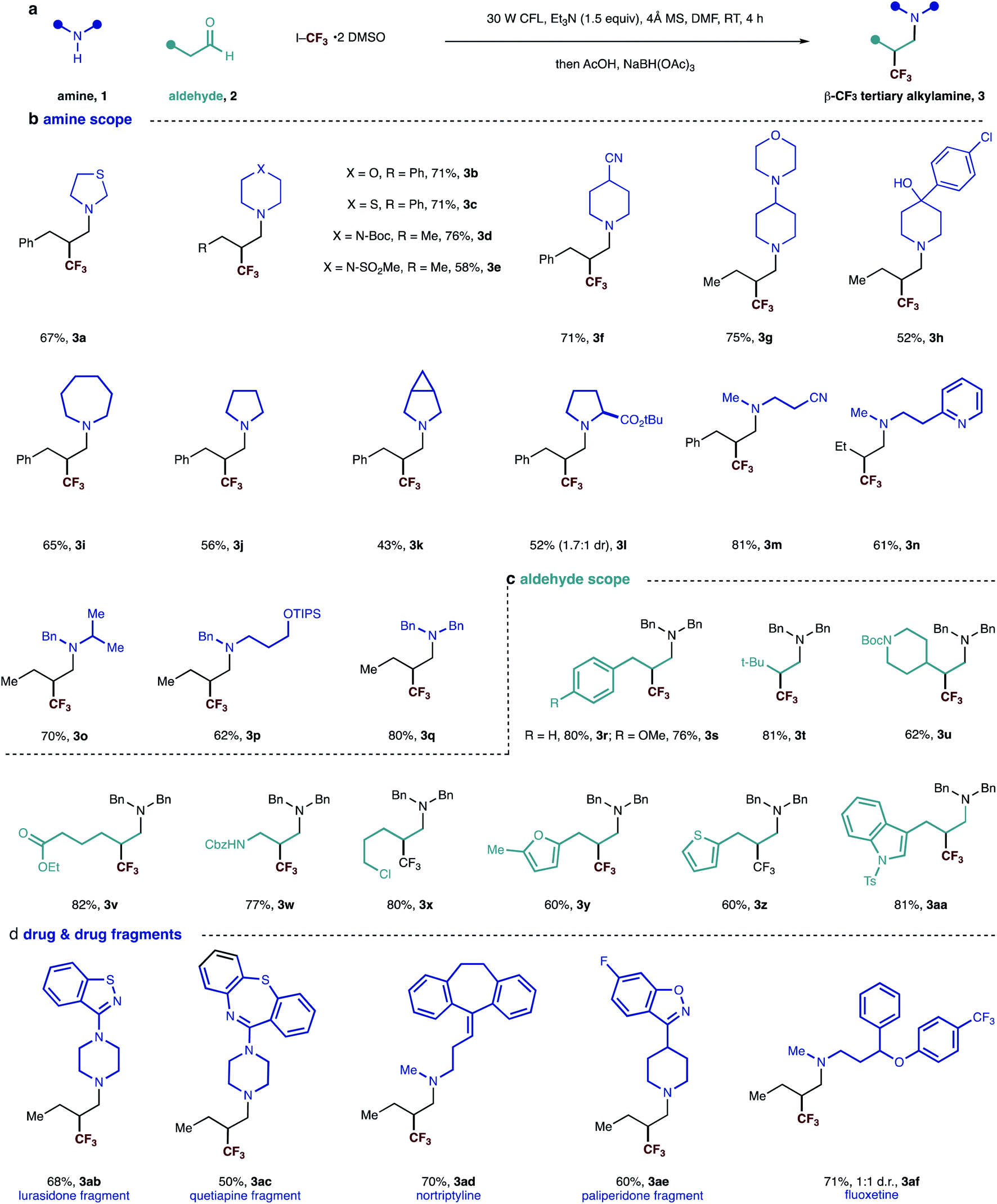 | ||
| Fig. 2 (a) General scheme and reaction conditions; (b–d) scope of visible-light mediated synthesis of β-trifluoromethylated tertiary alkylamine. | ||
We evaluated the synthetic potential of the visible light-mediated carbonyl trifluoromethylative amination reaction. A range of secondary alkylamines was successfully demonstrated in the reaction (Fig. 2). Cyclic amines – all of which are commonly found in a range of pharmaceuticals candidates – were found to be suitable substrates and delivered β-trifluoromethyl tertiary alkylamines 3a–3l in good yields. A variety of functional groups could also be incorporated, whose successful accommodation highlights the mild nature of the reaction conditions. The compatibility of the reaction with the presence of a tertiary amine motif (3g) is particularly noteworthy because these moieties typically undergo single-electron oxidation events under many photocatalytic conditions,15 demonstrating an important distinction of this process. Amines bearing branched and linear alkyl substituents as well as various functionality including cyano, pyridine and protected hydroxyl groups reacted well to give corresponding tertiary amines 3m–3q in good yields. In the case of example 3n, we did not observe Minisci-type additions into the pyridine motif, highlighting an important chemoselectivity feature of this process. Dibenzylamine worked well and afforded tertiary amine 3q in 80% yield, providing a pathway to primary amine derivatives after benzyl group cleavage.
