
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled hierarchical self-assembly of networked coordination nanocapsules via the use of molecular chaperones†
Xiangquan
Hu
a,
Sisi
Feng
 *b,
Jialei
Du
*c,
Li
Shao
a,
Jinxin
Lang
d,
Chen
Zhang
e,
Steven P.
Kelley
a,
Jian
Lin
f,
Scott J.
Dalgarno
g,
David A.
Atwood
h and
Jerry L.
Atwood
*a
*b,
Jialei
Du
*c,
Li
Shao
a,
Jinxin
Lang
d,
Chen
Zhang
e,
Steven P.
Kelley
a,
Jian
Lin
f,
Scott J.
Dalgarno
g,
David A.
Atwood
h and
Jerry L.
Atwood
*a
aDepartment of Chemistry, University of Missouri-Columbia, 601 S College Ave, Columbia, MO 65211, USA. E-mail: atwood@missouri.edu
bKey Laboratory of Chemical Biology and Molecular Engineering of Ministry of Education, Institute of Molecular Science, Shanxi University, Taiyuan 030006, P. R. China. E-mail: ssfeng@sxu.edu.cn
cInstitute for Advanced Interdisciplinary Research, University of Jinan, Jinan 250022, P. R. China. E-mail: ifc_dujl@ujn.edu.cn
dSchool of Chemistry, Xi'an Jiaotong University, Xi'an 710049, P. R. China
eDepartment of Chemical and Biomolecular Engineering, North Carolina Sate University, Raleigh, North Carolina 27695, USA
fDepartment of Mechanical and Aerospace Engineering, University of Missouri-Columbia, 601 S College Ave, Columbia, MO 65211, USA
gInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, UK
hDepartment of Chemistry, University of Kentucky, Lexington, KY 40506, USA
First published on 28th October 2020
Abstract
Supramolecular chaperones play an important role in directing the assembly of multiple protein subunits and redox-active metal ions into precise, complex and functional quaternary structures. Here we report that hydroxyl tailed C-alkylpyrogallol[4]arene ligands and redox-active MnII ions, with the assistance of proline chaperone molecules, can assemble into two-dimensional (2D) and/or three-dimensional (3D) networked 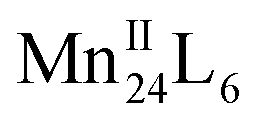 nanocapsules. Dimensionality is controlled by coordination between the exterior of nanocapsule subunits, and endohedral functionalization within the 2D system is achieved via chaperone guest encapsulation. The tailoring of surface properties of nanocapsules via coordination chemistry is also shown as an effective method for the fine-tuning magnetic properties, and electrochemical and spectroscopic studies support that the
nanocapsules. Dimensionality is controlled by coordination between the exterior of nanocapsule subunits, and endohedral functionalization within the 2D system is achieved via chaperone guest encapsulation. The tailoring of surface properties of nanocapsules via coordination chemistry is also shown as an effective method for the fine-tuning magnetic properties, and electrochemical and spectroscopic studies support that the 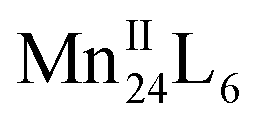 nanocapsule is an effective homogeneous water-oxidation electrocatalyst, operating at pH 6.07 with an exceptionally low overpotential of 368 mV.
nanocapsule is an effective homogeneous water-oxidation electrocatalyst, operating at pH 6.07 with an exceptionally low overpotential of 368 mV.
