
Sequential delivery of synergistic drugs by silica nanocarriers for enhanced tumour treatment†
Albane
Birault
a,
Simon
Giret
a,
Christophe
Théron
a,
Audrey
Gallud
b,
Afitz
Da Silva
b,
Denis
Durand
b,
Christophe
Nguyen
 b,
Nadir
Bettache
b,
Magali
Gary-Bobo
b,
John R.
Bartlett
cd,
Michel
Wong Chi Man
a and
Carole
Carcel
*a
b,
Nadir
Bettache
b,
Magali
Gary-Bobo
b,
John R.
Bartlett
cd,
Michel
Wong Chi Man
a and
Carole
Carcel
*a
aICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: carole.carcel@enscm.fr
bInstitut des Biomolécules Max Mousseron, UMR5247, Université de Montpellier, CNRS, ENSCM, Faculté de Pharmacie, 15 Avenue Charles Flahault, 34093, Montpellier, Cedex 05, France
cUniversity of the Sunshine Coast, 90 Sippy Downs Drive, Sippy Downs QLD 4556, Australia
dWestern Sydney University, Locked Bag 1797, Penrith, NSW 2751, Australia
First published on 20th January 2020
Abstract
Herein hybrid silica nanoparticles have been engineered to direct the sequential delivery of multiple chemotherapeutic drugs in response to external stimuli such as variations in pH. The nanocarriers consist of conventional MCM-41-type nanoparticles, which have been functionalised with an organic ligand (or stalk) grafted onto the external surface. The stalk is designed to “recognise” a complementary molecule, which serves as a “cap” to block the pores of the nanoparticles. First, camptothecin is introduced into the pores by diffusion prior to capping the pore apertures via molecular recognition. The cap, which is a derivative of 5-fluorouracil, serves as a second cytotoxic drug for synergistic chemotherapy. In vitro tests revealed that negligible release of the drugs occurred at pH 7.4, thus avoiding toxic side effects in the blood stream. In contrast, the stalk/cap complex is destabilised within the endolysosomal compartment (pH 5.5) of cancer cells, where release of the drugs was demonstrated. Furthermore, this environmentally responsive system exhibited a synergistic effect of the two drugs, where the pH-triggered release of the cytotoxic cap followed by diffusion-controlled release of the drug cargo within the pores led to essentially complete elimination of breast cancer cells.
Introduction
Combinatorial chemotherapy is widely employed as a primary treatment approach in cancer therapy to overcome the easily-developed defence of cancer cells against a single drug.1,2 However, conventional therapies involving the use of multiple free drugs often lead to well-known severe toxic side effects.2,3 One of the most effective approaches for overcoming such limitations is to load multiple therapeutic agents that exploit different anti-tumour mechanisms into a single effective nanocarrier with no drug leakage before reaching the target. Such smart nanocarriers could then increase the therapeutic efficiency and reduce toxic side effects by improving the bio-accessibility of drugs.4–8 In this sense, mesoporous silica nanoparticles (MSNPs) have been widely used, due to their versatility in forming the basis of stimuli-responsive drug delivery systems.9–13 Notably, pH-responsive MSNPs provide the advantage of being endogenously stimulated due to the difference in pH between cellular compartments.14–16 We recently reported17 the use of molecular recognition via H-bonding to design smart silica nanoparticles with a pH-activated cap, which exhibited slow release kinetics under slightly acidic conditions and good efficacy for cancer cells elimination.In order to avoid the well-known phenomenon of anti-cancer drug resistance and to optimize this earlier nanosystem, the current work explored the design of a multi-drug nanoplatform for combined chemotherapy. Our strategy, outlined in Scheme 1, involves coupling silica nanocarriers embodying molecular recognition sites with an active cap to enhance the anti-tumour activity.
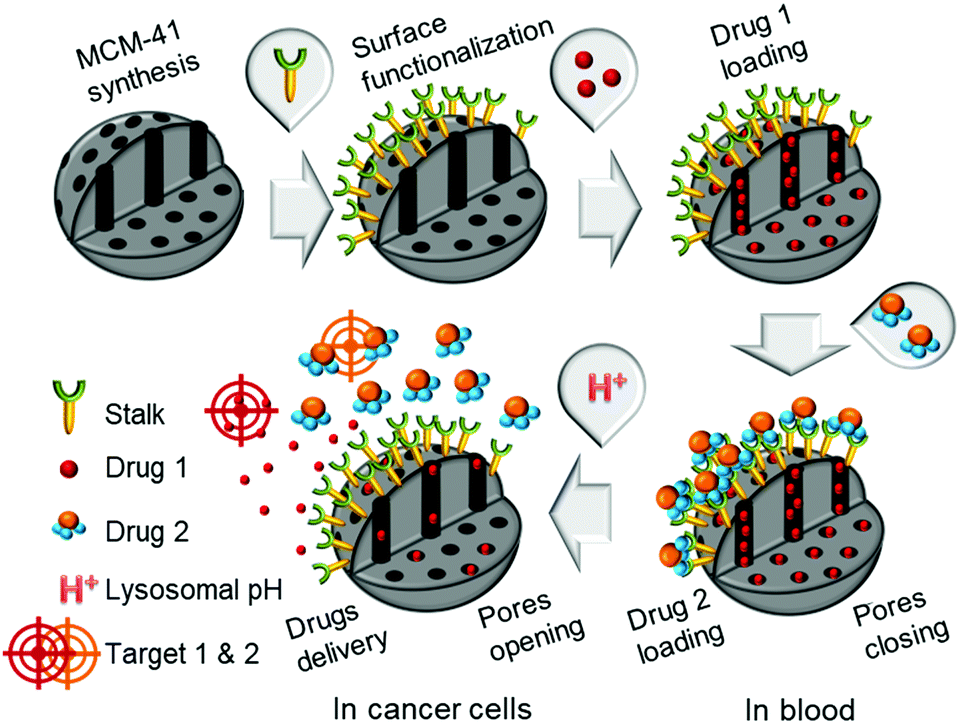 | ||
| Scheme 1 Conceptual combinatorial drug delivery MSNP nanocarriers with Drug 1 (camptothecin) encapsulated inside the pores and Drug 2 (5-FU derivative) as the capping agent. | ||
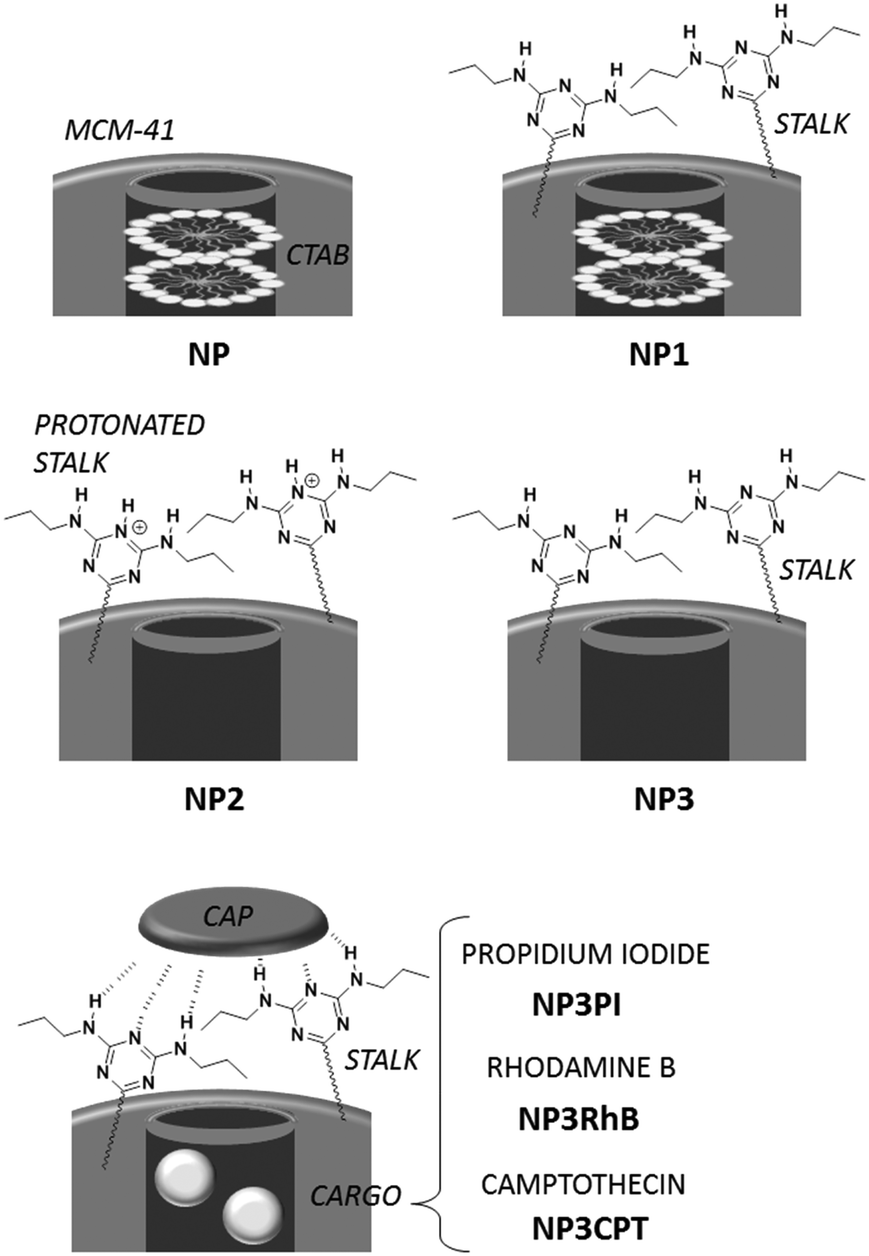 | ||
| Scheme 2 Synthesis of capped nanoparticles. | ||
After loading the functionalised nanoparticles with camptothecin (CPT, Drug 1), the pores are closed to avoid premature release under physiological conditions within the bloodstream (pH 7.4) using a 5-fluorouracil (5-FU) derivative (Drug 2), which can create stable H-bonds with Stalk located on the nanoparticles’ surfaces. The opening of the pores is then triggered at the acidic pH within lysosomes (pH 4.5–5.5), to deliver sequentially the two drugs within the cancer cells because of Stalk protonation followed by disruption of the Stalk–Cap bonds.
