An overview on the recent developments of Ag-based electrodes in the electrochemical reduction of CO2 to CO
Received
3rd August 2019
, Accepted 15th September 2019
First published on 16th September 2019
Abstract
Conversion of aqueous CO2 into CO through the electrochemical reduction reaction (ERR) suffers from poor selectivity and low current density with the classic Ag-based catalysts due to the inert nature of CO2 and the low activity of the active sites. Recently, it has been reported that nanostructured catalysts can display very distinctive behavior compared to their bulk counterparts. To explore the reasons behind this, the latest developments in Ag-based nanostructured materials in the CO2 ERR are summarized into six aspects in this review: (1) the size effect, (2) the morphology effect, (3) the porous effect, (4) the alloying effect, (5) the support effect, and (6) the surface modification effect. In particular, Ag-based alloy catalysts could exhibit a superior catalytic activity and long-term stability owing to the synergistic effect. Besides, the morphology of Ag-based alloy catalysts could also be controlled through adjusting their compositions. From the current literature survey we find that the maximum FE for CO obtained from Ag-based nanocatalysts reaches as high as 99.3%, which is a breakthrough over that from the traditional Ag foil. This review aims to offer a comprehensive understanding of Ag-based nanostructured catalysts in the CO2 ERR and consequently improve the catalyst design for active and selective electro-reduction of CO2 to CO.
 Samah A. Mahyoub | Samah A. Mahyoub is a Ph.D. candidate under the supervision of Dr Prof. Zhenmin Cheng in the School of Chemical Engineering at the East China University of Science and Technology. She obtained her Master's degree from the School of Chemical Engineering at Tianjin University in 2017. Her research is focused on CO2 electrochemical reduction. |
 Fahim A. Qaraah | Fahim A. Qaraah is a Ph.D. candidate under the supervision of Dr Prof. Guangli Xiu in the School of Environmental Science and Engineering at the East China University of Science and Technology. He obtained his Master's degree from the School of Chemical Engineering at Tianjin University in 2018. His research is focused on nanomaterials synthesis and their applications. |
 Chengzhen Chen | Chengzhen Chen received his Ph.D. degree under the supervision of Prof. Zhenmin Cheng in the Department of Chemical Engineering at the East China University of Science and Technology. His research is focused on catalyst materials synthesis and CO2 electrochemical reduction. |
 Fanghua Zhang | Fanghua Zhang is a Ph.D. candidate under the supervision of Prof. Zhenmin Cheng in the Department of Chemical Engineering at the East China University of Science and Technology. He obtained his Master's degree from the School of Chemical Engineering at the Ocean University of China in 2015. His research is focused on novel electrochemical reactor development for CO2 reduction. |
 Shenglin Yan | Shenglin Yan is a Ph.D. candidate under the supervision of Prof. Zhenmin Cheng in the School of Chemical Engineering at the East China University of Science and Technology. He received his B.S. degree from the College of Chemistry & Environmental Engineering, Yangtze University, in 2017. His research is highly focused on the synthesis of various metal nanostructures for CO2 electroreduction. |
 Zhenmin Cheng | Zhenmin Cheng has been a Professor of Chemical Engineering at the East China University of Science and Technology since 1999 and he received his Ph.D. degree from the same university in 1993. He was a visiting scholar in the Laboratoire des Sciences du Genie Chimique in France and the University of Queensland in 2001 to 2003. So far, he has published over 130 academic papers in SCI-indexed journals. His research covers multiphase chemical and electrochemical reaction engineering, transport process simulation, and interfacial engineering. |
1. Introduction
CO2 reduction by electrochemical catalysis has attracted considerable attention because of the following unique advantages: (1) CO2 is reduced by electrons instead of hydrogen; (2) the electrocatalytic process occurs under mild conditions; (3) the reduction products can be adjusted using the redox potential, reaction temperature, electrolyte, etc.; and (4) by improving the electrocatalysts, side products can be minimized.1–8 Electrochemical CO2 reduction could be achieved through multiple electron transfer in an aqueous solution with appropriate electrocatalysts to produce different products (Table 1) such as carbon monoxide (CO), methane (CH4), ethylene (C2H4), and even liquid products, for example, formic acid (HCOOH), methanol (CH3OH) and ethanol (C2H5OH).6,9 CO, as one of the valuable gases produced by CO2 electroreduction, can be efficiently separated from other reaction products for the production of a wide variety of chemicals such as acetic acid,10 phosgene,11 and aldehydes.12 Moreover, the mixture of H2 and CO (“syngas”) can be used to produce higher hydrocarbons to be used as vehicle fuels via the Fischer–Tropsch process.13,14 To date, Au, Ag, Zn, Pd, and Ga have been revealed to be active in the catalysis of the selective conversion of CO2 to CO.15–21 In particular, Ag has received the most particular consideration due to its excellent catalytic activity and moderate price. Consequently, the synthesis and characterization of Ag catalysts have been explored extensively.15,22–26 This review thus aims to summarize the recent progress in the CO2 electrochemical reduction reaction (CO2 ERR) to CO using nanostructured Ag catalysts.
Table 1 CO2 electrolysis products at different standard redox potentials
| Reaction |
E (V) vs. SHE |
| 2H+ + 2e− → H2 |
−0.41 |
| CO2 + 2H+ + 2e− → HCOOH |
−0.61 |
| CO2 + 2H+ + 2e− → CO + H2O |
−0.53 |
| CO2 + 4H+ + 4e− → C + 2H2O |
−0.20 |
| CO2 + 4H+ + 4e− → HCHO + H2O |
−0.48 |
| CO2 + 6H+ + 6e− → CH3OH + H2O |
−0.38 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O |
−0.24 |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O |
0.064 |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O |
0.084 |
2. Ag-based nanostructured electrode materials
In a typical CO2 electrolyzer, the anode and cathode are set in two chambers isolated with an ion-conducting membrane. At the anode, water is oxidized to molecular oxygen, whereas CO2 is reduced at the cathode, which is a solid electrocatalyst, while the electrolyte is normally an aqueous solution saturated with CO2 by bubbling. The CO2 ERR procedure on a catalyst surface involves three main steps: (1) CO2 adsorption and interaction with atoms on the catalyst surface; (2) CO2 activation and reduction through catalyst-initiated electron/proton transfer; and (3) product desorption and recovery of the catalyst surface for the next round of the reaction.8,27 Conversion of CO2 into CO over an Ag-based electrocatalyst in an aqueous electrolyte is accomplished through the following steps, as shown in eqn (1)–(4):28| | 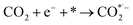 | (1) |
| | 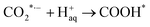 | (2) |
| | | COOH* + H+aq + e− → CO* + H2O | (3) |
where * represents the vacant catalytically active site. In the above reaction process, one electron transfers to CO2 in order to produce 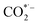 , which is considered to be the rate-controlling step for most transition metal-based catalysts owing to the high energy barrier required to start the process.8,27,29 Theoretically, the standard reduction potential for reducing CO2 in aqueous solution (i.e., pH 7, 1 M electrolyte, 25 °C, 1 atm) into CO is in the range of −0.2 V to 0.2 V (vs. the reversible hydrogen electrode (RHE)).22 The reason for such a high overpotential is often attributed to the high equilibrium potential associated with the formation of the radical intermediate
, which is considered to be the rate-controlling step for most transition metal-based catalysts owing to the high energy barrier required to start the process.8,27,29 Theoretically, the standard reduction potential for reducing CO2 in aqueous solution (i.e., pH 7, 1 M electrolyte, 25 °C, 1 atm) into CO is in the range of −0.2 V to 0.2 V (vs. the reversible hydrogen electrode (RHE)).22 The reason for such a high overpotential is often attributed to the high equilibrium potential associated with the formation of the radical intermediate 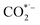 .30,31 Such a circumstance becomes serious in an aqueous electrolyte-based CO2 electrolyzer because the side reaction of hydrogen evolution also takes place at a similar potential (Table 1) and competes with CO2 reduction at the cathode surface. Thus, an efficient electrocatalyst should not only accelerate the CO2 ERR with significant productivity and selectivity but also suppress the competing HER.32
.30,31 Such a circumstance becomes serious in an aqueous electrolyte-based CO2 electrolyzer because the side reaction of hydrogen evolution also takes place at a similar potential (Table 1) and competes with CO2 reduction at the cathode surface. Thus, an efficient electrocatalyst should not only accelerate the CO2 ERR with significant productivity and selectivity but also suppress the competing HER.32
2.1 The size effect
A direct strategy to increase the number of active sites in the CO2 ERR is to use nanoparticles (NPs) as the catalyst. By applying crystal-growth theory to metal nanocrystals, particles with different sizes could be manufactured. The size of the particle directly affects the ratio of the atoms at the corner, edge, and surface positions. Atoms at the corner or edge positions have a low coordination number and high surface energy. Accordingly, NPs with a small size can exhibit remarkable performance in CO2 reduction.8,33,34 Consequently, it is, in principle, promising to adjust NP catalysts by controlling their surface structure and size. For example, the conversion rate of CO2 into CO was 10 times higher on 5 nm Ag NPS than on a bulk silver electrode in anionic liquid EMIM-BF4 solution.35 To understand the size effect of Ag nanocatalysts, Hwang et al. reported a facile one-pot synthesis of Ag NPS supported over carbon via employing a cysteamine anchoring agent to control the monodispersed particle sizes, as represented in Fig. 1.34 Among 3, 5 and 10 nm Ag NPs, the 5 nm Ag/C catalyst exhibits the best CO2 reduction reaction performance due to the smallest interaction between cysteamine and the COOH* intermediate.
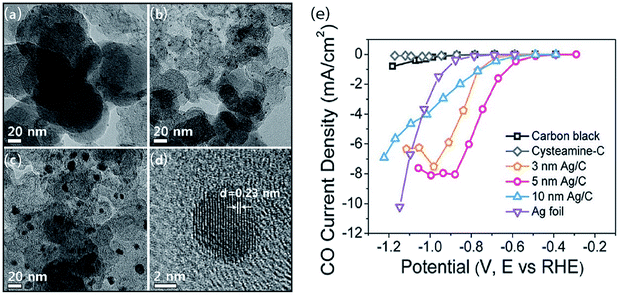 |
| | Fig. 1 TEM images of (a) 3 nm, (b) 5 nm and (c) 10 nm-sized Ag NPs/C. (d) HR-TEM image of 5 nm Ag/C and (e) CO partial current densities of the three typical Ag/C catalysts with different NP sizes and different potentials.34 Copyright 2015 American Chemical Society. | |
In another study, Zhang et al.36 synthesized a porous silver catalyst via electrochemical growth/reduction of an AgCl film on a silver substrate. The as-prepared catalyst comprised 30–50 nm particles and a considerable electrochemical surface area, displaying a size effect on the reduction of CO2 to CO.
