
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrochlore nanocrystals as versatile quasi-single-source precursors to lithium conducting garnets†
J. Mark
Weller
 a and
Candace K.
Chan
*ab
a and
Candace K.
Chan
*ab
aMaterials Science and Engineering, School for Engineering of Matter, Transport and Energy, Arizona State University, P.O. Box 876106, Tempe, AZ 85827, USA. E-mail: candace.chan@asu.edu
bDepartment of Heterogeneous Catalysis, Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
First published on 5th August 2020
Abstract
Lithium conducting garnets are attractive solid electrolytes for solid-state lithium batteries but are difficult to process, generally requiring high reaction and sintering temperatures with long durations. In this work, we demonstrate a synthetic route to obtain Ta-doped garnet (Li6.4La3Zr1.4Ta0.6O12) utilizing La- and Ta-doped lanthanum zirconate (La2.4Zr1.12Ta0.48O7.04) pyrochlore nanocrystals as quasi-single-source precursors. Via molten salt synthesis (MSS) in a highly basic flux, the pyrochlore nanocrystals transform to Li-garnet at reaction temperatures as low as 400 °C. We also show that the pyrochlore-to-garnet conversion can take place in one step using reactive sintering, resulting in densified garnet ceramics with high ionic conductivity (0.53 mS cm−1 at 21 °C) and relative density (up to 94.7%). This approach opens new avenues for lower temperature synthesis of lithium garnets using a quasi-single-source precursor and provides an alternative route to highly dense garnet solid electrolytes without requiring advanced sintering processes.
Garnet-type lithium lanthanum zirconate (Li7La3Zr2O12, LLZO) and its doped analogues are actively being explored as electrolytes for solid-state lithium batteries due to their beneficial properties (high ionic conductivity, electrochemical stability, inertness, etc.).1 However, the synthesis and processing of LLZO is challenging, requiring high reaction temperatures (>900 °C and reaction times in excess of 8 h) when using conventional solid-state reaction (SSR) methods and binary oxide precursors (e.g., La2O3, ZrO2).2,3 Recently, many synthesis strategies, such as sol–gel,4,5 combustion,6,7 and molten salt synthesis,8–10 have focused on reducing the LLZO formation temperature. These methods often result in garnet powders with comparable properties to those synthesized via conventional SSR, but still generally require synthesis temperatures above 700 °C.2,3
One common feature of LLZO synthesis is the nearly ubiquitous presence of pyrochlore type lanthanum zirconate (La2Zr2O7, LZO) as an intermediate phase before garnet formation, signalling incomplete reaction.4–7,9,11 Its presence after extended calcination or sintering of LLZO indicates decomposition of the garnet structure due to evaporation of Li at high temperatures.12 LZO beneficially contains the majority of the non-Li components of LLZO in a single crystalline phase. For this reason, some researchers have used it as a LLZO precursor. In one case, LLZO was synthesized from Li2CO3, La(OH)3, and La2Zr2O7,13 but still required a temperature of 800 °C and only formed the low-conductivity14 tetragonal (I41/acd) phase of garnet due to the absence of dopants needed to stabilize the high-conductivity1 cubic (Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d) phase. Deviannapoorani et al. used La2Zr2O7 along with mixtures of nominal composition Li7LaO5 or Li6.28Al0.24LaO5 to synthesize undoped and Al-doped cubic LLZO, but the resulting products were not phase-pure even with calcination at 1200 °C, which may have led to their lower ionic conductivity compared to LLZO synthesized using conventional precursors.11 We speculate that the challenge in using LZO as a precursor for garnet synthesis is due in part to the difference in La
d) phase. Deviannapoorani et al. used La2Zr2O7 along with mixtures of nominal composition Li7LaO5 or Li6.28Al0.24LaO5 to synthesize undoped and Al-doped cubic LLZO, but the resulting products were not phase-pure even with calcination at 1200 °C, which may have led to their lower ionic conductivity compared to LLZO synthesized using conventional precursors.11 We speculate that the challenge in using LZO as a precursor for garnet synthesis is due in part to the difference in La![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr stoichiometry between LZO and LLZO (1:1 and 3:2 moles per formula unit, respectively). One option to resolve this challenge is to obtain a La-excess pyrochlore phase that can also incorporate the dopants needed to stabilize the cubic garnet phase.
