
Disulfide chemistry in responsive aggregation of amphiphilic systems
Raju
Bej†
,
Pradip
Dey†
 and
Suhrit
Ghosh
*
and
Suhrit
Ghosh
*
School of Applied and Interdisciplinary Sciences, Indian Association for the Cultivation of Science, 2A and 2B Raja S. C. Mullick Road, Kolkata, 700032, India. E-mail: psusg2@iacs.res.in
First published on 18th November 2019
Abstract
The dynamic nature of the disulfide bond has enhanced the potential for disulfide based amphiphiles in the emerging biomedical field. Disulfide containing amphiphiles have extensively been used for constructing wide ranging soft nanostructures as potential candidates for delivery of drugs, proteins and genes owing to their degradable nature in the presence of intracellular glutathione (present in a many fold excess compared to the extracellular milieu). This degradable nature of amphiphiles is not only useful to deliver therapeutics but it also eliminates the toxicity issues associated with the carrier after delivery of such therapeutics. Therefore, these bioreducible and biocompatible nano-aggregates inspired researchers to use them as vehicles for therapeutic delivery and as a result the literature of disulfide containing amphiphiles has been intensified. This review article highlights the structural diversity in disulfide containing amphiphilic small molecule and polymeric systems, structural effects on their aqueous aggregation, redox-responsive disassembly and biological applications. Furthermore, the use of disulfide chemistry towards the design of cell penetrating polymers has also been discussed. Finally a brief perspective on some future opportunities of these systems is provided.
 Raju Bej | Mr Raju Bej is working as a Senior Research Fellow (SRF) under the supervision of Professor Suhrit Ghosh at the Indian Association for the Cultivation of Science (IACS), Kolkata. He received his MSc degree in Chemistry from the Indian Institute of Technology, Madras in 2014. His research interests are in the synthesis and biological evaluation of redox responsive polymeric nanocarriers in anticancer drug delivery. |
 Pradip Dey | Dr Pradip Dey has been working as a research associate under the mentorship of Professor Suhrit Ghosh at the Indian Association for the Cultivation of Science Kolkata, India since 2017. Before that he received his PhD from Freie Universität Berlin, Germany in 2015 under the guidance of Professor Rainer Haag. He completed his MSc (Chemistry) at the Indian Institute of Technology Kanpur in 2010. His current research efforts focus on understanding the self-assembly of supramolecularly engineered amphiphilic macromolecules and design of degradable amphiphilic polymers based on water soluble oligoglycerol dendrons for drug delivery and related applications. |
 Suhrit Ghosh | Professor Suhrit Ghosh was born in 1976 in India. He completed his MS (Chemistry) in 2000 at the Indian Institute of Science, Bangalore, and continued for a PhD until 2005. He then moved to the University of Massachusetts, Amherst, USA, as a postdoctoral research associate during 2005–2007 and subsequently worked in the University of Würzburg, Germany, as an Alexander von Humboldt Postdoctoral Fellow. He returned to India in 2008 and joined the Indian Association for the Cultivation of Science Kolkata as an Assistant Professor where he currently holds the position of Professor in the School of Applied and Interdisciplinary Sciences. The research interest of his group is focused on the supramolecular assembly of π-systems and macromolecules by directional interaction and stimuli-responsive amphiphilic polymers. |
Introduction
Amphiphilic small molecules and macromolecules exhibit immiscibility driven aggregation in water producing wide-ranging nanostructures including micelles, vesicles, fibrils, tubes and others1 depending on the chemical structures, hydrophilic–lipophilic balance and the concentration of the amphiphiles. These aggregated structures work as nano-containers to encapsulate guest molecules, which makes them promising candidates as delivery vehicles of therapeutics.2 Polymeric amphiphiles3 are particularly attractive over small molecules as their aggregates generally exhibit low critical aggregation concentrations (CAC) (in the range of 10−5–10−6 M), slow exchange dynamics,4 structural diversity, appropriate particle sizes (suitable for the EPR effect)5 and other physical properties which are distinctly different from small molecule surfactants. In the recent past there has been sustained effort in molecular engineering of amphiphiles so that the resulting aggregates can be made to be responsive to external stimuli such as temperature,6 light,7 pH,8 redox,9 enzymes10 or some of their combinations.11 Stimuli responsiveness makes these materials more appealing for potential applications in drug delivery, tissue engineering, smart optical systems, coatings, textiles and other areas. Among various stimuli-responsive materials, redox responsive amphiphilic aggregates9 are highly relevant for drug delivery to cancer cells, because it is well known that in the cellular environment glutathione (GSH) can function as a reducing agent.12GSH is a tripeptide consisting of glutamic acid, cysteine, and glycine. It is the most abundant nonprotein thiol-containing reducing agent in biological systems. GSH is synthesized in the cytoplasm of a cell and from there it is transported to the various intracellular compartments. In cells, the abundance of GSH is maintained by the glutathione redox couple (GSSG/GSH),13 which is responsible for controlling the intra-cellular oxidation–reduction balance. The redox status of various cellular compartments can be understood from different redox potential values of the GSSG/GSH couple,14 which can be quantitatively calculated using the Nernst equation:
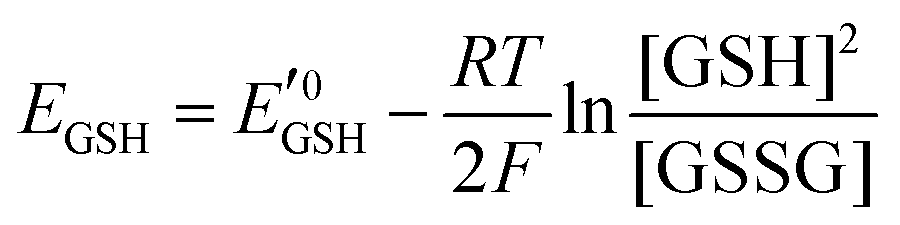
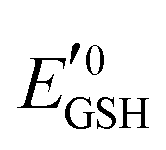 is the standard half cell reduction potential (
is the standard half cell reduction potential (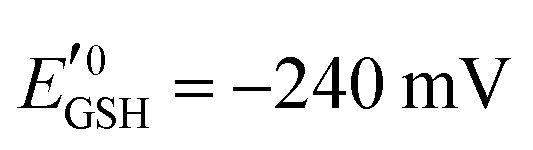 ), R = 8.315 J K−1 mol−1 is the gas constant, T = 298 K is the temperature, and F = 96
), R = 8.315 J K−1 mol−1 is the gas constant, T = 298 K is the temperature, and F = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1 is the Faraday constant. From this equation it is evident that the reduction potential depends on the ratio of GSH/GSSG and the concentration of GSH. Although in the cellular environment thiol and disulfide exchange is continuous, at a particular time there is a balance between the oxidized and reduced forms of GSH. More than 90% GSH remains in the reduced form whereas ∼10% GSH is present in the oxidized form (GSSG). In mammalian cells, the majority of reduced GSH (∼90%) is present in the cytoplasm while the mitochondria contain ∼10% GSH and the endoplasmic reticulum contains a very small percentage of GSH. In the human body, the intracellular GSH concentration generally varies in the range of 1–10 mM, which is significantly higher than extracellular common fluids (20–40 μM).15 It is also well known that because of up-regulation and down-regulation of various biological pathways in tumor cells, the concentration of GSH in their cytoplasm is four times higher than that of normal cells.16
485 C mol−1 is the Faraday constant. From this equation it is evident that the reduction potential depends on the ratio of GSH/GSSG and the concentration of GSH. Although in the cellular environment thiol and disulfide exchange is continuous, at a particular time there is a balance between the oxidized and reduced forms of GSH. More than 90% GSH remains in the reduced form whereas ∼10% GSH is present in the oxidized form (GSSG). In mammalian cells, the majority of reduced GSH (∼90%) is present in the cytoplasm while the mitochondria contain ∼10% GSH and the endoplasmic reticulum contains a very small percentage of GSH. In the human body, the intracellular GSH concentration generally varies in the range of 1–10 mM, which is significantly higher than extracellular common fluids (20–40 μM).15 It is also well known that because of up-regulation and down-regulation of various biological pathways in tumor cells, the concentration of GSH in their cytoplasm is four times higher than that of normal cells.16
This significant variation of the GSH concentration in the extracellular and intracellular compartments and moreover in tumor cells and normal cells provides an opportunity for designing target specific drug delivery systems in the tumor environment. For this purpose disulfide (S–S) containing amphiphiles are appropriate as the disulfide bond can be easily reduced to the thiol group in the presence of GSH. The disulfide bond exhibits a longer bond length than the C–C bond, low bond dissociation energy and low rotational barrier along the S–S axis, which make it dynamic in nature.17 In fact the disulfide bond is present in many biological systems. The thiol group of the cysteine residue is the source of sulfur in disulfide bond formation via oxidation in the cellular environment. The secondary and tertiary structures of many proteins contain the disulfide bond and its dynamic nature plays an important role in the folding and stability of proteins. For example, hair comprises a protein called keratin with very high disulfide content and depending on the disulfide percentage in hair, it may be soft or hard. Inspired by this, in the recent past a large number of amphiphilic systems containing the disulfide bond have been studied for nanostructure assembly, guest encapsulation and intra-cellular drug delivery application.18 In this review article, representative examples of nanostructure assemblies containing disulfide linkages, dynamics, GSH triggered disassembly and implications in drug delivery have been discussed.
Discussion
Disulfide containing small molecule amphiphiles
Disulfide based small molecule amphiphiles (SA) with various structural analogues have been studied in the context of redox responsive release, drug delivery and gene delivery. In this section we discuss a few representative examples of them.Regen and coworkers first introduced dynamicity in liposomes by introducing disulfide bonds in phospholipid based vesicles (PL-1–PL-3, Fig. 1), which could be polymerized (switched on) and depolymerized (switched off) via a thiol–disulfide redox cycle.19 They have performed photobleaching recovery experiments, using 3,3′-dioctadecyloxacarbocyanine perchlorate as a probe, which indicated that lateral diffusion within the vesicle membranes of PL-1 was reduced by approximately one order of magnitude upon polymerization, to a value of 2 × 10−9 cm2 s−1.19 Furthermore, they have examined the monomer exchange in phospholipid dimers containing disulfide bonds derived from saturated (DMPE, DPPE, and DSPE) and unsaturated (POPE and DOPE) phosphoethanolamines using nearest-neighbor recognition studies.20 The lipid–lipid interaction across the cholesterol rich phospholipid bilayers was also investigated by measuring nearest-neighbor preferences of exchangeable phospholipids derived from DMPE and DSPE (PL-4–PL-6, Fig. 1) in the presence of non-exchangeable dimers (i.e., templates) made from DMPE.21
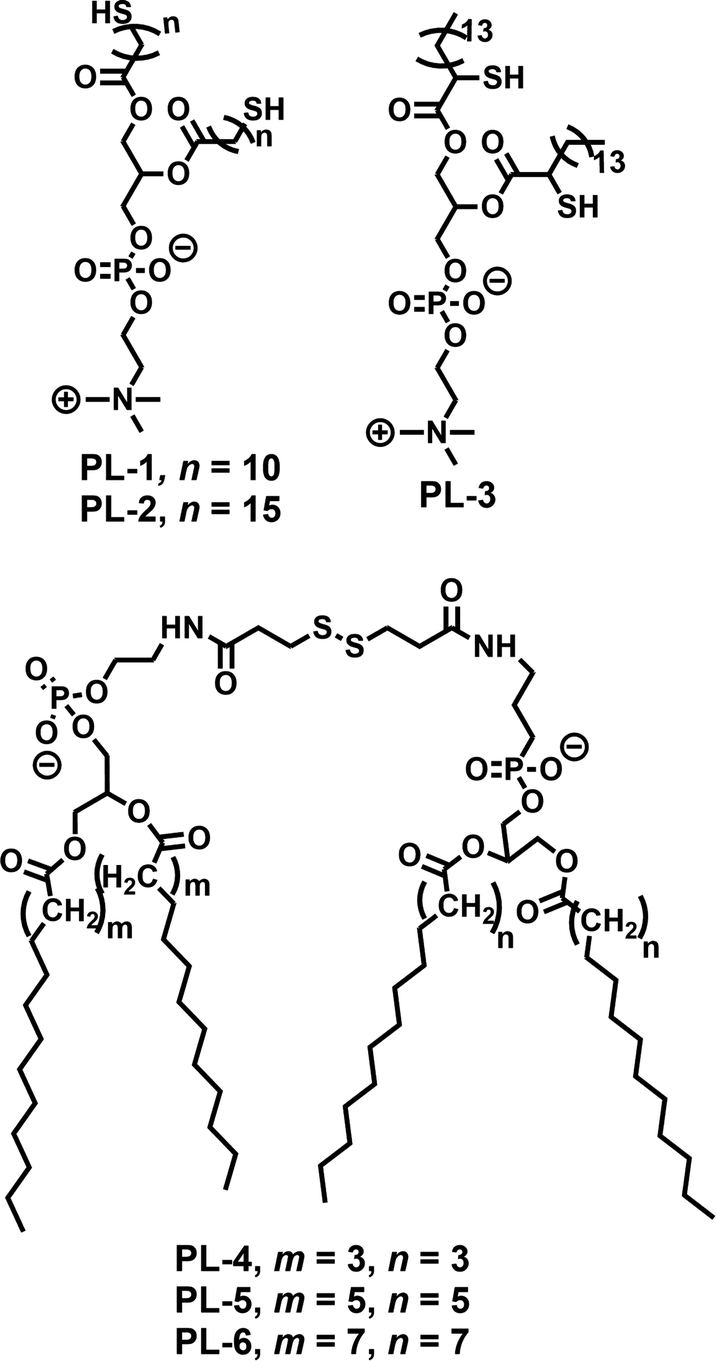 | ||
| Fig. 1 Structure of various phospholipids used for the preparation of disulfide containing liposomes as reported in ref. 19–21. | ||
Thyaumanavan and coworkers have reported a small molecule surfactant22 (SA-1) containing a redox responsive disulfide linkage between a hydrophilic carboxylate group and a hydrophobic alkyl chain (Fig. 2). In aqueous medium, SA-1 formed micellar aggregates which could encapsulate the hydrophobic guest Nile red (NR). The aggregates disassembled upon treatment with dithiothreitol (DTT) and released the entrapped dye molecules. They have studied the release of encapsulated Nile red by varying the concentration of DTT. In addition, tuning of the release kinetics was demonstrated by forming mixed-micelles containing SA-1 (disulfide-containing surfactant) and SA-2 (non-disulfide containing surfactant) in different proportions (Fig. 2).
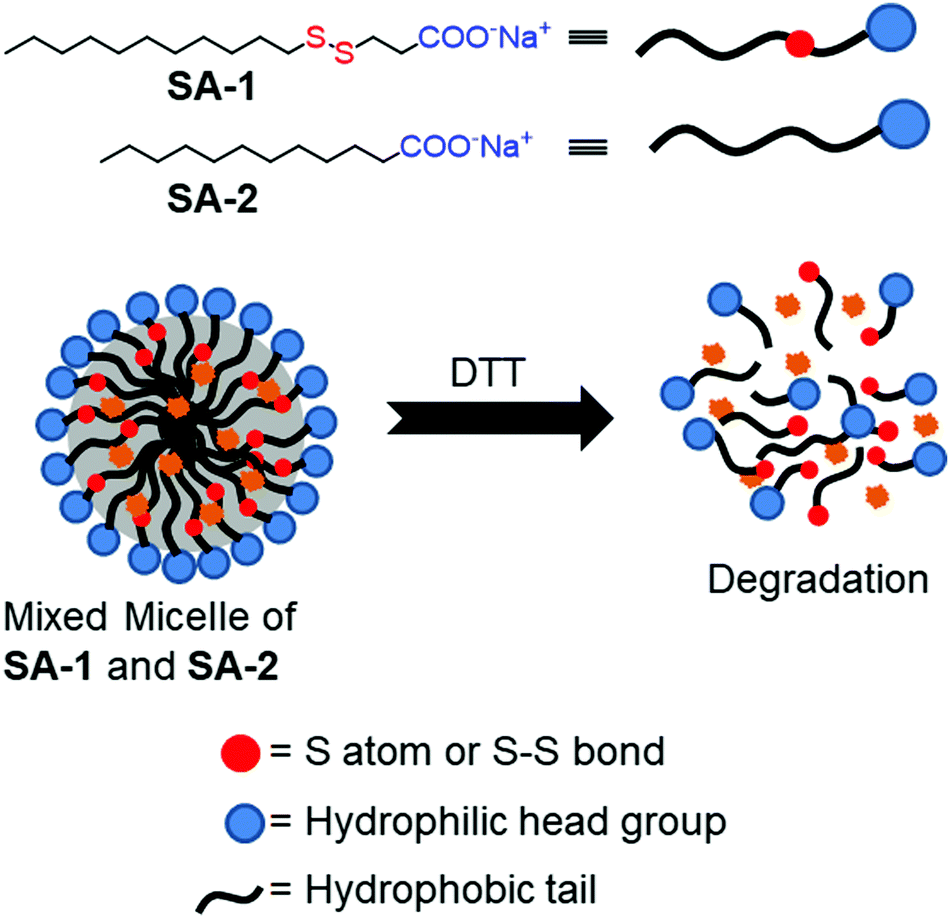 | ||
| Fig. 2 Aggregation and DTT induced disassembly of disulfide based surfactants (SA-1 and SA-2). Adapted from ref. 22. Copyright 2007, American Chemical Society. | ||
Gemini surfactants are a class of synthetic amphiphiles which possess, in sequence, a long hydrocarbon chain, an ionic group, a spacer, a second ionic group, and another hydrocarbon tail. Gemini surfactants with a redox cleavable linker were studied extensively (SA-3 to SA-7, Fig. 3a).23 In the presence of DTT or GSH, the disulfide linkages could be reduced to obtain the monomeric surfactant.23a–d Aggregation of these surfactants was studied in aqueous solution and the properties of the surfactants were compared to those of monomeric surfactants. Both the gemini and monomeric surfactant were able to form either vesicular or micellar aggregates in an aqueous medium but the critical aggregation concentration (CAC) was significantly increased for the monomeric surfactants. In addition, the monomeric surfactant could be oxidized to the original gemini surfactant by air oxygen or by addition of H2O2 (Fig. 3b).