2.2 The morphology effect
Besides the effects of size and local electric field, the morphological effect of metal NPs on the CO2 ERR is another factor in increasing the activity of Ag for CO2 electrochemical reduction to CO. Previous studies on single crystal Ag electrodes showed that CO2 conversion to CO is favorable over Ag (110) compared to Ag (111) or Ag (100).37 Density functional theory (DFT) simulation as represented in Fig. 2 shows that the Ag (110) or Ag (211) surface has a lower energy barrier for the first proton-coupled electron transfer for COOH˙ stabilization than Ag (111) or Ag (100), resulting in a better catalytic activity for the reduction of CO2 to CO on Ag (110) or Ag (211).25
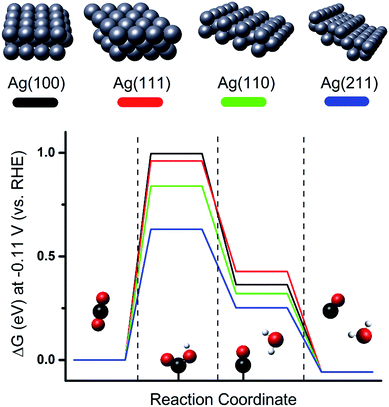 |
| | Fig. 2 Free energy diagrams for the electrocatalytic conversion of CO2 into CO on flat (Ag (100) and Ag (111)) and edge (Ag (221) and Ag (110)) surfaces at −0.11 V [RHE]. Proton-coupled electron transfer is involved in the first two reaction steps. Sphere colors: white, H; black, C; red, O; silver, Ag.25 Copyright 2015 American Chemical Society. | |
It is worth noting that preferentially oriented Ag nanoparticles were capable of attaining a very high faradaic efficiency of 96.7% at a low overpotential of 0.58 V, along with a very high areal CO partial current density of 4.4 mA cm−2. Since the preferentially oriented Ag nanoparticles are self-supported and produced in situ under the reaction conditions, it is likely to be possible to exclude utilization of ionic polymers for electrode manufacturing in the future, which would decrease the cost and difficulty for commercial application.38
Hsieh et al.23 synthesized a high-surface-area Ag nanocoral catalyst via an oxidation–reduction process in the presence of chloride anions in an aqueous medium. In the CO2 ERR to CO, the Ag nanocoral catalyst achieved a current efficiency of 95% at a potential below −0.6 V and a current density of 2 mA cm−2.
In contrast to Ag foil, the Ag nanocoral catalyst brought about a 32-fold improvement in surface-area normalized activity, at an overpotential of 0.49 V. They observed that the adsorbed chloride anions played a crucial role in the improved activity and selectivity of the Ag nanocoral electrocatalyst for CO2 reduction. Liu et al. prepared triangular silver nano-plate (Tri-Ag-NP) catalysts through a direct chemical reduction process for a highly active CO2 reduction reaction to CO. The as-prepared Tri-Ag-NPs showed a significantly improved faradaic efficiency (96.8%) and energy efficiency (61.7%), together with a substantial durability (7 days) compared to similarly sized Ag nanoparticles (SS-Ag-NPs) and bulk Ag. DFT simulation indicated that the considerably improved electrocatalytic activity and selectivity at decreased overpotentials are a result of both the optimum edge-to-corner ratio and the prevalent Ag (100) facet in Tri-Ag-NPs, which require less energy to initiate the rate-limiting step (Fig. 3).39
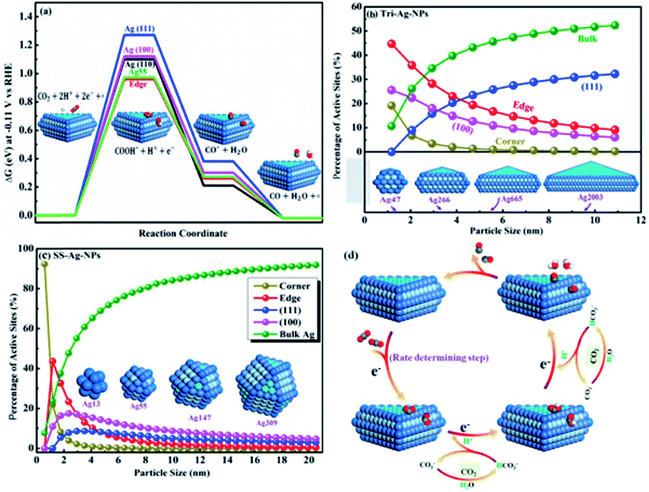 |
| | Fig. 3 (a) Free energy diagrams for CO2 to CO on different facets and the Ag55 cluster at −0.11 V; active adsorption site density on (b) Tri-Ag-NPs and (c) SS-Ag-NPs as a function of particle size; (d) proposed mechanism of the CO2 ERR to CO on Tri-Ag-NPs.39 Copyright 2017 American Chemical Society. | |
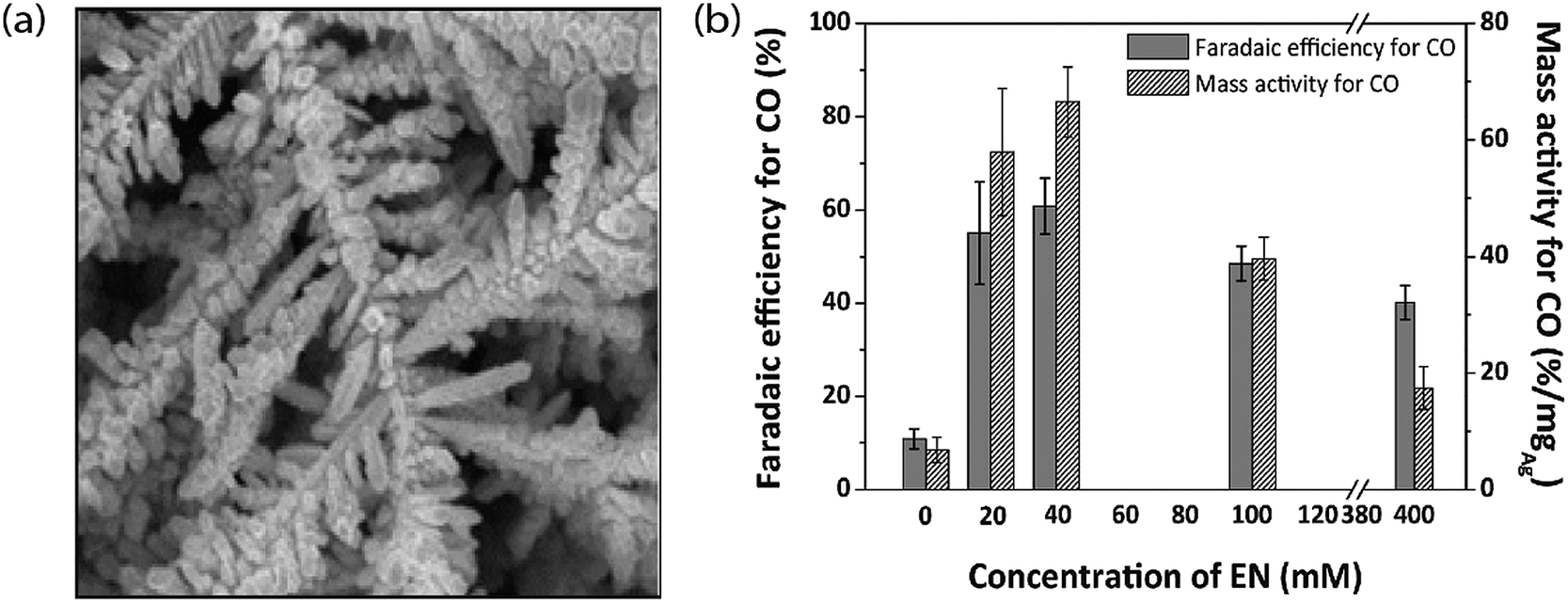
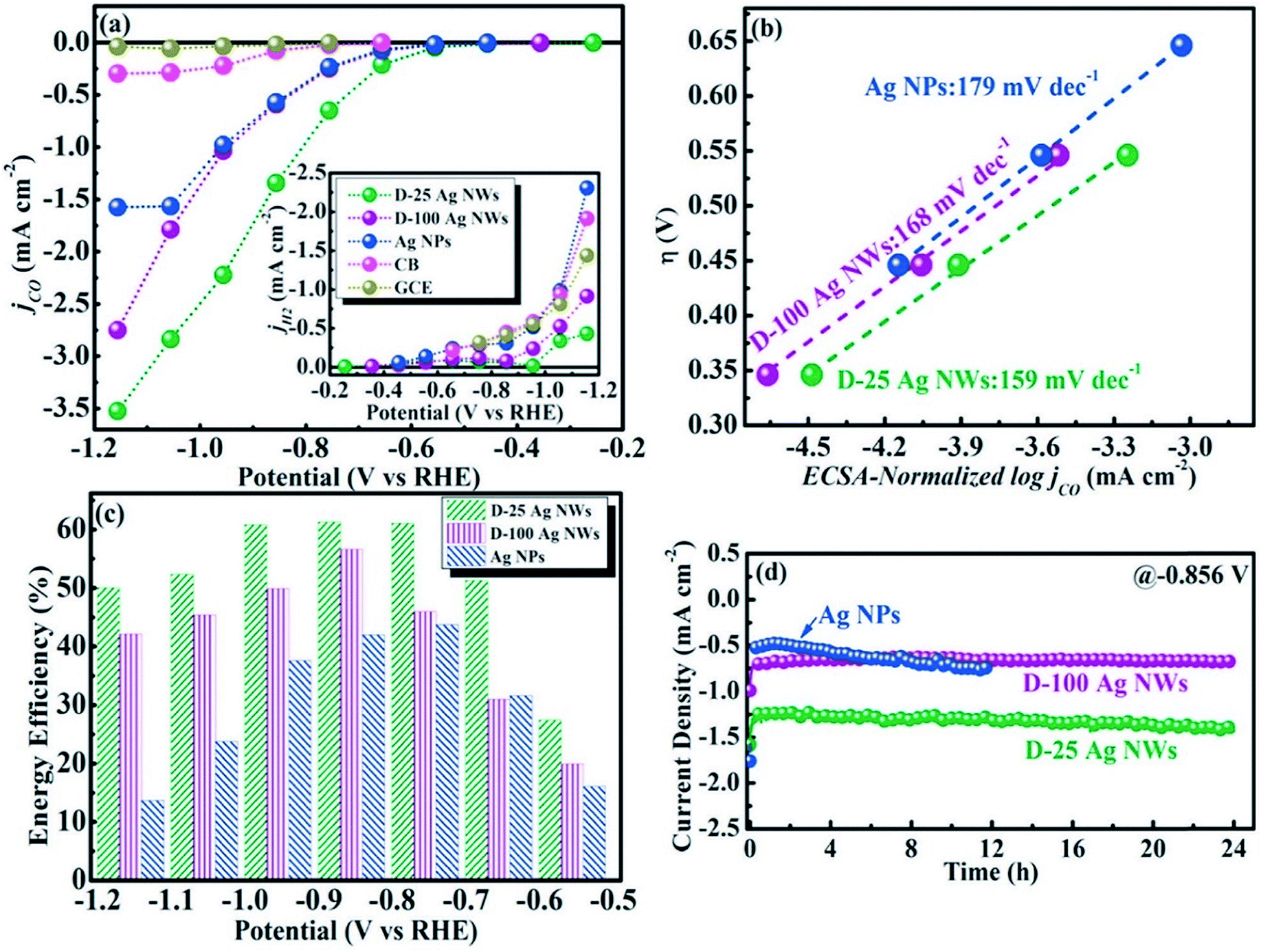
To understand the relation between the crystallographic structures of Ag catalysts and the CO formation efficiency, a dendrite-type Ag catalyst was synthesized through an electrodeposition process in an ammonia-based Ag deposition solution comprising ethylenediamine (EN) as the additive. The effect of electrodeposition variables on the features of this catalyst was established and linked with CO formation efficiency. The results showed that the addition of EN during Ag electrodeposition altered the diffraction peak intensity ratio of the (111) to (220) planes, along with their electronic states, which was also found to be related to the CO formation activity. The as-prepared catalysts exhibited high faradaic efficiency and activity for CO through CO2 electrolysis in 0.5 M KHCO3 under optimized deposition conditions (−0.45 V vs. Ag/AgCl, 40 mM EN) (Fig. 4).40 The one dimensional Ag nanowire (NW) is another specialized geometry that can have a large portion of specific facets exposed.41–43 5-fold twinned Ag nanowires (NWs) with diameters less than 25 nm (D-25) and 100 nm (D-100) were obtained using the bromide-mediated polyol technique. In contrast to the D-100 Ag NWs and Ag NPs, the D-25 Ag NWs were electrochemically more active in the CO2 ERR with higher selectivity and superior stability. Furthermore, a comparable low onset η in CO2 ERR initiation, together with a good stability for more than 24 hours, was accomplished, representing the outstanding performance of the D-25 Ag NWs in the CO2 ERR. DFT calculations show that the greater catalytic activity of the twinned Ag NWs mostly originates from the effects of diameter and length, in addition to the fully enclosed nanostructures (100) and the increased edge-to-corner ratio (Fig. 5).44 To exploit the CO2 ERR activity on an active crystal facet, ultrathin 2D nanosheets were synthesized and established as a new form of the Ag catalyst. The hierarchical structure of Ag nanosheets afforded an enriched surface area and promising gas transport/diffusion and exhibited improved FE (95%) at an overpotential as low as 0.29 V. The author thought that residual Cl− and a noteworthy number of grain boundaries might participate in the extremely efficient reduction of CO2 to CO using this material.45
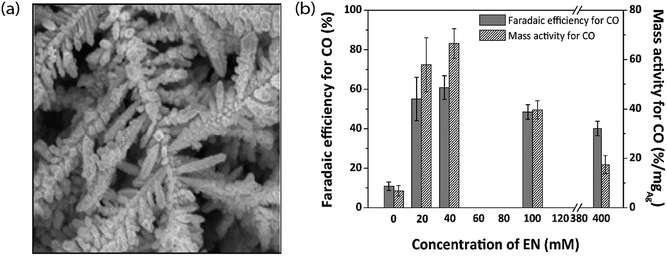 |
| | Fig. 4 (a) FESEM image of AgEN40 fabricated by electrodeposition at −0.45 V and (b) FE for CO and its conversion to mass activity for Ag catalysts fabricated at −0.45 V with various EN concentrations.40 Copyright 2017 Elsevier B.V. | |
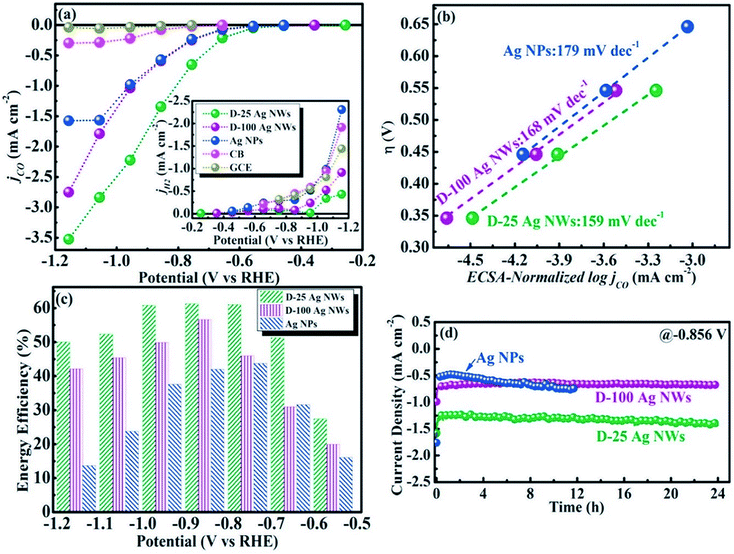 |
| | Fig. 5 (a) CO and H2 partial current densities of different catalysts; (b) Tafel plots of Ag NPs, D-25, and D-100 Ag NWs; (c) EFs of Ag NPs, D-25 and D-100 Ag NWs at various potentials; (d) long-term stabilities of Ag NPs, D-25 and D-100 Ag NWs at a potential of −0.9 V.44 Copyright 2018 Elsevier. | |
2.3 The nanoporous effect
In addition to the well-designed forms of the catalyst, nanostructures with porous or two-dimensional (2D) morphologies have also been developed to increase the atom utilization efficiency of the catalyst. For example, a monolithic nanoporous-Ag (np-Ag) catalyst is produced by a two-step dealloying of an Ag–Al precursor with an aqueous HCl solution. The as-prepared catalyst has a considerably large surface area (around 150 times larger) and high activity (about 20 times higher) compared to polycrystalline silver in CO2 electroreduction. The improved activity is due to the superior stabilization of COOH* intermediates on the extremely curved surfaces, which leads to a smaller overpotential for overcoming the kinetic barrier for producing CO.15
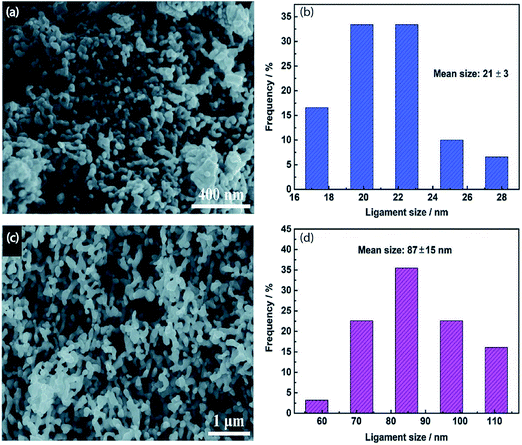 |
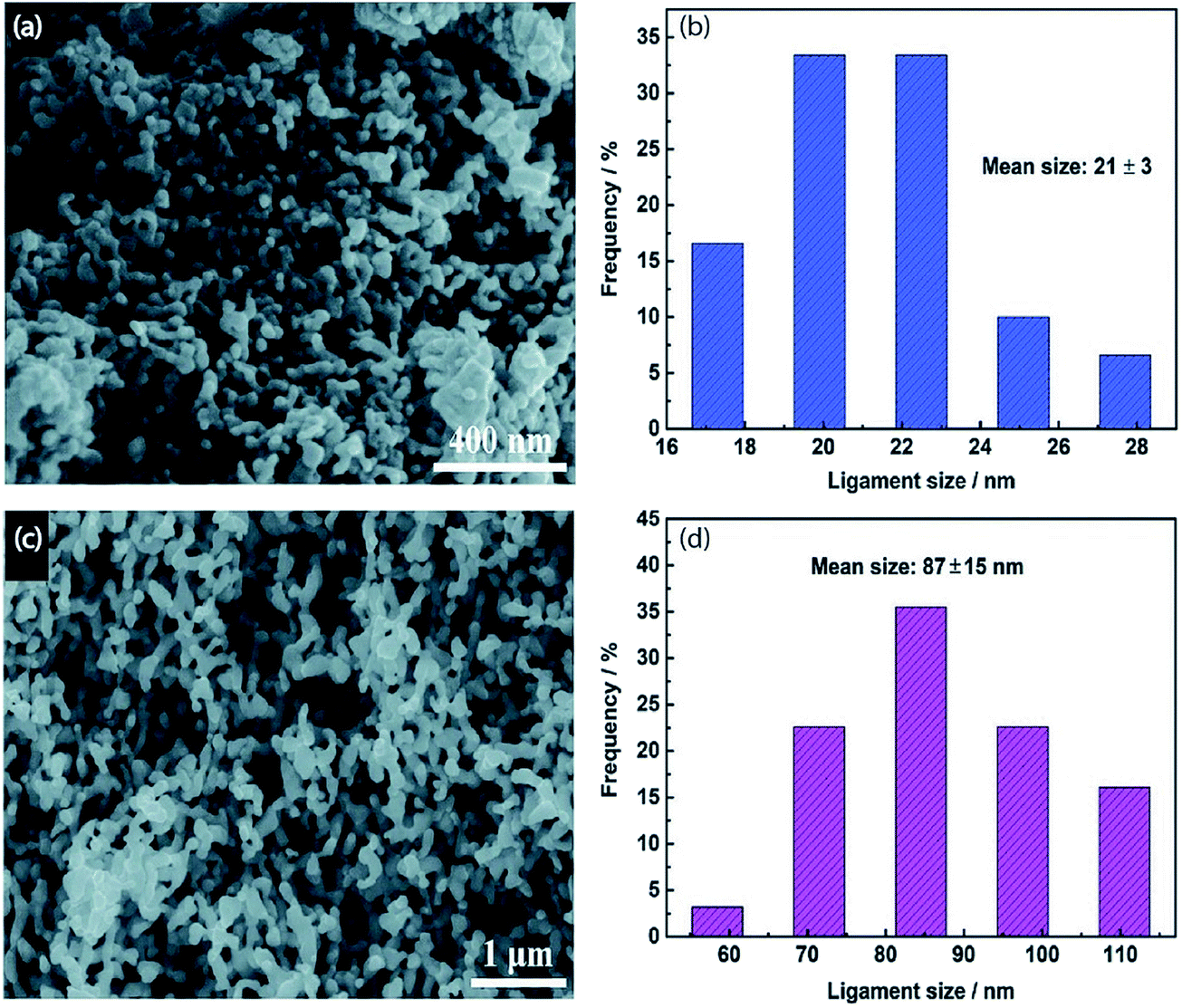
| | Fig. 6 (a) SEM image and (b) ligament size distribution of np-Ag (21 nm). (c) SEM image and (d) ligament size distribution of np-Ag (87 nm).46 Copyright 2018 Royal Society of Chemistry. | |
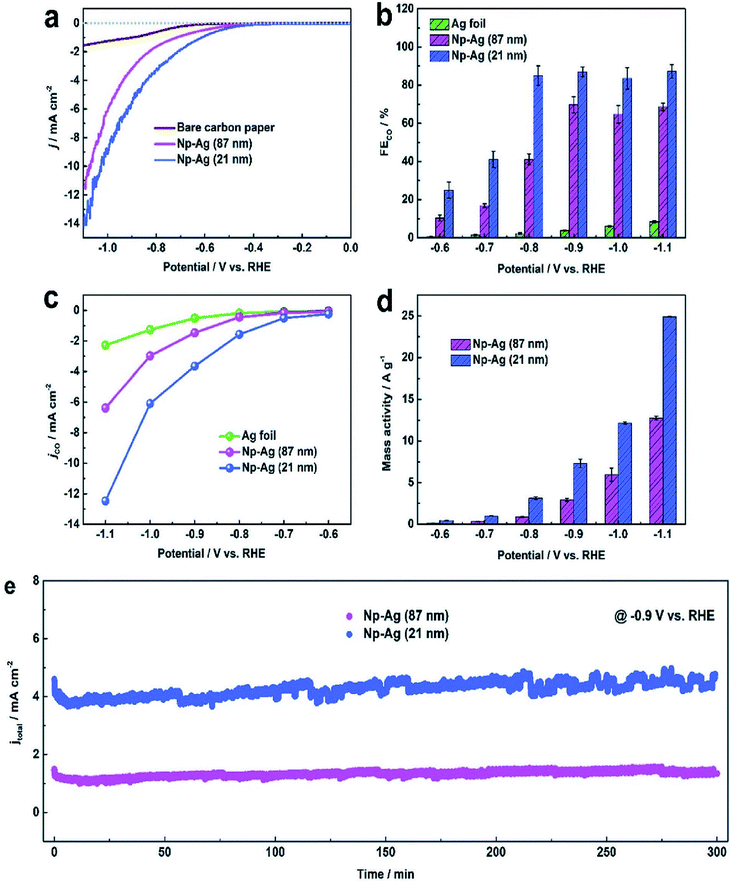 |
| | Fig. 7 (a) Cathodic LSV curves of bare carbon paper, Np-Ag (87 nm) and Np-Ag (21 nm). (b) FEs of CO, (c) CO partial current density, and (d) mass activity at different potentials of Ag foil, Np-Ag (87 nm) and Np-Ag (21 nm). (e) Stability of Np-Ag (87 nm) and np-Ag (21 nm) at −0.9 V vs. RHE.46 Copyright 2018 Royal Society of Chemistry. | |
Nanoporous Ag network catalysts (Np-Ag) with an average ligament size of 21 nm Ag (np-Ag 21) and 87 nm (np-Ag 87) were synthesized by dealloying of the precursor Mg80Ag20 (wt%) alloy ribbon with 1 wt% citric acid and 5 wt% phosphoric acid, separately. The results showed that the Np-Ag (21 nm) catalyst exhibited a considerably improved selectivity with a faradaic efficiency for CO formation of 85.0% at −0.8 V [RHE], about two times that (41.2%) of Np-Ag 87. The superior catalytic activity of Np-Ag 21 can be ascribed to the enriched electrochemical surface area and higher local pH resulting from the ligament size effect, in addition to more defect sites (i.e., grain boundaries) in ligaments (Fig. 6 and 7).46 In another study, an extremely active, selective, and stable Ag electrocatalyst for the reduction of CO2 to CO was fabricated through an easy and rapid anodic-etching process. The as-prepared catalyst showed a high FE > 92% at −0.4 V vs. RHE and stability for more than 100 h. The enhanced CO2 reduction achievement could be ascribed to the increased number of active sites and the better intrinsic CO2 reduction activity by fast initial electron transfer.47 Dutta et al.48 reported an additive-assisted metal-foam deposition method for preparing novel Ag-foam for CO2 electrochemical reduction. The as-prepared catalyst with a thickness of 17 μm in the Ag film shows that the pore sidewalls are composed of highly anisotropic, needle-shaped Ag on different length scales Fig. 8(e and f). The electrocatalytic CO2 reduction results showed remarkable activity in aqueous solution. For example, under low and moderate overpotentials these Ag foam samples offer a high activity and selectivity to CO production with a maximum Faraday efficiency of 90% in the potential range of −0.3 to −1.2 V versus the reversible hydrogen electrode (RHE). Most interesting is the ability of the novel Ag foam catalyst to produce hydrocarbons in noteworthy quantities, showing CH4 and C2H4 efficiencies of FECH4 = 51% and FEC2H4 = 8.6%, respectively, at <−1.1 V versus RHE. This notable Cu-like activity was justified in expression of the CO* binding energy, which is considerably improved on the novel Ag foam compared to the polycrystalline Ag-foil as the reference. Nonetheless, degradation is observed to be more rapid in the EC-CO2RR extended at −1.5 V versus RHE. FECH4 values were reduced to 32% within 5 hours of electrolysis. Likewise, the HR-SEM examination demonstrates morphological changes, mostly at higher applied over-potentials (−1.5 V) (Fig. 9).
 |
| | Fig. 8 Morphology of the Ag foam. (a) Side-view SEM image of the Ag foam. (b–d) Top-down SEM images showing the primary macroporosity of the Ag foam. (e and f) Top-down SEM images showing the secondary mesoporosity of the Ag foam.48 Copyright 2018 American Chemical Society. | |
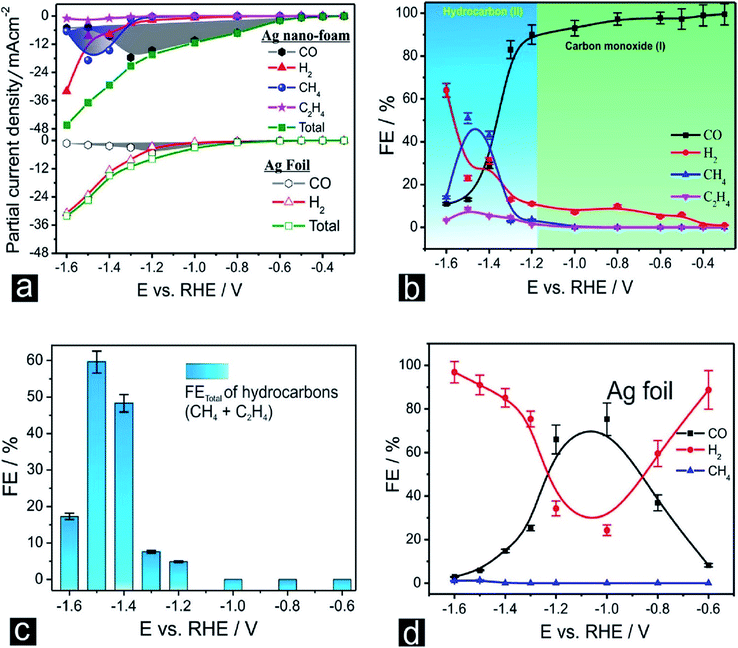 |
| | Fig. 9 (a) Potential (E)-dependent product distribution of the CO2 ERR displayed as partial current densities normalized to the geometric surface area (multi-catalyst approach, 1 h of potentiostatic CO2 ERR carried out at each potential applied using a freshly prepared Ag foam catalyst). (b) Corresponding plot of the faradaic efficiencies (FEs) as a function of the applied electrolysis potential (E). (c) Potential (E)-dependent FE plot with respect to the total hydrocarbon formation. (d) Potential (E)-dependent product distribution of the EC-CO2RR on an Ag foil electrode, displayed as faradaic efficiencies.48 Copyright 2018 American Chemical Society. | |
2.4 The alloying effect