As already mentioned, there are a plethora of studies reporting MSNPs' efficacy for cancer therapy applications.18,19 Although some of these have described the use of drugs as pore-blockers to form promising nanocarriers for combination therapy,20–22 to the best of our knowledge, this is the first time that a pH-responsive nanocarrier has been employed to exploit the synergistic toxicity of combining CPT and 5-FU for anti-cancer drug resistance applications.
To develop such systems, two separate syntheses are required: (1) the functionalisation of appropriate silica nanocarriers such as MCM-41, by Stalk grafting, to provide selective H-bonding recognition sites on the external surface; and (2) the synthesis of an antitumor derivative, with a complementary molecular recognition pattern, which, by pairing with the corresponding Stalks, would then cap the pores.
Organosilylated triazine derivatives, bearing donor–acceptor–donor recognition sites, were used as Stalks condensed onto the surface of the nanoparticles. The Stalk employed in this study is tetrasilylated, which enhances the functionality of the platform by providing additional degrees of freedom and binding sites for grafting the ligand onto the surface of the nanoparticles, compared to our previously described bisilylated system.17 The additional conformational flexibility of the tetrasilylated Stalk would also be expected to enable the amine sites on the ligand to orient correctly to form the desired H-bonds with the Cap. As drug candidates, we chose for the pore-blocker a commonly used anticancer drug, 5-fluorouracil (5-FU), which exhibits complementary molecular recognition properties to Stalk. Previously,23 we have demonstrated that a derivative such as Cap (Fig. 1) with a three-fold H-bonding pattern favours stable complexation at neutral pH in bulk hybrid silica materials. In contrast, the complex begins to dissociate at lower pH after triazine unit protonation, leading to the breakdown of H-bonds initially involved in stabilization of the capping complex, thus promoting removal of Cap at pH 5.5 and below and its associated release. The biological activity of Cap was previously evaluated23 by cytotoxic assay on MCF-7 human breast cancer cells, showing a relatively high efficiency (43% of breast cancer cell death at 50 μM) for this molecule as an anticancer drug compared with the parent 5-FU (68% of cell death at 50 μM).
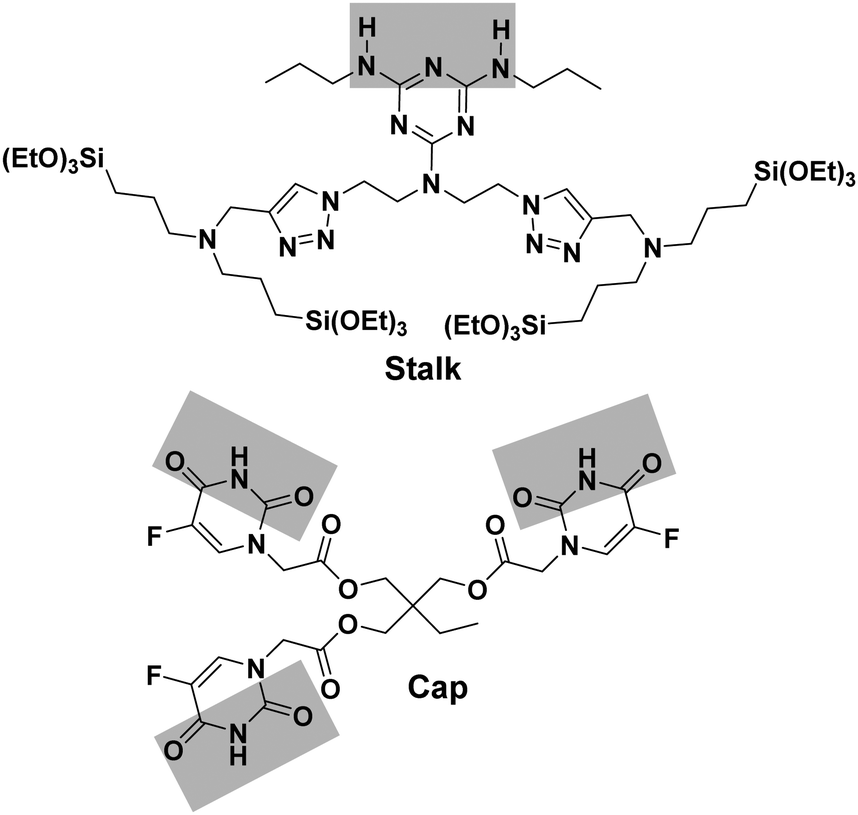 | ||
| Fig. 1 Structures of Stalk precursor and Cap. | ||
To investigate their viability and the combination therapeutic effect, we evaluated the cytotoxicity of these MSNPs towards human breast cancer cells (MCF-7). In a first study, the functionalised nanoparticles were loaded with fluorescent dyes, such as propidium iodide (PI) or rhodamine-B (RhB), to validate the opening of the pores in weakly-acidic lysosomal media and to assess the internalization of the MSNPs in the cells. In a second study, the MSNPs were loaded with CPT and the pores capped with 5-FU derivative Cap to examine the overall cytotoxicity by in vitro studies. CPT destroys cancer cells via a different biological pathway than 5-FU24,25 and the two drugs have been previously shown to demonstrate a strong synergistic effect when used together to treat cancer cells.26 The approach outlined in this study enables two cytotoxic drugs to be delivered autonomously and sequentially, which is typically the manner in which injection- or drip-based combination drug therapies are administered to maximise the efficacy of such treatment. Under in vitro conditions, the multi-drug payload is retained under normal physiological conditions and then released autonomously at the disease site, demonstrating nearly complete cell apoptosis within 72 h.
Experimental section
General
5-Fluorouracil, thionyl chloride, triethylamine and 1,1,1-tri(hydroxymethyl)propane were purchased from Sigma, USA and were used without purification. Solvents were dried by employing a MB SPS-800 apparatus.Characterization
FTIR spectra were obtained on a PerkinElmer FT-IR Spectrum BX spectrometer. CPMAS solid-state NMR spectra were obtained using a Bruker FT-AM 400 spectrometer and 1H- and 13C-NMR spectra were obtained using a Bruker AC-400 spectrometer, with CDCl3 or DMSO-d6 as solvent and tetramethylsilane (TMS) as a reference. N2 adsorption/desorption isotherms were obtained using a Micromeritics ASAP 2010 instrument at 77.15 K. Specific surface areas were calculated using the BET transform directly from the isotherms, using 0.162 nm2 as the cross-sectional area of N2. Electron micrographs were obtained with a JEOL 1200 EXII microscope for TEM and with a Hitachi S4800 30 kV apparatus for the SEM data. 29Si solid-state NMR spectra were obtained using a Bruker DSX 300 MHz spectrometer. Spectra were recorded using 4000 scans, with a relaxation delay of 3 s, pulse duration of 6 μs and acquisition time of 0.04 s.Thermogravimetric analyses (TGA) were performed on a Netzsch TG 209 C apparatus employing a heating rate of 10 °C min−1 under an air flow of 20 mL min−1 up to 585 °C. The small and wide-angle X-ray scattering (SWAXS) experiments were conducted using a Guinier Mering set up with a 2D image plate detector.
Precursor synthesis
1H NMR (400 MHz, DMSO-d6): δ = 11.93 (s, 1H), 8.09 (d, 1H), 4.37 ppm (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ): δ = 168.6, 158.2, 150.8, 140.1, 128.2, 53.4; HRMS (ESI) m/z: [M + H]+ calcd for C6H6N2O4F: 189.0312; found: 189.0313.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 90:10). Yield: 72%; m.p. decomposition after 180 °C.
:2 90:10). Yield: 72%; m.p. decomposition after 180 °C.