Next, we evaluated the scope of the aldehyde component using dibenzylamine as a representative coupling partner. A selection of functionalized linear aldehydes could be converted into their corresponding β-trifluoromethylated tertiary alkylamines in good yields. The use of γ-branched aldehydes resulted in products 3t and 3u in good yield, the success of which highlights that the reaction is not adversely influenced by neighboring steric effects of the reacting enamine intermediate.
Aldehydes containing a range of functional groups were found to be competent partners to deliver compounds 3r–3aa. Aldehydes bearing electron rich heterocycles (3y, 3z and 3aa) were tolerated under the reaction conditions and no trifluoromethylation on the electron-rich ring was observed.16 Unfortunately, aldehydes with branching adjacent to the carbonyl gave no product. Trifluoromethyl radical addition onto tri-substituted enamines seems to be very slow due to the steric hindrance.
Having examined the scope of both reaction partners, we evaluated the method's further synthetic potential with regards to modifying more structurally complex, drug-like, secondary amines. Given that secondary dialkylamines are present in a range of small-molecule drugs and pre-clinical candidates, the ability to modify these molecules with feedstock aldehydes and trifluoromethyl iodide would represent a useful strategy for the construction of trifluoromethylated tertiary amines for new medicinal applications. Five drugs and drug fragments containing secondary amine motifs underwent smooth carbonyl trifluoromethylative amination to furnish tertiary alkylamines 3ab–3af. Given that these fragments are populated with additional nitrogen-containing functional groups,17 the successful synthesis of the corresponding β-trifluoromethylated tertiary alkylamines highlight the ability of the visible light-mediated and metal-free strategy to accommodate commonly encountered pharmaceutically privileged scaffolds, which could aid the synthesis of new drug candidates.2
Finally, we sought to show the utility of our trifluoromethylation protocol for the rapid production of analogues of already biologically-active small-molecules in order to evidence the reactions potential utility in a discovery setting. A series of small-molecule tertiary amines, including fenpropiomorph (produced 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per annum) and fenpropidin display a methyl group in the β-position to the tertiary amine center are societally-relevant targets due to their global usage in the protection of cereal crops.18 Furthermore, while fenpropiomorph analogues (most notably amorolfine) have found clinical usage as topical drugs against nail infection, in vivo performance against invasive fungal infections (IFIs) has been severely limited due to their rapid metabolism within host systems.1b Thus, the improvement of the metabolic stability of these compounds is an active area of research for combating IFIs.18a,19 By using different hydrocinnamaldehyde derivatives with 2,6-dimethylmorpholine and piperidine as the secondary amine substrates, the carbonyl trifluoromethylative amination effectively replaces the methyl group of these moleules with a trifluoromethyl group. Trifluoromethyl analogues of fenpropiomorph and fenpropidin (4a) and (4b–d) were readily accessed in good yields under the reaction conditions, demonstrating the methods potential application in lead optimization (Fig. 3).
000 tons per annum) and fenpropidin display a methyl group in the β-position to the tertiary amine center are societally-relevant targets due to their global usage in the protection of cereal crops.18 Furthermore, while fenpropiomorph analogues (most notably amorolfine) have found clinical usage as topical drugs against nail infection, in vivo performance against invasive fungal infections (IFIs) has been severely limited due to their rapid metabolism within host systems.1b Thus, the improvement of the metabolic stability of these compounds is an active area of research for combating IFIs.18a,19 By using different hydrocinnamaldehyde derivatives with 2,6-dimethylmorpholine and piperidine as the secondary amine substrates, the carbonyl trifluoromethylative amination effectively replaces the methyl group of these moleules with a trifluoromethyl group. Trifluoromethyl analogues of fenpropiomorph and fenpropidin (4a) and (4b–d) were readily accessed in good yields under the reaction conditions, demonstrating the methods potential application in lead optimization (Fig. 3).