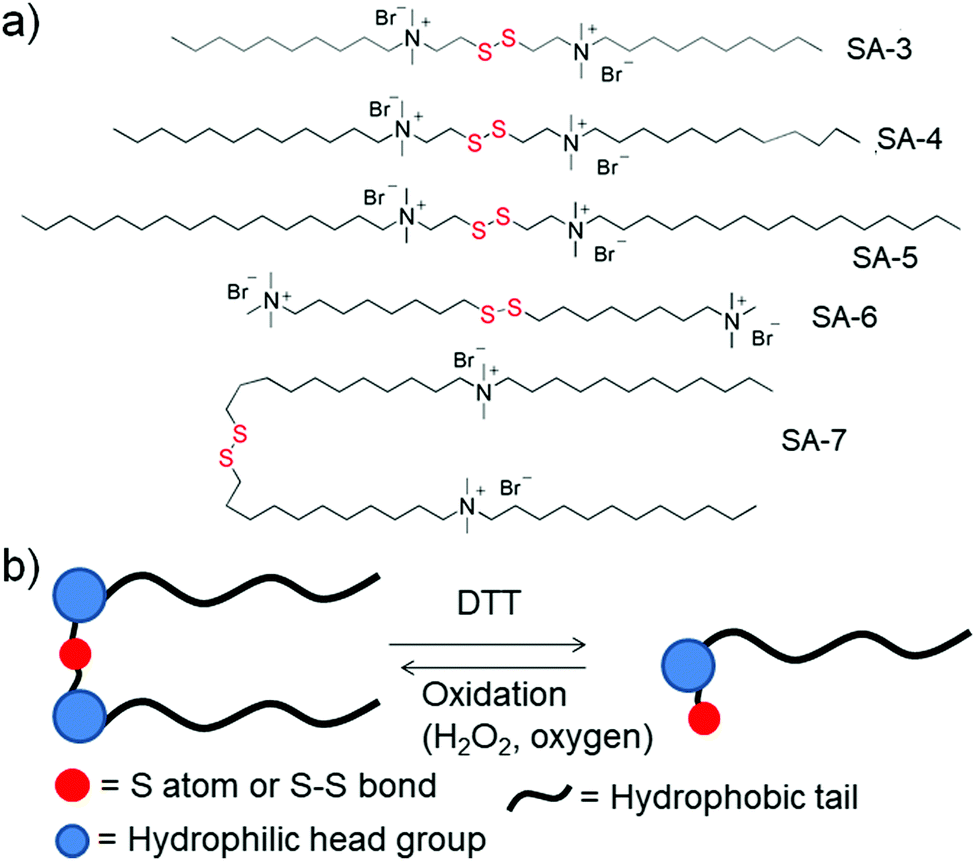 | ||
| Fig. 3 (a) Some representative examples of disulfide based gemini surfactants; (b) schematic representation of the equilibrium between gemini surfactants and their monomeric analogues on treatment with reducing agents. | ||
Cleavage of disulfide linkages with oxidation in gemini surfactants has also been reported in the literature.23e Here, the disulfide was oxidized to disulfone followed by cleavage of the disulfone bond, which happened in the presence of an oxidizing agent such as sodium hypochloride (NaOCl). Using one such kind of surfactant (SA-6, Fig. 3a), Abbott and coworkers have studied its surface active properties in the presence of various concentrations of oxidizing agent (NaOCl).23e
Cationic lipids bearing disulfide bonds (SA-8–SA-20, Fig. 4) have been explored as bio-responsive gene carriers.24 A series of such lipid amphiphiles have been studied by comparing the results with their nondegradable analogues. All the surfactants formed a stable complex with plasmid DNA via electrostatic interactions and have shown GSH triggered degradation and release of the complexed DNA. The transfection efficiency of these degradable complexes was found to be higher compared to their non-degradable analogues.
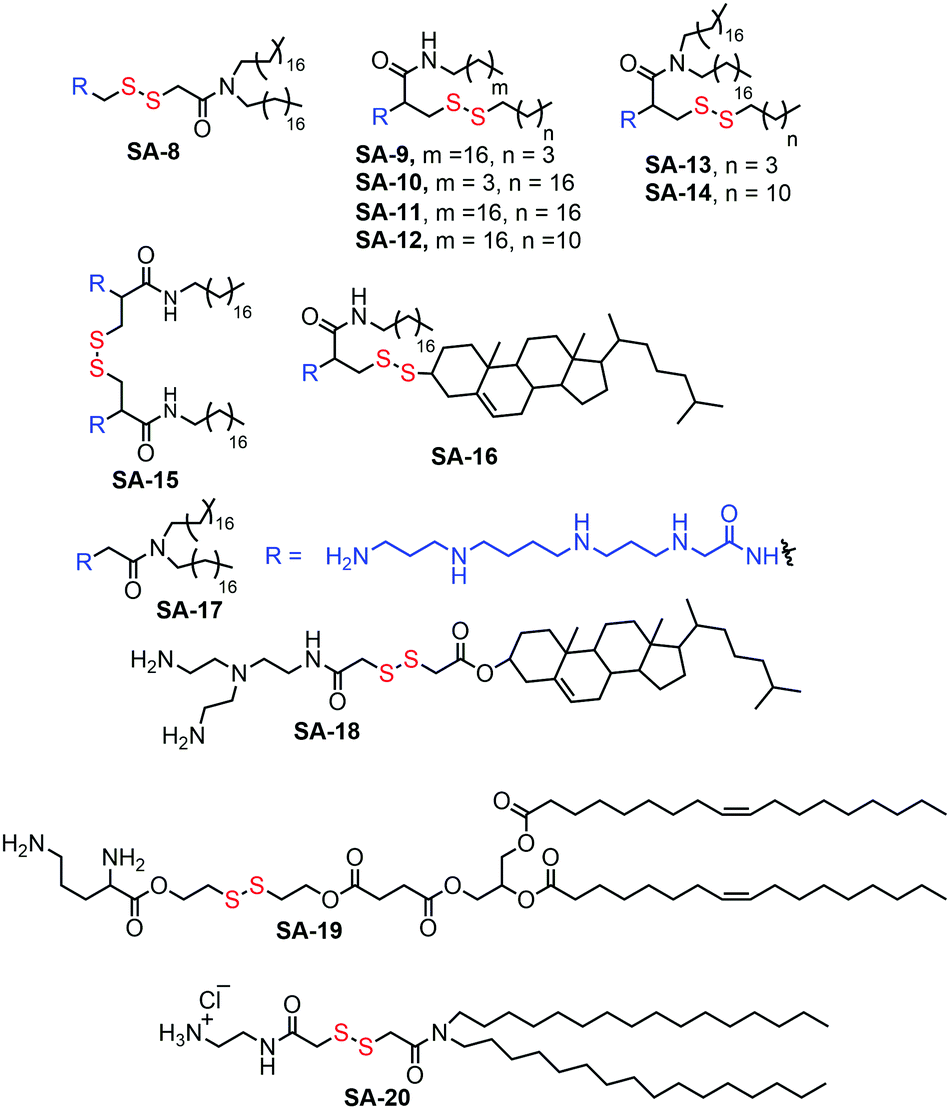 | ||
| Fig. 4 Various disulfide based cationic lipids used in gene delivery. | ||
The biocompatibility of the cationic lipids could be improved by replacing the cationic amine groups with glutathione (negatively charged) or cysteine residues (zwitterionic) by a disulfide exchange reaction. The surfaces were further decorated with a cell penetrating peptide to improve the accumulation of the polyplexes inside cells.25 However, the stability of this kind of amphiphilic nanovectors is questionable when considered for in vivo applications. Therefore, Haag and coworkers have synthesized disulfide-based temporarily fixed micelles (SA-21, Fig. 5) that can be degraded in reductive conditions, thus leading to efficient cargo release.26 The authors have conjugated lipoic acid to the hydrophobic tail of dendritic amphiphiles and upon complexation with RNA the hydrophobic core could be crosslinked, leading to enhanced stability of the polyplex. In the presence of the reducing intracellular environment the polyplex could be degraded, releasing the siRNA efficiently.26 As a result, potent gene silencing was achieved by the reduction-triggered disassembly. However, the non-crosslinked structurally similar constructs exhibited poor performance under similar tested assay conditions.
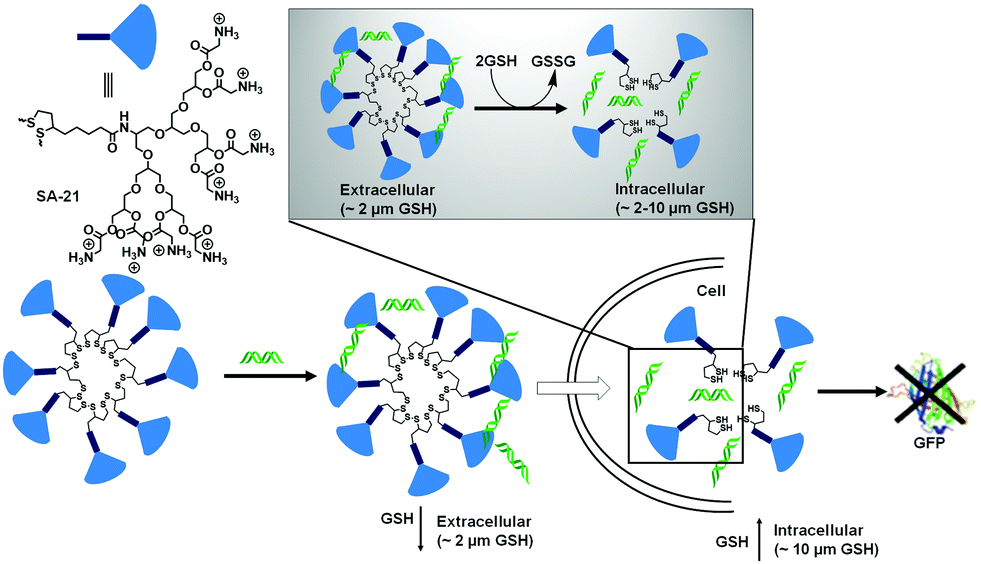 | ||
| Fig. 5 Structure of thioctic acid functionalized dendritic amphiphiles. Schematic representation of self-assembly (SA-21) in water and disassembly in the intracellular reducing environment. Adapted with permission from ref. 26. Copyright Wiley-VCH 2016. | ||
Disulfide based lipid amphiphiles have been explored for anticancer drug delivery.27 Recently, Bhattacharya and coworkers have reported27a redox responsive nanovesicles from two disulfide containing amphiphiles (SA-22 and SA-23) derived from tocopheryl–lipoic acid conjugates (Fig. 6). These liposomes could successfully encapsulate the anticancer drug doxorubicin (DOX) and release the drug in the presence of either DTT or GSH. DOX-loaded nanovesicles delivered the anticancer drug efficiently both in DOX-sensitive and DOX-resistant HeLa (HeLa-DOXR) cell lines. They concluded that the nanovesicle mediated DOX treatment showed significantly higher cell death compared to DOX alone in the HeLa-DOXR cell line. Furthermore, a few disulfide containing prodrugs have been reported based on camptothecin28 and paclitaxel29 which can form different nano-assemblies and under a reducing environment the potent drug could be released intracellularly leading to selective cell death.
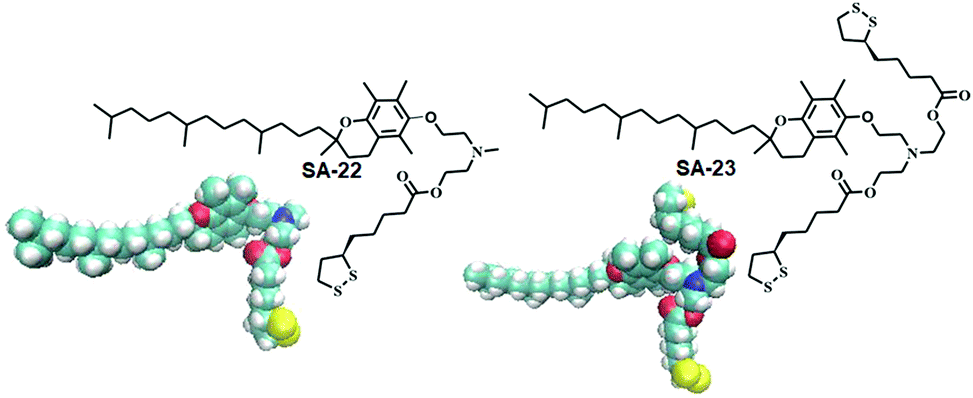 | ||
| Fig. 6 Lipoic acid containing amphiphiles used in anticancer drug delivery (reprinted from ref. 27a). Copyright 2018, American Chemical Society. | ||
Redox responsive supramolecular assemblies based on host–guest interactions have been reported which can disassemble in a reductive environment. Ravoo and coworkers have introduced30 disulfide groups in cyclodextrin based amphiphiles (SA-24 and SA-25) in which the disulfide bond is placed in between the hydrophobic substituents and hydrophilic macrocycle (cyclodextrin) (Fig. 7). SA-24 and SA-25 could form vesicles or nanoparticles in water and were capable of encapsulating hydrophobic guest molecules. Both the assemblies could be disassembled in the presence of DTT upon cleavage of the disulfide bond and thus the entrapped dye molecules could be released. They have also used a disulfide based crosslinker to prepare redox-responsive polymer nanocontainers comprising a cyclodextrin vesicle core and a thin reductively cleavable polymer shell anchored via host–guest recognition on the vesicle surface.31 The crosslinked nanocontainers were applied for live cell imaging experiments and cytoplasmic delivery of encapsulated hydrophilic payloads under a reductive environment, such as the pH-probe pyranine, and the fungal toxin phalloidin.31 In addition they have prepared similar disulfide crosslinked vesicles for studying multivalent binding using two widely different recognition motifs (mannose–Concanavalin A and biotin–streptavidin) and the impact of the ligand density on the nanocontainer surface was studied.32 Recently they have utilized similar CD vesicles for preparing nanocontainers to deliver amphiphilic phospholipids into cells.33 These nanocontainers were prepared in two steps.33 First the cargo lipids and reductively cleavable β-cyclodextrin amphiphiles (β-CDSS) were co-assembled into liposome-like cyclodextrin vesicle templates. Then they were decorated by host–guest chemistry with a reductively cleavable polymer shell.33
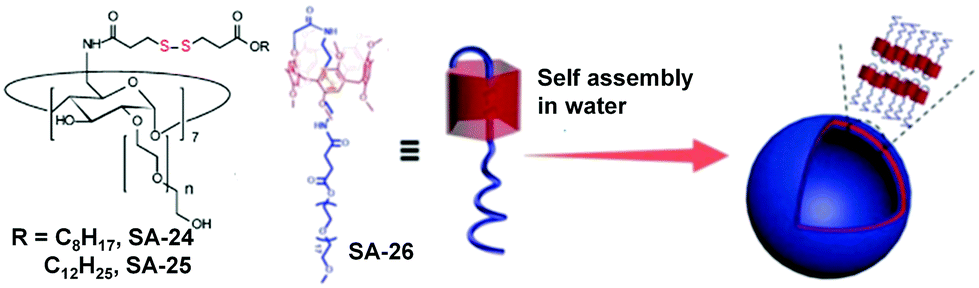 | ||
| Fig. 7 Disulfide containing cyclodextrin and pillar[5]arene based amphiphiles (adapted from ref. 30 and 34). Copyright 2003, American Chemical Society and the Royal Society of Chemistry 2019. | ||
Host guest interactions between pillar[5]arene and oligoethyleneglycol could also be utilized for the design of redox-responsive nanoassemblies by introduction of the disulfide bond in the guest PEGs. Using this principle, Yang and coworkers have fabricated a GSH-responsive delivery system from a pseudo[1]rotaxane macrocyclic amphiphile based on a PEG-modified pillar[5]arene.34 Disulfide bonds were introduced into the cavity of the pillar[5]arene, which was completely shielded and protected (SA-26, Fig. 7). The stable pillarene-based pseudo[1]rotaxane acted as a hydrophobic core component with the additionally attached PEG chain as a potential hydrophilic shell entity for self assembly. Furthermore, the amphiphile formed a supramolecular vesicle in aqueous medium with a very high loading of DOX.