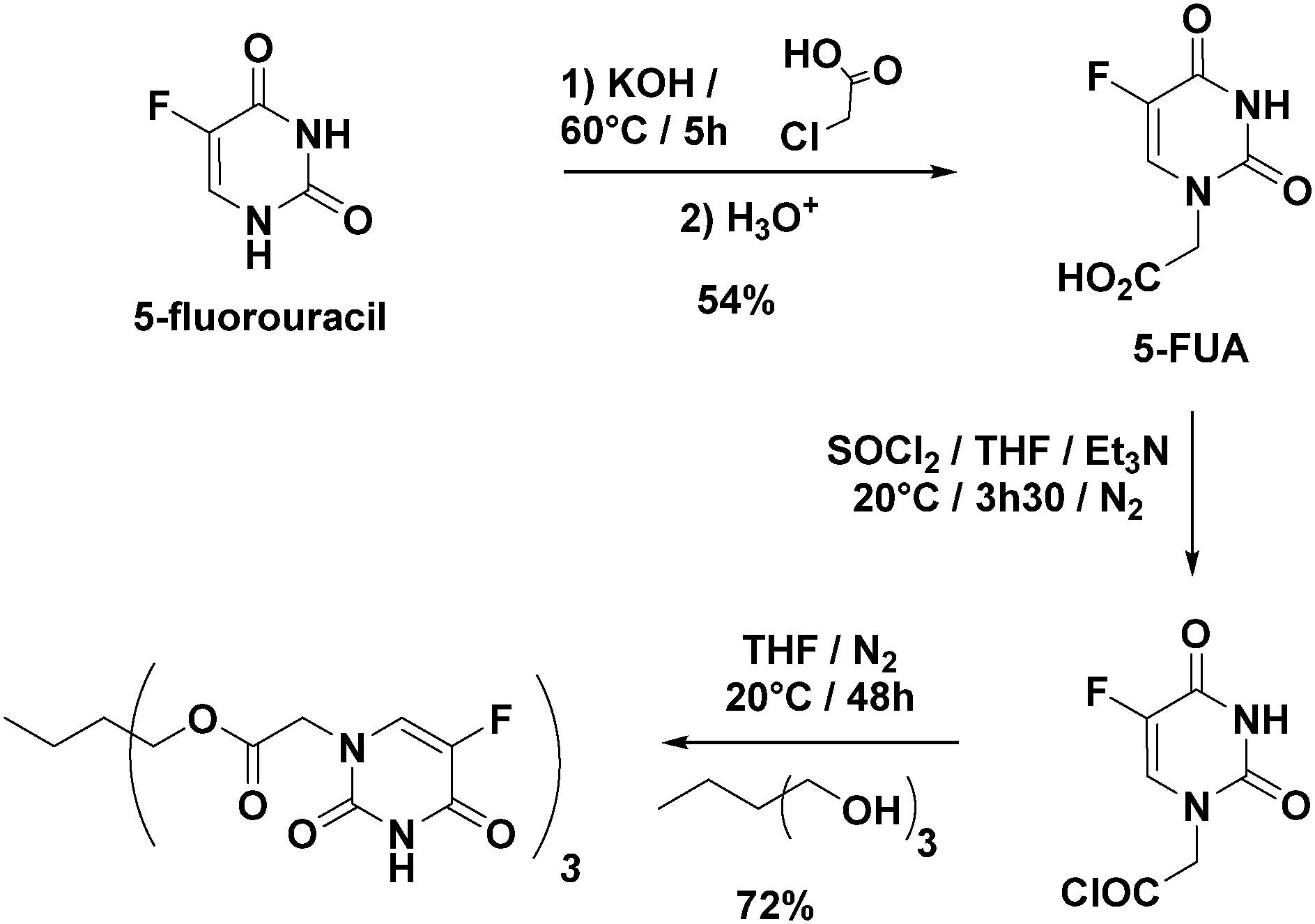 | ||
| Fig. 2 Optimized synthesis of Cap. | ||
1H NMR (400 MHz, DMSO-d6): δ = 12.00 (s, 3H), 8.04 (d, 3H), 4.50 (s, 6H), 4.05 (s, 6H), 1.36 (q, 2H), 0.79 ppm (t, 3H); 13C NMR (100 MHz, DMSO-d6): δ = 20.9, 34.7, 48.7, 63.4, 130.1, 138.1, 140.5, 149.6, 157.2, 167.6 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C24H24N6O12F3: 645.1404; found: 645.1412.
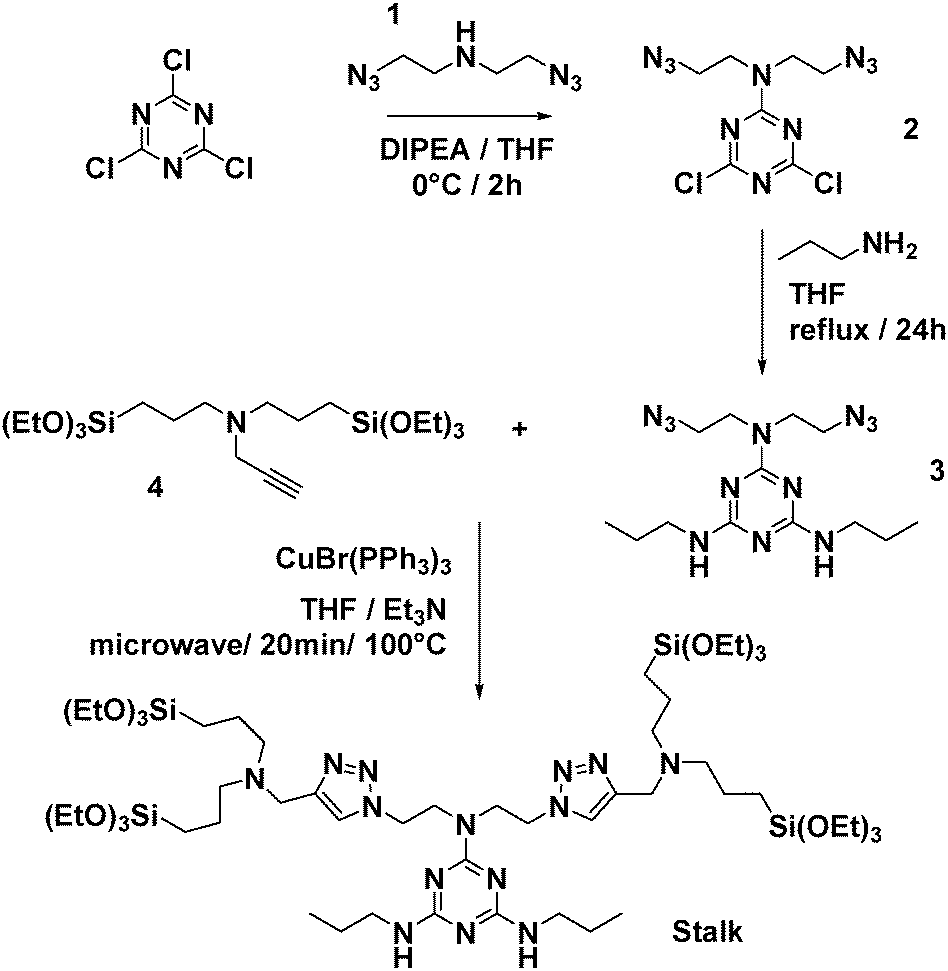 | ||
| Fig. 3 Synthesis of Stalk. | ||
1H NMR (400 MHz, CDCl3, Fig. S1, ESI†): δ = 0.56 (t, 8H), 0.95 (t, 6H), 1.22 (t, 36H), 1.58 (m, 12H), 2.39 (t, 8H), 3.29 (q, 4H), 3.64 (m, 8H), 3.80 (q, 24H), 4.47 (t, 4H), and 7.32 (s, 2H). 13C NMR (100 MHz, CDCl3): δ = 7.86 (s), 11.50 (s), 18.28 (s), 20.26 (s), 23.05 (s), 42.47 (s), 45.90 (s), 48.16 (s), 48.60 (s), 56.46 (s), 58.29 (s), 123.11 (s), 128.51 (s), and 132.01 (s). HRMS (ESI) m/z: [M + H]+ calcd for C55H115N14O12Si4 (MH): 1275.7896; found: 1275.7910.
Nanoparticles preparation (Scheme 2)
Loading, capping and release
NP3 nanoparticles were suspended in a solution of cargo molecules as described below. The suspension was then sonicated for 20 min and stirred for 18 h to promote filling of the pores with the cargo molecules. To ensure retention of the drug, the pore apertures were capped by addition of Cap (3 mg; 4.7 × 10−3 mmol) and the suspension was stirred for an additional 48 h. After centrifugation, the particles were washed with DMSO around 5–6 times following by washing with water and EtOH.NP3PI (5 mM): PI (45 mg, 6.7 × 10−2 mmol) in water (13.5 mL), and nanoparticles (90 mg).
NP3RhB (5 mM): RhB (32 mg, 6.7 × 10−2 mmol) in water (13.5 mL), and nanoparticles (90 mg).
NP3CPT (3 mM): CPT (15 mg, 4.3 × 10−2 mmol) in DMSO (15 mL), and nanoparticles (90 mg).
The CPT loadings obtained under the conditions used to impregnate the NP3 nanoparticles were 4 wt%, on the basis of visible absorbance spectroscopy studies.
The release of the CPT from NP3CPT mesopores was evaluated by UV-Vis absorbance at 380 nm at either pH 2, 5.5 or 7.4 for different periods of time over a 24 h period. For these studies, 3 mg of NP3CPT was dispersed into 1 mL of aqueous solution at the defined pH and then 100 μL aliquots of the nanoparticle dispersion were poured into Eppendorf tubes. The tubes were stirred during release of the drug and were then centrifuged (15 × 103 min−1, 15 min). The resulting supernatant was placed into a microreader plate and the concentration of drug was determined from an appropriate calibration curve.