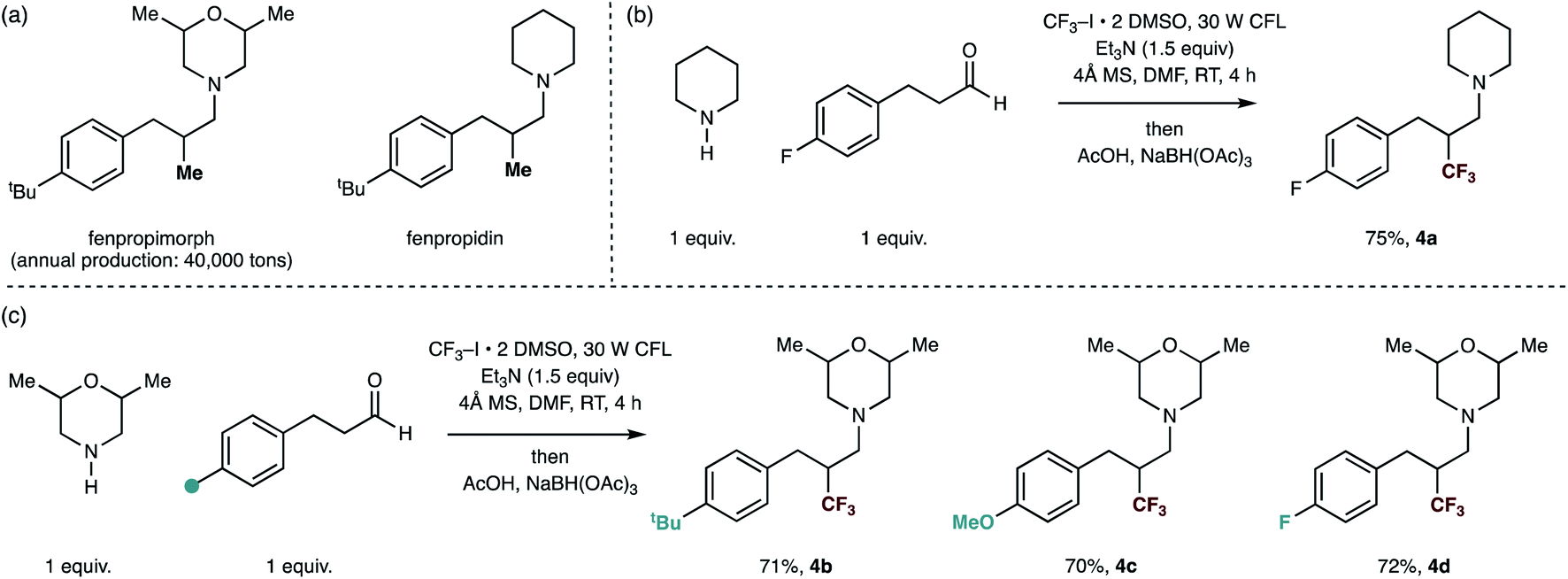 | ||
| Fig. 3 Straightforward syntheses of trifluoromethylated analogues of marketed fungicides by carbonyl trifluoromethylative amination reaction. | ||
Conclusions
In summary, we have developed a metal-free strategy for the rapid construction of β-trifluoromethylated tertiary amines from feedstock aldehydes, secondary amines and a readily available source of F3C radical. The mild and operationally straightforward conditions of the β-carbonyl trifluoromethylative amination reaction allow for its adoption in a complex setting as well as facile construction of fluorinated tertiary amine fragments that will be of interest in medicinal chemistry. We anticipate that the convergent nature of this method will allow for facile evaluation of trifluoromethyl effects with respect to tertiary amine motifs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Pomona College and Downing College for a Downing Scholarship (K. K.), the Swiss National Science Foundation (R. K.), and the Royal Society (for Wolfson Merit Award, M.J.G.).Notes and references
- (a) M. Morgenthaler, E. Schweizer, A. Hoffmann-Röder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Müller, ChemMedChem, 2007, 2, 1100–1115 CrossRef CAS; (b) P. G. Hartman and D. Sanglard, Curr. Pharm. Des., 1997, 3, 177–208 CAS; (c) Cytochrome P450: Structure, mechanism, and biochemistry, ed. O. de Montellano and R. Paul, Springer, 2005 Search PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS; (d) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS; (e) Q. Liu, C. Ni and J. Hu, Natl. Sci. Rev., 2017, 4, 303–325 CrossRef CAS.
- (a) M. B. van Niel, I. Collins, M. S. Beer, H. B. Broughton, S. K. F. Cheng, S. C. Goodacre, A. Heald, K. L. Locker, A. M. MacLeod, D. Morrison, C. R. Moyes, D. O'Connor, A. Pike, M. Rowley, M. G. N. Russell, B. Sohal, J. A. Stanton, S. Thomas, H. Verrier, A. P. Watt and J. L. Castro, J. Med. Chem., 1999, 42, 2087–2104 CrossRef; (b) C. Jamieson, E. M. Moir, Z. Rankovic and G. Wishart, J. Med. Chem., 2006, 49, 5029–5046 CrossRef CAS.
- (a) J. A. Sikorski, J. Med. Chem., 2006, 49, 1–22 CrossRef CAS; (b) World Intellectual Property Organization, WO2014072325AI, 2014; (c) T. Borisova, N. Pozdnyakova, E. Shaitanova, I. Gerus, M. Dudarenko, R. Mironets, G. Haufe and V. Kukhar, Bioorg. Med. Chem., 2015, 23, 4316–4323 CrossRef CAS; (d) C. R. Hopkins, ACS Chem. Neurosci., 2012, 3, 3–4 CrossRef CAS; (e) B. C. Buer, J. Chugh, H. M. Al-Hashimi and E. N. G. Marsh, Biochemistry, 2010, 49, 5760–5765 CrossRef CAS; (f) C. Jäckel, M. Salwiczek and B. Koksch, Angew. Chem., Int. Ed., 2006, 45, 4198–4203 CrossRef; (g) H.-P. Chiu, B. Kokona, R. Fairman and R. P. Cheng, J. Am. Chem. Soc., 2009, 131, 13192–13193 CrossRef CAS.
- (a) R. R. Karimov, A. Sharma and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 715–724 CrossRef CAS; (b) C.-L. Zhu, C. Wang, Q.-X. Qin, S. Yruegas, C. D. Martin and H. Xu, ACS Catal., 2018, 8, 5032–5037 CrossRef CAS; (c) F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2014, 53, 1881–1886 CrossRef CAS; (d) Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136–2139 CrossRef CAS; (e) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Org. Lett., 2014, 16, 1240–1243 CrossRef CAS; (f) S. Kawamura, H. Egami and M. Sodeoka, J. Am. Chem. Soc., 2015, 137, 4865–4873 CrossRef CAS; (g) H. Egami, S. Kawamura, A. Miyazaki and M. Sodeoka, Angew. Chem., Int. Ed., 2013, 52, 7841–7844 CrossRef CAS.
- H. Xiao, H. Shen, L. Zhu and C. Li, J. Am. Chem. Soc., 2019, 141, 11440–11445 CrossRef CAS.
- R. Kumar, N. J. Flodén, W. G. Whitehurst and M. J. Gaunt, Nature, 2020, 581, 415–420 CrossRef CAS.
- M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS.
- C. P. Andrieux, L. Gelis, M. Medebielle, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1990, 112, 3509–3520 CrossRef CAS.
- (a) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC; (b) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS; (c) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS; (d) G. Filippini, M. Silvi and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 4447–4451 CrossRef CAS; (e) Ł. Woźniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678–5681 CrossRef; (f) H. Matsui, M. Murase and T. Yajima, Org. Biomol. Chem., 2018, 16, 7120–7123 RSC.
- M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511–9515 CrossRef CAS.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS.
- (a) D. Cantacuzène and R. Dorme, Tetrahedron Lett., 1975, 16, 2031–2034 CrossRef; (b) D. Cantacuzène, C. Wakselman and R. Dorme, J. Chem. Soc., Perkin Trans., 1977, 1, 1365–1371 RSC.
- (a) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS; (b) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986–4987 CrossRef CAS.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS.
- (a) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS; (b) S. A. Forsyth, H. Q. N. Gunaratne, C. Hardacre, A. McKeown and D. W. Rooney, Org. Process Res. Dev., 2006, 10, 94–102 CrossRef CAS.
- G. R. Jachak, R. Ramesh, D. G. Sant, S. U. Jorwekar, M. R. Jadhav, S. G. Tupe, M. V. Deshpande and D. S. Reddy, ACS Med. Chem. Lett., 2015, 6, 1111–1116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04853d |
| This journal is © The Royal Society of Chemistry 2020 |