Introduction
Hierarchical self-assembly via metal coordination is a ubiquitous process for constructing sophisticated supramolecular structures in nature.1 As an example of its use in biological systems, metal coordination or bridging plays a crucial role in folding and assembling multiple protein subunits into precise, complex and functional quaternary structures (such as viral metalloproteins).2,3 Metallosupramolecular assemblies such as metal–organic nanocapsules (MONCs) and/or nanocages are potentially useful models for such complex biological processes,4,5 and are also promising with regard to energy storage,6–10 molecular encapsulation,11–15 catalytic,16–18 and biomedical applications.19,20 To date, synthetic chemists have been able to isolate discrete cages consisting of more than 100 precisely designed units through metal coordination.21 A long-standing challenge, however, is the rational combination of simple components to form hierarchical superstructures with a similar level of assembly complexity as proteins.22,23 Another challenge that has seldom been addressed in the literature is redox-controlled metal-directed assembly. Albeit at a higher level of complexity, living organisms are able to rapidly select the oxidation state of metal ions such as Cu, Mn and Fe, with regard to protein subunit folding and assembly of quaternary structure, often with the aid of supramolecular chaperones.24–26 These metallochaperones are typically employed to capture, protect and insert the highly active metal ions into the specific coordination sites before elements of the quaternary structure have formed through subunit self-assembly.26 The powerful self-assembly approach utilised by biological systems may thus provide access to new hierarchical superstructures (HSSs) with unique properties.Our group (and others) have used C-alkyl-pyrogallol[4]arenes (PgCn, where n is the number of carbon atoms in the pendant alkyl chains), bowl-shaped polydentate macrocycles, to synthesise MONCs via metal insertion.13,27,28 This approach gives rise to large, discrete cages which typically have one of two highly conserved structures: a dimeric cage composed of 2 PgCns seamed/bridged by 8 metal ions, or a hexameric cuboctahedral analog comprising 6 PgCns and 24 metal ions (the latter of which form 6 triangular faces). These MONCs are readily accessible via ambient or solvothermal syntheses using redox stable metal ions such as ZnII, NiII, GaIII.28–30 Variations in structure are also possible, for instance by replacing some pyrogallol rings with resorcinol in the PgCn framework, giving mixed macrocycles that cause ‘defects’ in the perfect MONC structure.29 Despite the fact that these two general supramolecular architectures accommodate metals of different size and charge, the controlled assembly of redox-active transition metals has proven difficult. For instance, it has been shown that the reaction of FeII or MnII ions with PgCns rapidly yielded MONCs with metal ions in mixed oxidation states.31,32 Indeed, the assembly of mixed-valence MONCs, such as MnII/MnIII, should be more kinetically favored than solely MnII-based analogs since MnII is more thermodynamically stable and kinetically labile than MnIII for coordination.26 We only recently achieved the assembly of CoII hexameric MONCs by using a route inspired by zinc-finger proteins (ZNFs).33 In that case the ZnII ion was used to direct assembly of hexameric MONCs that were spontaneously transmetallated with CoII ions to afford the target assembly. Such results indicate that new MONCs with redox-active functionality may (as can be the case with biological systems) require additional templates or chaperones to control their assembly into the correct state.
In this context, we are encouraged to challenge the synthesis of HSSs constructed from C-propan-3-ol-pyrogallol[4]arene (PgC3OH) and coordination-inert but redox-active MnII ions; the hydroxyl group on PgC3OH can link MONCs to obtain HSSs.30 This may not only help to develop a better understanding of the redox-based self-assembly of metalloproteins, but also the construction of HSSs with emergent properties, such as magnetism and catalysis, based on the oxidation state distribution of the metal ions.34–36 Several reaction conditions and methodologies have been investigated to this end, yet all failed to deliver the selective assembly of any anticipated HSSs (see ESI† for details). We hypothesised that in situ redox reactions may prevent the formation of such highly intricate structures.
Herein, we present a design strategy for the construction of such otherwise unobtainable HSSs that uses a reaction system consisting of PgC3OH, MnII ions, and proline. The use of proline was inspired by the MnII coordination sphere in manganese-based proteins, which may effectively capture and stabilise the free metal ion, as well as modulating its weak coordination ability with regard to metal insertion.26,37,38 We propose a system in which PgC3OH is assembled into hexameric hydrogen-bonded nanocapsules (MONCs), whilst proline molecules act as the molecular chaperones to capture, protect and insert the MnII ions into the framework (Scheme 1). Once formed, the thermodynamically and kinetically very stable MONCs serve as subunits (secondary structures) and organise into more complex HSSs through the formation of intermolecular metal–hydroxyl coordination bonds. Using this approach, we obtained 2D and 3D HSSs consisting of MnII-seamed MONC subunits (1, [Mn24(PgC3OH)6(H2O)44] and 2, [Mn24(PgC3OH)6(H2O)44(CH3CN)2]), structurally controlled by subtle changes in reaction conditions.