In vitro studies
Cytotoxicity
MCF-7 cells were seeded into 96-well plates at 104 cells per well in 200 μL culture medium and allowed to grow for 24 h. The three batches of nanoparticles (NP, NP3RhB, and NP3CPT) were dispersed in ethanol at a concentration of 10−2 M and sonicated in an ultrasonic bath until completely dispersed. Then, cells were incubated for 72 h with different nanoparticle concentrations (from 1 to 100 μg mL−1). For the NP3CPT batch, the experiment was also carried out for shorter lengths of time (6, 16 and 24 h). At the end of the incubation time, a MTT assay was performed to evaluate the toxicity. Briefly, cells were incubated for 4 h with 0.5 mg mL−1 of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide; Promega) in culture media. The MTT/media solution was then removed, and the precipitated crystals were dissolved in EtOH/DMSO (1:1). The solution absorbance was read at 540 nm.
Confocal imaging
MCF-7 cells were seeded one day prior to nanoparticle exposure at 106 cells per cm2 in glass-bottomed culture dishes from Ibidi Biovalley®. Cells were then exposed for 20 h to 40 μg mL−1 of NPs. Prior to imaging, the cells were stained with 50 nM LysoTracker Green DND-26 for 30 min and 5 μg mL−1 Hoechst 33342 was added during the last 10 min of incubation. Confocal images were acquired on a Zeiss Axio Observer confocal microscope equipped with an oil-immersion Plan-Apochromat 63×/1.40 objective.Flow cytometry
MCF-7 cells were seeded into a 6-well plate (Nunc; 106 cells per well) and allowed to grow for 24 h. The cells were harvested at 5, 18 and 22 h following NP exposure (40 μg mL−1) and re-suspended in DMEM-F12 phenol red-free medium. Dead cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 0.5 μg mL−1). The percentage of positive living cells for NP uptake was determined on a FACS Canto II flow cytometer. The data were analysed with Win MDI software v2.8.Results and discussion
Preparation of precursors
The approach used to prepare the Stalk (Fig. 3 and Fig. S1, ESI†) was based on a previously reported method.28Similarly, the Cap was prepared according to an optimised protocol based on a previously reported synthesis.23 The reaction was performed in homogeneous medium and employed an eco-friendly room-temperature approach for the second and third steps, which led to an increased yield and a reduction in the overall reaction time (Fig. 2).
Nanoparticle synthesis, functionalisation, loading and capping
MCM-41 nanoparticles (NP) were prepared by classical sol–gel processing in alkaline aqueous solution. CTAB used to template the compact hexagonal porosity (Fig. S2, ESI†) in the MSNPs was retained in the pores, and the Stalk was then covalently grafted onto the MSNP surface by condensation. After washing with water, the resulting nanoparticles NP1 were characterised by IR spectroscopy and solid-state 29Si NMR to demonstrate successful functionalisation.As shown in Fig. 4(A), bands arising from the Stalk in the spectrum of NP1 are observed at 1557 and 1498 cm−1, which are associated with –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching vibrations.29 Notably, these vibrations are not evident in the corresponding NP spectrum. In the region from 2700 to 3100 cm−1 (Fig. 4(B)) the spectra of NP and NP1 are similar and are dominated by bands associated with CTAB at 2854 and 2924 cm−1. Hence the spectral features of Stalk are not clearly observed, except for a shoulder at 2972 cm−1. CTAB, initially retained within the pores to maximize functionalisation on the external surface, was then extracted with HCl to afford NP2. Excess HCl remaining after extraction of CTAB was neutralised by suspending NP2 in an aqueous solution of Et3N and stirring the resulting mixture for 48 h at 70 °C, before centrifugation and washing to give NP3. CTAB elimination was confirmed by the disappearance of its associated bands at 2854 and 2924 cm−1 in the IR spectrum of NP3. In addition, the loss of bands associated with Stalk ethoxy species at 1388 and 1439 cm−130 in the spectrum of NP3 (Fig. 4(A)) is consistent with successful grafting of Stalk onto the surface of NP3. Similarly, the decrease in the relative intensities of the Stalk bands at 2883 and 2972 cm−1, which include contributions from νs(CH3) and νas(CH3), respectively, of ethoxy groups,30 is consistent with loss of ethoxy species following grafting.
N and CC stretching vibrations.29 Notably, these vibrations are not evident in the corresponding NP spectrum. In the region from 2700 to 3100 cm−1 (Fig. 4(B)) the spectra of NP and NP1 are similar and are dominated by bands associated with CTAB at 2854 and 2924 cm−1. Hence the spectral features of Stalk are not clearly observed, except for a shoulder at 2972 cm−1. CTAB, initially retained within the pores to maximize functionalisation on the external surface, was then extracted with HCl to afford NP2. Excess HCl remaining after extraction of CTAB was neutralised by suspending NP2 in an aqueous solution of Et3N and stirring the resulting mixture for 48 h at 70 °C, before centrifugation and washing to give NP3. CTAB elimination was confirmed by the disappearance of its associated bands at 2854 and 2924 cm−1 in the IR spectrum of NP3. In addition, the loss of bands associated with Stalk ethoxy species at 1388 and 1439 cm−130 in the spectrum of NP3 (Fig. 4(A)) is consistent with successful grafting of Stalk onto the surface of NP3. Similarly, the decrease in the relative intensities of the Stalk bands at 2883 and 2972 cm−1, which include contributions from νs(CH3) and νas(CH3), respectively, of ethoxy groups,30 is consistent with loss of ethoxy species following grafting.
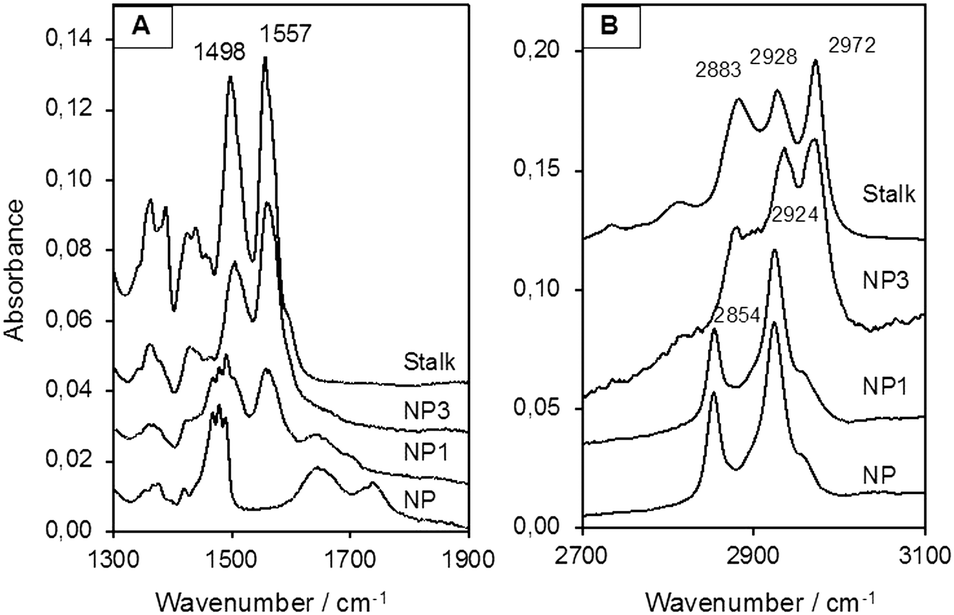 | ||
| Fig. 4 IR spectra (A) from 1300 to 1900 cm−1 and (B) from 2700 to 3100 cm−1 of NPs and the Stalk precursor. | ||
The solid-state 29Si NMR spectrum of NP1 (Fig. 5(B)) also demonstrated the successful grafting of the Stalk, with two sets of chemical shifts being evident. The first set, at −47, −57 and −67 ppm, are attributed to T1, T2 and T3 sites, respectively, arising from the Stalk and are not observed in the spectrum of NP (Fig. 5(A)), as expected. The relatively high intensity of the T2 signal compared to that of the T3 signal indicates that condensation of the ethoxy/hydroxy species on the Stalk is incomplete after grafting, with some of the four Si sites in the grafted Stalk moieties being bound to the surface of the nanoparticles via two oxo bonds instead of three. The second set of peaks in the 29Si-NMR spectra, at −95, −100 and −109 ppm, are assigned to Q2, Q3 and Q4 species, respectively, within the MCM-41 framework. The increase in the relative intensity of the Q4 signal for NP1, and the corresponding decrease in the –OH:Si ratio for the Qn species following grafting (from 0.75 to 0.65), is associated with conversion of Q2 and Q3 species to Q4 species during functionalisation. Thermogravimetric analysis (TGA) confirmed the presence of the organic Stalk on the surface of the MCM-41 nanoparticles (Fig. S3, ESI†).