:Zr stoichiometry between LZO and LLZO (1:1 and 3:2 moles per formula unit, respectively). One option to resolve this challenge is to obtain a La-excess pyrochlore phase that can also incorporate the dopants needed to stabilize the cubic garnet phase.
Pyrochlore-structured oxides generally display a high degree of flexibility in composition and substituents.15 Pyrochlores adopt the general formula A2B2O6X, where A = di- or trivalent cations, B = tetra- or pentavalent cations, and X = O2−, OH− or F−.15 The ease with which chemical substitutions can occur in pyrochlores is leveraged in this work, where we demonstrate the synthesis of ultrafine, nanocrystalline, multiply-doped LZO as a precursor for obtaining doped LLZO. Herein, we show that La2+yZr2−x−yTaxO7+(x−y)/2 nanocrystals can be synthesized with various amounts of Ta and excess La. The pyrochlore La2.4Zr1.12Ta0.48O7.04 (corresponding to a mole ratio of La:Zr:Ta = 3:1.4:0.6) is prepared and utilized as a quasi-single-source-precursor for the garnet (Li6.4La3Zr1.4Ta0.6O12), requiring only a lithium source for the transformation. These pyrochlores can convert to garnets at reaction temperatures as low as 400 °C using molten salt synthesis (MSS) in a highly basic flux. Additionally, densified garnet ceramics can be formed directly from pyrochlore powders via in situ reactive sintering with LiOH.
The synthesis of pyrochlore nanocrystals was adapted from the molten hydroxide method16,17 used for complex oxides. Several syntheses were performed at 400 °C (see ESI†) to determine the feasibility of the extensive doping required to convert LZO to LLZO with only the addition of a Li-source. Fig. 1a shows synchrotron X-ray diffraction (XRD) patterns of La-doped LZO, Ta-doped LZO, and La/Ta co-doped LZO of increasing doping levels. The broad features in the XRD data are due to the small crystallite18 size of these pyrochlores. These results show that phase-pure Ta-doped pyrochlore of nominal composition La2Zr1.5Ta0.5O7.25 could be prepared without any noticeable impurities, indicating that Ta readily incorporates into LZO using this method. For La-doped LZO, a small amount of La(OH)3 was observed in the XRD pattern, suggesting that fully doping LZO to a ratio of 3 La:2 Zr is not feasible. Minor La(OH)3 (also likely nanocrystalline based on the breadth of the Bragg peaks observed in Fig. 1a), was also observed in co-doped LZO of nominal compositions La2.2Zr1.575Ta0.225O7.0125 and La2.4Zr1.12Ta0.48O7.04. Synchrotron X-ray pair distribution function (PDF) analysis (Fig. 1b) of the La2.4Zr1.12Ta0.48O7.04 sample showed a better fit to the pyrochlore (Fdm) rather than the defect fluorite (Fmm) structure.19 The PDF refinement also indicates that, besides minor amounts of La(OH)3 (∼9 wt%), all other constituents, including Ta and the rest of the excess La, are contained in a single pyrochlore phase. Transmission electron microscopy (TEM) characterization confirmed that the pyrochlore particles are on the order of 10–20 nm in size and highly crystalline (Fig. 1c and d). Fig. 1e shows a high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of these pyrochlores with a composite energy dispersive X-ray spectroscopy (STEM-EDS) composition map and elemental maps for La, Zr, and Ta (see Fig. S1† for average EDS spectrum), demonstrating good uniformity of each species in the pyrochlores. A high resolution HAADF-STEM image (Fig. 1f) also confirms the high crystallinity of these pyrochlores.