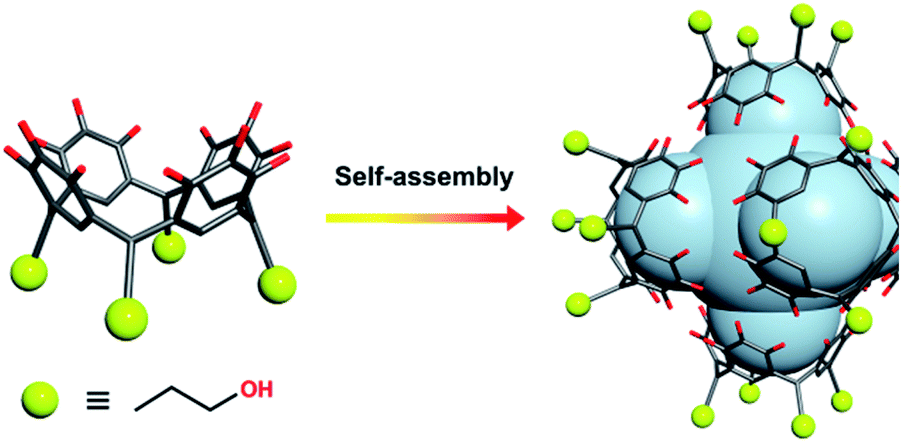 | ||
| Scheme 1 Pre-assembly strategy of MnII-seamed MONC subunits used in this study. Color codes: carbon, grey; oxygen, red; MnII, purple. | ||
Results and discussions
Compound 1 has been studied and characterised by scanning electron microscopy images (Fig. S1†), single-crystal X-ray diffraction (SC-XRD, Fig. S2†), FT-IR (Fig. S3†), elemental analysis (EA), MALDI-TOF MS (Fig. S4†), thermogravimetric analysis (TGA, Fig. S5†) and differential scanning calorimetry (DSC, Fig. S6†), details of which can be found in the ESI.† Compound 1 crystallises in the monoclinic space group P21/n. The crystal structure of 1 shows a 2D framework constructed from infinite MONCs subunits, with each MONC being assembled from 30 components: six PgC3OH molecules and 24 metal ions (Fig. 1). The overall geometry of the MONC subunit corresponds to that of a truncated octahedron, which is similar to the previously reported hexameric MONCs.28 Each hexagonal face of the MONC is capped by one [Mn3O3] trimetallic cluster with Mn–O distances in the range of 2.03–2.11 Å, Mn–O–Mn angles in the range of 133.17–137.50°, and O–Mn–O angles in the range of 99.34–105.66°. All MnII ions adopt an octahedral ligand field, where the equatorial positions are coordinated with oxygen atoms from the upper-rim of PgC3OH units. Bond-valence sum (BVS) analysis, coupled with examination of bonding energy reveals that all Mn ions are in the +2 oxidation state (Table S1 and Fig. S7†). Interestingly, each MONC encapsulates two proline chaperone ligands that coordinate in a bridging mode between two MnII ions (metal–carboxyl distances in the range of 2.24–2.27 Å). This suggests that the proline molecules perform the critical function of a molecular chaperone, capturing, protecting and inserting MnII ions into MONCs via ligand exchange during the assembly process. The extended view of 1 shows that each MONC is connected to four adjacent symmetry equivalents via double manganese–hydroxyl coordination (M–O distances: 2.26–2.35 Å). One MONC provides a hydroxyl tail and a metal coordination site for another, and the other axial positions are occupied by water molecules.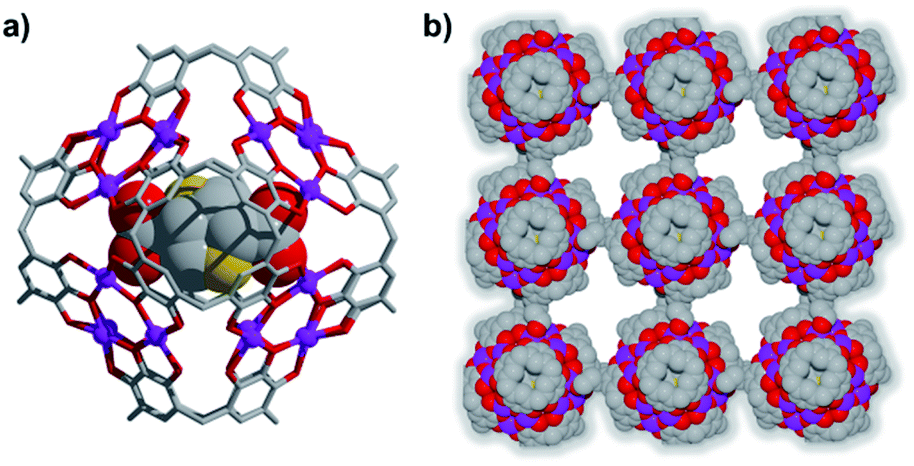 | ||
| Fig. 1 (a) Side and (b) extend views of the single crystal X-ray structure of 1. Inspection shows 2D HSSs composed of MnII-MONC subunits, each of which encapsulates two proline chaperone molecules via metal coordination. Color codes: manganese (purple), carbon (grey), oxygen (red), nitrogen (yellow). Hydrogen atoms, axial water ligands and hydroxyl tail alkyl chains not involved in metal–ligand coordination to adjacent MONC subunits were removed for clarity. | ||
Introduction of a greater amount of water to similar reaction conditions as those used in the synthesis of 1 changed both the internal and external properties of the MnII-seamed MONCs, resulting in the formation of a 3D HSSs which crystallises in an orthorhombic system (structure solution in space group Pccn, 2, Fig. 2, Table S2 and Fig. S8–S10†). On the internal surface, all axial positions at the metal centres are occupied by water molecules, whilst inspection of the exterior reveals that each MONC subunit is linked to eight symmetry equivalents via single manganese–hydroxyl coordination bonds (two crystallographic M–O distances: 2.276 and 2.279 Å, respectively), the result being assembly into a cubic tertiary structure (Fig. 2b). This supramolecular nanocube is assembled from 216 MnII ions and 54 PgC3OH macrocyclic ligands and has an edge of 4.5 nm. Within the nanocube there are two types of MONC subunits with different orientation in the solid lattice, highlighted by the disparate colours in Fig. 2. This structural motif is similar to the unit cell of CsCl (Fig. 2c), and the extended view exhibits a hierarchical CsCl-like superstructure (Fig. 2d).