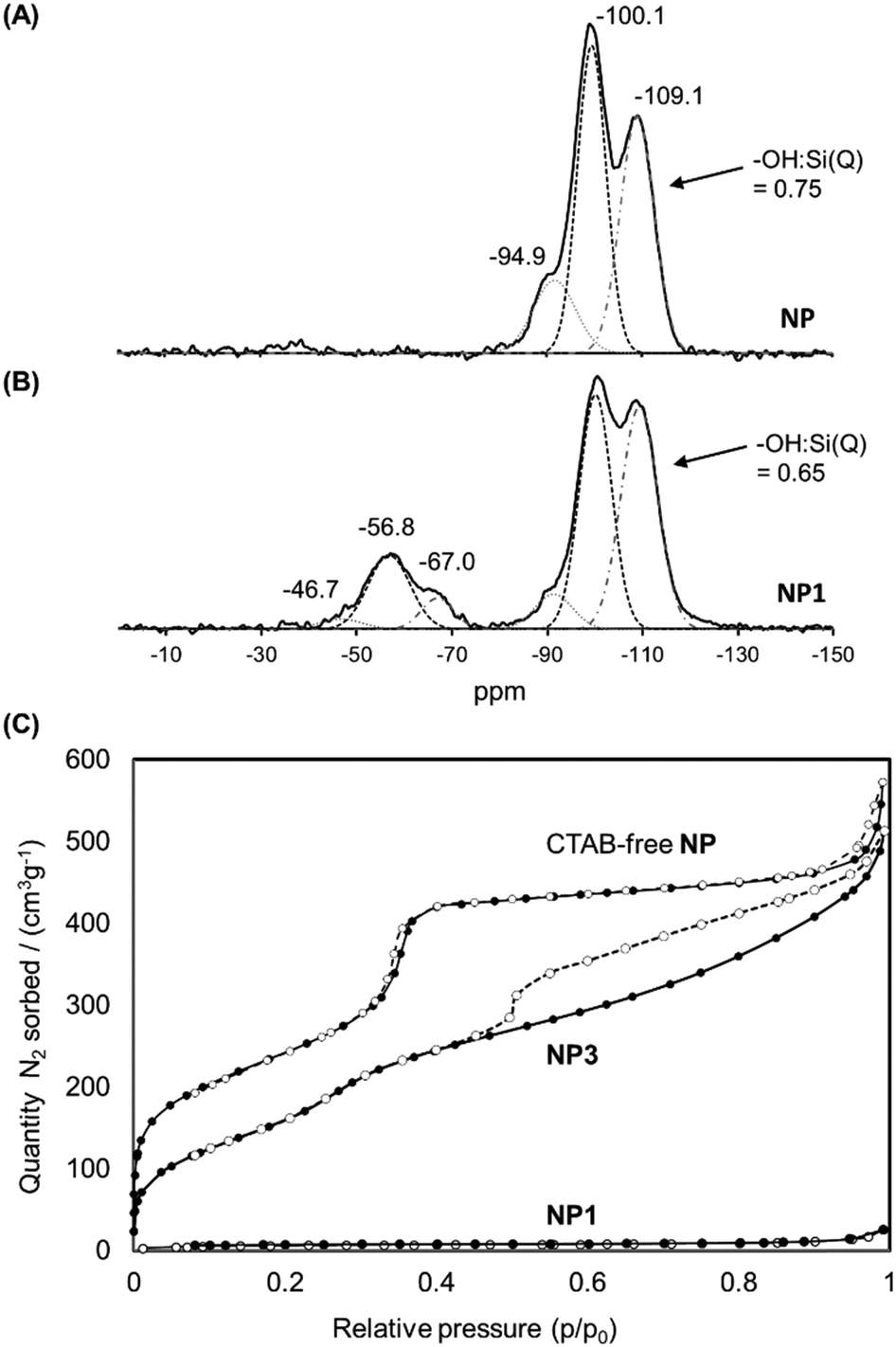 | ||
| Fig. 5 Solid state 29Si NMR spectra of NP (A) and NP1 (B). (C) Isotherms of CTAB-free NP, NP1 and NP3 obtained by nitrogen sorption analyses. | ||
The N2 adsorption/desorption isotherms of NP1 (grafted nanoparticles containing CTAB) are compared with those of CTAB-free NP and NP3 in Fig. 5(C). As expected, NP1 exhibits very low accessibility to small molecules such as N2, with a corresponding BJH adsorption pore volume and BET surface area of 0.03 cm3 g−1 and 21 m2 g−1, respectively, consistent with the presence of CTAB within the mesopores. In contrast, removal of CTAB by washing yields a mesoporous material (Type IV isotherm) with a pore volume of 0.92 cm3 g−1 and BET surface area of 902 m2 g−1. The isotherm of CTAB-free NP shows a steep increase near P/P0 = 0.35, consistent with the presence of ordered mesopores. The corresponding data for NP3 are also consistent with a highly porous material, albeit with a smaller pore volume (0.72 cm3 g−1) and surface area (675 m2 g−1) than the unfunctionalised material NP. A slight increase in the adsorption branch at p/p0 around 0.3 for NP3 is consistent with smaller pore size than in the CTAB-free NP. More significantly, the isotherm of NP3 has a distinct hysteresis with non-parallel branches on the adsorption and desorption arms. Such profiles are often observed for porous samples having ink-bottle type pores, consistent with modification of the external surface in the vicinity of the pore openings. These data suggest that functionalisation leads to some obstruction of the nanoparticle porosity, although the pore network is still highly accessible to small molecules such as N2.
To load the functionalised nanoparticles with a drug cargo, NP3 was suspended with several different cargos, including RhB, PI or CPT in water or DMSO. Cap was then added to the suspension to complex the Stalks on the surface and thus block the pores. The suspension was stirred at RT for a further 48 h and then centrifuged. The resulting nanoparticles were vigorously washed with DMSO (until a transparent supernatant was obtained) to remove adsorbed cargo that had not been incorporated within the pores prior to capping and residual physisorbed Cap. After additional washings with water and EtOH, the nanoparticles were dried under vacuum to give the NP3RhB, NP3PI or NP3CPT nanomaterials, depending on the cargo compound used.
The quantity of Cap complexed by Stalk in the NP3 system was assessed by monitoring the relative quantities of Si, N and F in the NP3CPT system via energy dispersive X-ray (EDX)-TEM analysis. The results obtained are illustrated in Fig. 6 and Fig. S4 (ESI†). The semi-quantitative EDX analysis revealed significant quantities of F atoms (Fig. S4, ESI†) arising from the presence of surface-complexed Cap. In addition, the relative quantities of Si, N and F atoms (Fig. S4, ESI†), together with the relative masses of SiO2 and Stalk determined by TGA (Fig. S3, ESI†), are consistent with a Stalk:Cap molar ratio of 3:1 and a CPT loading of 4.0 wt% in the NP3CPT system. The data also reveal the presence of three Stalk (and hence one Cap) per 100 Si atoms (see ESI†). This is consistent with the expected complexation between the complementary Stalk and Cap and the associated obstruction of the MSNP pore apertures in NP3CPT.
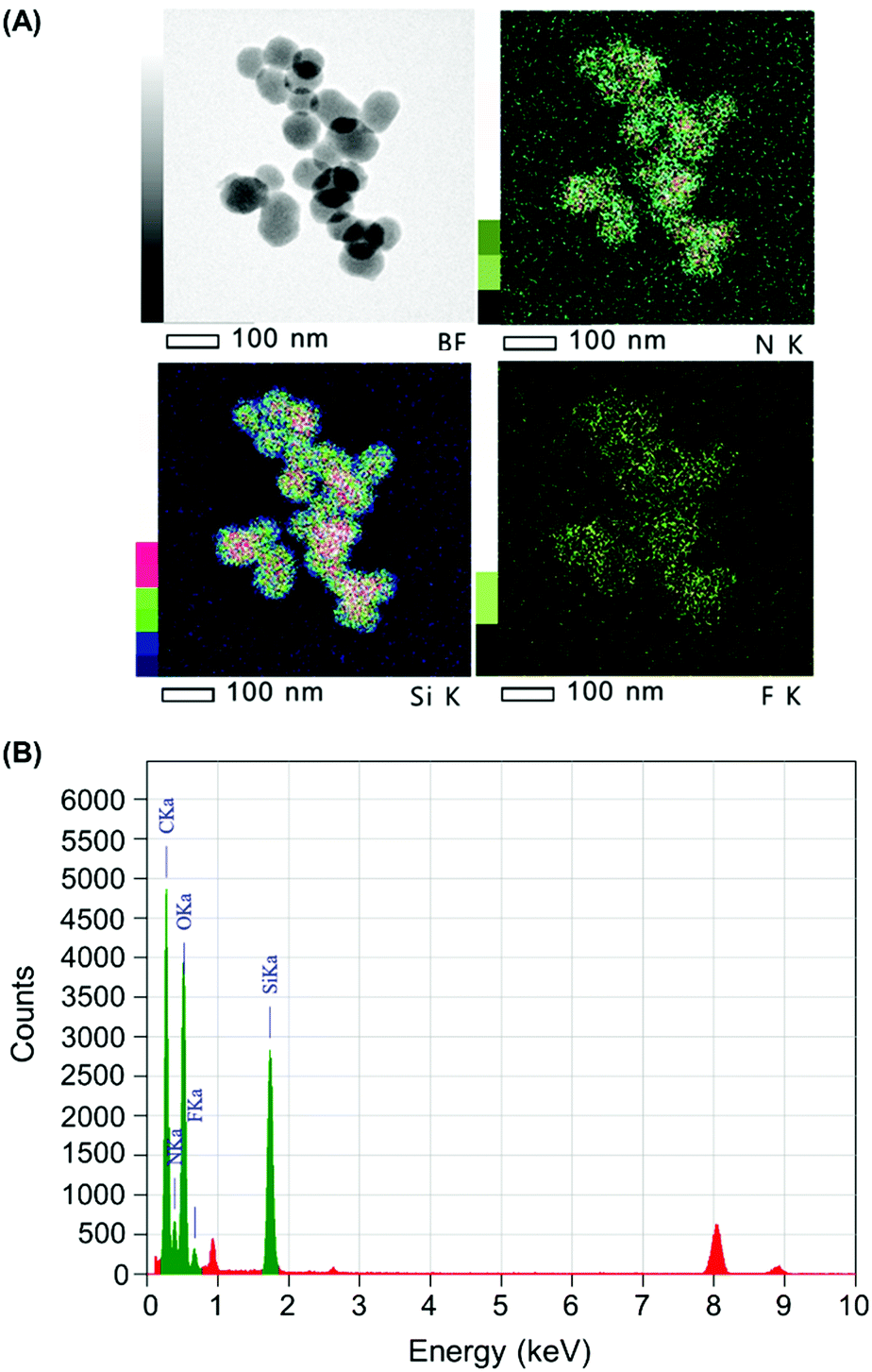 | ||
| Fig. 6 (A) Bright field (BF) and elemental mapping (Si, N and F) and (B) EDX spectrum of NP3CPT by EDX-TEM. | ||
In addition, the size and morphology of the nanoparticles were characterized at different stages during the synthesis to verify their uniformity. SEM and TEM micrographs of NP, NP3 and NP3CPT (Fig. 7A) showed relatively mono-dispersed nanoparticles with diameters ranging from 110 to 200 nm. The TEM images also revealed the expected transverse porosity in the materials. A comparison of the nanoparticle size obtained by SEM with that observed in solution via DLS studies (Fig. 7B) indicates that the functionalised nanoparticles are essentially unaggregated after being dispersed in solution, with relatively small polydispersity indices. As expected, the data indicate that functionalisation of NP with Stalk, to form NP3, results in a small increase in the hydrodynamic diameter of the nanoparticles (from 140 to ∼190 nm) without any increase in polydispersity, consistent with functionalisation of the particles by Stalk. The size and low polydispersity of NP3 remain unchanged after impregnation with CPT (to form NP3CPT).