 | ||
| Fig. 1 Characterization of pyrochlore precursors. (a) Synchrotron XRD patterns of La/Ta co-doped LZO pyrochlores synthesized in molten hydroxides; (b) synchrotron X-ray PDF analysis of La2.4Zr1.12Ta0.48O7.04 showing the structure is a better fit to pyrochlore vs. defect fluorite with minimal La(OH)3 as a secondary phase; (c) TEM electron diffraction ring pattern with major pyrochlore reflections indicated (low magnification TEM image in inset) and corresponding (d) high resolution TEM image of La2Zr1.5Ta0.5O7.25 nanocrystals; (e) HAADF-STEM image of La2.4Zr1.12Ta0.48O7.04 pyrochlores with EDS map from the region outlined in cyan showing distribution of La, Zr, and Ta throughout the nanocrystals; (f) high resolution HAADF-STEM image of pyrochlores in (e). | ||
These results show that the La2.4Zr1.12Ta0.48O7.04 pyrochlores contain the necessary La, Zr, and Ta to be used as quasi-single-source precursors for the synthesis of garnets of composition Li6.4La3Zr1.4Ta0.6O12 (LLZTO). To obtain LLZTO particles, a ternary mixture of LiNO3–LiOH–Li2O2 was used as the reaction medium in which to synthesize the garnet from the pyrochlores via MSS. This ternary mixture is characterized by high Lux-Flood basicity,20,21 which is expected to enable a low oxide formation temperature22,23 while also serving as the lithium source for the reaction and also has the advantage of mitigating10 proton-exchange in the as-synthesized powder compared to neutral molten salt media such as eutectic LiCl–KCl. The reaction time (1–5 h), temperature (400–550 °C), and ratio of Li2O2:LiOH ([0, 0.5, or 1]:3.2) in the melt were varied to understand the minimum conditions (time, temperature, amount of Li2O2) that enable garnet formation from the pyrochlores (see ESI† for detailed procedures, Fig. S2† for XRD patterns, Table S1† for specific experimental conditions used). These experiments reveal that pyrochlore nanocrystals convert to garnet crystals at temperatures as low as 400 °C in 5 h, which to our knowledge is the lowest Li-garnet synthesis temperature to be reported. For comparison, our previous work showed that garnet synthesis in neutral LiCl–KCl molten salts required a reaction temperature of 850 (ref. 10) or 900 °C.9 Full conversion of the pyrochlore precursor to the garnet phase was observed with increased reaction time, temperature, or Lux-Flood basicity of the melt (i.e., increased amounts of Li2O2 for a fixed ratio of LiNO3/LiOH of 1.1:3.2). The minimum conditions in which phase-pure garnet were obtained at each temperature are summarized in Table 1.
Interestingly, it appears that the Lux-Flood basicity of Li2O2 is not the sole driving force for the reduced garnet formation temperature using this method, as a comparable melt with Na2O2 replacing Li2O2 did not result in garnet formation (see Fig. S2c†). This indicates that the Li2O2 likely first decomposes into reactive Li2O,24 which in turn promotes formation of the garnet phase at low temperatures, indicating a potentially broader use of Li2O2 as a reactive Li-source for LLZO synthesis. Indeed, the primary difference between low (0.5 moles) and high (1 mole) Li2O2 compositions in Table 1 is that generally less reaction time is required for higher Li2O2 content, indicating that Li2O2 modulates the reactivity/reaction rate. These results also indicate that garnet synthesis from pyrochlores is kinetically faster than using conventional reagents. For comparison, garnet of the same composition was synthesized from La(NO3)3, ZrOCl2, and Ta2O5 using MSS in the same melt; phase-pure garnet was obtained at 550 °C in 4 hours as in our previous work10 (Fig. S3†). With the pyrochlore reagents, phase-pure garnet adopting the high-conductivity cubic (Iad)1 structure formed in the same melt composition at 550 °C with only 1 h of reaction time (Fig. 2a and Table 1). We surmise that the presence of all major cationic components (La, Zr, and Ta) other than Li as well as the high surface area of the pyrochlore nanocrystals enables this rapid conversion to the garnet phase. These particles are generally submicrometer in size from scanning electron microscopy (SEM) observation (Fig. 2b) and highly crystalline based on TEM analysis (Fig. 2c and d). The HAADF-STEM and EDS analysis (Fig. S4†) also shows the uniform distribution of La, Zr, and Ta throughout the particles. These results confirm that this approach can enable the preparation of submicron garnet powders from doped pyrochlore nanocrystals. SEM images from a subset of other reaction conditions are shown in Fig. S5,† showing that in general, when only the minimum reaction time required to form garnet is used, the particle size of the resultant garnet is comparable to that in Fig. 2b regardless of reaction temperature. For lower synthesis temperatures (e.g. 450 °C, Fig. S5a–d†), increasing the reaction time from 3 to 5 hours does not appreciably increase the particle size with most particles being <1 μm. However, particle sizes coarsen substantially for longer reaction times at higher temperature (e.g. 500 °C) where primary particles go from mostly <1 μm for a shorter reaction time of 3 h (Fig. S5e and f†) to mostly >1 μm for a longer reaction time (Fig. S5g and h†) of 5 h.