Pyridinium groups are known to form host–guest complexes with pillar[5]arene. Zhang and coworkers have introduced the disulfide bond in the linker containing the host pyridinium group (Fig. 8).35 They have prepared spherical micelles in aqueous solution with good colloidal stability for preparing a responsive nano-assembly between a poly(ethylene glycol)-functionalized pillar[5]arene (PEG-P[5]A) and a pyridinium-terminated porphyrin derivative bearing a disulfide bond (TPPC6-SS-Py, SA-27, Fig. 8).35 In addition, they have demonstrated rapid release of porphyrin photosensitizers in a reducing environment.
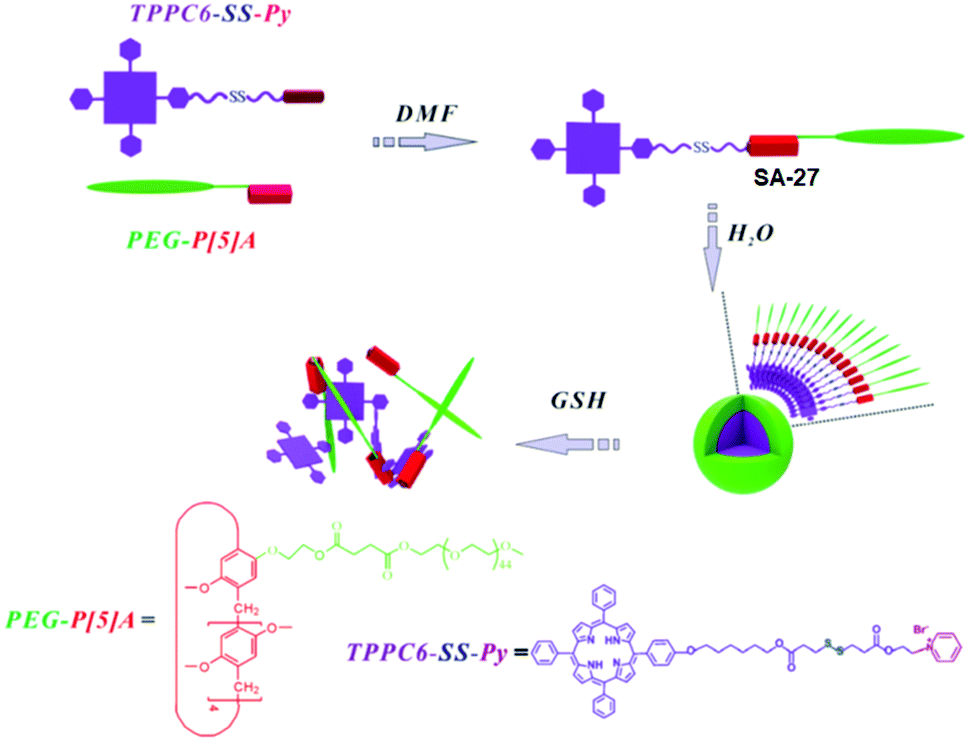 | ||
| Fig. 8 Illustration of the formation of PEG-P[5]A/TPPC6-SS-Py supramolecular micelles and their GSH-responsive drug release. Reprinted from ref. 35. Copyright 2016, the Royal Society of Chemistry 2019. | ||
Disulfide based amphiphilic polymers
To overcome the limitations of small molecule based amphiphiles, amphiphilic polymers have been explored to a great extent in the last 2–3 decades for drug delivery and other biological applications. In this context disulfide containing amphiphilic polymers have been studied for redox-responsive disassembly. Thayumanavan and co-workers have designed an amphiphilic polymer (P1, Fig. 9) containing a single disulfide linkage between the hydrophobic polystyrene (PS) and hydrophilic poly ethyleneoxide (PEO) block.36 They utilized the nanostructure assembly of this block copolymer to produce a nanoporous (with pore diameter ∼20 nm) thin film by the removal of PEO units after DTT-mediated cleavage of the disulfide linker.36 In addition, the available free thiol groups inside the porous structure were utilized for developing a gold nanostructure inside the polymeric cage (Fig. 9).36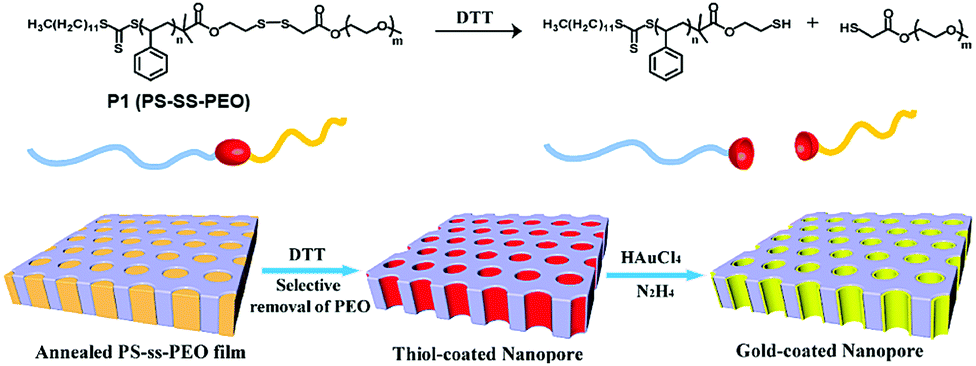 | ||
| Fig. 9 Structure of the block copolymer and schematic representation of the preparation of the nanoporous thin film and gold-coated nanopore. Adapted with permission from ref. 36. Copyright 2009, American Chemical Society. | ||
Similarly, different block copolymers containing single or multiple disulfides have been developed and used for preparing nanostructures and delivery of therapeutics such as chemotherapeutics like doxorubicin (DOX), paclitaxel (PTX), camptothecin (CPT), DNA or RNA, therapeutic proteins and so on.
Hubbell and co-workers reported the synthesis of a shell sheddable polymersome from a disulfide linked amphiphilic block copolymer (P2, Fig. 10)37 containing PEG as the hydrophilic block and polypropylenesulfides (PPS) as the hydrophobic block. They have shown the release of calcein from the polymersome due to disintegration of the shell using cysteine as the reducing agent.37 Kataoka and coworkers have extended this detachable shell micelle to a disulfide containing double hydrophilic block copolymer which can form a polyplex with DNA (P3, Fig. 10).38 The authors have synthesized mPEG-poly(aspertamide) which can complex with DNA. They showed that the introduction of the disulfide bond in the block copolymer led to a three-fold enhancement in gene transfection.38
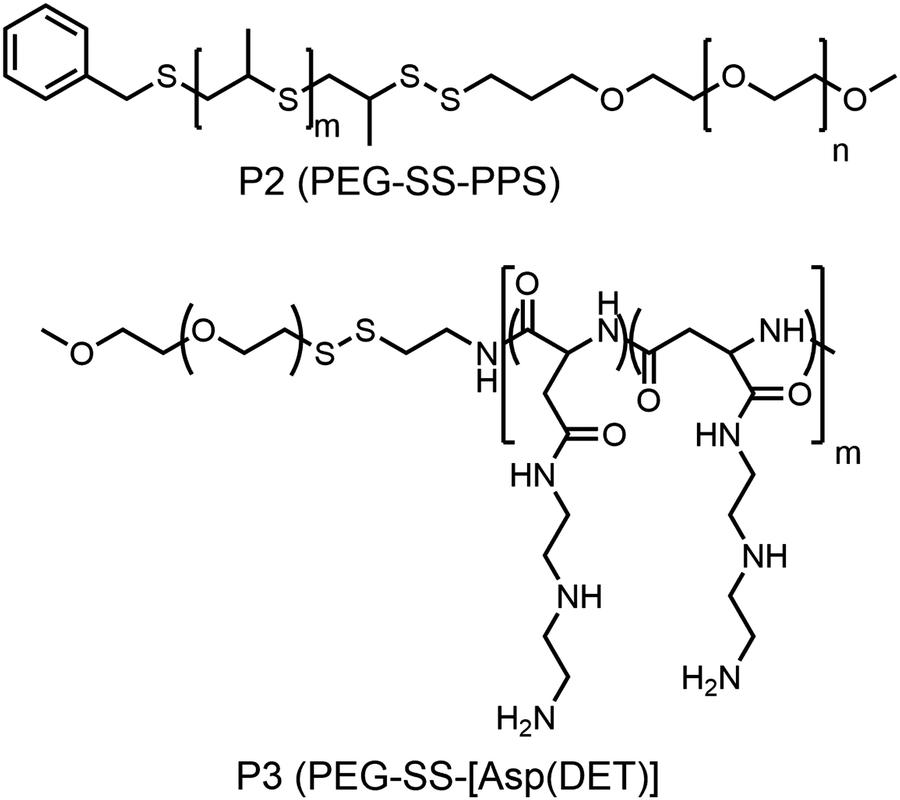 | ||
| Fig. 10 Structure of single disulfide containing polymers as reported in ref. 37 and 38. | ||
Furthermore, several groups have prepared single disulfide containing shell sheddable block copolymers based on polycaprolactone (PCL),39 polylactide (PLA) (P7)40 and polycarbonates (P8)41 where different hydrophilic blocks were used such as PEG (P4),39a poly(ethyl ethylene phosphate) (PEEP) (P5),42 dextran (P6)39cetc. (Fig. 11). Wang and coworkers have demonstrated that introduction of PEEP in the PCL chain enhanced the cellular uptake of the nano-carrier (P5).42b On a similar note Zhong and coworkers have prepared a PCL based diblock copolymer (P9, dPGS-SS-PCL) containing a disulfide bond in between the hydrophobic and hydrophilic shell where the anionic dendritic polyglycerol sulfate (dPGS) was used as a hydrophilic shell instead of commonly used PEG as a micellar shell to overcome the limitations of using PEG such as modest tumor cell targeting and retention (Fig. 12).43 This polymer has shown high DOX loading and low DOX leakage with faster DOX release inside tumor cells.43 Furthermore, the conjugation of an anti-inflammatory polymer (dPGS) to the PCL chain via a disulfide bond led to a better tolerated dose and therapeutic index without any major adverse effects as studied in MCF-7 human mammary carcinoma-bearing micelle.43
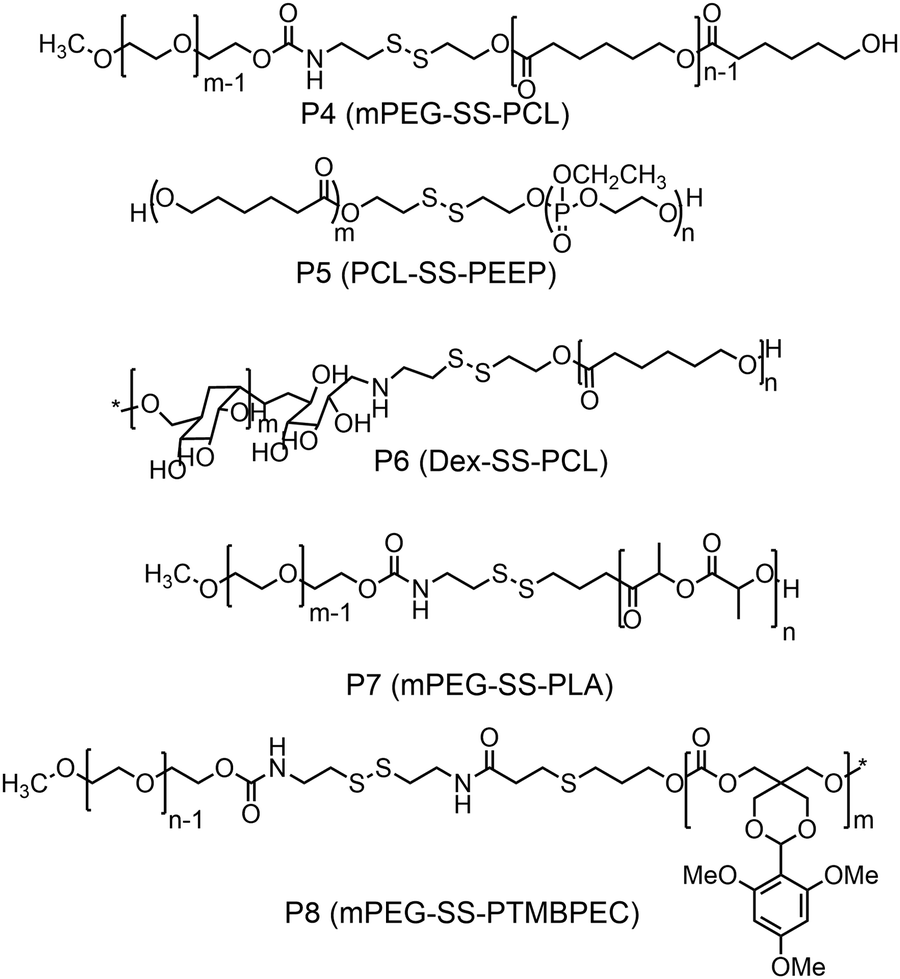 | ||
| Fig. 11 Structure of PCL, PLA and polycarbonate based amphiphilic polymers. | ||
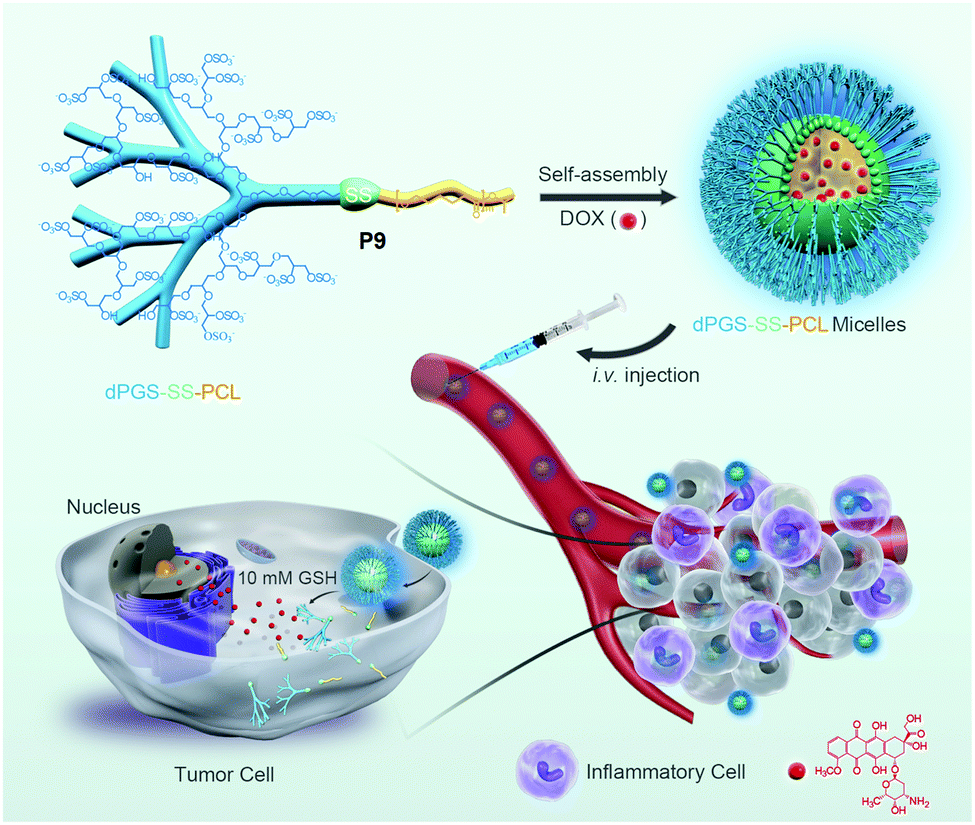 | ||
| Fig. 12 Schematic illustration of dendritic polyglycerol sulfate-SS-poly(ε-caprolactone) (P9, dPGS-SS-PCL) micelles for active targeting of inflammation-related tumor tissues and triggered cytoplasmic drug release within tumor cells. Reprinted with permission from ref. 43. Copyright 2016, American Chemical Society. | ||
Several other disulfide bridged bio-reducible block copolymers have been reported with varying hydrophilic and hydrophobic blocks, which are shown in Table 1 along with relevant information.44–48
| Block copolymer | Hydrophilic block | Hydrophobic block | Structure formed | Application | Ref. |
|---|---|---|---|---|---|
| PEG-g-DSMEG | mPEG | Poly(β-amino ester) | Micelle | DOX delivery | 44 |
| PEG-PPMD, PEG-PCMD | PEG | Poly(ω-pentadecalactone-co-butylene-co-3,3′-dithiodipropionate), poly(ω-pentadecalactone-co-N-methyldiethyleneamine-co-sebacate) | Micelle | Docetaxel delivery | 45 |
| PEG-SS-PBLG | PEG | Poly(γ-benzyl-L-glutamate) | Micelle | Campothecin delivery | 46 |
| mPEG-SS-PzLL | mPEG | Poly(ε-benzyloxycarbonyl-L-lysine) | Micelle | DOX | 47 |
| mPEG-ss-PNLG | mPEG | Poly[2(dibutylamino)ethylamine-L-glutamate] | Micelle | DOX delivery | 48 |
In many of these aggregated systems, the non-covalent encapsulation stability plays a key role in controlling the premature release of drugs or therapeutics at undesired locations. To tackle this issue primarily two approaches have been explored. One is crosslinking the core after micellization and the second approach is reducing the critical aggregation concentrations (CAC) by using star shape or hyperbranched core containing polymers instead of linear polymers. In this line Li and co-workers have reported a six arm star PCL-PEG based block copolymer containing single disulfides in each arm between the hydrophobic and hydrophilic block (P10, Fig. 13).49 The authors showed that the star shaped polymer aggregate had a very low CAC and was stable to high dilution which mimics the in vivo situation.