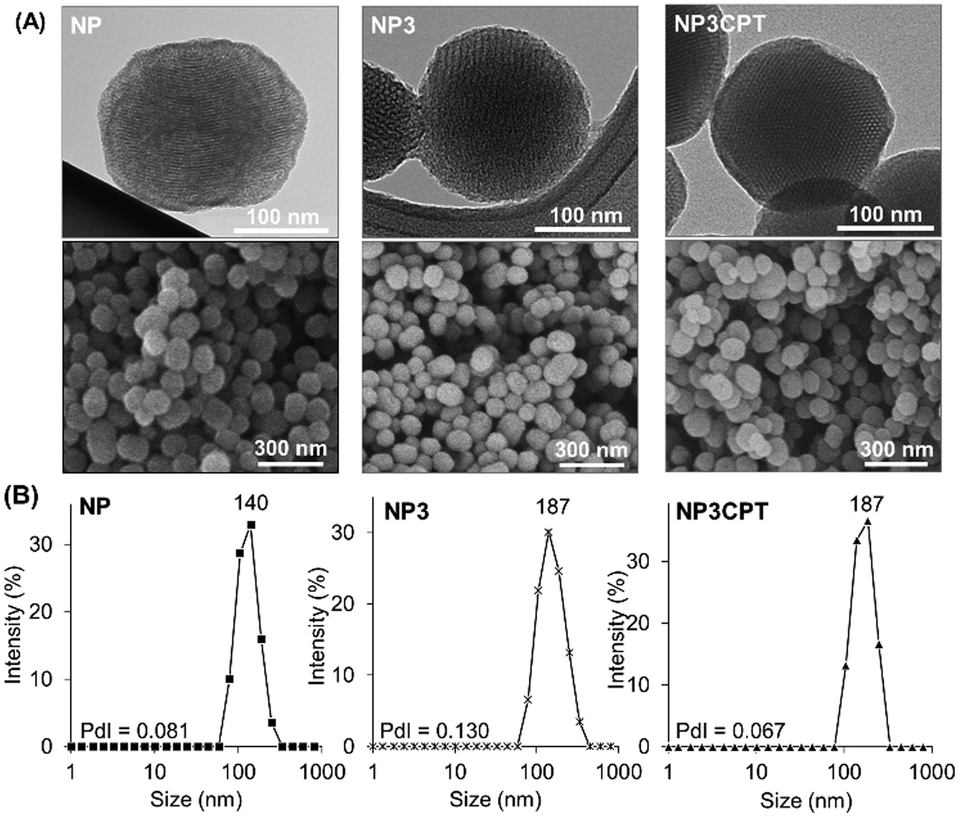 | ||
| Fig. 7 (A) TEM and SEM images and (B) DLS measurements of NP, NP3 and NP3CPT, nanomaterials. The polydispersity indices (PdI, measured by DLS) are included. | ||
Consequently, the resulting drug loaded NP3CPT have a suitable size for nanomedicine applications.
Thereafter, the stability of the Stalk–Cap complex as a function of pH (2, 5.5 or 7.4) was investigated in the case of NP3CPT by monitoring CPT release in solution via UV-Vis absorbance at 380 nm (Fig. 8). The data indicated that only about 5% of the drug is released at neutral pH, confirming that sufficient Stalk–Cap bonds remain intact to ensure that the nanoparticles can efficiently transport the chemotherapeutic agents within the blood stream without premature release of the payload.
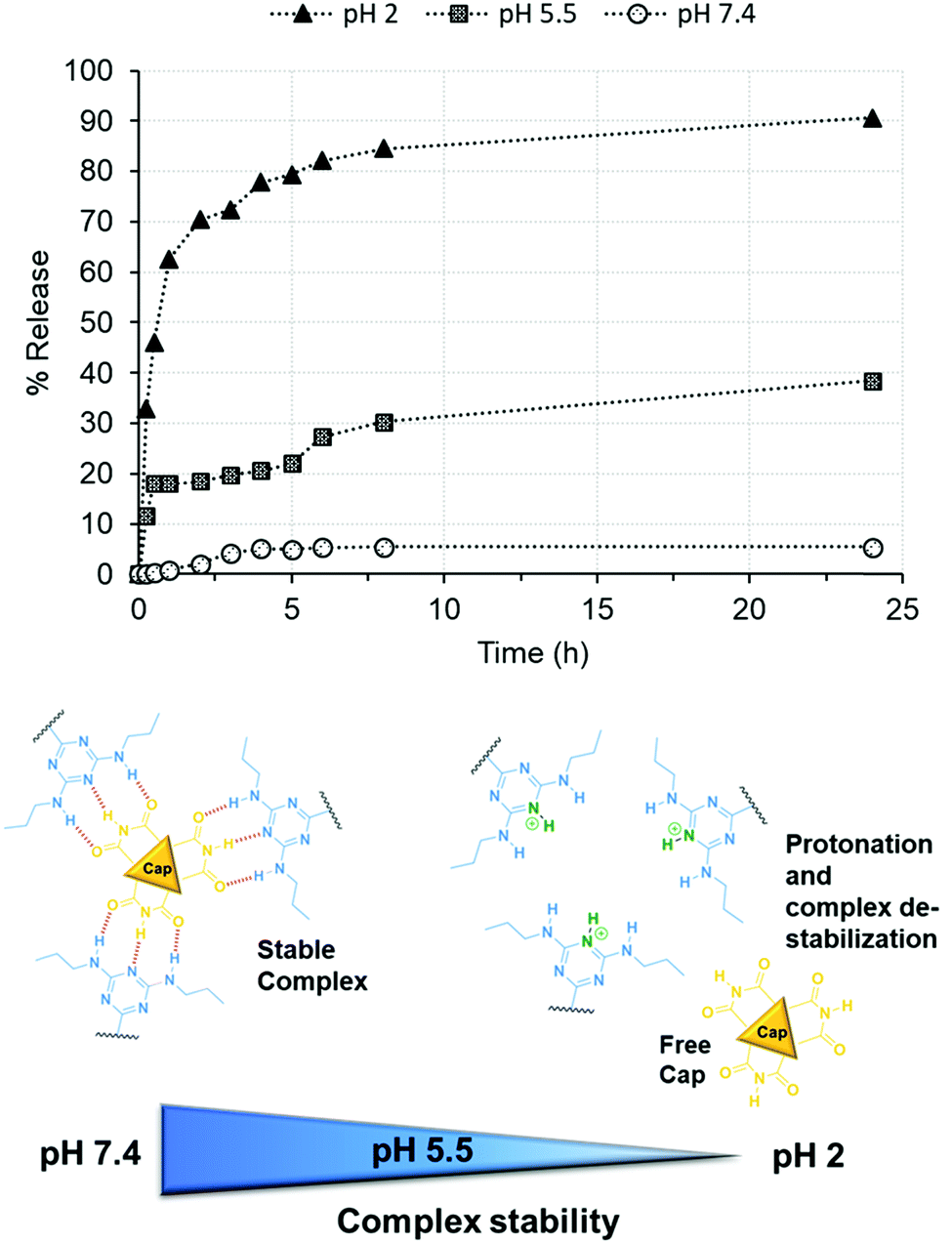 | ||
| Fig. 8 Release of CPT from NP3CPT mesopores (determined by UV/Vis absorbance spectroscopy) as function of pH. | ||
In contrast, around 38% of CPT was released at pH 5.5 after 24 h. This pH was chosen to mimic that present within the endosomal compartments, and the results suggest that release of the cargo and Cap would be expected to occur within the endosomal regions. As expected, essentially quantitative release of Cap is observed at pH 2 after only a few hours. This result demonstrates that the removal of the chemotherapeutic Cap and subsequent release of the second drug loaded within the pores is controlled by environmental pH. Our earlier studies of the release of Cap confirm that its release concentration profile is similar to that of CPT.
In vitro studies on MCF-7 breast cancer cells
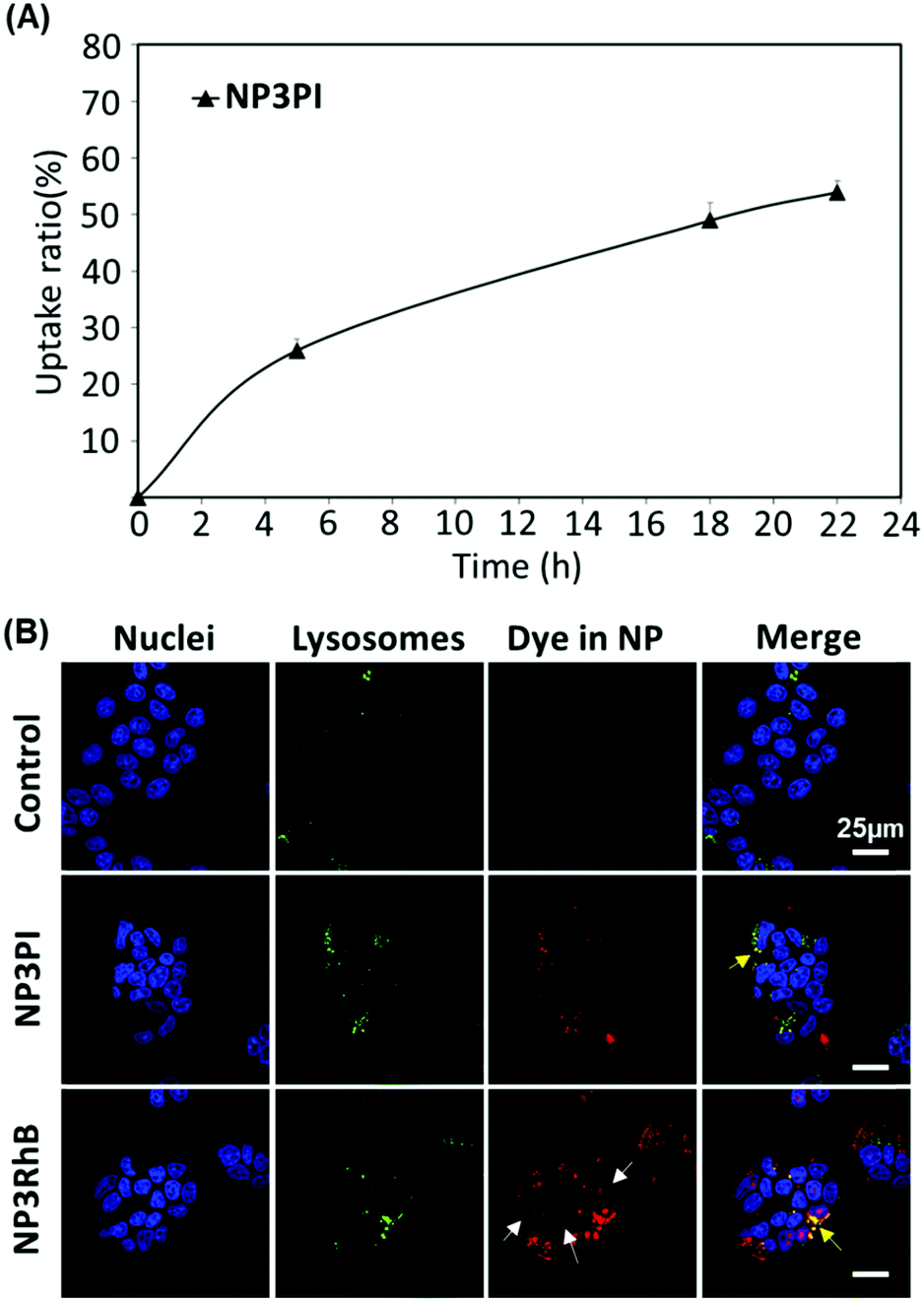 | ||
| Fig. 9 (A) Internalisation rate in MCF-7 cells using NP3PI. Error bars show standard deviation. (B) Confocal microscopy of control (top), NP3PI (middle) and NP3RhB (bottom) after 20 h of incubation in a culture medium of MCF-7 cells. | ||
Uptake of nanoparticles in MCF-7 cells – confocal microscopy
To assess the uptake of nanoparticles in MCF-7 breast cancer cells, the cells were incubated for 20 h with 40 μg mL−1 of NP3RhB and NP3PI (Fig. 9(B)). The endolysosomal compartments were labelled with the green lysotracker and the nuclei marked in blue with the Hoechst stain. The control images correspond to cells that have not been incubated with nanoparticles. In culture cells incubated with NP3PI, we can detect the presence of PI in red, and the yellow regions observed in the merged picture demonstrate the co-localisation of NP3PI with endo-lysosomal compartments (see yellow arrows in Fig. 9(B)). This validates the internalisation of nanoparticles by the endocytosis pathway. In addition, when culture cells were incubated with nanoparticles loaded with RhB (NP3RhB), which is known to cross the cell membranes (and lysosome), we observed that the red dye fully stained the cytoplasm (see white arrows in Fig. 9(B)), confirming the release of the cargo molecules once the nanocarriers have entered the lysosomes. Overall, these data confirm (1) the uptake of the nanomaterials by the cell via the endolysosomal pathway and (2) that the release of active drug molecules is triggered at lysosomal pH, consistent with the pH-sensitivity data included in Fig. 8.Cytotoxicity
The cytotoxicity of the new nanocarrier on MCF-7 breast cancer cells was then assessed via MTT assays. This study was performed with NP3RhB to determine the action of the cytotoxic 5-FU cap (RhB is known to be inactive31) and with NP3CPT to determine if the cytotoxicity is increased by the presence of two different drugs. As shown in Fig. 10(A), which illustrates cell death after 72 h of incubation with different batches of nanoparticles, NP exhibited essentially no cytotoxic activity (control). The corresponding data for NP3RhB confirmed that the active cap was released, with good cytotoxicity demonstrated against MCF-7 cells. Finally, NP3CPT showed excellent tumour elimination, with more than 90% of cell death from a concentration of 50 μg mL−1 and up to 99% of cell death at 100 μg mL−1. The IC50 of NP3CPT under these conditions is 4 μg mL−1, a particularly low value, which demonstrates the complementary and concomitant activity of the multidrug approach employed here, where each drug attacks the cells via different and complementary biological pathways.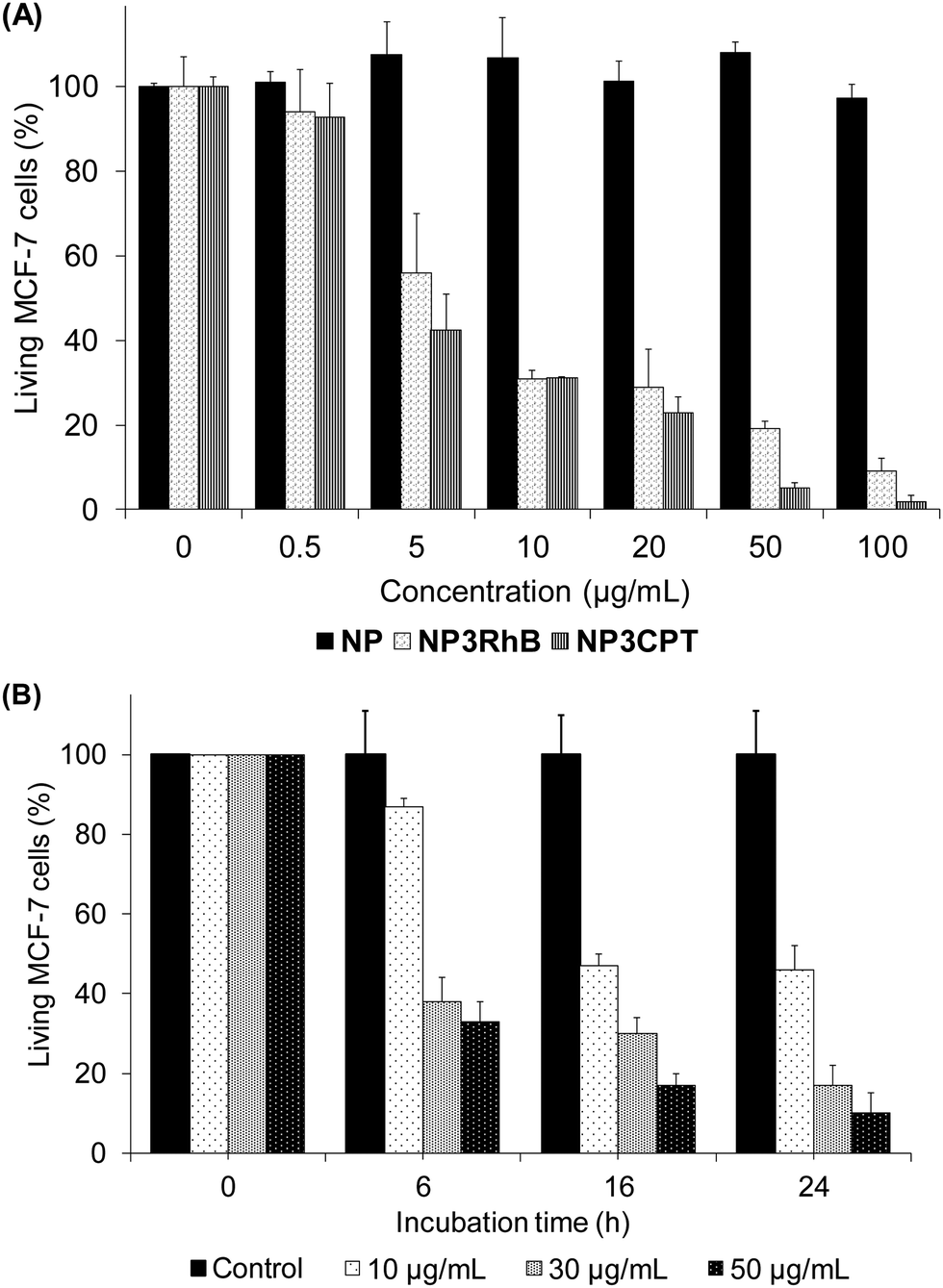 | ||
| Fig. 10 (A) Cytotoxicity test of NP, NP3RhB and NP3CPT on MCF-7 cells after 72 h of incubation. (B) Cytotoxic effect of NP3CPT on MCF-7 as a function of incubation time and concentration. Error bars show standard deviation. | ||
Following this promising result, the activity of NP3CPT on MCF-7 cells was determined at shorter times. For this, incubations with NP3CPT at 10, 30 and 50 μg mL−1 were performed and followed by MTT assays after 6, 16 and 24 h. The results presented in Fig. 10(B) revealed that the action of NP3CPT was relatively fast. Indeed, after only 6 h of incubation, MTT assay results already showed a significant inhibition of cell growth. Moreover, after 24 h the results obtained were close to those observed in the previous study carried out after 72 h of incubation. These data confirm that essentially complete apoptosis of the MCF-7 cells can be achieved, in vitro, with the NP3CPT system within 24 h.