 | ||
| Fig. 2 Characterization of garnet synthesized from pyrochlore precursors (a) XRD pattern of Li6.4La3Zr1.4Ta0.6O12 powder synthesized via MSS from pyrochlores (550 °C 1 h) with reference pattern (Logéat et al.),25 (b) SEM image showing primarily submicron, faceted LLZTO crystals, (c) HRTEM image of fused LLZTO particle (synthesized at 500 °C for 3 h, low magnification image in inset) in 〈135〉 zone-axis orientation and Fourier-filtered image (cyan bordered region) overlaid to remove signal from amorphous carbon grid, (d) Fourier-filtered HRTEM image of (c) with [135] LLZTO crystal motif overlaid (La: blue, Zr/Ta: red), and (e) 〈135〉 zone-axis electron diffraction pattern of particle in (c) with select reflections indicated. | ||
In addition to the aforementioned Li6.4La3Zr1.4Ta0.6O12 composition, we find that other garnet compositions are also accessible from doped pyrochlores using this method. The oxygen sublattice may be doped with fluorine by using NaF as pyrochlore dopant, resulting in cubic garnet with nominal composition Li6.375La3Zr2O11.375F0.625 (Fig. S6†). Additionally, a more highly doped garnet composition, Li6.025La2.75Ca0.25Zr1.4Ta0.6O11.375F0.625, was formed from pyrochlores co-doped with calcium, tantalum, and fluorine (Fig. S7†). These results show that doping with elements that readily incorporate into the pyrochlore structure,15,26 such as alkaline earths (e.g., Ca, Sr), lanthanides (e.g., La, Nd), tetra- and pentavalent transition metals (e.g., Ti, Zr, Nb, Ta), and F− or OH−, may be possible using this approach. The F and Ca/Ta/F doped compositions were investigated as a proof-of-concept for assessing wider applicability of this pyrochlore-to-garnet synthesis method, but further investigation of electrochemical performance was only pursued for Ta-doped garnets.
Li6.4La3Zr1.4Ta0.6O12 garnet powders synthesized from pyrochlores were pressed and sintered at 1200 °C for 2 or 3 h followed by SEM fracture surface imaging and ionic conductivity analysis via electrochemical impedance spectroscopy (EIS) (see Table S2† for circuit fitting parameters). Fig. 3a shows an SEM fracture surface image of a garnet pellet after conventional sintering, revealing predominately transgranular fracture and large (20–50 μm) grains. Additionally, the quasi-single-source feature of the pyrochlore nanocrystals was also exploited for an alternative approach to obtain dense garnet electrolytes. In this case, the pyrochlores were blended with LiOH (10 mol% excess relative to the target garnet stoichiometry of Li6.4La3Zr1.4Ta0.6O12) via ball-milling followed by pressing and sintering. In this reactive sintering approach, the reaction of LiOH with the pyrochlores during high temperature calcination results in simultaneous garnet formation and densification. Fig. 3b shows a fracture surface image of a reactively sintered pellet (1200 °C 3 h) revealing an extremely dense microstructure with small grains on the order of 2–5 μm and a relative density of 94.7%. Nyquist plots of symmetric cells using 20 wt% (1.5 mol%) Sn–Li alloy27 electrodes are shown in Fig. 3c for both conventionally and reactively sintered garnets (1200 °C for 2 h, relative densities of 88.0% and 94.1% respectively) possessing room temperature (∼21 °C) total ionic conductivities of 0.42 and 0.53 mS cm−1 respectively, comparable to those obtained from LLZTO prepared using standard SSR and other methods7,10,28–41 (see Table S3† for detailed comparisons). No obvious grain boundary impedance is noticeable in the Nyquist plots in Fig. 3c, indicating that the tight grain structure from both sintering methods allows easy intergranular ion conduction. Finally, the activation energy of Li-ion conduction was determined from the temperature dependence of ionic conductivity (Fig. 3d, also see Fig. S8† for Nyquist plots at each temperature) for each sample to be 0.42 and 0.38 eV per atom for the conventionally and reactively sintered garnets respectively. The two processes described here for preparing garnet powders and ceramics from pyrochlores are illustrated schematically in Fig. 3e.