Alloying is known as a widely used technique to alter the catalytic performance of nanomaterials. The addition of foreign atoms can alter not only the geometric but also the electronic structure of the parent nanostructures and induce synergistic effects. Ag-based alloy catalysts prepared by alloying some transition metals such as Cu,49–51 In,52–54 Zn,55 Co56 and Sn57 revealed that the composition has a substantial impact on the CO faradaic efficiency and partial current density. Theoretical studies manifest that alloying could modify the suboptimal linear relation between CO and COOH binding energies on transition metals and that the partial covalency imposed by some p-block atoms as dopants may decrease the onset overpotential for CO on Ag.58–61 Choj et al. synthesized dendritic Ag–Cu catalysts through an electrodeposition technique and investigated their activities in electrochemical CO2 reduction. Among the synthesized dendritic catalysts, the Ag100 dendritic catalyst exhibits a CO faradaic efficiency of 64.6% at a constant potential of −1.7 VSCE in a CO2-saturated 0.5 M KHCO3 solution. However, the catalytic activity of the Ag57Cu43 dendritic catalyst was 2.2 times higher than that of the Ag100 dendritic catalyst in terms of silver unit mass activity. These results showed that the direct production of syngas was possible by adjusting the composition of Ag and Cu.50 In addition, Ag/Cu foam with manageable pore sizes and wall thicknesses was synthesized through bubble-template Cu electrodeposition followed by Ag metal galvanic replacement for the formation of CO via the electrochemical conversion of CO2. At lower overpotentials, the foams demonstrated morphologically advantages that were created from their rough surfaces; nevertheless, this positive influence was missing at higher overpotentials due to the uncovered Cu.62 Furthermore, a recent study found that the combination of Ag with inexpensive metals like Cu not only led to a rise in the unit mass activity with regard to CO production but also reduced the overpotential of the reaction to a ∼300 mV lower value compared with bulk Ag and Ag foam, respectively (Fig. 10). The bimetallic Ag–Cu nanofoam also preserved a stable selectivity over 5 hours of testing. Raman study showed that the existence of Ag moved the peak intensity of the intramolecular CO band (CO surface coverage) to more positive potentials compared to that of Cu foam. This also illustrates the faster desorption of CO from the Ag–Cu surface at low overpotentials which enhanced the activity for CO2 reduction to CO.51 Park et al.53 studied the catalytic performance of the electrodeposited Ag and Ag–In dendrites in the electrochemical reduction of CO2 to CO. In the sample of electrodeposited Ag dendrites, the CO faradaic efficiency was intensely affected by the Ag (220)/Ag (111) ratio, whereas the CO partial current density exhibited a reliance on the roughness of the catalyst. The addition of an optimized amount of In into the Ag dendrites enhanced the efficiency of CO production. This is due to the increased Ag (220)/Ag (111) intensity ratio in the Ag–In catalyst and the suppression of the hydrogen evolution reaction (HER) versus the diminution in the quantity of active Ag with the increasing addition of In. So as to additionally improve catalytic performance, bubble-assisted Cu foam (CF) was utilized to provide a vast electrochemical surface area (ECSA) for the Ag/In catalyst. Compared with the reported activities, noteworthy performance was detected for Ag600/In/CF in terms of the CO faradaic efficiency and partial current density, particularly at a low overpotential which can be attributed to the supressed hydrogen evolution by In and morphological advantage of Cu foam.54 Polycrystalline Ag–Zn foil was also utilized for CO2 electroreduction in an aqueous bicarbonate electrolyte, and CO is the major product, likely because of the weak CO binding energy of the surface, with methane and methanol evolved as minor products. In contrast to pure silver and pure zinc foil, improvement in activity and selectivity for methane and methanol is observed which demonstrates the presence of a synergistic impact among silver and zinc at the surface of the composite.55
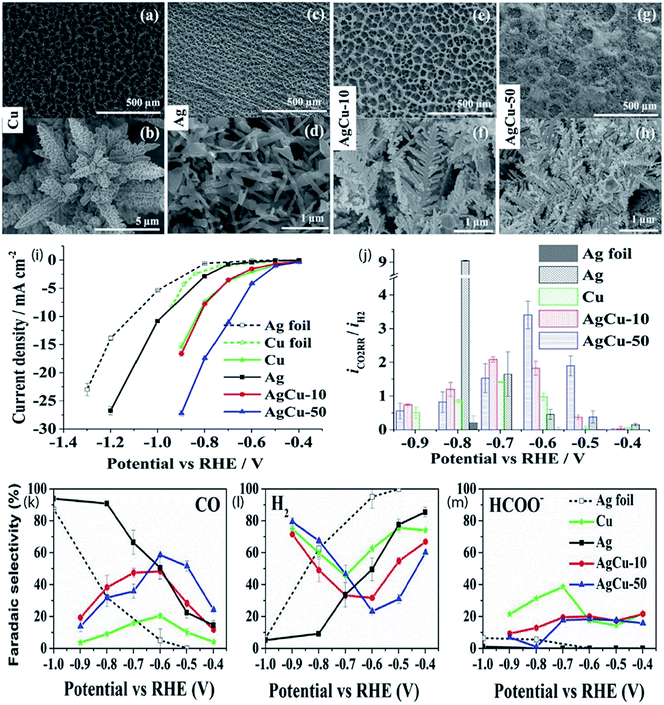 |
| | Fig. 10 SEM images of Cu (a and b), Ag (c and d), AgCu-10 (e and f) and AgCu-50 (g and h) foam electrodeposited at 1 A cm−2; (i) steady-state current as a function of applied potential during 1 h of electrolysis; (j) current ratio of CO2 reduction to the HER at different potentials; faradaic selectivities of the major products CO and formate along with H2 of electrodeposited Cu, electrodeposited Ag, AgCu-10, AgCu-50, and Ag foil as a function of applied potential vs. RHE (k–m). The error bars correspond to the average of two individual measurements on two different samples.51 Copyright 2019 American Chemical Society. | |
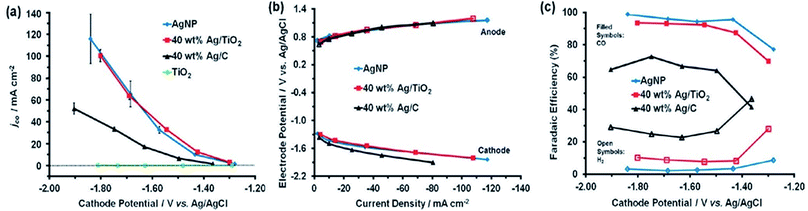 |
| | Fig. 11 (a) The partial current density for CO and (b) faradaic efficiency of Ag/TiO2 catalysts with different Ag loadings: 5, 10, 20, 40, and 60 wt%; (c) partial current densities for CO vs. cathode potential relative to the cathode Ag loading for different catalysts. The error bars represent the standard deviation of the average of three experiments (N = 3).63 Copyright 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
2.5 The support effect
The catalyst support can have a great effect on catalytic performance and can reduce the catalyst loading while improving its performance and durability.34,52,61,63 It was demonstrated that Ag supported on TiO2 outperforms Ag supported on black carbon in the reduction of CO2 to CO. However, Ag/TiO2 performs at the almost identical level as unsupported Ag nanoparticles, for which the faradaic productivity for CO surpasses 90% and the current density surpasses 100 mA cm−2. Compared to carbon black, TiO2 is an excellent support for Ag catalysts for the electrochemical reduction of CO2 to CO. By utilizing TiO2 as the support, the Ag loading can be minimized without sacrificing performance to selective CO formation. 40 wt% Ag/TiO2 can generate a similar quantity of CO as unsupported Ag NPs but at 2.5 fold less Ag loading. In view of the cyclic voltammetry data, a reaction pathway is suggested that involves the contribution of (Ti+4/Ti+3) from the support material, which acts as the redox couple and stabilizes the reaction intermediates (Fig. 11).63 Larrazábal et al.52 fabricated catalysts with different silver loadings (5–40 wt% Ag) supported on In(OH)3 and In2O3, respectively, for electroreduction of CO2 to CO. The Ag/In2O3 and Ag/In(OH)3 systems exhibited an improved current efficiency for CO at a modest overpotential compared both to the pristine supports and to catalysts with analogous Ag loadings on a carbon black support. This finding suggests the presence of a synergistic effect between Ag and the In2O3 support, which was especially apparent between Ag and In(OH)3 at higher Ag loadings. CO2-TPD measurements showed a rise in the CO2 uptake capacity of In(OH)3 and In2O3 originating from the interaction of Ag with the supports. In a recent study, three-dimensional copper foam was chosen as the substrate for coating silver dendrites through electrodeposition. The cyclic voltammograms showed that the coupling of Ag dendritic catalysts with the high active surface area of the Cu foam effectively raised the electrocatalytic performance toward CO formation and resulted in an increase to a stable maximum faradaic efficiency of 95.7% at −0.8 V [RHE] compared to flat Cu foil with the Ag catalyst and a flat Ag foil cathode. This great achievement was ascribed to mainly two advantages of the Cu foam-based cathode: (i) reduced mass transport restriction due to the enlarged 3D electrochemically active contact area between the homogeneously coated Cu foam and the electrolyte, and (ii) the high selectivity of the deposited Ag catalyst to CO formation (Fig. 12).61
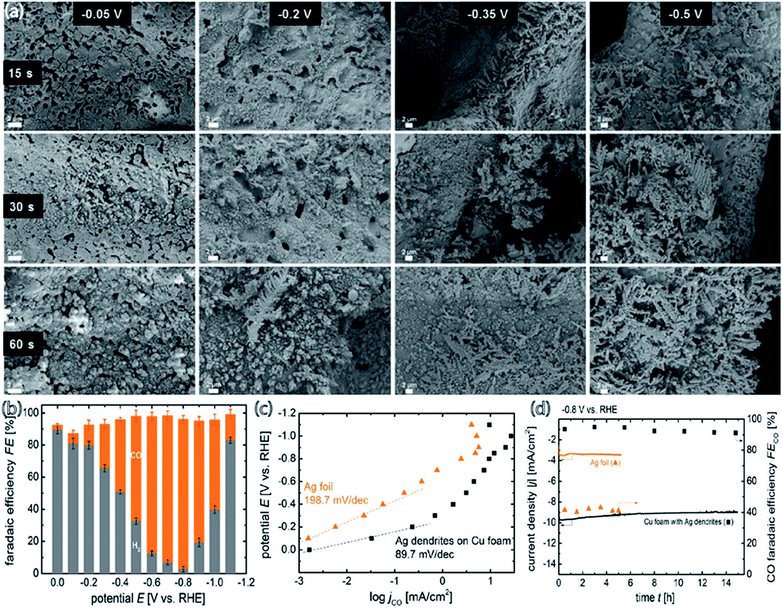 |
| | Fig. 12 (a) SEM images of the Ag dendritic catalyst electrodeposited on Cu foam for 15, 30, and 60 s at constant potentials of −0.05, −0.2, −0.35, and −0.5 VRHE, respectively, in a 0.1 M AgNO3 containing plating solution; (b) faradaic efficiency of CO and H2 production as a function of the applied potential for the cathodes prepared at −0.2 V [RHE] with Ag electrodeposition at 60 s; (c) applied potential versus CO production partial current density for Ag dendrites and Ag foil; (d) stability of total CO2 reduction current density (left ordinate) for Ag dendrites and Ag foil at −0.8 V [RHE] in 0.5 M KHCO3. The CO faradaic efficiency of both tested cathodes as a function of operating time is plotted on the right ordinate.61 Copyright 2018 American Chemical Society. | |
2.6 The surface modification effect
Surface property modification is an effective approach to improve the performance of the catalyst. Generally, the surface properties of catalysts can be adjusted by improvement with functional molecules. For instance, in an early study the authors observed the covalency-aided electrochemical reaction (CAER) mechanism in which p-block dopants have a significant impact on modifying the reaction energetics via enforcing partial covalency in the metal catalysts, thus boosting their catalytic activity well beyond modifications originating from d-block dopants. In particular, sulfur or arsenic doping can efficiently reduce the overpotential with the desired structure and electrochemical durability.58 Additionally, another study showed that particular interactions among Ag nanoparticles and the attaching agents (Ag–S) affected the durability of the intermediate COOH*, leading to boosted catalytic activity. These studies suggested that functional groups of the organic molecules can notably minimize the overpotential or enhance CO selectivity for the electrochemical CO2 reduction.34 To discover the direct relationship among the functional groups and CO2 reduction efficiency of the Ag nanoparticle, Kim et al. established a strategy to adjust CO2 reduction activity by modifying the binding energies of the intermediates on the electrocatalyst surfaces with the support of molecules that have functional groups. Therefore, they synthesized three kinds of Ag nanoparticles through utilizing various surface capping agents including oleylamine, oleic acid, and dodecanethiol having amine, carboxyl, and thiol functional groups. The amine capped Ag/C exhibited higher selectivity with 94.2% CO faradaic efficiency owing to the efficient inhibition of the HER and appropriate activity of the CO2 reduction reaction. Density functional theory (DFT) calculations indicated that the amine-capped Ag nanoparticles stabilized the COOH* intermediate whereas a reverse effect was found with thiol molecules. Furthermore, ethylenediamine exhibited a higher selectivity towards CO formation, in contrast with cysteamine, when it was applied as an anchoring agent to immobilize the Ag NPs on the carbon support (Fig. 13).64 Recently, in order to understand the relation between the catalytic performance and the N/S doping ratio in Ag-based electrocatalysts. An all-water-based solution method was established to synthesize extremely active Ag electrocatalytic films through a seed-mediated metal growth technique on aminopropyltriethoxysilane (APTES)-deposited substrates. The obtained Ag films showed excellent durability throughout electrochemical CO2 reduction as the Ag film was robustly attached to the APTES layer. The N- and S-doped Ag film with an adjusted N/S doping ratio of 1.75 achieved a CO FE of 75.7% at a low overpotential of 0.19 V [RHE] which is greater than those before reported for Ag-based electrocatalysts.65
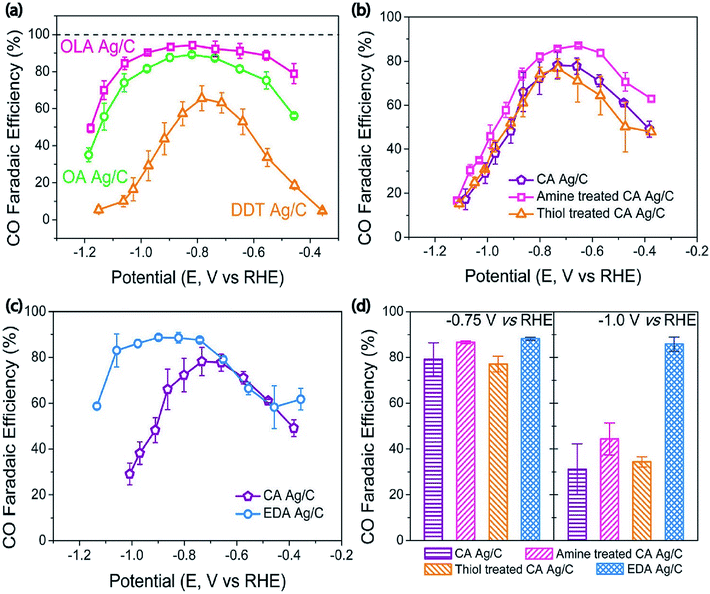 |
| | Fig. 13 (a) CO faradaic efficiency depending on the applied potential. (b) CO faradaic efficiency depending on the applied potential for CA Ag/C, amine-treated CA Ag/C, and thiol-treated CA Ag/C. (c) CO faradaic efficiency depending on the applied potential for CA Ag/C and EDA Ag/C. (d) CO faradaic efficiency at fixed potentials of −0.70 V and −1.0 V (vs. RHE).64 Copyright 2017, American Chemical Society. | |
3. Outlook on surface engineering
The surface science of catalysts is fundamental to heterogeneous catalysis. Great research efforts have contributed to the surface designing of catalysts to improve the reaction performance by increasing the surface area and catalytically active sites. Zhou et al. reported a simple and efficient method by anodization to modify a polycrystalline Ag electrode. The anodized Ag electrode exhibited a great improvement in the electroreduction activity toward CO under a moderate potential (0.5 V, 92.8%) as shown in Fig. 14. These results indicate that a thin silver oxide layer is produced on the electrode surface. These results indicate that the production of a thin silver oxide layer on the electrode surface and then reducing this oxide layer display an important role in enhancing carbon dioxide reduction.66 Surface-sensitive evaluation systems were used to understand the mechanism of the improvement of the electrocatalytic CO2 reduction to CO on electrochemically treated Ag surfaces where the CO2ERR efficiencies changed depending on the electrochemical treatment of the Ag surface. The samples, CVAg-5, and pulsed Ag showed the highest improvement in CO2 reduction efficiency for selective CO formation in contrast to the bare Ag foil and CVAg-50. The excessive work function exists at the nanostructure boundaries of the effective Ag electrode, which strongly indicates localized O concentration on the nanostructured Ag surface. The stable surface O adjusts the electronic territory of Ag to have stronger collaboration with COOH* and CO* intermediates.67 Recent studies showed that the total CO FE of oxide-derived silver was positively shifted by more than 0.4 volts compared to the polycrystalline silver electrode. Electrokinetics analyses demonstrate that the boosted catalytic activity is attributed to the improved stabilization of the COOH* intermediate. Moreover, it is very likely that extremely nanostructured Ag was able to create a high local pH close to the catalyst surface, which may also simplify the catalytic activity towards the reduction of CO2 with curbed H2 evolution (Fig. 15).68 The variances in the surface structure between a pure silver film and oxide-derived silver catalysts were investigated by operando extended X-ray absorption fine structure (EXAFS) analysis. The results showed the existence of a long Ag–O bond where oxygen may be the main contributor to the reduction of CO2 to CO on oxide-derived silver catalysts (Fig. 16). The silver needs a long and consequently atomic oxygen bond in order to change its reactivity and enhance its tendency to CO2 binding. Both the existence of oxygen and nanostructuring of the catalyst which raise the local pH boosting the activity and selectivity for CO formation on silver electrocatalysts are consequently crucial factors of an active and selective CO2 reduction catalyst.69 Oxygen plasma-treated Ag exhibited more than 90% faradaic efficiency for CO production at −0.6 V versus RHE. The secret behind the upgraded reactivity was discovered through DFT calculations, which suggested that the greatly defective surface produced by plasma oxidation in the presence of local electric fields was able to decrease the thermodynamic barriers, permitting CO2 reduction to CO at a low overpotential. DFT calculations suggested that the defect-rich surface of the plasma-oxidized silver foil in the presence of local electric fields radically decreased the overpotential of CO2 electroreduction.70 Furthermore, it has been observed that halide anions adsorbed onto the surfaces of the electrode can increase the capture capacity of CO2 and suppress the adsorption of protons. Zhang et al.71 found that an iodide derived nanostructured silver (ID-Ag) catalyst is proficient in reducing CO2 to CO with a faradaic efficiency (FE) of 94.5% at a potential of −0.7 V. It should be noted that the improved catalytic activity and selectivity are associated with a high surface area with a huge amount of electrochemically active sites for CO2 conversion. In addition, the combination of halide (X) anions on the catalyst surface plays a vital role in enhancing the selectivity for CO production by suppressing the competing hydrogen evolution reaction (HER). The activity of X–Ag catalysts is in the order Cl > Br > I, which is in agreement with the suggested model of the active site. The Tafel analysis of electrochemical CO2 reduction points to the sensitivity of the mechanism of electrocatalysts to the nature of X (Fig. 17).72
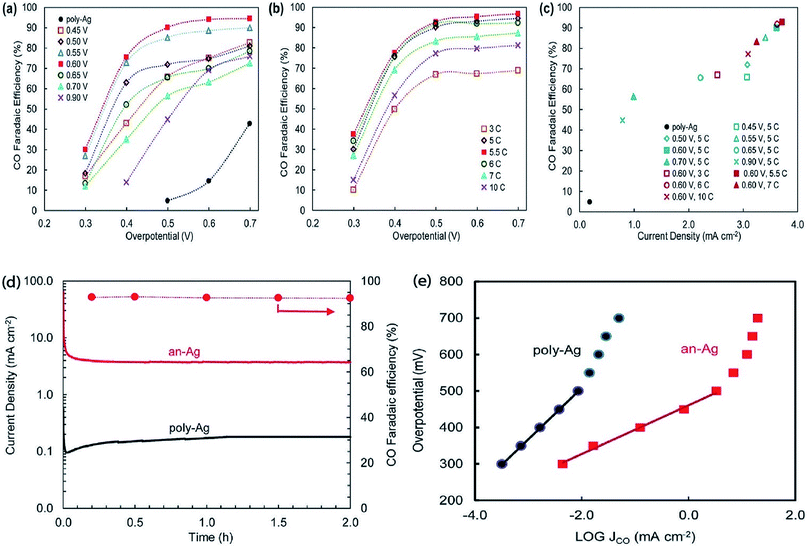 |
| | Fig. 14 Electrocatalytic performance of anodized Ag electrodes. (a) CO faradaic efficiency for Ag electrodes anodized at different potentials against Ag/AgCl. The anodization charge was fixed at 5 coulombs in a neutral (pH 7) 0.1 M NaNO3 electrolyte. (b) CO faradaic efficiency for Ag electrodes anodized at different charges. The anodization potential was fixed at 0.6 V against Ag/AgCl in a neutral 0.1 M NaNO3 electrolyte. (c) CO faradaic efficiency (at 0.5 V overpotential) and current density (when 10 coulombs of total charge were passed through) for Ag electrodes anodized under various conditions in a neutral 0.1 M NaNO3 electrolyte. (d) Current density during 2 hour electrocatalysis for the an-Ag electrode in comparison with poly-Ag (solid lines, left axis) and CO faradaic efficiency (at 0.5 V overpotential) versus time for the an-Ag electrode. (e) Tafel plots of the Ag electrode anodized at a potential of 0.6 V with 5.5 coulombs of charge in a neutral 0.1 M NaNO3 electrolyte and unanodized poly-Ag.66 Copyright 2015 Royal Society of Chemistry. | |
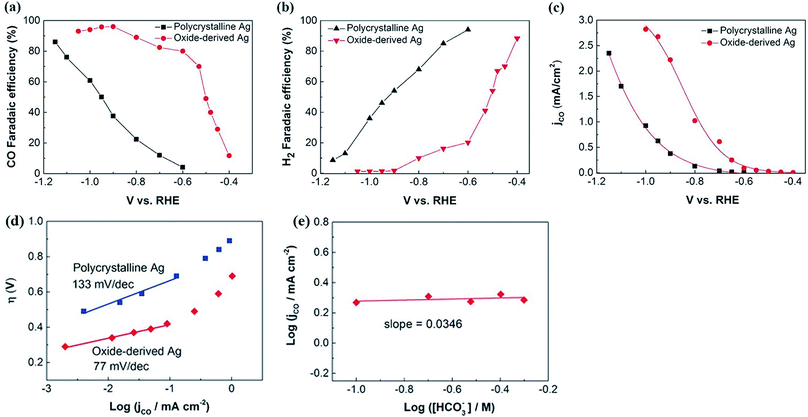 |
| | Fig. 15 Comparison of the electrocatalytic activity of polycrystalline Ag and oxide-derived Ag. Faradaic efficiency for CO (a) and H2 (b) on polycrystalline Ag and oxide-derived Ag at various potentials in CO2-saturated 0.1 m KHCO3 (pH 6.8). (c) The current density for CO at various potentials; (d) Tafel plots of the CO partial current density for polycrystalline Ag and oxide-derived Ag. (e) Bicarbonate concentration at a constant potential for oxide-derived Ag.68 Copyright 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
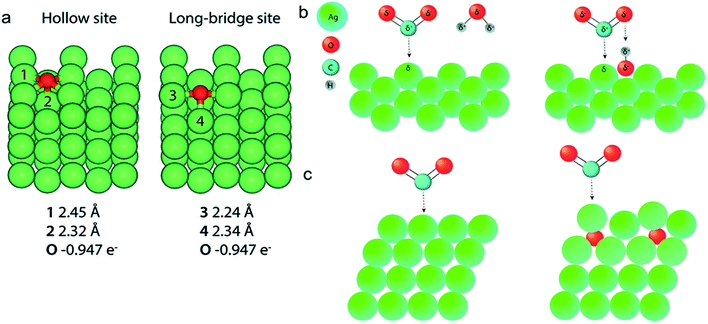 |
| | Fig. 16 (a) DFT calculations and Bader analysis for O on an Ag (110) slab give Ag–O bond lengths very similar to those found in the EXAFS data fitting. (b) Left: A CO2 molecule binds to an Ag (110) facet in the presence of a water molecule. Right: The presence of a water molecule. Right: The presence of oxygen in the cavities of a silver (110) facet can facilitate the binding of CO2 to silver through the presence of a shared proton. (c) Left: A CO2 molecule binding to an Ag (111) surface. Right: Subsurface O below Ag (111) facets shift the d-band of silver toward the Fermi level (indicated by a change in color of the affected Ag atoms), increasing the reactivity of the subsurface O–Ag (111) facet compared to Ag (111). Adapted from ref. 69. Copyright 2019 Royal Society of Chemistry. | |
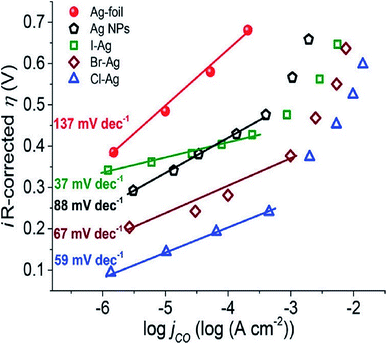 |
| | Fig. 17 Tafel plots (iR-corrected overpotentials η as a function with the CO partial current density jCO) for CO2 electroreduction on Ag foil, Ag nanoparticles, Cl–Ag, Br–Ag, and I–Ag.72 Copyright 2019 American Chemical Society. | |
Recently, a phosphate silver-derived silver (PD-Ag) electrocatalyst was synthesized through a low-cost and quite simple electrochemical technique, displaying a promising performance for the selective reduction of CO2 to CO. The high FE of PD-Ag could reach 97.3% with a potential of −0.7 V vs. RHE, and a current density of 2.93 mA cm−2. Moreover, the PD-Ag catalyst's activity can be maintained for a long period without noticeable deactivation for at least 10 h (Fig. 18).24 To better understand the recent developments of Ag-based nanostructured catalysts in the CO2 ERR, a summarization is given for some Ag-based nanocatalysts and their faradaic efficiencies in Table 2. At present, the maximum FEs of CO obtained with Ag-based nanocatalysts reach as high as 99.3%, which is a significant improvement over those with the traditional Ag foil.
Table 2 Summary of the performance of some representative Ag-based nanocatalysts in the CO2 ERR
| Electrocatalysts |
Potential |
Electrolytes |
CO FE [%] |
Ref. |
| 5 nm Ag/C |
−0.75 V [RHE] |
0.5 M KHCO3 |
84.4% |
34
|
| PONs Ag |
−0.69 V [RHE] |
0.5 M KHCO3 |
96.7% |
38
|
| Ag nanocorals |
<−0.6 V [RHE] |
0.1 M KHCO3 |
95% |
23
|
| Ag TNP |
−0.206[RHE] |
0.1 M KHCO3 |
96.8% |
39
|
| Dendritic Ag |
−1.5 V [SCE] |
0.5 M KHCO3 |
>60% |
40
|
| Ag-200 nm NWs |
−0.6 [RHE] |
0.5 M KHCO3 |
91% |
42
|
|
D-25 Ag NWs |
−0.95 V [RHE] |
0.1 M KHCO3 |
99.3% |
44
|
| Ag nanosheets |
−0.29 V [RHE] |
0.5 M NaHCO3 |
95% |
45
|
| Ag nanoporous |
−0.490 V [RHE] |
0.5 M KHCO3 |
92% |
15
|
| Ag nano foam |
−0.3–1.2 V [RHE] |
0.5 M KHCO3 |
90% |
48
|
| Dendritic Ag–Cu |
−1.5 V [SCE] |
0.5 M KHCO3 |
<40% |
50
|
| Ag dendrites on Cu |
−0.8 V [RHE] |
0.5 M KHCO3 |
95.7% |
61
|
| Ag/TiO2 |
−1.8 V [Ag/AgCl] |
1 M KOH |
<90% |
63
|
| OD-Ag |
−0.8 [RHE] |
0.1 M KHCO3 |
80% |
68
|
| ID-Ag |
−0.7 [RHE] |
0.5 M KHCO3 |
94.5% |
71
|
| PD-Ag |
−0.7 [RHE] |
0.5 M KHCO3 |
∼97.3% |
24
|
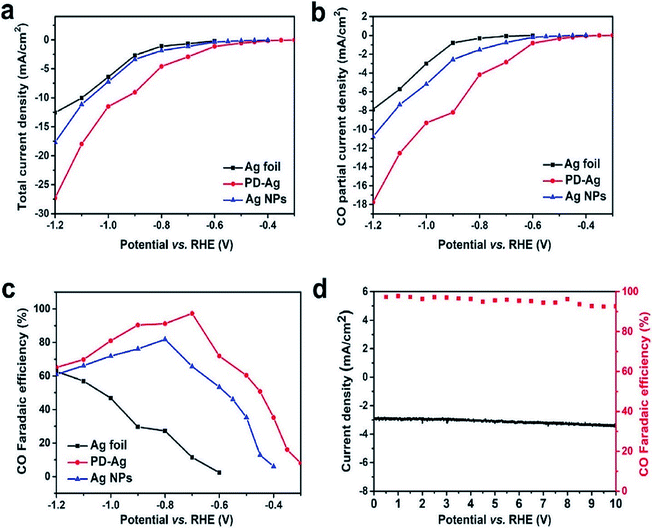 |
| | Fig. 18 (A) Total current density at different potentials on Ag foil (black trace), PD-Ag (red trace), and Ag nanoparticles (blue trace). (b) The current density for CO at different potentials on Ag foil (black trace), PD-Ag (red trace), and Ag nanoparticles (blue trace). (c) CO faradaic efficiencies versus applied potentials catalyzed by Ag foil (black trace), PD-Ag (red trace), and Ag nanoparticles (blue trace). (d) Stability performance of PD-Ag for CO2 reduction operated at a potentiostatic potential of −0.7 V vs. RHE for 10 h. Total current density vs. time and FE for CO vs. time.24 Copyright 2019 American Chemical Society. | |
4. Conclusion
In this article, we summarized the progress of recent silver-based electrocatalysts for CO2 reduction, focused on morphology control, composition manipulation, and support effects on the catalytic performance towards CO formation compared to that of Ag foil or bulk electrodes. Adjustment of precisely exposed facets and the surface electronic structure seems to be effective to improve catalytic activity and selectivity. A multi-metal system with synergistic effects from different metals can be optimized in terms of CO faradaic efficiency and partial current density, particularly in the low overpotential region. In addition, owing to the fast deactivation of the Ag-based catalysts, the competitive hydrogen evolution reaction (HER) causes a low conversion efficiency and product selectivity. The catalyst support has a remarkable effect on the catalyst performance and durability, which is a critical step in the enhancement of the commercial feasibility of this technology. It is essential to establish a new stabilization approach.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The financial support from the National Natural Science Foundation (21676085) is gratefully acknowledged.
References
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- J. L. Inglis, B. J. MacLean, M. T. Pryce and J. G. Vos, Coord. Chem. Rev., 2012, 256, 2571–2600 CrossRef CAS.
- X. Mao and T. A. Hatton, Ind. Eng. Chem. Res., 2015, 54, 4033–4042 CrossRef CAS.
- D. R. Kauffman, J. Thakkar, R. Siva, C. Matranga, P. R. Ohodnicki, C. Zeng and R. Jin, ACS Appl. Mater. Interfaces, 2015, 7, 15626–15632 CrossRef CAS PubMed.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050 RSC.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
-
W. Schneider and W. Diller, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
-
E. D. Bergmann, D. Ginsburg and R. Pappo, in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 179–556 Search PubMed.
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- J. Kim, J. T. Song, H. Ryoo, J.-G. Kim, S.-Y. Chung and J. Oh, J. Mater. Chem. A, 2018, 6, 5119–5128 RSC.
- C. Chen, B. Zhang, J. Zhong and Z. Cheng, J. Mater. Chem. A, 2017, 5, 21955–21964 RSC.
- Y. H. Li, P. F. Liu, C. Li and H. G. Yang, Chem.–Eur. J., 2018, 24, 15486–15490 CrossRef CAS PubMed.
- M. Valenti, N. P. Prasad, R. Kas, D. Bohra, M. Ma, V. Balasubramanian, L. Chu, S. Gimenez, J. Bisquert, B. Dam and W. A. Smith, ACS Catal., 2019, 9, 3527–3536 CrossRef CAS.
- C. Yan, L. Lin, D. Gao, G. Wang and X. Bao, J. Mater. Chem. A, 2018, 6, 19743–19749 RSC.
-
Y. Hori, in Modern Aspects of Electrochemistry, Springer New York, New York, NY, 1989, pp. 89–189 Search PubMed.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819 RSC.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- J. Gao, C. Zhu, M. Zhu, Y. Fu, H. Huang, Y. Liu and Z. Kang, ACS Sustainable Chem. Eng., 2019, 7, 3536–3543 CrossRef CAS.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- M. E. Hafez, H. Ma, Y.-Y. Peng, W. Ma and Y.-T. Long, J. Phys. Chem. Lett., 2019, 10, 3276–3281 CrossRef CAS PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- A. Klinkova, P. De Luna, C.-T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS.
- K. Chandrasekaran and L. O. Bockris, Surf. Sci., 1987, 185, 495–514 CrossRef CAS.
- H. A. Schwarz and R. W. Dodson, J. Phys. Chem., 1989, 93, 409–414 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- A. Salehi-Khojin, H.-R. M. Jhong, B. A. Rosen, W. Zhu, S. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- L. Zhang, Z. Wang, N. Mehio, X. Jin and S. Dai, ChemSusChem, 2016, 9, 428–432 CrossRef CAS PubMed.
- N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283–286 CrossRef CAS.
- X. Peng, S. G. Karakalos and W. E. Mustain, ACS Appl. Mater. Interfaces, 2018, 10, 1734–1742 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- Y. S. Ham, S. Choe, M. J. Kim, T. Lim, S.-K. Kim and J. J. Kim, Appl. Catal., B, 2017, 208, 35–43 CrossRef CAS.
- W. Qiu, R. Liang, Y. Luo, G. Cui, J. Qiu and X. Sun, Inorg. Chem. Front., 2018, 5, 2238–2241 RSC.
- C. Luan, Y. Shao, Q. Lu, S. Gao, K. Huang, H. Wu and K. Yao, ACS Appl. Mater. Interfaces, 2018, 10, 17950–17956 CrossRef CAS PubMed.
- W. Xi, R. Ma, H. Wang, Z. Gao, W. Zhang and Y. Zhao, ACS Sustainable
Chem. Eng., 2018, 6, 7687–7694 CrossRef CAS.
- S. Liu, X.-Z. Wang, H. Tao, T. Li, Q. Liu, Z. Xu, X.-Z. Fu and J.-L. Luo, Nano Energy, 2018, 45, 456–462 CrossRef CAS.
- C.-Y. Lee, Y. Zhao, C. Wang, D. R. G. Mitchell and G. G. Wallace, Sustainable Energy Fuels, 2017, 1, 1023–1027 RSC.
- W. Yang, W. Ma, Z. Zhang and C. Zhao, Faraday Discuss., 2018, 210, 289–299 RSC.
- M. Ma, K. Liu, J. Shen, R. Kas and W. A. Smith, ACS Energy Lett., 2018, 3, 1301–1306 CrossRef CAS PubMed.
- A. Dutta, C. E. Morstein, M. Rahaman, A. Cedeño López and P. Broekmann, ACS Catal., 2018, 8, 8357–8368 CrossRef CAS.
- Y. Wang, C. Niu and Y. Zhu, Nanomaterials, 2019, 9, 173 CrossRef CAS PubMed.
- J. Choi, M. J. Kim, S. H. Ahn, I. Choi, J. H. Jang, Y. S. Ham, J. J. Kim and S.-K. Kim, Chem. Eng. J., 2016, 299, 37–44 CrossRef CAS.
- T. Kottakkat, K. Klingan, S. Jiang, Z. P. Jovanov, V. H. Davies, G. A. M. El-Nagar, H. Dau and C. Roth, ACS Appl. Mater. Interfaces, 2019, 11, 14734–14744 CrossRef CAS PubMed.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérez-Ramírez, J. Catal., 2016, 343, 266–277 CrossRef.
- H. Park, J. Choi, H. Kim, E. Hwang, D.-H. Ha, S. H. Ahn and S.-K. Kim, Appl. Catal., B, 2017, 219, 123–131 CrossRef CAS.
- H. Lee, J. Kim, I. Choi and S. H. Ahn, Electrochim. Acta, 2018, 133102 Search PubMed.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram, J. T. Feaster, A. L. Jongerius, C. Hahn and T. F. Jaramillo, Energy Technol., 2017, 5, 955–961 CrossRef CAS.
- S. Singh, R. K. Gautam, K. Malik and A. Verma, J. CO2 Util., 2017, 18, 139–146 CrossRef CAS.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- H.-K. Lim, H. Shin, W. A. Goddard, Y. J. Hwang, B. K. Min and H. Kim, J. Am. Chem. Soc., 2014, 136, 11355–11361 CrossRef CAS PubMed.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS PubMed.
- A. S. Bandarenka and M. T. M. Koper, J. Catal., 2013, 308, 11–24 CrossRef CAS.
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, J. Arbiol and J. R. Morante, ACS Appl. Mater. Interfaces, 2018, 10, 43650–43660 CrossRef CAS PubMed.
- H. Lee, S.-K. Kim and S. H. Ahn, J. Ind. Eng. Chem., 2017, 54, 218–225 CrossRef CAS.
- S. Ma, Y. Lan, G. M. J. Perez, S. Moniri and P. J. A. Kenis, ChemSusChem, 2014, 7, 866–874 CrossRef CAS PubMed.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- S. M. Lee, H. Lee, J. Kim, S. H. Ahn and S. T. Chang, Appl. Catal., B, 2019, 259, 118045 CrossRef CAS.
- L. Q. Zhou, C. Ling, M. Jones and H. Jia, Chem. Commun., 2015, 51, 17704–17707 RSC.
- M. S. Jee, H. Kim, H. S. Jeon, K. H. Chae, J. Cho, B. K. Min and Y. J. Hwang, Catal. Today, 2017, 288, 48–53 CrossRef CAS.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- N. J. Firet, M. A. Blommaert, T. Burdyny, A. Venugopal, D. Bohra, A. Longo and W. A. Smith, J. Mater. Chem. A, 2019, 7, 2597–2607 RSC.
- H. Mistry, Y.-W. Choi, A. Bagger, F. Scholten, C. S. Bonifacio, I. Sinev, N. J. Divins, I. Zegkinoglou, H. S. Jeon, K. Kisslinger, E. A. Stach, J. C. Yang, J. Rossmeisl and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2017, 56, 11394–11398 CrossRef CAS PubMed.
- Y. Zhang, L. Ji, W. Qiu, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 2666–2669 RSC.
- Y.-C. Hsieh, L. E. Betancourt, S. D. Senanayake, E. Hu, Y. Zhang, W. Xu and D. E. Polyansky, ACS Appl. Energy Mater., 2019, 2, 102–109 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Fahim A.
Qaraah
b,
Chengzhen
Chen
a,
Fanghua
Zhang
a,
Shenglin
Yan
a and
Zhenmin
Cheng
a,
Fahim A.
Qaraah
b,
Chengzhen
Chen
a,
Fanghua
Zhang
a,
Shenglin
Yan
a and
Zhenmin
Cheng







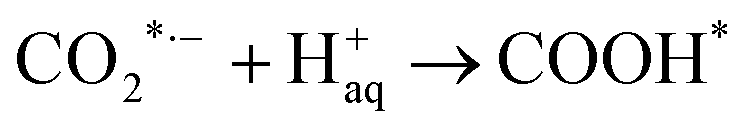
 , which is considered to be the rate-controlling step for most transition metal-based catalysts owing to the high energy barrier required to start the process.8,27,29 Theoretically, the standard reduction potential for reducing CO2 in aqueous solution (i.e., pH 7, 1 M electrolyte, 25 °C, 1 atm) into CO is in the range of −0.2 V to 0.2 V (vs. the reversible hydrogen electrode (RHE)).22 The reason for such a high overpotential is often attributed to the high equilibrium potential associated with the formation of the radical intermediate
, which is considered to be the rate-controlling step for most transition metal-based catalysts owing to the high energy barrier required to start the process.8,27,29 Theoretically, the standard reduction potential for reducing CO2 in aqueous solution (i.e., pH 7, 1 M electrolyte, 25 °C, 1 atm) into CO is in the range of −0.2 V to 0.2 V (vs. the reversible hydrogen electrode (RHE)).22 The reason for such a high overpotential is often attributed to the high equilibrium potential associated with the formation of the radical intermediate 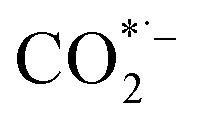 .30,31 Such a circumstance becomes serious in an aqueous electrolyte-based CO2 electrolyzer because the side reaction of hydrogen evolution also takes place at a similar potential (Table 1) and competes with CO2 reduction at the cathode surface. Thus, an efficient electrocatalyst should not only accelerate the CO2 ERR with significant productivity and selectivity but also suppress the competing HER.32
.30,31 Such a circumstance becomes serious in an aqueous electrolyte-based CO2 electrolyzer because the side reaction of hydrogen evolution also takes place at a similar potential (Table 1) and competes with CO2 reduction at the cathode surface. Thus, an efficient electrocatalyst should not only accelerate the CO2 ERR with significant productivity and selectivity but also suppress the competing HER.32