Conclusions
Herein, a silica-based nanocarrier system in which CPT was loaded within the pores and a 5-FU analogue complexed on the surface as a pore-capping agent has been shown to be a viable system for environmentally-triggered, sequential release of these anti-cancer drugs for successful eradication of breast cancer cells. The nanosystem has been shown to respond to changes in environmental pH, thus enabling the delivery of a multi-drug payload within the lysosomal compartment of cancer cells. In contrast, almost no release of the cytotoxic compounds was observed under normal physiological conditions, thus minimising toxic side-effects in normal tissue and within the blood stream.The efficacy of this approach was demonstrated through in vitro trials, involving the consecutive delivery of the 5-FU derivative (rapid release) and CPT (diffusion-limited release) in MCF-7 breast cancer cells.
Under the conditions used in this study, essentially complete breast cancer cell apoptosis was observed within 72 h. This result highlights the attractive synergistic effect of the two drugs acting in tandem against tumoral cells. Additionally, the loading of the two synergistic drugs into silica nanocarriers offers the significant advantage of preventing the debilitating side effects usually associated with combination cancer therapy.
It is envisaged that the approach developed in this work could also be extended to multi-combinatorial therapy, involving the sequential release of three or more drugs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the CNRS and the French Higher Education and Research Ministry. This work was partially funded with a young researcher grant from the French National Research agency (ANR-2010-JCJC-1006-01). We acknowledge the imaging facility MRI, member of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04, “Investments for the future”). We thank Prof. Fuyuhiko Tamanoi for access to EDX-HRTEM at Kyoto University and help and support in pH-release testing.Notes and references
- Q. Hu, W. Sun, C. Wang and Z. Gu, Adv. Drug Delivery Rev., 2016, 98, 19–34 CrossRef CAS PubMed.
- R. Bayat Mokhtari, T. S. Homayouni, N. Baluch, E. Morgatskaya, S. Kumar, B. Das and H. Yeger, Oncotarget, 2017, 8, 38022–38043 Search PubMed.
- P. Sharma and James P. Allison, Cell, 2015, 161, 205–214 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Adv. Drug Delivery Rev., 2006, 58, 1456–1459 CrossRef CAS PubMed.
- N. Kolishetti, S. Dhar, P. M. Valencia, L. Q. Lin, R. Karnik, S. J. Lippard, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17939–17944 CrossRef CAS PubMed.
- A. Jhaveri, P. Deshpande and V. Torchilin, J. Controlled Release, 2014, 190, 352–370 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. d. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- R. K. Singh, J. C. Knowles and H.-W. Kim, J. Tissue Eng., 2019, 10, 2041731419877528 Search PubMed.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S.-Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS.
- I. I. Slowing, J. L. Vivero-Escoto, C.-W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- J. M. Rosenholm, V. Mamaeva, C. Sahlgren and M. Lindén, Nanomedicine, 2012, 7, 111–120 CrossRef CAS PubMed.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang, T. Jiang and S. Wang, Nanomedicine, 2015, 11, 313–327 CrossRef CAS PubMed.
- J. Wen, K. Yang, F. Liu, H. Li, Y. Xu and S. Sun, Chem. Soc. Rev., 2017, 46, 6024–6045 RSC.
- M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef PubMed.
- C.-H. Lee, S.-H. Cheng, I.-P. Huang, J. S. Souris, C.-S. Yang, C.-Y. Mou and L.-W. Lo, Angew. Chem., Int. Ed., 2010, 49, 8214–8219 CrossRef CAS PubMed.
- L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CrossRef CAS.
- C. Théron, A. Gallud, S. Giret, M. Maynadier, D. Grégoire, P. Puche, E. Jacquet, G. Pop, O. Sgarbura, V. Bellet, U. Hibner, J. I. Zink, M. Garcia, M. Wong Chi Man, C. Carcel and M. Gary-Bobo, RSC Adv., 2015, 5, 64932–64936 RSC.
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. Wang, B. Xing and Y. Zhao, Adv. Funct. Mater., 2012, 22, 5144–5156 CrossRef CAS.
- A. Rahikkala, S. A. P. Pereira, P. Figueiredo, M. L. C. Passos, A. R. T. S. Araújo, M. L. M. F. S. Saraiva and H. A. Santos, Adv. Biosyst., 2018, 2, 1800020 CrossRef.
- Z.-Y. Li, Y. Liu, X.-Q. Wang, L.-H. Liu, J.-J. Hu, G.-F. Luo, W.-H. Chen, L. Rong and X.-Z. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 7995–8001 CrossRef CAS PubMed.
- F. Muhammad, M. Guo, A. Wang, J. Zhao, W. Qi, Y. Guo and G. Zhu, J. Colloid Interface Sci., 2014, 434, 1–8 CrossRef CAS PubMed.
- M. C. Llinàs, G. Martínez-Edo, A. Cascante, I. Porcar, S. Borrós and D. Sánchez-García, Drug Delivery, 2018, 25, 1137–1146 CrossRef PubMed.
- S. Giret, C. Théron, A. Gallud, M. Maynadier, M. Gary-Bobo, M. Garcia, M. Wong Chi Man and C. Carcel, Chem. – Eur. J., 2013, 19, 12806–12814 CrossRef CAS PubMed.
- L. F. Liu, S. D. Desai, T.-K. Li, Y. Mao, M. E. I. Sun and S.-P. Sim, Ann. N. Y. Acad. Sci., 2000, 922, 1–10 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- S. Guichard, I. Hennebelle, R. Bugat and P. Canal, Biochem. Pharmacol., 1998, 55, 667–676 CrossRef CAS PubMed.
- Q. Luo, P. Wang, Y. Miao, H. He and X. Tang, Carbohydr. Polym., 2012, 87, 2642–2647 CrossRef CAS.
- C. Théron, A. Birault, M. Bernhardt, L. M. A. Ali, C. Nguyen, M. Gary-Bobo, J. R. Bartlett, M. Wong Chi Man and C. Carcel, J. Sol-Gel Sci. Technol., 2019, 89, 45–55 CrossRef.
- M. K. Trivedi, R. M. Tallapragada, A. Branton, D. Trivedi, G. Nayak, R. K. Mishra and S. Jana, J. Mol. Pharm. Org. Process Res., 2015, 3, 128–134 Search PubMed.
- M. A. Mondragón, V. M. J. Castaño, M. C. A. Garcia and S. Téllez, Vib. Spectrosc., 1995, 9, 293–304 CrossRef.
- P. Fisher, Wildl. Soc. Bull., 1999, 27, 318–329 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tb02225b |
| This journal is © The Royal Society of Chemistry 2020 |