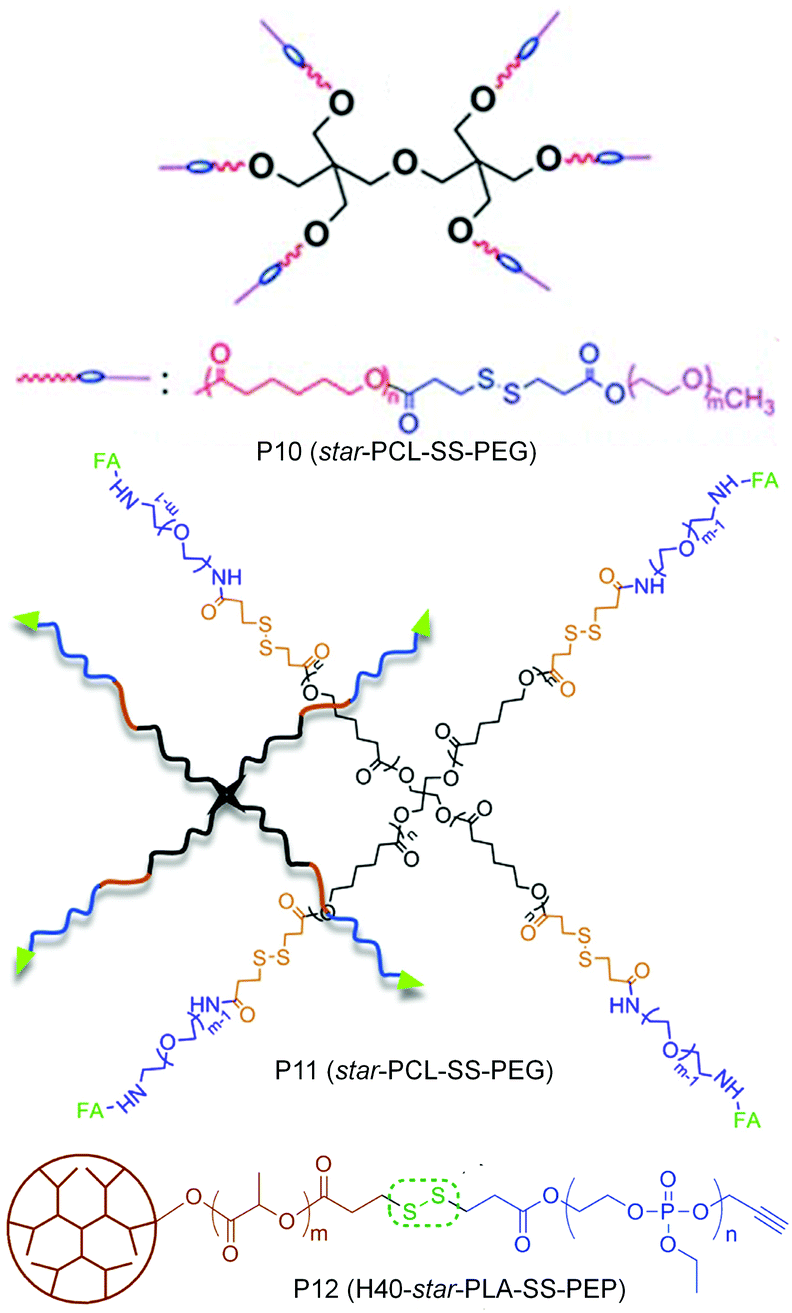 | ||
| Fig. 13 Structure of star amphiphilic polymers containing disulfide bonds. Adapted from ref. 49–51. Copyright 2011, Royal Society of Chemistry, 2014, Elsevier Ltd and 2011, American Chemical Society. | ||
Yan and coworkers have prepared a tri block copolymer H40-star-PLA-SS-PEP where PLA was grafted to a hyperbranched polyester core (P12, Fig. 13).50 Then the single disulfide was introduced in between the PLA and the hydrophilic poly(2-ethoxy-2-oxo-1,3,2-dioxaphospholane) (PEP).50 The effect of a reducing environment (0.1 mM DTT) on the degradation of the micelles as well as release of DOX was investigated. Zhou and coworkers have extended the use of a star (four arm) block copolymer PCL-SS-PEG to the delivery of DOX by introducing a targeting folic acid group at the terminal of the hydrophilic PEG chain (P11, Fig. 13).51 The authors showed the effect of active targeting and reduction sensitivity of the micelles on the efficient delivery of DOX in vivo in solid tumors.
While these examples demonstrate redox-responsive assemblies from polymers containing a single disulfide bridge, there have been also many reports on amphiphilic polymers containing multiple disulfide linkages (Fig. 14). Thayumanavan and coworkers have synthesized a block copolymer where disulfide bonds were present in the hydrophobic part (P13, Fig. 14), which can be cleaved under a reducing environment, leading to disassembly of the micelle.52
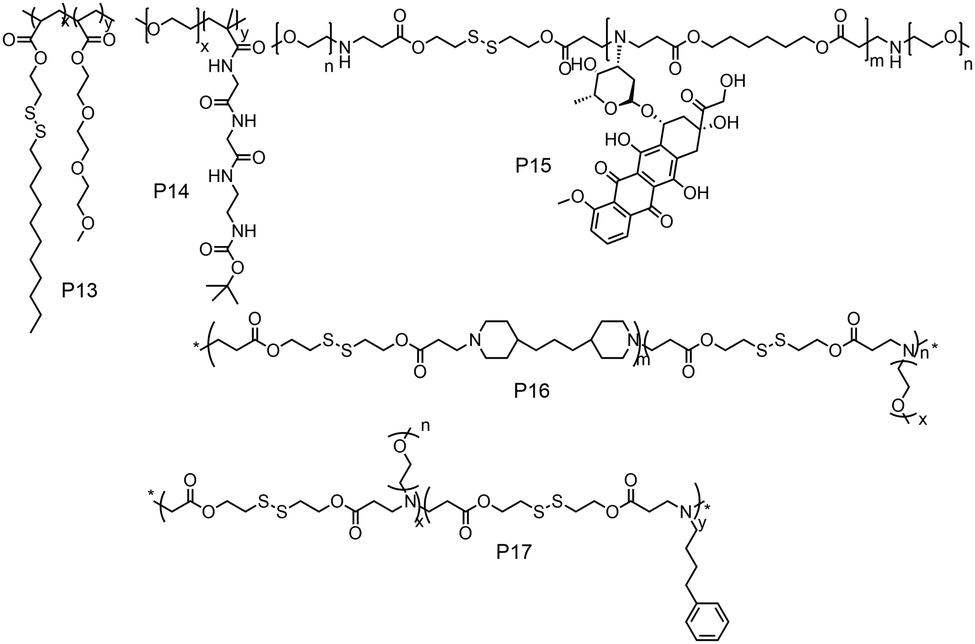 | ||
| Fig. 14 Structure of amphiphilic polymers with disulfide bonds present in the backbone or on the grafted hydrophobic chains as reported in ref. 52, and 54–57, respectively. | ||
They have prepared the polymer by polymerizing disulfide containing alkyl chains and OEG acrylate by free radical polymerization.52 They have shown reduction sensitive dye release in the presence of high GSH concentration, in vitro DOX release and antitumor activity.52 De and coworkers studied amphiphilic diblock copolymers consisting of polymerized segments of a pendent disulfide-labeled chlorambucil (CBL, loading content >40 wt%) prodrug monomer and hydrophilic 2-(dimethylamino)ethyl methacrylate monomer via reversible addition–fragmentation chain transfer polymerization.53 They have demonstrated CBL release and complete disassembly of micelles under a reductive environment.53
Gan and coworkers have developed poly(ethylene oxide)-b-poly(N-methacryloyl-N′-(t-butyloxycarbonyl)cystamine) (P14, PEO-b-PMABC) block copolymer by RAFT polymerization of a PEO based macro chain transfer agent.54 They have shown DTT mediated in vitro DOX release and antitumor activity, whereas the control polymer lacking pendant disulfides did not show any anti-tumor activity and drug release.54 Kong and coworkers have developed an amphiphilic polymer (P15, Fig. 14) containing multiple disulfide bonds where doxorubicin was polymerized with disulfide based diacrylate and mPEG amine in one pot.55 In this way the authors could synthesize a polymer containing 25.5% DOX loading and could efficiently release the drug in a reductive environment in the presence of GSH.55
Xing and coworkers have developed a multiple disulfide containing amphiphilic polymer (P16) by copolymerizing mPEG amine, 4,4′-trimethylene dipiperidine and 2,2′-dithiodiethanol diacrylate where disulfide groups are part of the main chain (Fig. 14).56 They have shown both reduction and pH responsive disassembly of these aggregates and release of doxorubicin.56 Similarly, the same group have synthesized another poly(β-amino ester) based amphiphilic polymer (P17, Fig. 14) using mPEG amine and 4-phenyl butyl amine groups as side chains.57 The incorporation of the 4-phenyl butyl amine group has increased the hydrophobicity, which increased the encapsulation efficiency of DOX in the micelles.
On the other hand, Park and co-workers have developed an amphiphilic copolymer by conjugating lithocholic acid to carboxymethyl dextran (P18, Fig. 15).58 The hydrophobic content could be varied by varying the amount of lithocolic acid to dextran. The conjugated polymer could form micelles driven by the hydrophobicity of lithocolic acid and able to encapsulate DOX which showed GSH mediated release in contrast to micelles formed from a polymer having no disulfides.58 Similarly, Zhang and coworkers have conjugated deoxycholic acid to hyaluronic acid (P19, Fig. 15),59 which showed a high loading capacity of paclitaxel (PTX) (34.1%) with a very high entrapment efficiency of 92%.59
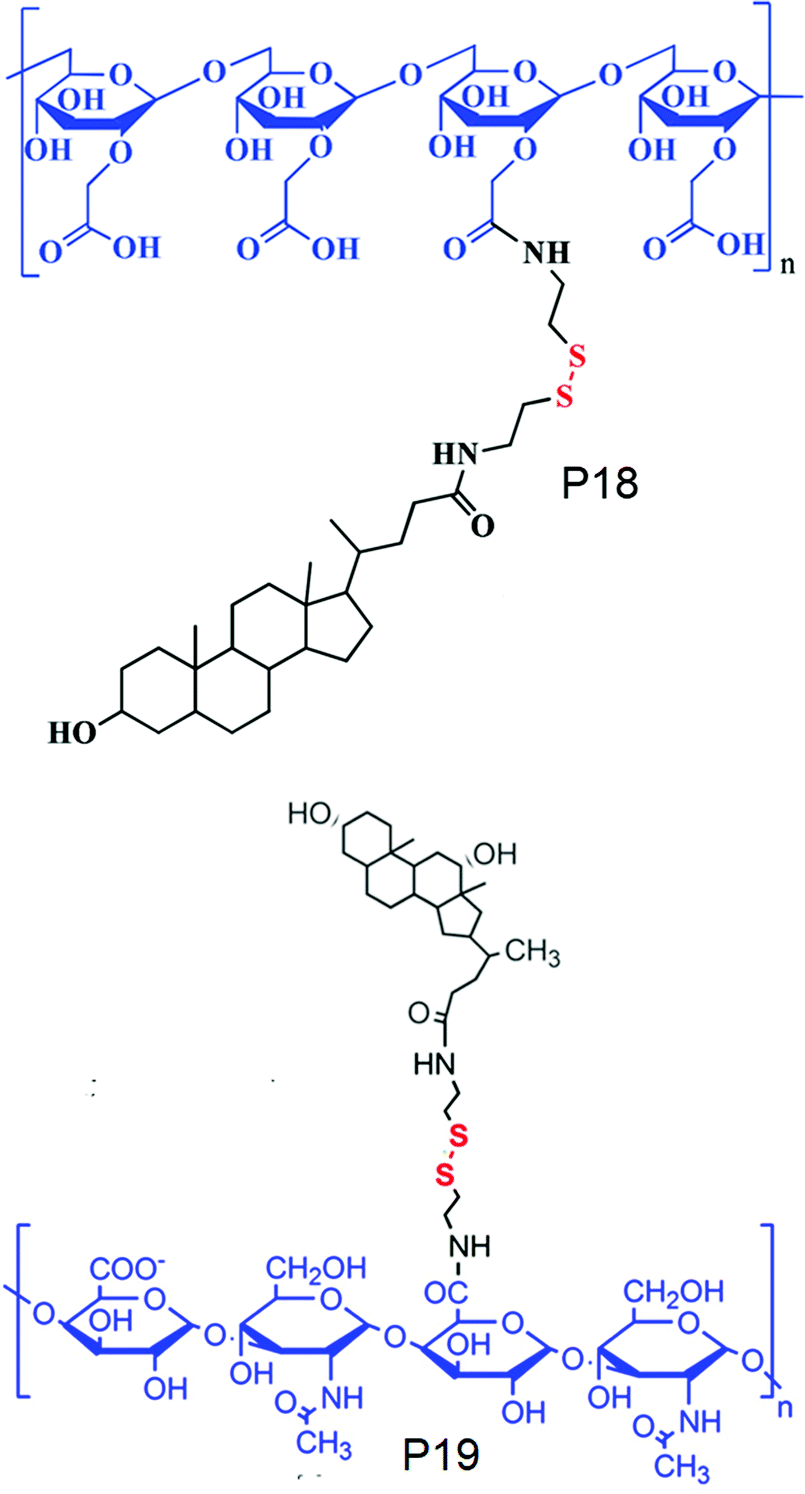 | ||
| Fig. 15 Structure of cholesterol conjugated carboxy dextran (P18) and hyaluronic acid (P19). Adapted from ref. 58 and 59 respectively. Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim and 2011, Elsevier Ltd. | ||
The previous discussion showed various examples of disulfide containing amphiphilic polymer aggregates. While conceptually they appear promising for drug delivery application, the exchange dynamics in such aggregates is a potential drawback for actual application. To address this issue, cross-linking the core of these aggregates by reversible or cleavable bonds is a promising method if the bond can be cleaved in response to physical or chemical stimuli given in the environment at the site of the drug action. Katoaka and co-workers utilized a disulfide based crosslinking agent to stabilize siRNA in the micellar core (Fig. 16).60 They introduced pyridyldisulfide groups (L1) in the polylysine block by reacting it with an NHS activated pyridyldisulfide (PyDS) linker (P20, Fig. 16). A PEG–P(Lys) based amphiphilic polymer was used for stabilizing poly(α,β-aspartic acid) (P(Asp)), a model for physiologically active polyelectrolytes including an oligonucleotide, in the micellar core by the oxidation of thiols introduced in the side chains of lysine units of PEG–P(Lys) (P20).60a Moreover, the authors have demonstrated micellar stability under a high salt concentration and the dissociation behavior of the crosslinked micelle after the addition of a reducing reagent by dynamic light scattering measurements.60a
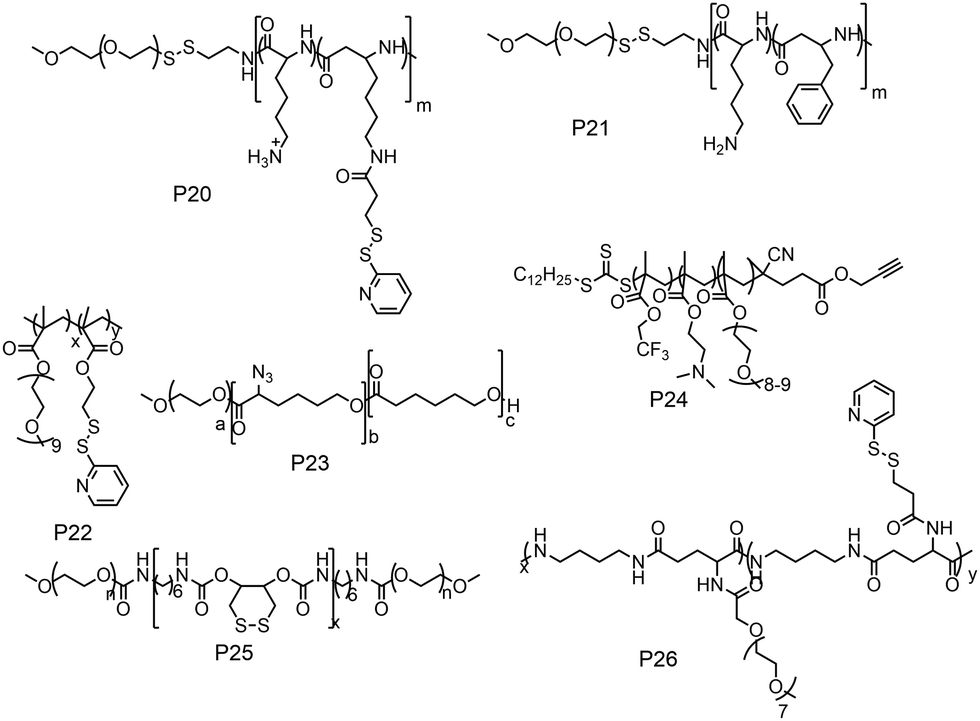 | ||
| Fig. 16 Amphiphilic polymers used for the preparation of crosslinked nanogels. | ||
The same group demonstrated encapsulation of antisense oligonucleotide (ODN) in the micelle. They have mixed the reducing agent DTT, which led to formation of crosslinked micelles keeping the ODN in the core.60b Upon dialysis, 2-thiopyridone groups could be removed. Similarly, by reacting with 2-imino thiolane the authors could generate free thiol groups from the poly L-lysine.61 By introducing such free thiol groups, they could perform crosslinking of the core and stabilization of the encapsulated cargo inside the micelle compared to the non-crosslinked one.61,62
In this regard, Lee and co-workers have developed a polylysine and phenyl alanine based triple block copolymer (P21, Fig. 16) which can assemble to form spherical micelles in water. Subsequent addition of a sulfo-NHS linker (L2, Fig. 17) led to the crosslinking of the polylysine layer, i.e., the middle block.63 In this way they could encapsulate methotrexate (MTX) and showed GSH triggered release. The non-crosslinked micelles have shown immature MTX release.63
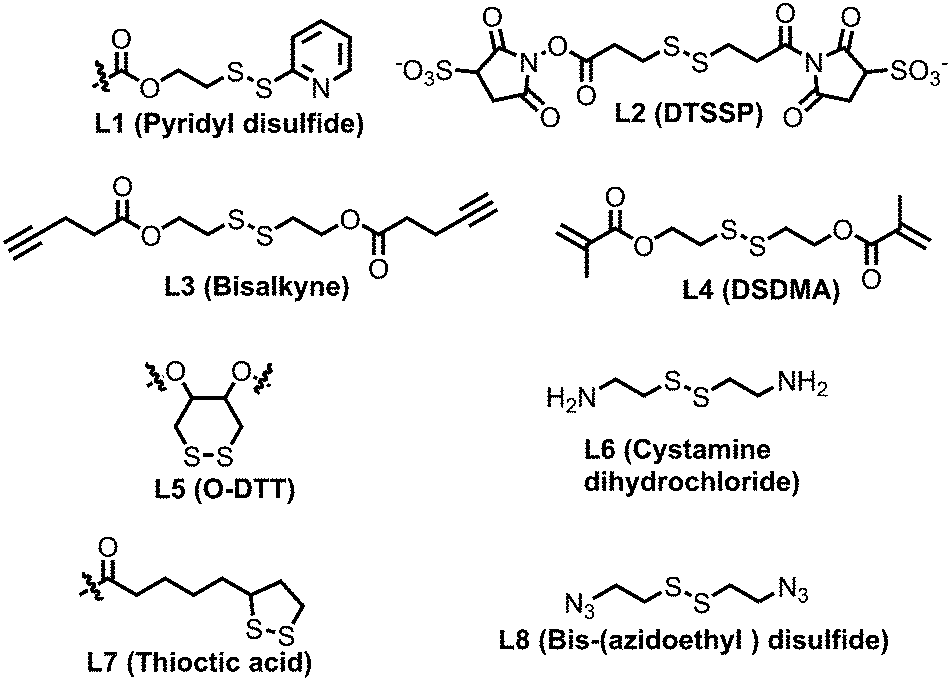 | ||
| Fig. 17 Structure of crosslinkers used for the preparation of the cross-linked nano-assembly. | ||