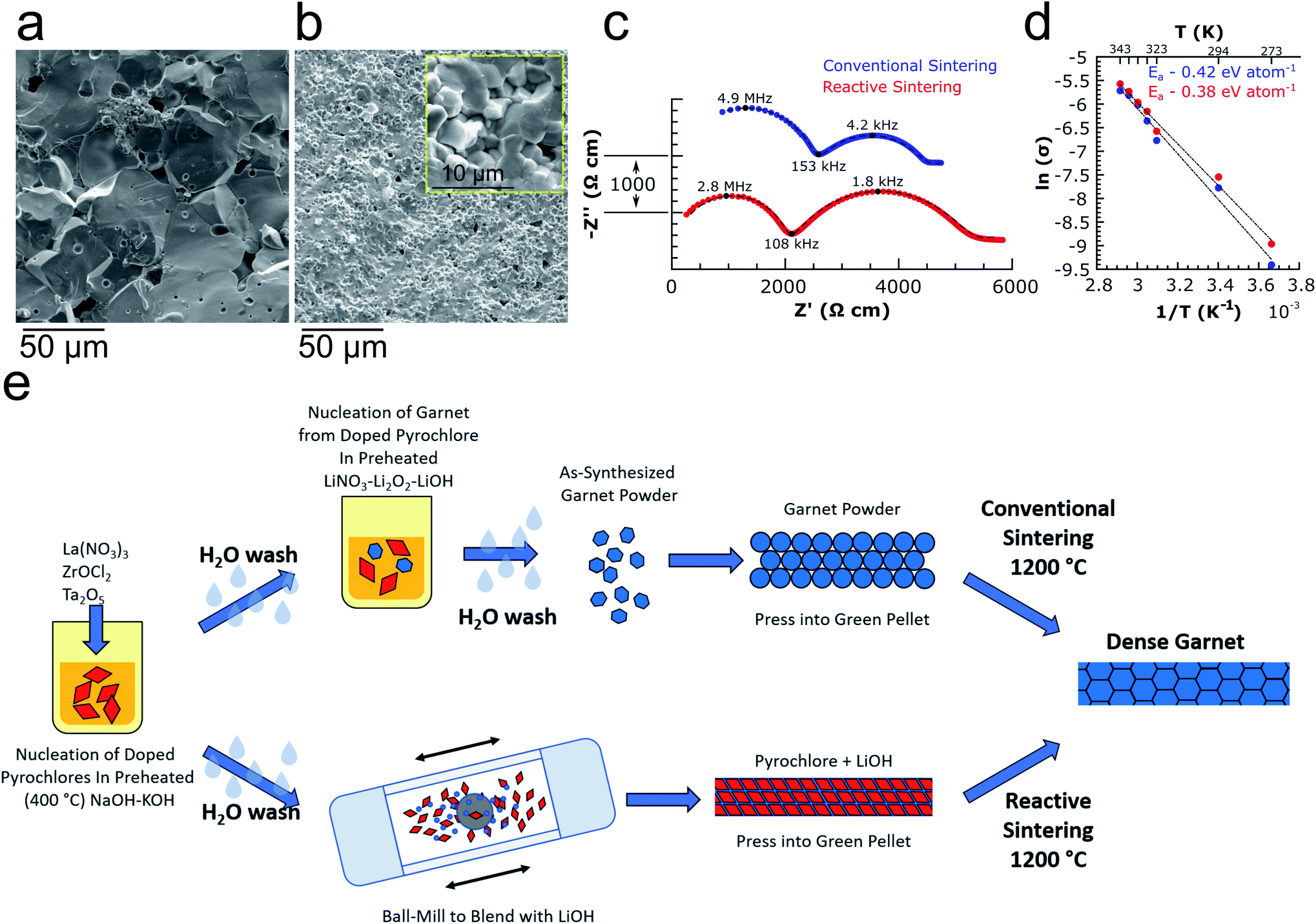 | ||
| Fig. 3 Properties of garnet solid electrolytes resulting from the pyrochlore-to-garnet processes (a) SEM fracture surface image of a pellet sintered from LLZTO garnet powder synthesized from pyrochlore precursors (1200 °C, 3 h sintering time), (b) SEM fracture surface image of a reactively sintered pellet from pyrochlores + LiOH (1200 °C 3 h sintering time, higher magnification image in inset), (c) room temperature EIS spectra of conventionally and reactively sintered pellets (1200 °C, 2 h sintering time) with 1.5 mol% Sn–Li alloy electrodes (measured between 1 Hz and 7 MHz, impedance normalized to pellet dimensions, spectra are vertically offset for clarity with vertical axis scale shown), and (d) Arrhenius plots of ionic conductivity measured between 273–343 K for samples in (c) with activation energies determined to be 0.42 and 0.38 eV per atom for conventional and reactive sintering respectively. (e) Graphical depiction of workflow for using MSS to convert pyrochlores to garnets followed by conventional sintering (top), and for using in situ reactive sintering to form dense garnet ceramics directly from pyrochlores and a Li source such as LiOH (bottom). | ||
An XRD pattern of an LLZTO pellet formed from the reactive sintering approach is shown in Fig. S9† confirming that phase-pure garnet can be prepared from the pyrochlore powders directly using in situ reactive sintering without requiring an initial synthesis step. Another benefit to using pyrochlores and LiOH in this way is the inherent stability of both components with water, which eliminates challenges associated with Li+/H+ exchange42,43 and subsequent carbonate formation,42–46 which are also seen when using aqueous solutions9,29,47 for processing garnets. Additionally, the microstructure and high relative density of the reactively sintered LLZTO resembles that of garnet sintered using hot-pressing,30 spark-plasma sintering,48 or Joule-heating induced rapid sintering.49 This indicates that pyrochlore-to-garnet reactive sintering may enable comparable performance and properties without the more complex equipment required for these advanced sintering methods. Furthermore, reactive sintering is more generally applicable to processing of garnet solid-electrolytes in other forms such as thin films, where hot-pressing or spark-plasma sintering are not applicable. Such experiments are planned as future work.