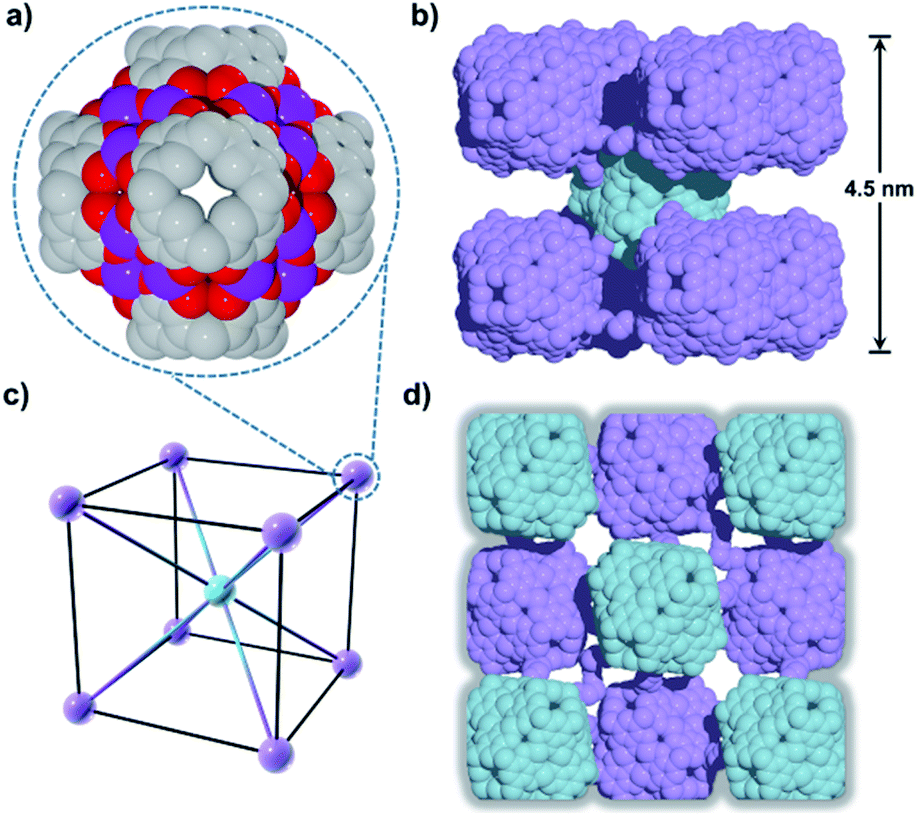 | ||
| Fig. 2 (a) MnII-seamed MONC subunits (secondary structure). (b) Supramolecular nanocubes (tertiary structure). (c) CsCl unit cell and (d) the 3D hierarchical CsCl-like superstructure (quaternary structure). Hydrogen atoms, axial ligands and hydroxyl tail alkyl chains not involved in metal–ligand coordination to adjacent MONC subunits were removed for clarity. | ||
Magnetic susceptibility data for 1 and 2 were recorded in the temperature range of 2.0–300 K in an applied magnetic field of 1000 Oe. The χm, χmT vs. T plots for the complexes are shown in Fig. 3, where χm is the molar magnetic susceptibility. For supramolecular assemblies 1 and 2, the values of χmT at 300 K are 81.8 and 92.2 cm3 mol−1 K, respectively, but lower than that of expected for the sum of the Curie constants for 24 non-interacting MnII (s = 5/2) ions, with g = 2.00 (105.0 cm3 mol−1 K). Upon cooling, χmT first gradually decreases to a value of 76.1 cm3 K mol−1 at 100 K, and then decreases more rapidly on further cooling to 27.3 cm3 K mol−1 at 2.0 K for 1, however, χmT decreases to the minimum value of 29.4 cm3 mol−1 K at 2.0 K for 2, indicating antiferromagnetic coupling within the MnII ions. Above 50 K, the temperature dependence of χm−1 obeys the Curie–Weiss law with C = 90.91 cm3 K mol−1 and θ = −12.8 K above 2.0 K for 1 and C = 102.88 cm3 mol−1 K and θ = −41.8 K for 2 (see Fig. 3, inset). The negative θ values confirm the antiferromagnetic coupling within the MnII ions and the antiferromagnetic coupling in 2 is stronger than that in 1. Furthermore, the shapes of the M/H plots are quite like that of the antiferromagnet, in which the M values increase rapidly at low fields, with no obvious saturation observed up to 70 kOe (Fig. S11 and 12†).