Thayumanavan and co-workers demonstrated a unique approach to synthesize redox-responsive nanogels. They used a random copolymer based on OEG and PyDS pendant groups (P22, Fig. 16)64 which can assemble to form a micelle with the PyDS groups located in the core. In the presence of a small amount of DTT, part of the PyDS groups are converted to free thiols which subsequently react with the remaining PyDS groups to cross link the core of the particle by a bio-reducible disulfide linkage. The authors have varied the crosslinking density by varying the amount of reducing agent (DTT), which was shown to have a significant impact on the exchange dynamics and release of the entrapped cargo.60 By encapsulating two hydrophobic dyes DiO and DiI (FRET pair) in the nanogels, the authors have demonstrated the stability of dye encapsulation inside the nanogels and tunability in the release of the guest molecules through in vitro fluorescence resonance energy transfer (FRET) experiments.64 The noncovalently encapsulated dye molecules were released in response to a redox trigger DTT as a consequence of degradation of the disulfide linkage. In addition, the authors have shown that hydrophobic chemotherapeutic drug molecules could be encapsulated in these polymeric nanogels and released in the intra-cellular environment to achieve drug-induced cytotoxicity.64
The same group has shown that the surface of the nanogels could be accessible for further modification.65 By treating the nanogels with cell penetrating Tat-SH (C-terminal cysteine), they could demonstrate successful tagging of the Tat peptide leading to facile and fast internalization of the surface modified nanogels compared to the non-tagged control nanogels.61 Jerome and co-workers have used a PEG–PCL based block copolymer (P23, Fig. 16) where polycaprolactone was functionalized with azide groups.66 Then, it was crosslinked by adding a hydrophobic bis-alkyne (L3, Fig. 17) crosslinker which acted through a 1, 3 dipolar cycloaddition reaction using Cu(I) as a catalyst. The authors have demonstrated the stability of the crosslinked particle by the lack of any disassembly when diluting the sample with DMF, which is good solvent for both the mPEG and PCL blocks.66 On the other hand, the non-crosslinked micelles were dissociated at a similar concentration.
Whittaker and coworkers have prepared a block copolymer (P24, Fig. 16) by RAFT polymerization of OEG methacrylate, 2,2,2-trifuoroethyl methacrylate (TFEMA) and 2-(dimethylamino)ethyl methacrylate units.67 They crosslinked the core via RAFT polymerization using bis(2-methacryloyl)oxyethyl disulfide (L4, Fig. 17) to have a star polymer.67 The authors have utilized this core crosslinked star polymer for 19F MRI contrast agents for selective imaging.
Chen and coworkers have explored disulfide mediated crosslinking using DTT in a polyurethane (P25, Fig. 16) prepared by polymerizing O-DTT (L5, Fig. 17), hexane diisocyanate and mPEG. After forming the micelle, they could crosslink the micelles by the reducing agent. They utilized this crosslinked micelle for the delivery of DOX.68 Similarly, Thayaumanavan and coworkers have prepared polyamide based self-crosslinked polyamide based nanogels from an amphiphilic random copolymer (P26, Fig. 16)69 and demonstrated degradation of the nanogel in the presence of GSH.
Recently the same group has shown that engineering of PyDS crosslinking can be useful for entrapping RNA.70 They have prepared a polymer containing quaternized PyDS groups (P27, Fig. 18).71 After complexation with RNA, DTT was introduced to convert part of the PyDS groups to free thiol, which initiated core crosslinking by reacting with the rest of the PyDS groups. In this process charged 2-thiopyridone groups could also be removed,70,71 producing a non-cationic (and thus non-toxic) RNA-polymer nanogel system. In the presence of excess reducing agent the crosslinking was destroyed, resulting in removal of the entrapped RNA in the nanogel. The same group also utilized PyDS mediated crosslinking for synthesizing a CF3 group containing nanogel which was applied as an MRI imaging probe.72 A few other systems on disulfide containing crosslinked nanoassemblies for different applications are shown in Table 2.73–80
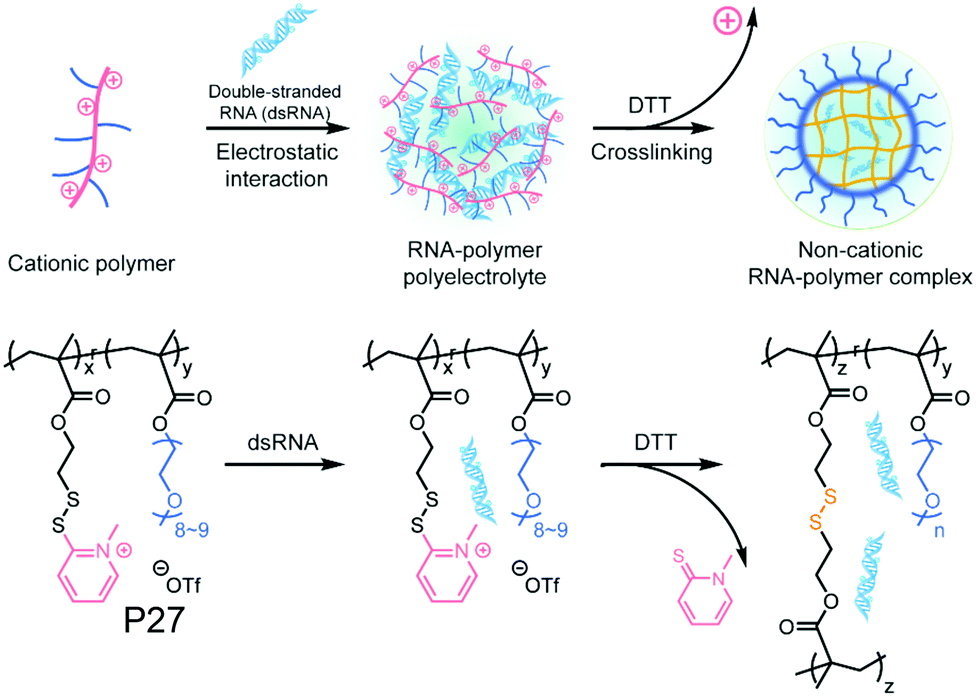 | ||
| Fig. 18 Schematic illustration of formation of the non-cationic RNA–polymer complex. Reprinted from ref. 70. Copyright 2019, American Chemical Society. | ||
| Block copolymer | Hydrophilic block | Hydrophobic block | Structure formed | Crosslinking chemistry | Application | Ref. |
|---|---|---|---|---|---|---|
| P(GMA65-stat-GlyMA1.7)-PHMA140 | Glycerol methacrylate | HMPA | Worm like micelle | Cystamine (L6) | — | 73 |
| MPC-co-GMA-co-SMA | MPC | Stearyl methacrylate | Micelle | Cystamine (L6) | — | 74 |
| PEG-P(HEMA-co-AC) | PEG | Polyhydroxethyl methacrylate and Acrolyl carbonate | Micelle | Cystamine (L6) | Cytochrome C delivery | 75 |
| PEG-Pglu(EDA-LA)-PPhe | PEG | Phenylalanine co L-glutamate | Micelle | Thiotic acid (L7) | DOX delivery | 76 |
| PEG-b-(MPC)n | PEG | poly(5-methyl-5-propargylxycarbonyl-1,3-dioxane-2-one) | Micelle | Bis-(azidoethyl) disulfide (L8) | DOX delivery | 77 |
| PEG-b-PTyr-LA | PEG | poly(L-tyrosine)-lipoic acid | Micelle | Lipoic acid (L7) | DOX delivery | 78 |
| PEG-D-poly(L-cysteine) | PEG | Poly(L-cysteine) | Micelle | Cysteine, photodegradation | CPT delivery | 79 |
| HA-P9TMC-DTC) | Hyaluronic acid | Poly(trimethylene carbonate-co-dithiolane trimethylene carbonate | Micelle | Thiolane | Docetaxel delivery | 80 |
Amphiphilic polydisulfides
As discussed in the previous sections, disulfide containing amphiphilic polymers have been widely used in the field of biomedical application as a delivery vehicle for drugs, genes, siRNA, proteins and so on owing to the dynamic and redox degradable nature of the disulfide bond. However in the majority of the examples, the disulfide linkage has been embedded into a polymeric scaffold which itself is not degradable.This may have adverse implications in biological applications. In this regard, fully bio-reducible polydisulfide (PDS) scaffolds may be attractive as the entire backbone in such cases may be degradable under a reducing environment.
In fact PDSs have been known for a long time and mostly have been synthesized using oxidative polymerization,81 which does not endow structural diversity and limits the scope for sophisticated biological application. We have developed a versatile methodology for synthesis of functional PDSs by a step-growth polymerization between the commercially available pyridyl disulfide (commonly known as aldrithiol-2) and a di-thiol under mild conditions.82 Extending this methodology we could prepare amphiphilic PDS with a hydrophobic backbone and pendant hydrophilic carboxylate groups (P28, Fig. 19),83 which under basic conditions showed aqueous self assembly, generating micelle-like structures with hydrophobic guest encapsulation ability and glutathione responsive sustained release.
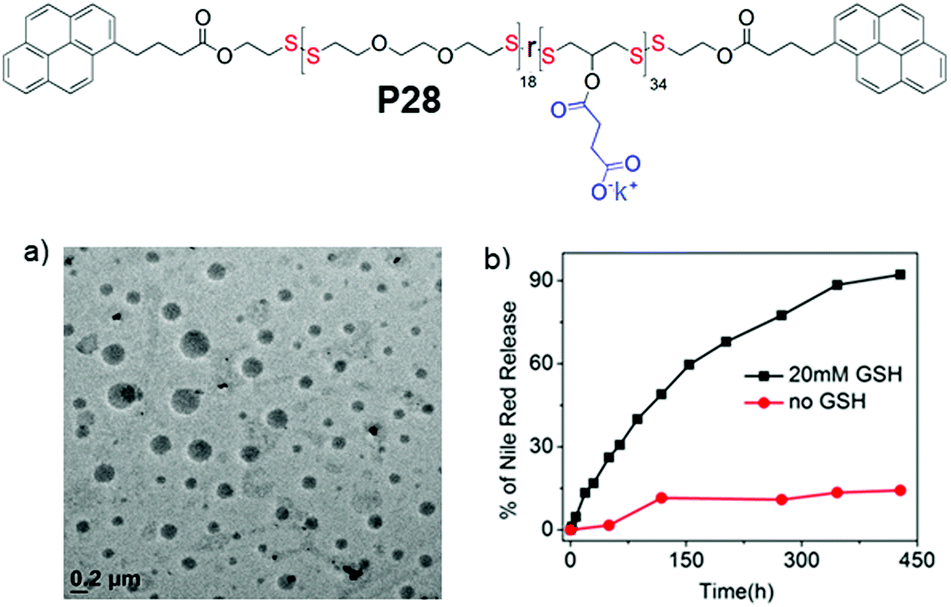 | ||
| Fig. 19 Top: Chemical structure of carboxylic acid pendant amphiphilic poly(disulfide) P28. (a) HRTEM image of an aqueous dispersion of P28 (C = 100 μM); (b) release profile of encapsulated Nile red in P28 in the absence (0.0 mM) and presence of 20 mM GSH. Adapted from ref. 83. Copyright 2018 Wiley and Sons. | ||
We further extended this strategy to synthesize telechelic hydrophobic PDSs, which were used as a macroinitiator to grow hydrophilic polymers from both terminals, producing two ABA type amphiphilic block copolymers with a fully degradable PDS middle block with tunable hydrophobicity (P29, P30, Fig. 20).84 In aqueous medium, both the polymers formed micellar aggregates with an average diameter of ∼15 nm. To check the impact of such structural changes on the micellar disassembly and drug release kinetics, Nile red (NR) was encapsulated as a hydrophobic probe in the interior of the micelles. For P29 micellar assembly with a relatively more hydrophobic interior, slower disintegration of the backbone and dye-release kinetics were observed compared to the micelles with a less hydrophobic core (P30), owing to slow diffusion of the polar GSH toward the more hydrophobic micellar core.