In conclusion, a new synthesis approach is presented wherein doped La2Zr2O7 pyrochlore nanocrystals are synthesized with a composition that will result in the correct stoichiometry to form Li-conducting garnets based on Li7La3Zr2O12. La and Ta co-doped pyrochlores with a La:Zr:Ta stoichiometry of 3:1.4:0.6 are demonstrated to readily form garnet-type Li6.4La3Zr1.4Ta0.6O12 between 400–550 °C in a ternary mixture of molten LiNO3–LiOH–Li2O2, with unprecedentedly low reaction temperatures. Pyrochlores can also be used as quasi-single-source precursors and blended with a Li source and reactively sintered in 2 hours at 1200 °C to form highly dense, highly conducting garnet ceramics with a microstructure similar to that obtained through advanced sintering techniques, providing a unique and successful approach to dense garnet electrolytes that can be easily extended to more advanced ceramic forming techniques such as tape-casting or additive manufacturing, potentially improving processability of garnet solid-electrolytes for solid-state lithium batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF CAREER award DMR 1553519. J. M. W. also acknowledges support from an ASU Fulton Schools of Engineering Dean's Fellowship and C. K. C. acknowledges support from the Max Planck Society as well as the Alexander von Humboldt Foundation for a Humboldt Research Fellowship. The authors gratefully acknowledge the use of facilities within the Eyring Materials Center at Arizona State University supported in part by NNCI-ECCS-1542160. The authors also thank Diamond Light Source (Didcot, UK) for access to beamline I15-1 (proposal no. CY23152, AP24) and D. Keeble for assistance with synchrotron measurements.References
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- S. Ramakumar, C. Deviannapoorani, L. Dhivya, L. S. Shankar and R. Murugan, Prog. Mater. Sci., 2017, 88, 325–411 CrossRef CAS.
- C. K. Chan, T. Yang and J. Mark Weller, Electrochim. Acta, 2017, 253, 268–280 CrossRef CAS.
- J. Sakamoto, E. Rangasamy, H. Kim, Y. Kim and J. Wolfenstine, Nanotechnology, 2013, 24, 424005 CrossRef PubMed.