 | ||
| Fig. 3 Temperature dependence of χm, χmT, and χm−1 (inset) collected in applied field of 1000 Oe for (a) 1 and (b) 2. Red solid line represents best fits. | ||
Water oxidation (WO, 2H2O → O2 + 4H+ + 4e−) is regarded as a key half-reaction for solar fuel production.39 The rational design and synthesis of cheap, efficient and stable water-oxidising catalysts are significant challenges in science and technology.40 In nature, the oxygen-evolving complex (OEC, a CaMn4O5 cluster) in photosystem II (PS II) can efficiently oxidize water.41 It has been shown that the MnIV–O–MnIII–H2O motif plays a crucial role in the activity of the OEC and its mimics.42 Inspired by the OEC, several Mn clusters have been used as structural mimics. In particular, the presence of high oxidation state +3 and +4 Mn ions and four water binding sites have been applied for electrocatalytic oxidation of water, examples such as Mn12O12(OAc)16−xLx(H2O)4 (L = acetate, benzoate, benzenesulfonate, diphenylphosphonate, and dichloroacetate).43–45 However, the catalytic activity of these biomimetic Mn-based clusters for water oxidation was shown to be hindered by either high overpotentials (ranging from 640–820 mV) or low structural stability.40 Kinetically and thermodynamically very stable Mn clusters assembled with exclusively MnII ions may solve one of such problem even though a series of mononuclear manganese complexes [(Py2NR2)MnII(H2O)2]2+ (R = H, Me, tBu) were reported to be active in electrocatalytic water oxidation with an relatively high overpotential of approximately 800 mV (FTO working electrode).34 However, to the best of our knowledge it remains unknown whether polynuclear MnII clusters are capable of being highly active with respect to water oxidation.
This long-standing question has been examined with 1 and 2 using electrochemical techniques. Crystals of 1 and 2 were dissolved in 0.1 M aqueous acetate buffer at pH 6.07 via sonochemistry, the pH at which the OEC within PSII shows optimal catalytic performance.46 The resulting solutions of 1 and 2 were subjected to UV-Vis spectroscopy and showed two broad absorption bands at around λmax = 262 and 315 nm for 1 and λmax = 260 and 312 nm for 2, which can be assigned to the π–π* transition and ligand-to-metal charge transfer transition, respectively (Fig. S13†). The redox peaks associated with manganese of 1 and 2 in aqueous acetate buffer have been detected via cyclic voltammetry (Fig. 4a, b and S14†). These corresponded to the oxidation of Mn2+ to Mn4+ (E = 0.87 V) and the reduction of Mn4+ to Mn3+ (E = 0.83 V), Mn4+ to Mn2+ (E = 0.55 V), and Mn3+ to Mn2+ (E = 0.26 V).47 The solution stability of the coordination structures was investigated using dynamic light scattering (DLS) techniques. It was shown that sonication of these solutions resulted in the formation of species in the size range of 2–3 nm, corresponding to the molecular hydrodynamic diameter of discrete MONCs (Fig. S15†),13 and implying that HSSs converted into discrete MONCs; we envisage that some metal-coordinated hydroxyl groups of PgC3OH moieties on axial positions may be displaced by water molecules. Interestingly, upon evaporation of an aqueous acetate buffer solution of 1 and 2, spherical, micron-scale metallosuperstructures were observed by SEM (Fig. 4c, d and S16†). FT-IR and small angle X-ray scattering studies further supported that they were composed of many MONC subunits (Fig. S17 and 18†). We propose that the hierarchical metal–organic micron spheriods (MOMSs) may be stabilized by a large number of van der Waals interactions between neighboring alkyl chains and hydrophilic regions of the discrete MONCs.