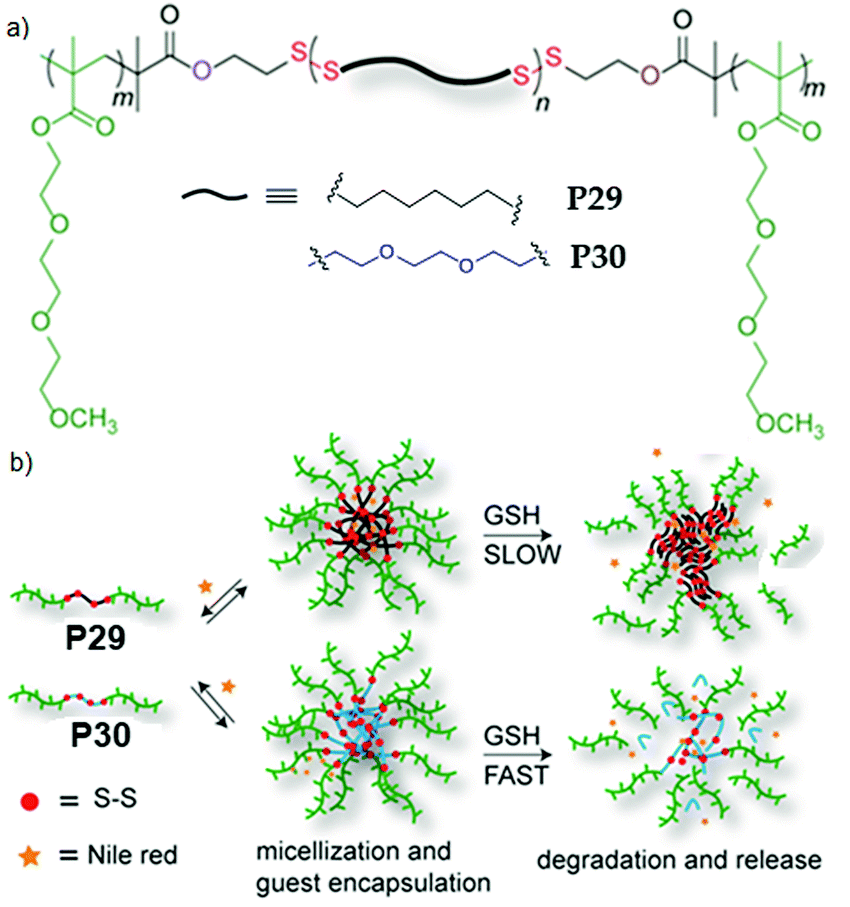 | ||
| Fig. 20 (a) Chemical structures of amphiphilic poly(disulfide) P29 and P30. (b) Aqueous aggregation and glutathione (GSH) induced backbone degradation and release of encapsulated Nile red in the micellar core of P29 and P30. Adapted from ref. 84. Copyright 2015 Royal Society of Chemistry. | ||
Subsequently we have compared the aggregation and drug delivery performance of P31 and P32 (Fig. 21),85 which have the same hydrophobic PDS block but different hydrophilic blocks such as poly-N-isopropylacrylamide (PNIPAM) or poly(triethyleneglycol) moomethylether-methacrylate (PTEGMA). In contrast to P32, P31 formed a polymersome structure, which can be rationalized by the volume occupied by the hydrophilic blocks in these two polymers (Fig. 21).85 The PNIPAM block possibly occupies less volume than the PTEGMA block and therefore the packing of the hydrophobic PDS block is influenced, resulting in two different morphologies, which enabled us to examine the impact on intra-cellular drug-delivery. Both of the polymers were found to be remarkably biocompatible and enabled encapsulation of an anticancer drug doxorubicin (DOX).
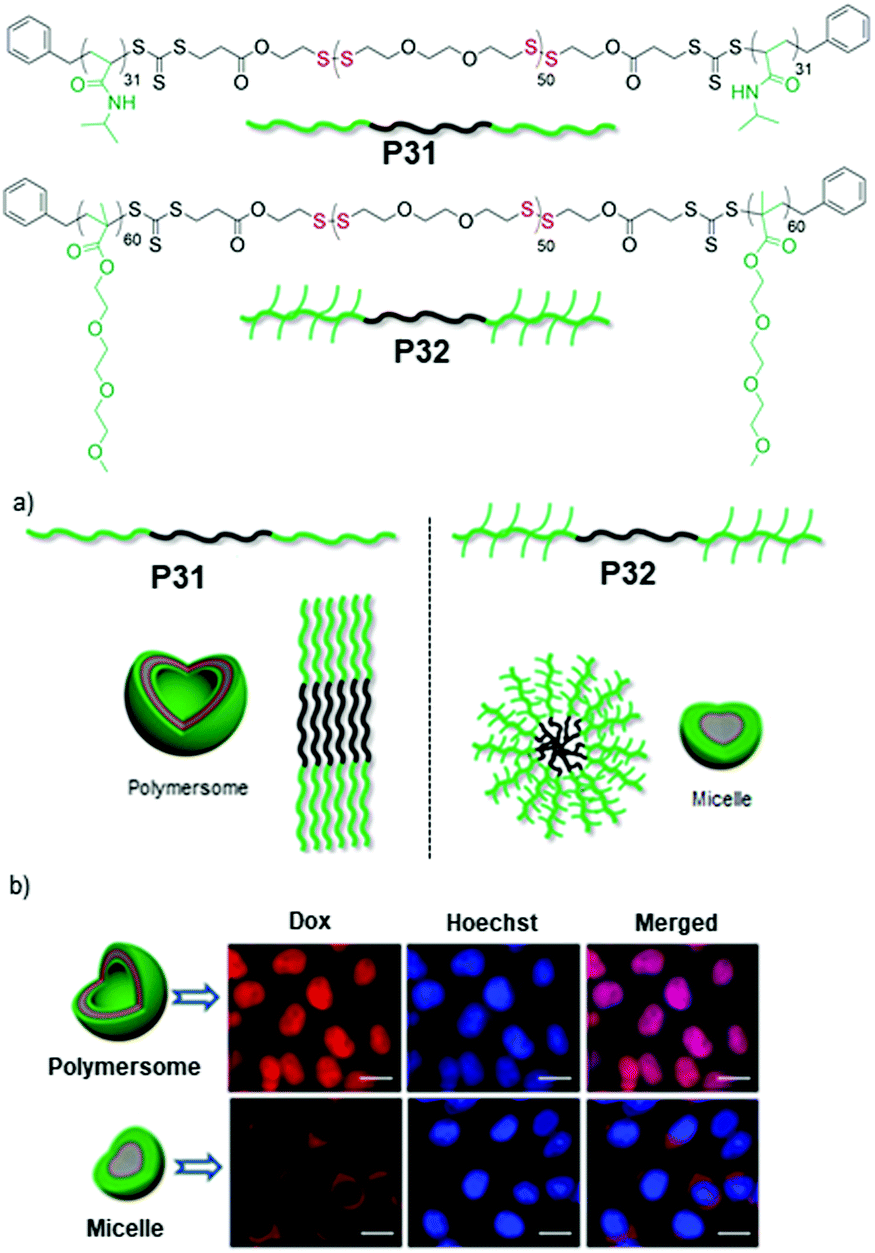 | ||
| Fig. 21 Top: Chemical structures of amphiphilic poly(disulfide) P31 and P32. (a) Schematic showing the different packing model for aqueous aggregates of P31 and P32. (b) Fluorescence microscopy images of HeLa cells after 48 h of treatment with the Dox loaded polymersome (P31) and micelle (P32). (Scale bar = 20.0 μm in all images). Adapted with permission from ref. 85. Copyright 2018 Wiley and Sons. | ||
But glutathione (GSH) triggered degradation and release of the entrapped drug appeared much faster from the polymersome than the micelle, which was further reflected in their cell killing efficiencies. The DOX encapsulating polymersome (P31, Fig. 21) showed excellent killing efficiency of HeLa cells (cancer cells) compared to the DOX encapsulating micelle (P32, Fig. 21). Time dependent cellular uptake experiments revealed that the polymersome entrapped drug escaped from the cargo and reached the nucleus effectively, while the drug-loaded micelle still remained trapped in the perinuclear zone, explaining the significant difference in the drug delivery performance of the polymersome and micelle.85
We have recently reported conjugation of a chemotherapeutic agent, camptothecin (CPT), with redox-destructible amphiphilic poly(disulfide)s (PDS) by a labile carbonate linker (P33, Fig. 22).86 This PDS-CPT conjugate exhibits a fluorescent polymersome assembly in water which could be disassembled in the presence of GSH. GSH triggered cleavage of the backbone disulfide bond generated free thiol which intra-molecularly attacked the carbonate group linking the CPT moiety, resulting in release of the active drug. The drug-appended polymersome entered into cells within 4 h and showed excellent killing efficiency (IC50 = 3.1 μg mL−1) selectively of cancer cells, whereas the non-drug-conjugated PDS analogue appeared to be biocompatible, confirming the nontoxic nature of the polymer backbone and also the degradation products.86
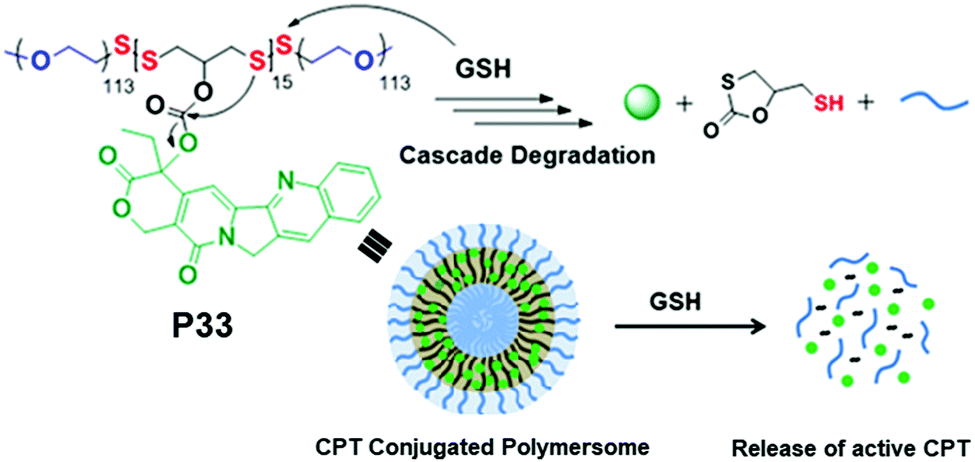 | ||
| Fig. 22 Chemical structure, self-assembly and GSH triggered cascade degradation of CPT conjugated amphiphilic poly(disulfide) P33. Reproduced with permission from ref. 86. Copyright 2019 American Chemical Society. | ||
Cell-penetrating polydisulfides
In recent years cell penetrating polydisulfides (CPDs) have emerged as a promising alternative to other existing cell penetrating molecules like cell penetrating peptides (CPPs).87 The main difference lies in the backbone where the polypeptides in CPPs were replaced with polydisulfides (CPDs),88 which are believed to be advantageous as the endogenous glutathione (GSH) reduces the CPDs intracellularly upon cell uptake, leading to spontaneous cargo release (Fig. 23).89 The main problems of CPPs are associated with toxicity whereas CPDs are nontoxic due to such cytosolic degradation. The other major advantage of CPDs is that they undergo cellular uptake via an endocytosis independent pathway (Fig. 23) by specific interaction with cell surface thiols.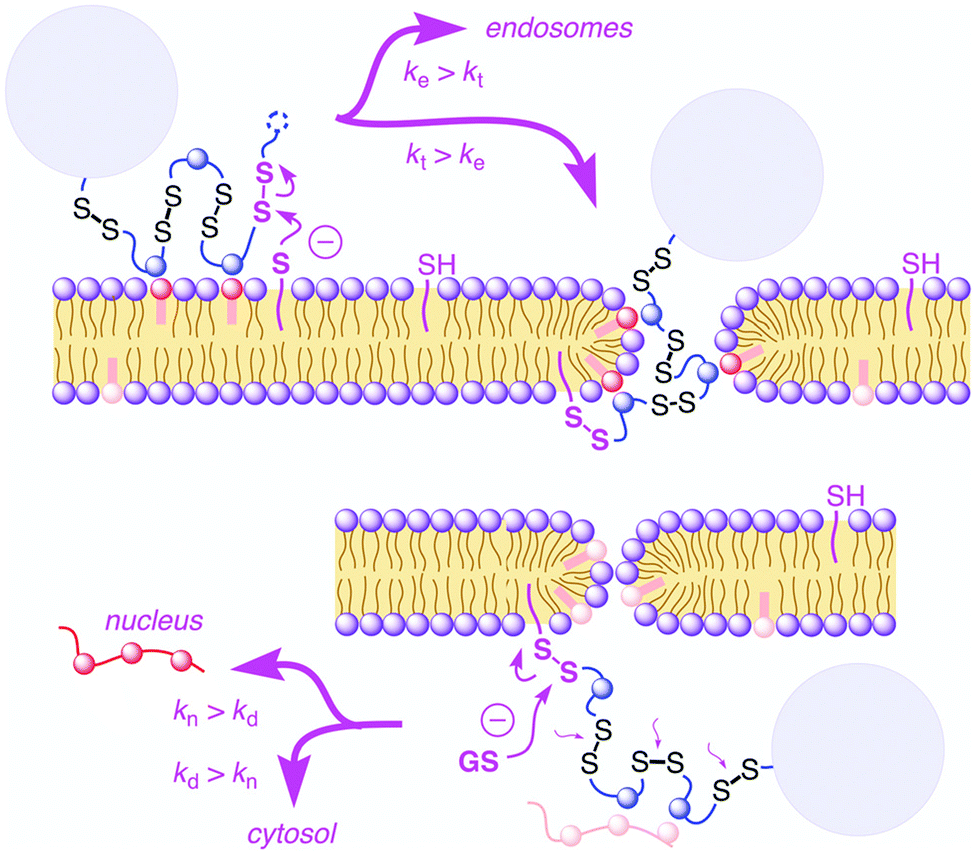 | ||
| Fig. 23 Cellular uptake of cell-penetrating poly(disulfide)s: supported by ion pairing of CPDs (blue) with anionic lipids (red), CPDs are attached covalently to the cell surface by disulfide exchange with exofacial thiols (magenta, top left). Reprinted from ref. 89a. Copyright the Royal Society of Chemistry 2016. | ||
In 2013, Matile and coworkers introduced the concept of substrate initiated cell penetrating polydisulfides (siCPDs) where strained disulfide (thioctic acid) based guanidine containing cyclic monomers were polymerized in situ under mild conditions using disulfide ring opening exchange polymerization (Fig. 24).90 These CPDs entered cells through a distinct pathway using the dynamic covalent disulfide bonds present in the backbone (Fig. 23). At first the CPDs approached the cell surface due to electrostatic interactions of ion pairs of CPDs with anionic lipids in the membranes by the powerful proximity effect known from CPPs and other polycationic oligomers.91 In addition, disulfides present on the CPDs were engaged in dynamic covalent chemistry with exofacial thiols present on the cell surface. This rapid thio-disulfide exchange resulted in covalent attachment of the CPDs to the cell surface (Fig. 23). The blocking of free thiols present on the cell surface using Ellman's reagent led to a reduction of the uptake of CPDs which in turn supported the cell uptake mechanism of CPDs (Fig. 23).91
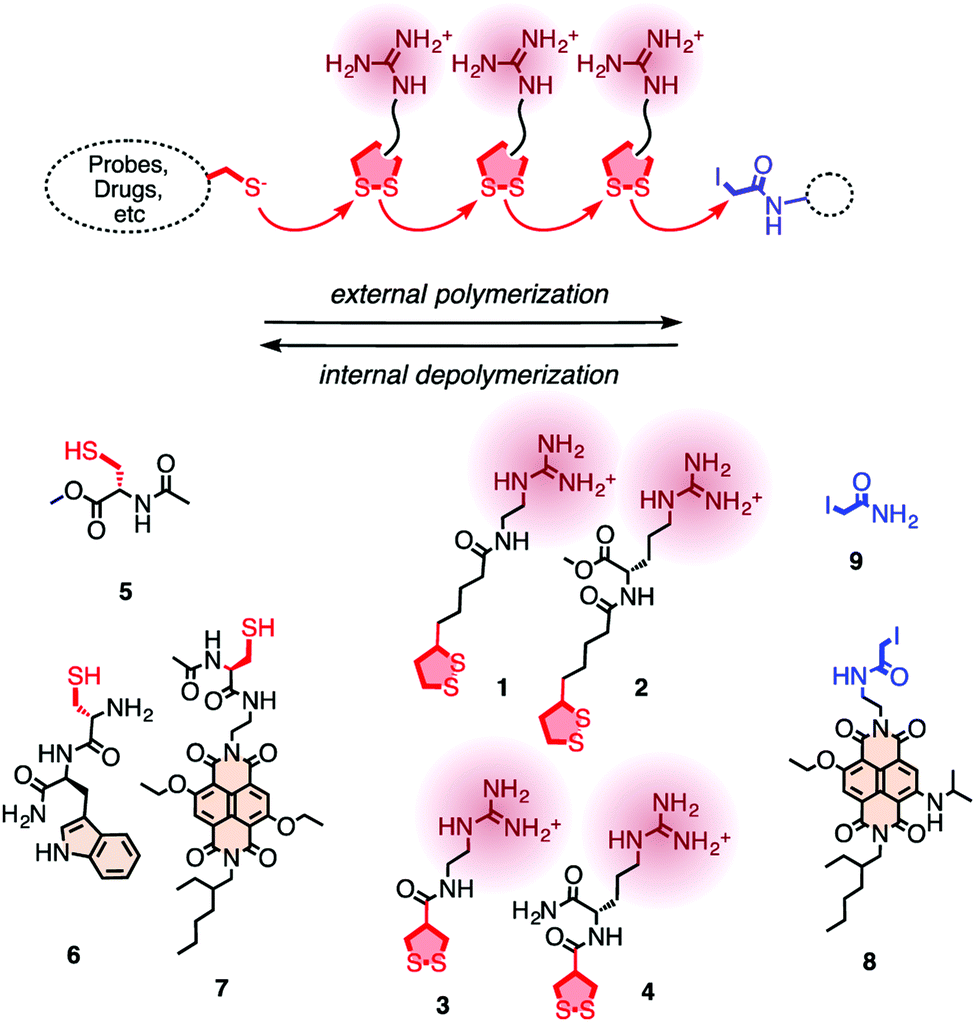 | ||
| Fig. 24 For the covalent delivery of unmodified probes, guanidinium-containing propagators (1–4) are polymerized on thiolated substrates (e.g., 5–7) and terminated with iodoacetamides (8, 9). Reprinted from ref. 90. Copyright 2013 American Chemical Society. | ||
Furthermore, utilizing these concepts the authors have shown delivery of proteins like streptavidin and quantum dots in the cytosol.92 Yao and coworkers have also used a similar strategy to deliver antibodies and silica nanoparticles to cancer cells via endocytosis independent pathways.93 To overcome the limitation of using iodoacetamides as a terminator, Matile and coworkers further introduced ethynyl benziodoxolones where a click reaction could be performed to introduce functional groups such as aromatic dyes.89a The incorporation of π-basic, hydrophobic side chains besides guanidinium cations was found to produce longer polymers with increased activity.94 They have also demonstrated that the ring tension played the crucial role in the cellular uptake compared to the inactivated or activated linear disulfides or with thiols.95
Recently, Abe and coworkers have introduced disulfide groups at the terminus of oligonucleotide strands by phosphoramidite chemistry.96 They have modified oligonucleotides with two different disulfides: a tertiary butyl disulfide group or a cyclic disulfide unit (Fig. 25). Both of them were functional and showed cellular uptake in an endocytosis independent pathway96 with similar efficiency of transport to the cytoplasm.