- N. Janani, S. Ramakumar, L. Dhivya, C. Deviannapoorani, K. Saranya and R. Murugan, Ionics, 2011, 17, 575–580 CrossRef CAS.
- S. Afyon, F. Krumeich and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 18636–18648 RSC.
- J. M. Weller, J. A. Whetten and C. K. Chan, ACS Appl. Mater. Interfaces, 2020, 12, 953–962 CrossRef CAS PubMed.
- M. V. Reddy and S. Adams, J. Solid State Electrochem., 2017, 21, 2921–2928 CrossRef CAS.
- J. M. Weller, J. A. Whetten and C. K. Chan, ACS Appl. Energy Mater., 2018, 1, 552–560 CrossRef CAS.
- J. M. Weller and C. K. Chan, ACS Appl. Energy Mater., 2020, 3, 6466–6475 CrossRef CAS.
- C. Deviannapoorani, S. Ramakumar, N. Janani and R. Murugan, Solid State Ionics, 2015, 283, 123–130 CrossRef CAS.
- E. Rangasamy, J. Wolfenstine and J. Sakamoto, Solid State Ionics, 2012, 206, 28–32 CrossRef CAS.
- T. Kimura, Y. Yamada, K. Yamamoto, T. Matsuda, H. Nomura and T. Hirayama, J. Am. Ceram. Soc., 2017, 100, 1313–1319 CrossRef CAS.
- J. Awaka, N. Kijima, H. Hayakawa and J. Akimoto, J. Solid State Chem., 2009, 182, 2046–2052 CrossRef CAS.
- M. A. Subramanian, G. Aravamudan and G. V. S. Rao, Prog. Solid State Chem., 1983, 15, 55–143 CrossRef CAS.
- H. Liu, C. Hu and Z. L. Wang, Nano Lett., 2006, 6, 1535–1540 CrossRef CAS PubMed.
- H. Jin, D. Huang, Q. Gao, L. Li, N. Wang, Y. Wang and S. Hou, Mater. Res. Bull., 2012, 47, 51–53 CrossRef CAS.
- B. D. Cullity and S. R. Stock, Elements of X-RAY DIFFRACTION, 2nd edn, 1978 Search PubMed.
- B. Paul, K. Singh, T. Jaroń, A. Roy and A. Chowdhury, J. Alloys Compd., 2016, 686, 130–136 CrossRef CAS.
- H. Lux, Z. Elektrochem., 1939, 45, 303 CAS.
- H. Flood and T. Förland, Acta Chem. Scand., 1947, 592–604 CrossRef CAS PubMed.
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237 RSC.
- Y. Du and D. Inman, J. Mater. Sci., 1996, 31, 5505–5511 CrossRef CAS.
- K. P. C. Yao, D. G. Kwabi, R. A. Quinlan, A. N. Mansour, A. Grimaud, Y. Lee, Y. Lu and Y. Shao-horn, J. Electrochem. Soc., 2013, 160, 824–831 CrossRef.
- A. Logéat, T. Köhler, U. Eisele, B. Stiaszny, A. Harzer, M. Tovar, A. Senyshyn, H. Ehrenberg and B. Kozinsky, Solid State Ionics, 2012, 206, 33–38 CrossRef.
- A. A. Yaroshevskii and Y. A. Bagdasarov, Geochem. Int., 2008, 46, 1245–1266 CrossRef.
- C. Wang, H. Xie, L. Zhang, Y. Gong, G. Pastel, J. Dai, B. Liu, E. D. Wachsman and L. Hu, Adv. Energy Mater., 2018, 8, 1701963 CrossRef.
- X. Huang, Y. Lu, Z. Song, K. Rui, Q. Wang, T. Xiu, M. E. Badding and Z. Wen, Energy Storage Materials, 2019, 22, 207–217 CrossRef.
- X. Huang, Y. Lu, J. Jin, S. Gu, T. Xiu, Z. Song, M. E. Badding and Z. Wen, ACS Appl. Mater. Interfaces, 2018, 10, 17147–17155 CrossRef CAS PubMed.