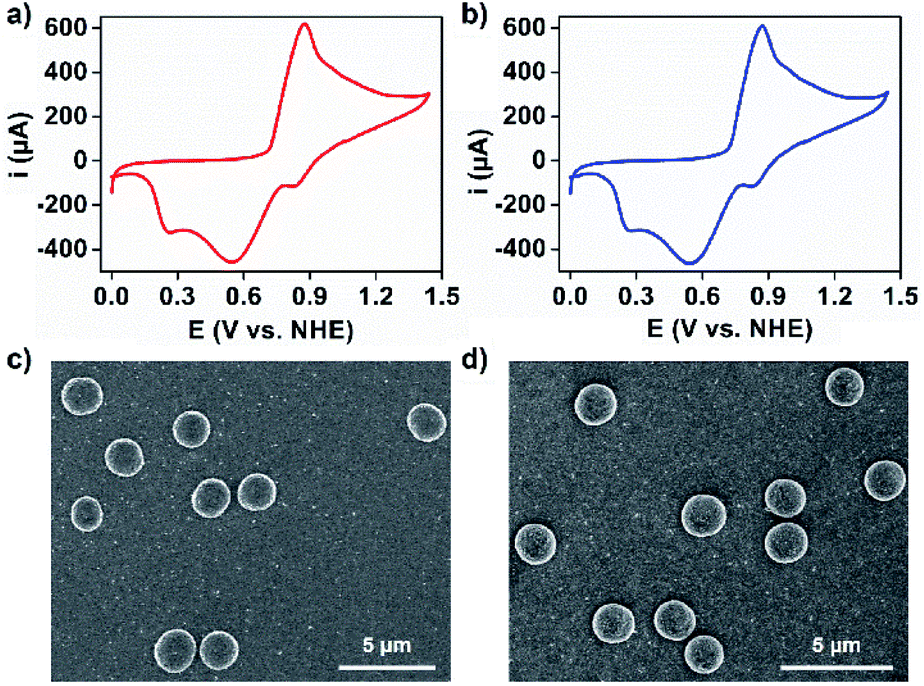 | ||
| Fig. 4 Cyclic voltammograms (CVs) of (a) 1 and (b) 2 (0.5 mM) in 0.1 M acetate buffer at pH 6.07 using an FTO (S = 1 cm2) working electrode. Scan rate is 50 mV s−1. SEM images of hierarchical micron spheroids formed from an aqueous acetate buffer of (c) 1 and (d) 2. | ||
Furthermore, cyclic voltammograms (CVs) clearly indicated that water oxidation can be catalyzed by both 1 and 2 (Fig. 5).43,47 Water oxidation occurs at an exceptionally low overpotential of only 368 mV. This is higher than that of the current state-of-art Ru-bda complex (bda = 2,2′-bipyridine-6,6′-dicarboxylate, 180 mV at pH 7), illustrating that there is still room for further improvements.48 Continuous CV scan experiments and bulk electrolysis of 1 and 2 demonstrated that these electrocatalysts have high catalytic activity and stability toward water oxidation (Fig. S19 and 20†). UV-Vis and DLS measurements taken after electrolysis of 1 and 2 showed that the waves and particle size are retained (Fig. S21 and 22†). Moreover, the MOMSs re-formed and could be detected upon evaporation of the catalyst solution in subsequent SEM studies (Fig. S23†). Collectively, these measurements suggest that the MONC subunit is a homogeneous water oxidation electrocatalyst. This result may thus provide a new strategy for the design and synthesis of cheap, efficient and stable water-oxidizing catalysts since it first suggests that soluble MnII clusters may be used to effectively facilitate the oxidation of water, despite the enormous efforts made to mimic the CaMn4O5 cluster to date. We further envision that improvements of catalyst stability and activity may be possible. This may be achieved through (for example) attaching appropriate axial ligands to the constituent metal ions, or functionalizing alkyl chains on the MONC surface. In addition, other soluble metal-seamed dimeric or hexameric MONCs, such as those formed with FeII, CoII and CuII ions, are also promising with regard to electrocatalytic water oxidation.36
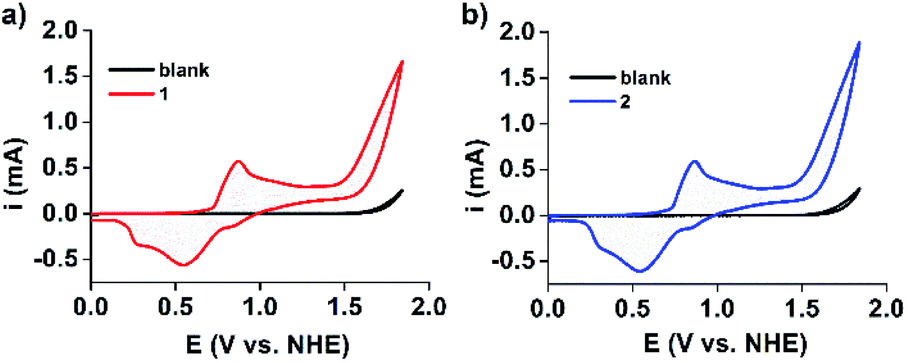 | ||
| Fig. 5 CV scans of (a) 1 and (b) 2 (0.5 mM, 50 mV s−1 scan rate) in 0.1 M acetate buffer at pH 6.07. For comparison, CVs of the blank buffer are also shown. FTO (S = 1 cm2) was used as the working electrode. | ||
Conclusions
In summary, we have developed a new strategy for the rational construction of HSSs using biomimetic self-assembly as the synthetic methodology. Akin to the self-assembly behaviours of protein subunits with redox-active metal ions, the assembly of these sophisticated supramolecular architectures has been accomplished by employing proline molecules as molecular chaperones to selectively insert redox-active MnII ions into the coordination sites of a pre-assembled ONC skeleton, which further directs the MnII-seamed MONC subunits to fold and assemble into more complex HSSs across different dimensionality. This is achieved via control of both interior and/or exterior surface properties of the MONC subunits, through coordination and host–guest chemistries, also allowing for the fine-tuning of magnetic properties. The catalytic activity and stability of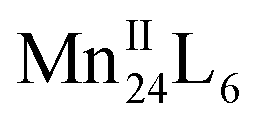 MONCs toward homogeneous water oxidation at pH 6.07 with an exceptionally low overpotential of only 368 mV is noteworthy.
MONCs toward homogeneous water oxidation at pH 6.07 with an exceptionally low overpotential of only 368 mV is noteworthy.
Overall, this approach represents an important advancement in supramolecular chemistry by design. Further efforts will be invested in the design and synthesis of extremely challenging and complex HSSs with other redox-active or coordinatively inert metal ions (e.g. CrII/CrIII and FeII/FeIII), as well as inserting suitably functionalized guest molecules for potential application in the areas of molecular electronics/magnets and catalysis, all of which may be modulated by appropriate molecular chaperones. Finally, this strategy may be widely exploited in the rational design and synthesis of other metal–organic systems, such as metal–organic cages (MOCs), polyhedra (MOPs) or artificial metalloproteins, the properties and/or functions of which can be subsequently tailored accordingly.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank University of Missouri for financial and research facility support of this work. We thank the U.S. National Science Foundation for support of this work (1825352). This work was also partially supported by the Natural Science Foundation of Shandong Province (ZR2018BB035, 2019GSF109013), Shanxi Province (201701D121039).Notes and references
- T. Zhou, D. H. Hendrickson, Q. J. Sattentau and P. D. Kwong, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14575–14580 CrossRef CAS.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS.
- R. A. Bilbeisi, T. K. Roson and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 9027–9030 CrossRef CAS.
- J. L. Atwood, L. J. Barbour, S. J. Dalgarno, M. J. Hardie, C. L. Raston and H. R. Webb, J. Am. Chem. Soc., 2004, 126, 13170–13171 CrossRef CAS.
- T. R. Cook, Y. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS.
- Z. Niu, S. Feng, X. Liu, J. Ma, S. Ma and P. Cheng, J. Am. Chem. Soc., 2015, 137, 14873–14876 CrossRef CAS.
- J. J. Perry IV, J. A. Perma and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- T.-F. Liu, P.-Y. Chen, A. A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 17358–17361 CrossRef CAS.
- H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650–11661 CrossRef CAS.
- D. Zhang, T. K. Roson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS.
- H. Kumari, C. A. Deakyne and J. L. Atwood, Acc. Chem. Res., 2014, 47, 3080–3088 CrossRef CAS.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- M. Yoshizawa and L. Catti, Acc. Chem. Res., 2019, 52, 2392–2404 CrossRef CAS.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS.
- Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349–358 CrossRef CAS.
- Y. Yu and J. Rebek, Acc. Chem. Res., 2018, 51, 3031–3040 CrossRef CAS.
- H. Sepehrpour, X. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS.
- Y. Fang, X. Lian, Y. Huang, G. Fu, Z. Xiao, Q. Wang, B. Nan, J.-P. Pellois and H.-C. Zhou, Small, 2018, 14, 1802709 CrossRef.
- Q. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS.
- P. F. Damasceno, M. Engel and S. C. Glotzer, Science, 2012, 337, 453–457 CrossRef CAS.
- L. Peng, N. A. Vermeulen, C. D. Malliakas, D. A. Gomez-Gualdron, A. J. Howarth, B. L. Mehdi, A. Dohnalkova, N. D. Browning, M. O'Keeffe and O. K. Farha, Science, 2017, 356, 624–627 CrossRef.
- L. A. Finney and T. V. O'Halloran, Science, 2003, 300, 931–936 CrossRef CAS.
- A. C. Rosenzweig, Chem. Biol., 2002, 9, 673–677 CrossRef CAS.
- J. W. Whittaker, Biochim. Biophys. Acta, 2010, 2, 298–307 CrossRef.
- K. Su, M. Wu, D. Yuan and M. Hong, Nat. Commun., 2018, 9, 1–6 CrossRef CAS.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- D. A. Fowler, A. S. Rathnayake, S. Kennedy, H. Kumari, C. M. Beavers, S. J. Teat and J. L. Atwood, J. Am. Chem. Soc., 2013, 135, 12184–12187 CrossRef CAS.
- C. Zhang, R. S. Patil, C. Liu, C. L. Barnes, T. Li and J. L. Atwood, J. Am. Chem. Soc., 2017, 139, 2920–2923 CrossRef CAS.
- A. S. Rathnayake, H. W. L. Fraser, E. K. Brechin, S. J. Dalgarno, J. E. Baumeister, J. White, P. Rungthanaphatsophon, J. R. Walensky, C. L. Barnes, S. J. Teat and J. L. Atwood, Nat. Commun., 2018, 9, 1–6 CrossRef CAS.
- A. S. Rathnayake, H. W. L. Fraser, E. K. Brechin, S. J. Dalgarno, J. E. Baumeister, J. White, P. Rungthanaphatsophon, J. R. Walensky, S. P. Kelley, C. L. Barnes and J. L. Atwood, J. Am. Chem. Soc., 2018, 140, 15611–15615 CrossRef CAS.
- X. Hu, J. Chai, C. Zhang, J. Lang, S. P. Kelley, S. Feng, B. Liu, D. A. Atwood and J. L. Atwood, J. Am. Chem. Soc., 2019, 141, 9151–9154 CrossRef.
- G. Karotsis, S. J. Teat, W. Wernsdorfer, S. Pilligkos, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 8285–8288 CrossRef CAS.
- P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40, 3249–3265 RSC.
- W.-T. Lee, S. B. Munoz, D. A. Dickie and J. M. Smith, Angew. Chem., Int. Ed., 2014, 53, 9856–9859 CrossRef CAS.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS.
- J. R. Calhoun, F. Nastri, O. Maglio, V. Pavone, A. Lombardi and W. F. DeGrado, J. Pept. Sci., 2005, 80, 264–278 CrossRef CAS.
- K. J. Young, B. J. Brenan, R. Tagore and G. W. Brudvig, Acc. Chem. Res., 2015, 48, 567–574 CrossRef CAS.
- A. I. Nguyen, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11630–11639 CAS.
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS.
- A. K. Poulsen, A. Rompel and C. J. McKenzie, Angew. Chem., Int. Ed., 2005, 44, 6916–6920 CrossRef CAS.
- Y. Mousazade, M. R. Mohammadi, P. Chernev, R. Bikas, R. Bagheri, Z. Song, T. Lis, H. Dau and M. M. Najafpour, Catal. Sci. Technol., 2018, 8, 4390–4398 RSC.
- Y. Yan, L. S. Lee and A. A. Ruddy, Inorg. Chem., 2015, 54, 4550–4555 CrossRef CAS.
- G. Maayan, N. Gluz and G. Christou, Nat. Catal., 2018, 1, 48–54 CrossRef CAS.
- H. Schiller and H. Dau, J. Photochem. Photobiol., B, 2000, 55, 138–144 CrossRef CAS.
- Y. Du, Z. Fu and L. Bi, Dalton Trans., 2018, 47, 7282–7289 RSC.
- B. Zhang and Li. Sun, J. Am. Chem. Soc., 2019, 141, 5565–5580 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1981690 and 1981691. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05002d |
| This journal is © The Royal Society of Chemistry 2020 |