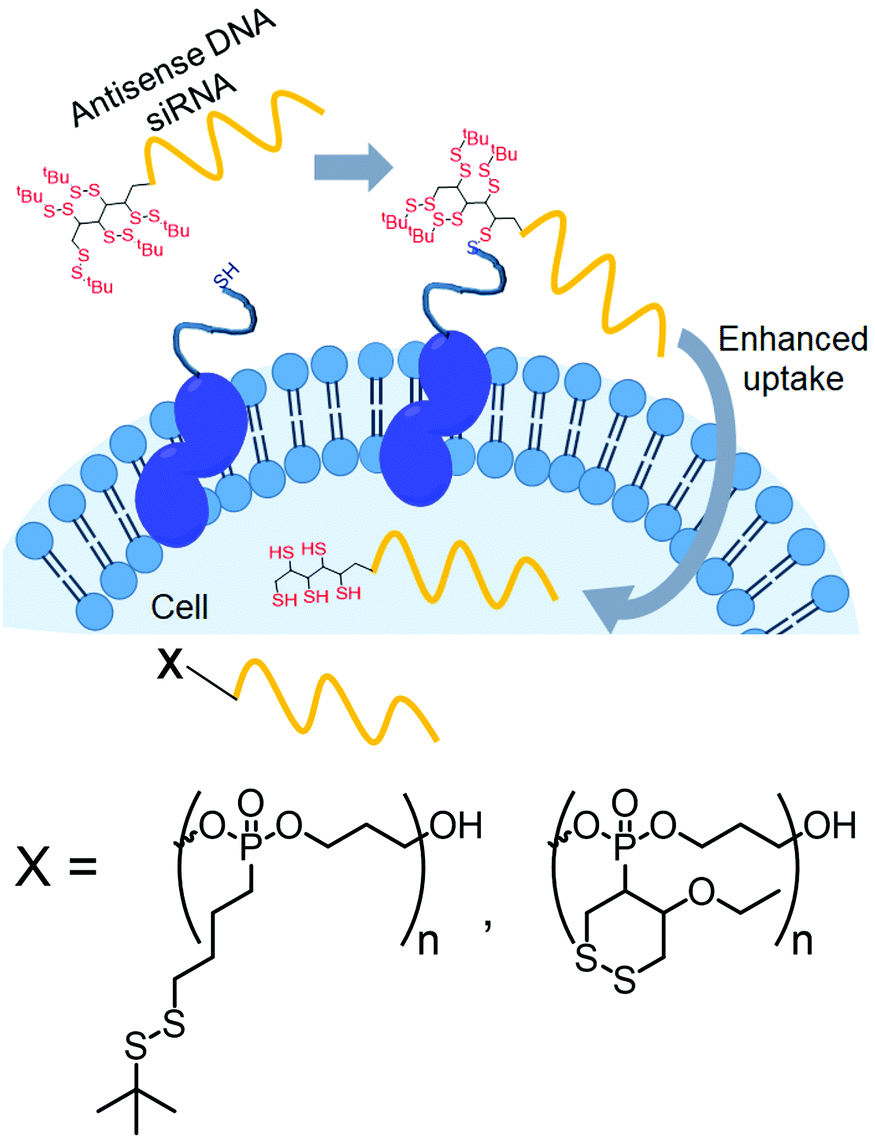 | ||
| Fig. 25 Conjugation with disulfide units enhanced the permeability of oligonucleotides. Structure of disulfide containing blocks used for the preparation of the cell penetrating polymer. Adapted from ref. 96. Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
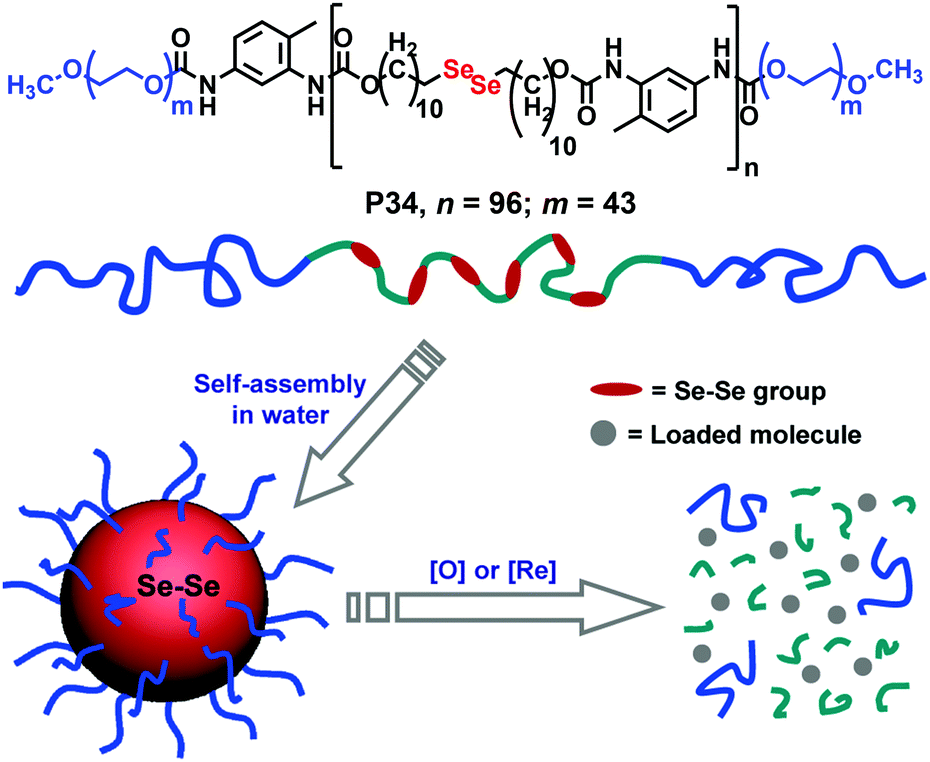 | ||
| Fig. 26 Structure of the diselenide containing amphiphilic block copolymer and redox responsive degradation in the presence of GSH. Reproduced from ref. 97b. Copyright 2010 American Chemical Society. | ||
Conclusions
In this review article we have highlighted various examples of disulfide containing amphiphilic small molecules and macromolecules, their aqueous aggregation generating wide ranging soft nanostructures with redox responsive properties. Amphiphilic polymers were discussed based on the number and location of disulfides present in the polymer backbone. In addition disulfide based crosslinkers to impart stability and stimuli responsiveness in the micellar core have been discussed. However, in the majority of these examples the disulfide group cleavage does not ensure total degradation of the polymer, which has been addressed by the fully bio-reducible amphiphilic polydisulfides. Recent studies demonstrate structure dependent morphology variation in these materials and the strong impact on their drug delivery application. Finally, we have demonstrated cell penetrating polydisulfides which appear promising for future application. Overall it is evident that disulfide chemistry has much to offer in biological applications of synthetic polymers and small molecule amphiphiles, which has been firmly established by in vitro experiments. However, further understanding on intra-cellular organelle targeting and pharmacokinetics and in vivo studies are essential to recognize the true potential of these disulfide based soft materials for biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RB thanks CSIR for a scholarship and PD thanks IACS for a post-doctoral fellowship.Notes and references
- (a) Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC; (b) Y. Wang and S. M. Grayson, Adv. Drug Delivery Rev., 2012, 64, 852 CrossRef CAS; (c) T. H. Epps and R. K. O’Reilly, Chem. Sci., 2016, 7, 1674 RSC; (d) N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295 RSC; (e) J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525 RSC; (f) M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323 CrossRef CAS; (g) T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38 CrossRef CAS.
- (a) X.-B. Xiong, A. Falamarzian, S. M. Garg and A. Lavasanifar, J. Controlled Release, 2011, 155, 248 CrossRef CAS; (b) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS; (c) R. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836 CrossRef CAS; (d) R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449 RSC.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343 CrossRef CAS.
- (a) G. Fleischer, J. Phys. Chem., 1993, 97, 517 CrossRef CAS; (b) I. Goldmints, J. F. Holzwarth, K. A. Smith and T. A. Hatton, Langmuir, 1997, 13, 6130 CrossRef CAS; (c) B. Michels, G. Waton and R. Zana, Langmuir, 1997, 13, 3111 CrossRef CAS; (d) A. G. Denkova, E. Mendes and M. O. Coppens, Soft Matter, 2010, 6, 2351 RSC; (e) P. Rajdev and S. Ghosh, J. Phys. Chem. B, 2019, 123, 327 CrossRef CAS; (f) G. Waton, B. Michels and R. Zana, Macromolecules, 2001, 34, 907 CrossRef CAS.
- (a) H. Maeda and Y. Matsumura, Cancer Res., 1986, 46, 6387 Search PubMed; (b) J. Fang, H. Nakamura and H. F. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136 CrossRef CAS; (c) F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135 CrossRef CAS.
- M. I. Gibson and R. K. O’Reilly, Chem. Soc. Rev., 2013, 42, 7204 RSC.
- J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117 RSC.
- C. Y. Zhang, Y. Q. Yang, T. X. Huang, B. Zhao, X. D. Guo, J. F. Wang and L. J. Zhang, Biomaterials, 2012, 33, 6273 CrossRef CAS.
- (a) J. Zhuang, M. R. Gordon, J. Li, L. Ventura and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421 RSC; (b) L. Zhai, Chem. Soc. Rev., 2013, 42, 7148 RSC.
- (a) L. C. Glangchai, M. Caldorera-Moore, L. Shi and K. J. Roy, J. Controlled Release, 2008, 125, 263 CrossRef CAS; (b) Z. Gu, M. Yan, B. Hu, K.-I. Joo, A. Biswas, Y. Huang, Y. Lu, P. Wang and Y. Tang, Nano Lett., 2009, 9, 4533 CrossRef CAS; (c) M. A. Azagarsamy, P. Sokkalingam and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 14184 CrossRef CAS.
- (a) S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS; (b) P. Couvreur, E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866 CrossRef; (c) D. A. Torres, M. Garzoni, A. V. Subrahmanyam, G. M. Pavan and S. Thayumanavan, J. Am. Chem. Soc., 2014, 136, 5385 CrossRef; (d) E. Cabane, X. Zhang, K. Langowska, C. G. Palivan and W. Meier, Biointerphases, 2012, 7, 1 CrossRef.
- F. G. Hopkins and E. J. Morgan, Biochem. J., 1936, 30, 1446 CrossRef CAS.
- E. Q. Rosenthal-Kim and J. E. Puskas, Pure Appl. Chem., 2012, 84, 2121 CAS.
- (a) F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191 CrossRef CAS; (b) D. Montero, C. Tachibana, J. R. Winther and C. Appenzeller-Herzog, Redox Biol., 2013, 1, 508 CrossRef CAS.
- (a) K. M. Halprin and A. Ohkawara, J. Invest. Dermatol., 1967, 48, 149 CrossRef CAS; (b) D. P. Jonesa, J. L. Carlsona, P. S. Samieca, P. Sternberg Jr., V. C. Mody Jr., R. L. Reed and L. A. S. Brown, Clin. Chim. Acta, 1998, 275, 175 CrossRef.
- (a) A. Russo, W. DeGraff, N. Friedman and J. B. Mitchell, Cancer Res., 1986, 46, 2845 CAS; (b) P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307 CAS.
- (a) W. J. Leesand and G. M. Whitesides, J. Org. Chem., 1993, 58, 642 CrossRef; (b) S. Ulrich, Acc. Chem. Res., 2019, 52, 510 CrossRef CAS; (c) P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682 CrossRef CAS.
- (a) H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef; (b) J. K. Oh, Polym. Chem., 2019, 10, 1554 RSC; (c) R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2 CrossRef CAS; (d) J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 97 RSC; (e) M. H. Lee, Z. Yang, C. W. Lim, Y. Hak Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071 CrossRef CAS; (f) S. Pottanam-Chali and B. J. Ravoo, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201907484; (g) F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180 CrossRef CAS; (h) P. Kumar, B. Liu and G. Behl, Macromol. Biosci., 2019, 19, 1900071 CrossRef.
- N. K. P. Samuel, M. Singh, K. Yamaguchi and S. L. Regen, J. Am. Chem. Soc., 1985, 107, 42 CrossRef CAS.
- T. Dewa, S. J. Vigmond and S. L. Regen, J. Am. Chem. Soc., 1996, 118, 3435 CrossRef CAS.
- J. Zhang, B. Jing, N. Tokutake and S. L. Regen, Biochemistry, 2005, 44, 3598 CrossRef CAS.
- S. Ghosh, K. Irvin and S. Thayumanavan, Langmuir, 2007, 23, 7916 CrossRef CAS.
- (a) V. M. J. Säily, S. J. Ryhänen, H. Lankinen, P. Luciani, G. Mancini, M. J. Parry and P. K. J. Kinnunen, Langmuir, 2006, 22, 956 CrossRef; (b) Y. Takano, T. Asakawa, M. Inai, A. Ohta and H. Asakawa, J. Oleo Sci., 2017, 66, 1321 CrossRef CAS; (c) T. Asakawa, Y. Shimizu, T. Ozawa, A. Ohta and S. Miyagishi, J. Oleo Sci., 2008, 57, 243 CrossRef CAS; (d) A. Pinazo, M. Diz, C. Solans, M. A. Pes, P. Erra and M. R. Infante, J. Am. Oil Chem. Soc., 1993, 70, 1 CrossRef; (e) L. I. Jong and N. L. Abbott, Langmuir, 2000, 16, 5553 CrossRef CAS; (f) H. Fan, F. Han, Z. Liu, L. Qina, Z. Li, D. Liang, F. Ke, J. Huang and H. Fu, J. Colloid Interface Sci., 2008, 321, 227 CrossRef CAS; (g) H. Fan, X. Zhu, L. Gao, Z. Li and J. Huang, J. Phys. Chem. B, 2008, 112, 10165 CrossRef CAS.
- (a) B. Wetzer, G. Byk, M. Frederic, M. Airiau, F. Blanche, B. Pitard and D. Scherman, Biochem. J., 2001, 356, 747 CrossRef CAS; (b) G. Byk, B. Wetzer, M. Frederic, C. Dubertret, B. Pitard, G. Jaslin and D. Scherman, J. Med. Chem., 2000, 43, 4377 CrossRef CAS; (c) A. Hirko, F. Tang and J. A. Hughes, Curr. Med. Chem., 2003, 10, 1185 CrossRef CAS PubMed; (d) F. Tang and J. A. Hughes, Bioconjugate Chem., 1999, 10, 791 CrossRef CAS; (e) V. V. Kumar and A. Chaudhuri, FEBS Lett., 2004, 571, 205 CrossRef CAS PubMed.