- T. Thompson, A. Sharafi, M. D. Johannes, A. Huq, J. L. Allen, J. Wolfenstine and J. Sakamoto, Adv. Energy Mater., 2015, 5, 1–9 Search PubMed.
- Y. Wang and W. Lai, Electrochem. Solid-State Lett., 2012, 15, A68 CrossRef CAS.
- M. Yi, T. Liu, X. Wang, J. Li, C. Wang and Y. Mo, Ceram. Int., 2019, 45, 786–792 CrossRef CAS.
- N. Janani, S. Ramakumar, S. Kannan and R. Murugan, J. Am. Ceram. Soc., 2015, 98, 2039–2046 CrossRef CAS.
- R. Inada, K. Kusakabe, T. Tanaka, S. Kudo and Y. Sakurai, Solid State Ionics, 2014, 262, 568–572 CrossRef CAS.
- R. Inada, S. Yasuda, M. Tojo, K. Tsuritani, T. Tojo and Y. Sakurai, Front. Energy Res., 2016, 4, 1–12 Search PubMed.
- L. Dhivya and R. Murugan, ACS Appl. Mater. Interfaces, 2014, 6, 17606–17615 CrossRef CAS PubMed.
- M. Huang, M. Shoji, Y. Shen, C. W. Nan, H. Munakata and K. Kanamura, J. Power Sources, 2014, 261, 206–211 CrossRef CAS.
- C.-L. L. Tsai, V. Roddatis, C. V. Chandran, Q. Ma, S. Uhlenbruck, M. Bram, P. Heitjans and O. Guillon, ACS Appl. Mater. Interfaces, 2016, 8, 10617–10626 CrossRef CAS PubMed.
- X. Chen, T. Cao, M. Xue, H. Lv, B. Li and C. Zhang, Solid State Ionics, 2018, 314, 92–97 CrossRef CAS.
- X. Huang, T. Xiu, M. E. Badding and Z. Wen, Ceram. Int., 2018, 44, 5660–5667 CrossRef CAS.
- Y. Li, J. T. Han, C. A. Wang, H. Xie and J. B. Goodenough, J. Mater. Chem., 2012, 22, 15357–15361 RSC.
- K. Hofstetter, A. J. Samson, S. Narayanan and V. Thangadurai, J. Power Sources, 2018, 390, 297–312 CrossRef CAS.
- G. Larraz, a. Orera and M. L. Sanjuán, J. Mater. Chem. A, 2013, 1, 11419 RSC.
- L. Cheng, M. Liu, A. Mehta, H. L. Xin, F. Lin, K. A. Persson, G. Chen, E. J. Crumlin and M. M. Doeff, ACS Appl. Energy Mater., 2018, 1, 7244–7252 CrossRef CAS.
- H. Huo, J. Luo, V. Thangadurai, X. Guo, C. W. Nan and X. Sun, ACS Energy Lett., 2020, 5, 252–262 CrossRef CAS.
- L. Cheng, C. H. Wu, A. Jarry, W. Chen, Y. Ye, J. Zhu, R. Kostecki, K. Persson, J. Guo, M. Salmeron, G. Chen and M. Doeff, ACS Appl. Mater. Interfaces, 2015, 7, 17649–17655 CrossRef CAS PubMed.
- R. Ye, C. Tsai, M. Ihrig, S. Sevinc, M. Rosen, E. Dashjav, Y. J. Sohn, E. Figgemeier and M. Finsterbusch, Green Chem., 2020, 22, 4952–4961 RSC.
- H. Yamada, T. Ito and R. Hongahally Basappa, Electrochim. Acta, 2016, 222, 648–656 CrossRef CAS.
- C. Wang, W. Ping, Q. Bai, H. Cui, R. Hensleigh, R. Wang, A. H. Brozena, Z. Xu, J. Dai, Y. Pei, C. Zheng, G. Pastel, J. Gao, X. Wang, H. Wang, J. Zhao, B. Yang, J. Luo, Y. Mo, B. Dunn and L. Hu, Science, 2020, 526, 521–526 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for EIS fitting, tabulated data, XRD patterns, SEM images, S/TEM images and spectroscopic analyses, pair distribution function analysis, descriptions of synthesis and sintering conditions, and EIS data. See DOI: 10.1039/d0ta05842d |
| This journal is © The Royal Society of Chemistry 2020 |