- Z. Huang, W. Li, J. A. MacKay and F. C. Szoka Jr., Mol. Ther., 2005, 11, 409 CrossRef CAS.
- A. Tschiche, B. N. S. Thota, F. Neumann, A. Schäfer, N. Ma and R. Haag, Macromol. Biosci., 2016, 16, 811 CrossRef CAS.
- (a) B. Maiti, K. Kumar, P. Moitra, P. Kondaiah and S. Bhattacharya, Bioconjugate Chem., 2018, 29, 255 CrossRef CAS; (b) M. H. Lee, E.-J. Kim, H. Lee, H. M. Kim, M. J. Chang, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 16380 CrossRef CAS PubMed.
- (a) Z. Xu, M. Hou, X. Shi, Y.-E. Gao, P. Xue, S. Liu and Y. Kang, Biomater. Sci., 2017, 5, 444 RSC; (b) S. Dongbang, H. M. Jeon, M. H. Lee, W. S. Shin, J. K. Kwon, C. Kang and J. S. Kim, RSC Adv., 2014, 4, 18744 RSC; (c) S. Dong, J. He, Y. Sun, D. Li, L. Li, M. Zhang and P. Ni, Mol. Pharmaceutics, 2019, 16, 3770 CrossRef CAS.
- Q. Pei, X. Hu, S. Liu, Y. Li, Z. Xie and X. Jing, J. Controlled Release, 2017, 254, 23 CrossRef CAS PubMed.
- D. Nolan, R. Darcy and B. J. Ravoo, Langmuir, 2003, 19, 4469 CrossRef CAS.
- W. C. de Vries, D. Grill, M. Tesch, A. Ricker, H. Ngsse, J. Klingauf, A. Studer, V. Gerke and B. J. Ravoo, Angew. Chem., Int. Ed., 2017, 56, 9603 CrossRef CAS.
- W. C. de Vries, M. Tesch, A. Studer and B. J. Ravoo, ACS Appl. Mater. Interfaces, 2017, 9, 41760 CrossRef CAS.
- W. C. de Vries, S. Kudruk, D. Grill, M. Niehues, A. L. L. Matos, M. Wissing, A. Studer, V. Gerke and B. J. Ravoo, Adv. Sci., 2019, 1901935, DOI:10.1002/advs.201901935.
- Y.-H. Cui, R. Deng, Z. Li, X.-S. Du, Q. Jia, X.-H. Wang, C.-Y. Wang, K. Meguellati and Y.-W. Yang, Mater. Chem. Front., 2019, 3, 1427 RSC.
- Y. Chen, L. Rui, L. Liu and W. Zhang, Polym. Chem., 2016, 7, 3268 RSC.
- J.-H. Ryu, S. Park, B. Kim, A. Klaikherd, T. P. Russell and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 9870 CrossRef CAS.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966 CrossRef CAS.
- S. Takae, K. Miyata, M. Oba, T. Ishii, N. Nishiyama, K. Itaka, Y. Yamasaki, H. Koyama and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 6001 CrossRef CAS PubMed.
- (a) H. Sun, B. Guo, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2009, 30, 6358 CrossRef CAS; (b) H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848 CrossRef CAS; (c) Y. Zhong, W. Yang, H. Sun, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 3723 CrossRef CAS PubMed; (d) W. Wang, H. Sun, F. Meng, S. Ma, H. Liub and Z. Zhong, Soft Matter, 2012, 8, 3949 RSC.
- N. Songa, W. Liua, Q. Tua, R. Liua, Y. Zhanga and J. Wang, Colloids Surf., B, 2011, 87, 454 CrossRef.
- W. Chen, P. Zhong, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2013, 169, 171 CrossRef CAS PubMed.
- (a) L.-Y. Tang, Y.-C. Wang, Y. Li, J.-Z. Du and J. Wang, Bioconjugate Chem., 2009, 20, 1095 CrossRef CAS PubMed; (b) Y.-C. Wang, F. Wang, T.-M. Sun and J. Wang, Bioconjugate Chem., 2011, 22, 1939 CrossRef CAS PubMed.
- Y. Zhong, M. Dimde, D. Stöbener, F. Meng, C. Deng, Z. Zhong and R. Haag, ACS Appl. Mater. Interfaces, 2016, 8, 27530 CrossRef CAS PubMed.
- Q. N. Bui, Y. Li, M.-S. Jang, D. P. Huynh, J. H. Lee and D. S. Lee, Macromolecules, 2015, 48, 4046 CrossRef CAS.
- Y. Chen, M. Su, Y. Li, J. Gao, C. Zhang, Z. Cao, J. Zhou, J. Liu and Z. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 30519 CrossRef CAS PubMed.
- T. Thambi, H. Y. Yoon, K. Kim, I. C. Kwon, C. K. Yoo and J. H. Park, Bioconjugate Chem., 2011, 22, 1924 CrossRef CAS PubMed.
- H.-Y. Wen, H.-Q. Dong, W.-J. Xie, Y.-Y. Li, K. Wang, G. M. Paulettic and D.-L. Shi, Chem. Commun., 2011, 47, 3550 RSC.
- H. Y. Yang, M.-S. Jang, G. H. Gao, J. H. Leeb and D. S. Lee, Polym. Chem., 2016, 7, 1813 RSC.
- T.-B. Ren, Y. Feng, Z.-H. Zhang, L. Li and Y.-Y. Li, Soft Matter, 2011, 7, 2329 RSC.
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567 CrossRef CAS PubMed.
- C. Shi, X. Guo, Q. Qu, Z. Tang, Y. Wang and S. Zhou, Biomaterials, 2014, 35, 8711 CrossRef CAS PubMed.
- J.-H. Ryu, R. Roy, J. Ventura and S. Thayumanavan, Langmuir, 2010, 26, 7086 CrossRef CAS PubMed.
- B. Saha, S. Bhattacharyya, S. Mete, A. Mukherjee and P. De, ACS Appl. Polym. Mater., 2019, 1, 2503 CrossRef CAS.
- P. Sun, D. Zhou and Z. Gan, J. Controlled Release, 2011, 155, 96 CrossRef CAS.
- X. Duan, T. Bai, J. Du and J. Kong, J. Mater. Chem. B, 2018, 6, 39 RSC.
- J. Chen, X. Qiu, J. Ouyang, J. Kong, W. Zhong and M. M. Q. Xing, Biomacromolecules, 2011, 12, 3601 CrossRef CAS PubMed.
- J. Chen, F. Zehtabi, J. Ouyang, J. Kong, W. Zhong and M. M. Q. Xing, J. Mater. Chem., 2012, 22, 7121 RSC.
- T. Thambi, D. G. You, H. S. Han, V. G. Deepagan, S. M. Jeon, Y. D. Suh, K. Y. Choi, K. Kim, I. C. Kwon, G.-R. Yi, J. Y. Lee, D. S. Lee and J. H. Park, Adv. Healthcare Mater., 2014, 3, 1829 CrossRef CAS.
- J. Li, M. Huo, J. Wang, J. Zhou, J. M. Mohammad, Y. Zhang, Q. Zhu, A. Y. Waddad and Q. Zhang, Biomaterials, 2012, 33, 2310 CrossRef CAS PubMed.
- (a) Y. Kakizawa, A. Harada and K. Kataoka, J. Am. Chem. Soc., 1999, 121, 11247 CrossRef CAS; (b) Y. Kakizawa, A. Harada and K. Kataoka, Biomacromolecules, 2001, 2, 491 CrossRef CAS.
- (a) K. Miyata, Y. Kakizawa, N. Nishiyama, A. i. Harada, Y. Yamasaki, H. Koyama and K. Kataoka, J. Am. Chem. Soc., 2004, 126, 2355 CrossRef CAS PubMed; (b) K. Miyata, Y. Kakizawa, N. Nishiyama, Y. Yamasaki, T. Watanabe, M. Kohara and K. Kataoka, J. Controlled Release, 2005, 109, 15 CrossRef CAS PubMed.
- S. Matsumoto, R. J. Christie, N. Nishiyama, K. Miyata, A. Ishii, M. Oba, H. Koyama, Y. Yamasaki and K. Kataoka, Biomacromolecules, 2009, 10, 119 CrossRef CAS.
- A. N. Koo, H. J. Lee, S. E. Kim, J. H. Chang, C. Park, C. Kim, J. H. Park and S. C. Lee, Chem. Commun., 2008, 6570 RSC.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227 CrossRef CAS PubMed.
- J.-H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246 CrossRef CAS PubMed.
- S. Cajot, N. Lautram, C. Passirani and C. Jérôme, J. Controlled Release, 2011, 152, 30 CrossRef CAS PubMed.
- K. Wang, H. Peng, K. J. Thurecht, S. Puttick and A. K. Whittaker, Polym. Chem., 2014, 5, 1760 RSC.
- S. Yu, J. Ding, C. He, Y. Cao, W. Xu and X. Chen, Adv. Healthcare Mater., 2014, 3, 752 CrossRef CAS PubMed.
- P. Prasad, M. R. Molla, W. Cui, M. Canakci, B. Osborne, J. Mager and S. Thayumanavan, Biomacromolecules, 2015, 16, 3491 CrossRef CAS PubMed.
- Z. Jiang, w. Cui, P. Prasad, M. A. Touve, N. C. Gianneschi, J. Mager and S. Thayumanavan, Biomacromolecules, 2019, 20, 435 CrossRef CAS PubMed.
- Z. Jiang, W. Cui, J. Mager and S. Thayumanavan, Ind. Eng. Chem. Res., 2019, 58, 6982 CrossRef CAS.
- O. Munkhbat, M. Canakci, S. Zheng, W. Hu, B. Osborne, A. A. Bogdanov and S. Thayumanavan, Biomacromolecules, 2019, 20, 790 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, K. J. Bentley, R. Wehr, N. J. Warren, B. R. Saunders and S. P. Armes, Polym. Chem., 2017, 8, 5962 RSC.
- L.-P. Lv, J.-P. Xu, X.-S. Liu, G.-Y. Liu, X. Yang and J. Ji, Macromol. Chem. Phys., 2010, 211, 2292 CrossRef CAS.
- W. Chen, M. Zheng, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Biomacromolecules, 2013, 14, 1214 CrossRef CAS PubMed.
- Y. Li, S. Wang, D. Zhu, Y. Shen, B. Du, X. Liu and Y. Zheng, RSC Adv., 2015, 5, 20025 RSC.
- Y. Xia, H. He, X. Liu, D. Hu, L. Yin, Y. Lu and W. Xu, Polym. Chem., 2016, 7, 6330 RSC.
- S. Xue, X. Gu, J. Zhang, H. Sun, C. Deng and Z. Zhong, Biomacromolecules, 2018, 19, 3586 CrossRef PubMed.
- Y. Ding, C. Du, J. Qian, L. Zhou, Y. Su, R. Zhang and C.-M. Dong, Polym. Chem., 2018, 9, 3488 RSC.
- Y. Zhu, J. Zhang, F. Meng, L. Cheng, J. Feijen and Z. Zhong, J. Mater. Chem. B, 2018, 6, 3040 RSC.
- (a) J. C. Patrick, US Pat., 1890, 191, Issued 6 Dec, 1932 Search PubMed; (b) J. C. Patrick and N. M. Mnookin, US Pat., 1996, 486, Issued 2 April, 1935 Search PubMed; (c) K. Kishore and K. Ganesh, Adv. Polym. Sci., 1995, 121, 83 CrossRef; (d) C. S. Marvel and L. E. Olson, J. Am. Chem. Soc., 1957, 79, 3089 CrossRef; (e) R. L. Whistler and D. J. Hoffman, J. Polym. Sci., Part A-1: Polym. Chem., 1967, 5, 2111 CrossRef CAS; (f) W. Choi, F. Sanda, N. Kihara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 79 CrossRef CAS; (g) E. J. Goethals and C. Sillis, Macromol. Chem., 1968, 119, 249 CrossRef CAS; (h) Y. Lee, H. Koo, G. Jin, H. Mo, M. Y. Cho, J.-Y. Park, J. S. Choi and J. S. Park, Biomacromolecules, 2005, 6, 24 CrossRef CAS PubMed; (i) E. Q. Rosenthal, J. E. Puskas and C. Wesdemiotis, Biomacromolecules, 2012, 13, 154 CrossRef CAS PubMed; (j) T. Tatsuma, Y. Yokoyama, D. A. Buttry and N. Oyama, J. Phys. Chem. B, 1997, 101, 7556 CrossRef CAS; (k) Y. Ding and A. S. Hay, Macromolecules, 1996, 29, 6386 CrossRef CAS.
- D. Basak, R. Kumar and S. Ghosh, Macromol. Rapid Commun., 2014, 35, 1340 CrossRef CAS PubMed.
- R. Bej, J. Sarkar and S. Ghosh, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 194 CrossRef CAS.
- D. Basak, R. Bej and S. Ghosh, Polym. Chem., 2015, 6, 6465 RSC.
- R. Bej, J. Sarkar, D. Ray, V. K. Aswal and S. Ghosh, Macromol. Biosci., 2018, 18, 1800057 CrossRef PubMed.
- R. Bej and S. Ghosh, Bioconjugate Chem., 2019, 30, 101 CrossRef CAS PubMed.
- (a) E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752 RSC; (b) G. Gasparini, E.-K. Bang, J. Montenegro and S. Matile, Chem. Commun., 2015, 51, 10389 RSC.
- S. Du, S. S. Liew, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2018, 140, 15986 CrossRef CAS PubMed.
- (a) P. Morelli, X. Martin-Benlloch, R. Tessier, J. Waser, N. Sakaia and S. Matile, Polym. Chem., 2016, 7, 3465 RSC; (b) G. S. Pulcu, N. S. Galenkamp, Y. Qing, G. Gasparini, E. Mikhailova, S. Matile and H. Bayley, J. Am. Chem. Soc., 2019, 141, 12444 CrossRef CAS PubMed.
- E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088 CrossRef CAS PubMed.
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069 CrossRef CAS PubMed.
- E. Derivery, E. Bartolami, S. Matile and M. Gonzalez-Gaitan, J. Am. Chem. Soc., 2017, 139, 10172 CrossRef CAS.
- (a) J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153 CrossRef CAS PubMed; (b) C. Yu, L. Qian, J. Ge, J. Fu, P. Yuan, S. C. L. Yao and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef.
- E.-K. Bang, S. Ward, G. Gasparini, N. Sakai and S. Matile, Polym. Chem., 2014, 5, 2433 RSC.
- G. Gasparini, G. Sargsyan, E.-K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328 CrossRef CAS PubMed.
- Z. Shu, I. Tanaka, A. Ota, D. Fushihara, N. Abe, S. Kawaguchi, K. Nakamoto, F. Tomoike, S. Tada, Y. Ito, Y. Kimura and H. Abe, Angew. Chem., Int. Ed., 2019, 58, 6611 CrossRef CAS PubMed.
- (a) J. Xia, T. Li, C. Lu and H. Xu, Macromolecules, 2018, 51, 7435 CrossRef CAS; (b) N. Ma, Y. Li, H. Xu, Z. Wang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 442 CrossRef CAS PubMed; (c) M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519 RSC; (d) H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647 CrossRef CAS PubMed; (e) W. Zhou, L. Wang, F. Li, W. Zhang, W. Huang, F. Huo and H. Xu, Adv. Funct. Mater., 2017, 27, 1605465 CrossRef; (f) C. Wei, Y. Zhang, H. Xu, Y. Xu, Y. Xu and M. Lang, J. Mater. Chem. B, 2016, 4, 5059 RSC; (g) X. Zeng, X. Zhou, M. Li, C. Wang, J. Xu, D. Ma and W. Xue, J. Mater. Sci.: Mater. Med., 2015, 26, 234 CrossRef PubMed; (h) C. Wei, Y. Zhang, H. Xu, Y. Xu, Y. Xu and M. Lang, J. Mater. Chem. B, 2016, 4, 5059 RSC.
- (a) J. Yang, W. Liu, M. Sui, J. Tang and Y. Shen, Biomaterials, 2011, 32, 9136 CrossRef CAS PubMed; (b) Z. Zhang, Y. Li, J. Wan, P. Long, J. Guo, G. Chen and C. Wang, Polym. Chem., 2017, 8, 2410 RSC.
- W. Ong, Y. Yang, A. C. Cruciano and R. L. McCarley, J. Am. Chem. Soc., 2008, 130, 14739 CrossRef CAS PubMed.
- J. Liu, Y. Pang, J. Chen, P. Huang, W. Huang, X. Zhu and D. Yan, Biomaterials, 2012, 33, 7765 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |