
General bis(fluorophenyl azide) photo-crosslinkers for conjugated and non-conjugated polyelectrolytes†
Desmond W. Y.
Teo‡
a,
Zaini
Jamal‡
a,
Qiu-Jing
Seah
a,
Rui-Qi
Png
 b and
Lay-Lay
Chua
*ab
b and
Lay-Lay
Chua
*ab
aDepartment of Chemistry, National University of Singapore, Lower Kent Ridge Road, S117552, Singapore. E-mail: chmcll@nus.edu.sg
bDepartment of Physics, National University of Singapore, Lower Kent Ridge Road, S117550, Singapore
First published on 15th November 2019
Abstract
Crosslinking of semiconductors is critical towards building heterostructures to enable flexibility in the design of devices. Bis(fluorophenyl azide)s (FPAs) and their sterically-substituted derivatives (sFPAs) have been demonstrated to be able to photo-crosslink semiconductor polymers upon deep-UV exposure, stabilizing their morphologies in devices. Here, we report the development of both UV (254 nm) and i-line (365 nm) photo-activatable FPA and sFPA – FPA80 and sFPA82 – both incorporating 3,5-bis(methylene)-1,1-dimethyl-4-piperidonium, a conjugation-extending bridge that also bears an ionic group. These materials are useful for generic photo-crosslinking of both conjugated and non-conjugated polyelectrolytes. Good thermal stability and solubility in polar organic solvents are obtained. Good film retention (>80%) can be achieved with a crosslinker loading of less than 10 wt/wt% for high-molecular polymers (Mw > 100k), superior to alkyl bisazides. The photo-crosslinking efficiency of ca. 20% in model polyelectrolytes is limited by the occlusion of ionic crosslinkers from the polymer matrix where the C–H insertion reaction occurs, hence suppressing photo-crosslinking efficiency.
Introduction
Photopatterning and crosslinking of the polymer semiconductor, charge injection and/or transport layers are needed to improve the performance and/or functionality of next-generation organic semiconductor devices.1 The key crosslinking methodologies are based on photo, thermal and chemical activation mechanisms, of which photo and thermal reactive systems have been most actively investigated, particularly for polymer solar cells.2–4 Crosslinking of charge transport layers is also important, as it enables the finessing of multi-layer structures for light-emitting diodes, and other devices, without relying on orthogonal solvent processing.5Amongst the photo reactive chemistries, fluorinated phenyl azide (FPA) is the most interesting because of its efficient generation of singlet nitrene that can insert non-specifically into unactivated C–H bonds.6–10 C–H bonds are ubiquitously present in the alkyl side chains of semiconductor polymers; a crosslink is effected when photolyzed FPA bonds two polymer chains together. The undesirable nitrene attacks on the aromatic C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, or heteroatom of the polymer backbone can be suppressed by steric substitution of the FPA ring, to give what we call the sterically-substituted FPA methodology (sFPA).1 This can provide photo-crosslinking at near unity efficiency, i.e., the fraction of reactive moieties forming crosslinks, for a variety of semiconductor polymers with little detrimental effects on bandgap, exciton properties, carrier mobility and energetics.1
C, or heteroatom of the polymer backbone can be suppressed by steric substitution of the FPA ring, to give what we call the sterically-substituted FPA methodology (sFPA).1 This can provide photo-crosslinking at near unity efficiency, i.e., the fraction of reactive moieties forming crosslinks, for a variety of semiconductor polymers with little detrimental effects on bandgap, exciton properties, carrier mobility and energetics.1
The unfluorinated phenyl azides, on the other hand, give lower photo-crosslinking efficiencies, because of short singlet nitrene lifetimes, competing ketenimine expansion, and the crowding of the triplet nitrene crosslinking pathways by numerous side reactions.11,12 The alkyl azides are even less effective. They photolyze to imines, which do not usually give C–H insertion reactions,13,14 although alkyl azides can undergo thermal 1,3-dipolar cycloaddition reactions,15 which have been exploited to form crosslink polymer:fullerene systems.16–18 Diazirines are wholly ineffective in polymer films,19 presumably because the photolyzed singlet carbenes have lifetimes that are too short20 compared to the diffusion release of N2 from the carbene in the solid matrix. Therefore, the sFPA methodology remains the most promising one for non-specific photo-crosslinking of organic polymer semiconductors.
Photo-crosslinking of π-conjugated polyelectrolytes (CPEs), a special class of organic polymer semiconductors with ionizable groups tethered to the polymer backbone, will potentially enable the development of sophisticated structures for bioelectronics and neuromorphic devices. This will also be useful to block organic ‘electromigration’, for example in PEDT:PSSH,21 and to form robust charge-doped layers for carrier injection and collection.22 The ions present in CPEs, however, require the photo-crosslinker to be functionalized also with ionic groups for compatibilization. Sulfonated bis(phenyl azide)s can be used, but at low photo-crosslinking efficiencies,23 trapping a large fraction of undesired side products. The previously developed FPAs can provide higher efficiencies, but require deep UV activation.24 However photo-activation at longer wavelengths, in particular i-line (365 nm), is attractive to manufacturing, because of compatibility with existing tools. We have thus extended the FPA methodology to develop FPA photo-crosslinkers photo-activatable from the deep UV (254 nm) to i-line (365 nm) for polyelectrolytes. These photo-crosslinker additives can be readily formulated into the desired polyelectrolyte, obviating the need to synthesize new derivatives for each polyelectrolyte.
Results and discussion
Synthesis of photo-crosslinkers with an ionic linker
The chemical structures of the two new photo-crosslinkers synthesized here, (3E,5E)-3,5-bis(4-azido-2,3,5,6-tetrafluorobenzylidene)-1,1-dimethyl-4-oxopiperidin-1-ium trifluoromethanesulfonate (FPA80-TfO) and (3E,5E)-3,5-bis(4-azido-3,5-difluoro-2,6-di-isopropylbenzylidene)-1,1-dimethyl-4-oxopiperidin-1-ium trifluoromethanesulfonate (sFPA82-TfO), conjugation-extended by the 3,5-bis(methylene)-1,1-dimethyl-4-piperidonium bridging unit, and counterbalanced by trifluoromethanesulfonate (TfO), are depicted in Fig. 1. For reference, 3′-bis(4-azido-2,3,5,6-tetrafluorobenzamido)dipropyldimethylammonium trifluoromethanesulfonate (FPA70-TfO), with the bis(carbonyl-3-aminopropyl)dimethylammonium bridge unit, is also synthesized. This crosslinker is identical to AAA-X, where X = PF6, BF4, ClO4 and CF3COO, described earlier,24 except for the change in anion to TfO for better solubility in acetronitrile, which is the solvent of choice for a wide range of polyelectrolytes, conjugated polyelectrolytes and doped polymers.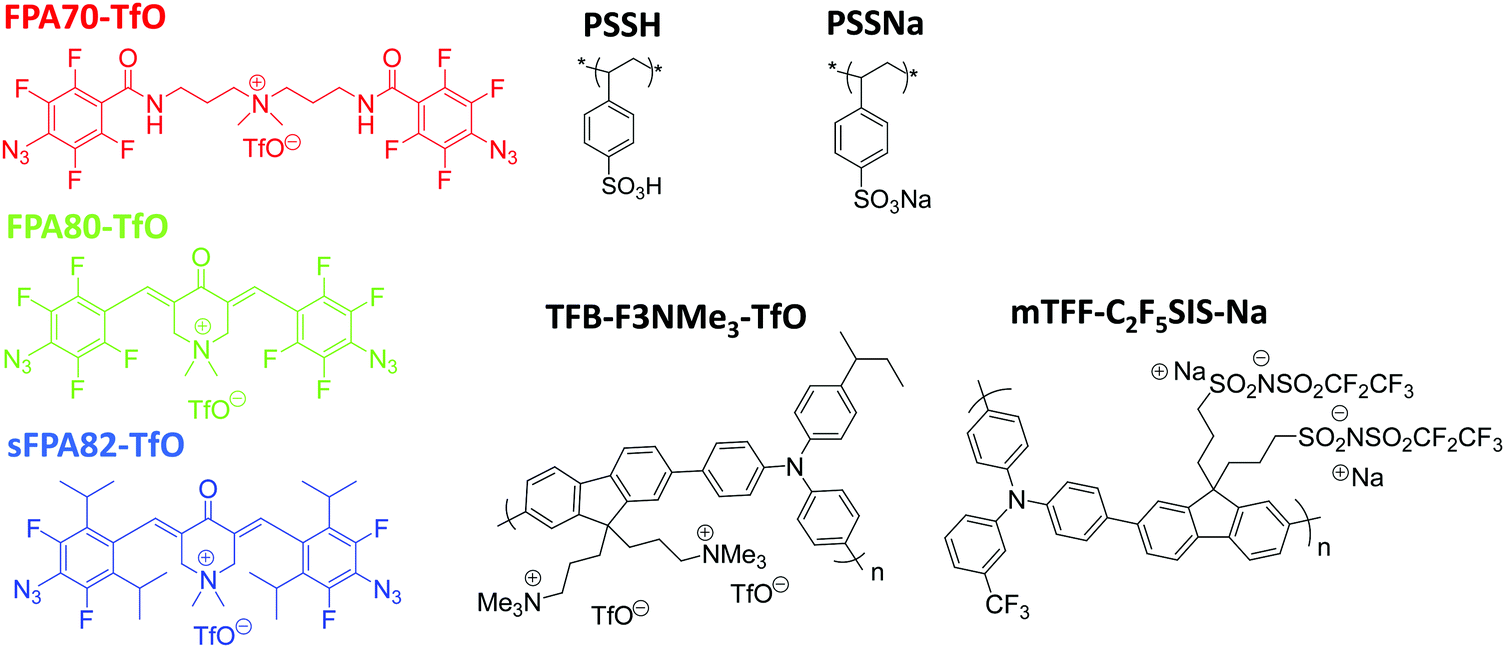 | ||
| Fig. 1 Chemical structures of photo-crosslinkers with ionic bridge units (FPA70-TfO, FPA80-TfO, and sFPA82-TfO), polyelectrolytes (PSSNa, and PSSH) and conjugated polyelectrolytes (TFB-F3NMe3-TfO, and mTFF-C2F5SIS-Na) used in this work. | ||
Briefly, for FPA80-TfO, azide nucleophilic aromatic substitution was performed at the para-positions of 2,3,4,5,6-pentafluorobenzaldehyde, which was then aldol-condensed with N-methyl-4-piperidone, and the amine was quaternized with methyl triflate (see Scheme 1a). For sFPA82-TfO, 3,4,5-trifluoro-1-bromobenzene was reacted with 2-chloropropane by Friedel–Crafts acylation, then formylated, azide nucleophilic aromatic substitution was then performed at the para-positions, and the aldehyde was then aldol-condensed with N-methyl-4-piperidone, and the amine quaternized with methyl triflate (see Scheme 1b). For FPA70-TfO, 3,3′-diamino-N-methyldipropylamine was first reacted with 2,3,4,5,6-pentafluorobenzoyl chloride, azide nucleophilic aromatic substitution was performed at the para-positions, and the amine was then quaternized with methyl triflate to yield FPA70-TfO (see Scheme 1c). This is superior to the previous method of using methyl iodide followed by AgI precipitation.
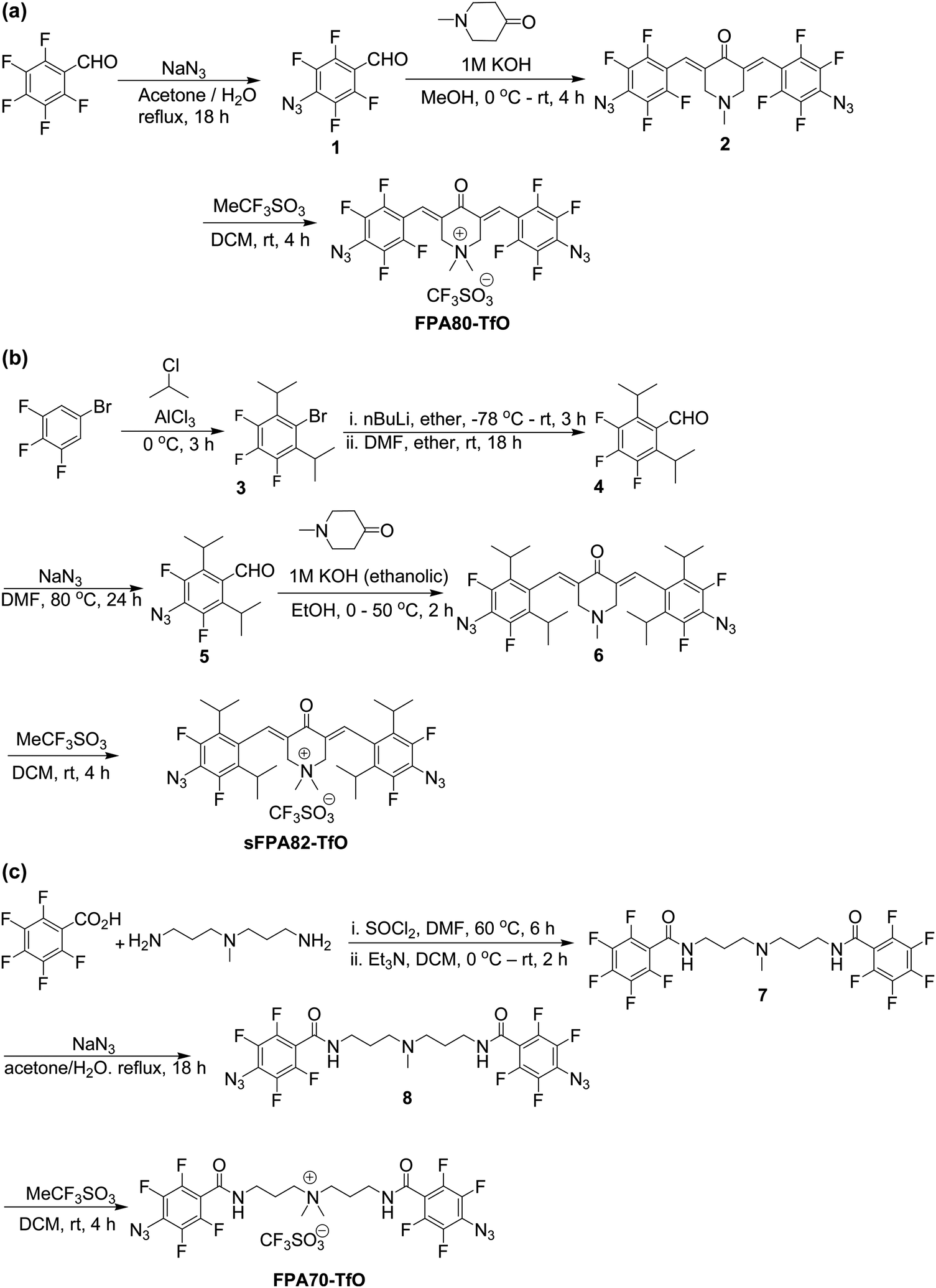 | ||
| Scheme 1 (a) Synthesis route of FPA80-TfO. (b) Synthesis route of sFPA82-TfO. (c) Synthesis route for FPA70-TfO. | ||
FPA80-TfO, FPA70-TfO and sFPA82-TfO are soluble in lower alcohols, acetone and aprotic polar solvents, including acetronitrile, hence they can be blended into polyelectrolytes that are soluble in a similar solvent system. Further, these solid photo-crosslinkers can be stored in the dark in the fridge for up to at least 1 year. UV-Vis and IR spectra show no observable change, indicating the good shelf-life of the crosslinker. These crosslinkers in the reference PSSNa films can still be photolyzed to give crosslinked PSSNa films.
Characterization of the crosslinkers
The solution-state UV absorption spectra of the three photo-crosslinkers are shown in Fig. 2a. FPA70-TfO shows one absorption maximum at 256 nm.24 Conjugation extension in both FPA80-TfO and sFPA82-TfO shifts the lowest-lying π–π* absorption band maximum (λmax) to ca. 320 nm with the tail extending into the visible spectrum. FPA80-TfO shows λmax at 330 nm with the tail extending to 430 nm, and molar absorptivity at 365 nm of ca. 1.3 × 104 cm−1 M−1, similar to 2,6-bis(4′-azidobenzylidene)methylcyclohexanone.12sFPA82-TfO shows λmax at 313 nm, with the tail extending to 410 nm, blue-shifted from FPA80 by inevitable ring twists introduced by the bulky isopropyl groups at the ortho-positions of the FPA rings. Nevertheless, its molar absorptivity at 365 nm, 3.5 × 103 cm−1 M−1, is still useable. All the crosslinkers have absorption bands extending into the deep UV region.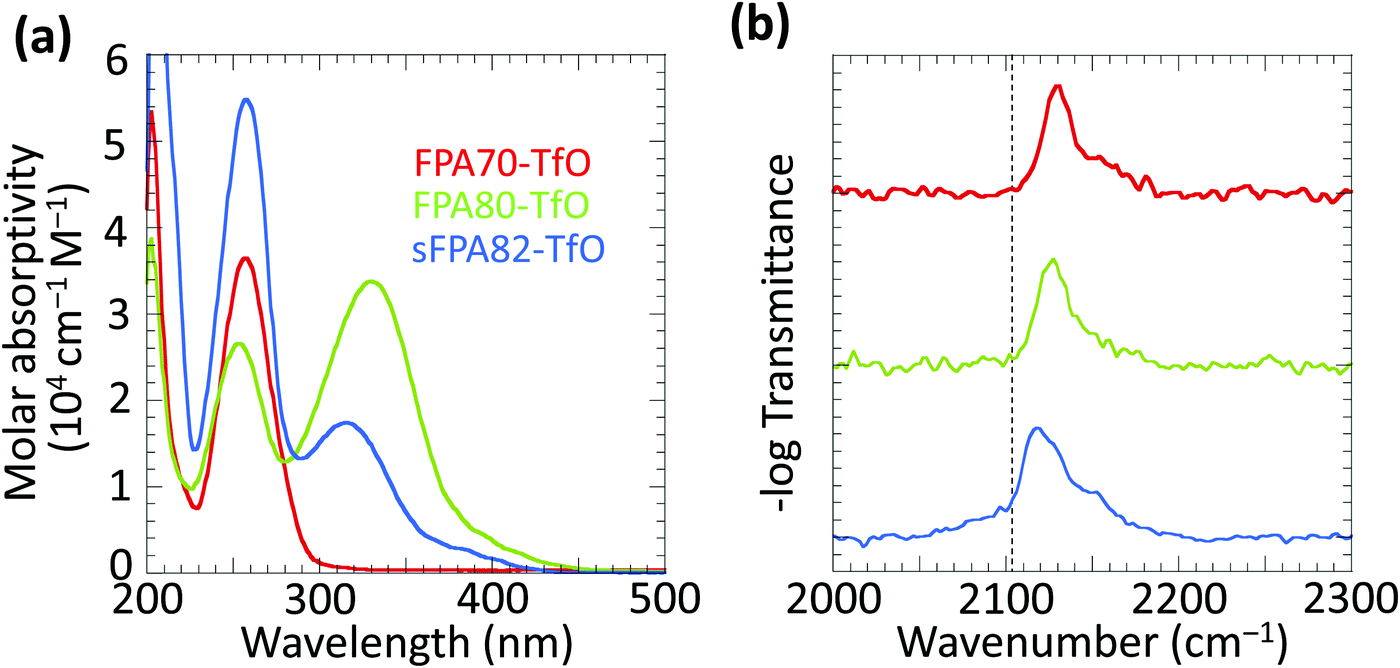 | ||
| Fig. 2 UV and FTIR spectroscopy of photo-crosslinkers. (a) Solution-state UV spectra in acetonitrile. (b) Solid-state FTIR spectra of 10 wt/wt% crosslinker in poly(styrenesulfonic acid) matrix, showing the N3 asymmetric stretching vibration band. Vertical dashed line marks the unfluorinated phenyl azide band position. | ||
FTIR spectra of 10 wt/wt% photo-crosslinkers in dry PSSH matrix (under N2) confirmed the presence of the azide group through the asymmetric stretching band νas(N3) in the 2080–2170 cm−1 region.25FPA70, FPA80 and sFPA82 bands appear at 2129, 2126, and 2118 cm−1, respectively (Fig. 2b). The unfunctionalised phenyl azide band is at 2103 cm−1.26 The blue shift here is due to the electron-withdrawing fluorine substitution, partially mitigated for sFPA82 by the electron-donating isopropyl substitution.
To compare the relative photo speed of the crosslinkers, we exposed 200 nm-thick films of PSSNa with 10 wt/wt% crosslinker to 254 nm DUV emitted by an 8 W lamp placed 3 cm away from the film, monitoring the intensity of νas(N3) as a function of exposure time (Fig. S1, ESI†). The optical density due to the crosslinker is less than 0.2, so self-absorption is small, and that of PSSNa is less than 0.1, so the matrix filter effect is also negligible, at this wavelength. The photo speed decreases as: FPA70 (0.073 s−1) > sFPA82 (0.038) > FPA80 (0.017). The ratio of sFPA82 to FPA80 is in agreement with the ratio of their molar absorptivities, but FPA70 is faster by a factor of ca. 3, indicating a correspondingly higher quantum yield. The photospeed when exposed to i-line (365 nm wavelength) decreases as: FPA80 (0.016 s−1) > sFPA82 (0.006 s−1). The decrease is in agreement with the decrease in their molar absorptivities.
To evaluate the thermolysis of these azides, we employed thermogravimetric analysis (TGA) and bake-temperature-dependent FTIR spectroscopy. For TGA, the crosslinker powders were heated at 5 °C min−1 under flowing nitrogen (Fig. 3a). The first weight loss step at 140–170 °C, 8–9% loss, corresponds cleanly to the step change expected for azide thermolysis with the evolution of N2 gas (theory: 7.2–8.2% loss). There is little difference between the onset decomposition temperatures of the three materials; there is also no water of hydration in the crystals. FTIR spectra of the crosslinkers dispersed in PSSH films (10 wt/wt%) and baked at increasing temperatures, 5 min at each step, confirm the thermolysis of the azides (Fig. 3b). The νas(N3) spectra are shown in Fig. S2 (ESI†). All three crosslinkers show a similar extrapolated onset thermolysis temperature of 115 °C, with the maximum loss rate at ca. 135 °C, broadly consistent with the TGA results. Therefore, these crosslinkers can be briefly processed in films at temperatures up to 110 °C before the photo exposure step.
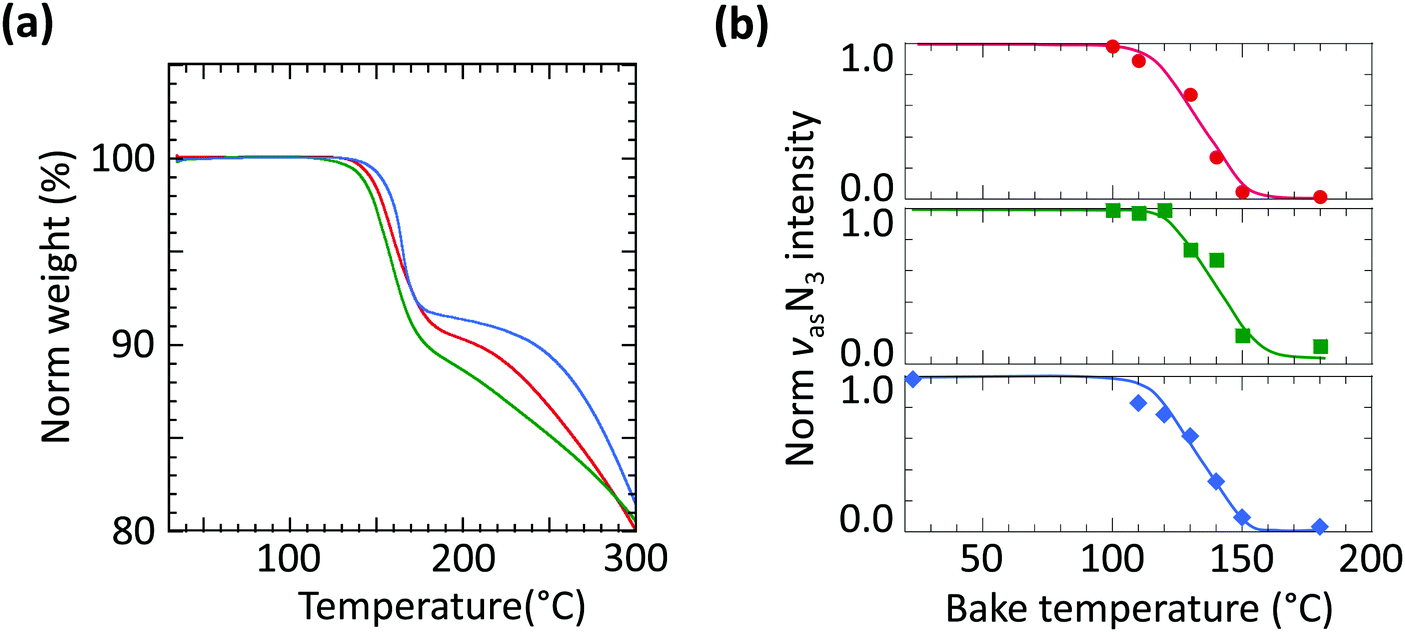 | ||
| Fig. 3 Thermolysis of crosslinkers. (a) Thermogravimetry characteristics. Conditions: equilibration, 35 °C, 10 min; ramp 5 °C min−1; flowing N2. (b) Bake-temperature dependence of the intensity of the νasN3 mode in the PSSH film matrix. Red, green, and blue data for FPA70-TfO, FPA80-TfO, and sFPA82-TfO, respectively. | ||
Crosslinking efficiency of the photo-crosslinkers
Fig. 4 shows that the film retention, i.e. gel characteristic, values of all three crosslinkers are similar for each of the four model polyelectrolytes tested, plotted against the crosslinker number concentration c. The selection of polyelectrolytes comprises an acidic polymer (PSSH, Mw 75k), a non-conjugated polyanion (PSSNa, Mw 75k), a conjugated polyanion (mTFF-C2F5SIS-Na, Mw 40k), and a conjugated polycation (TFB-F3NMe3-TfO, Mw 78k). These are important polyelectrolytes in widespread use.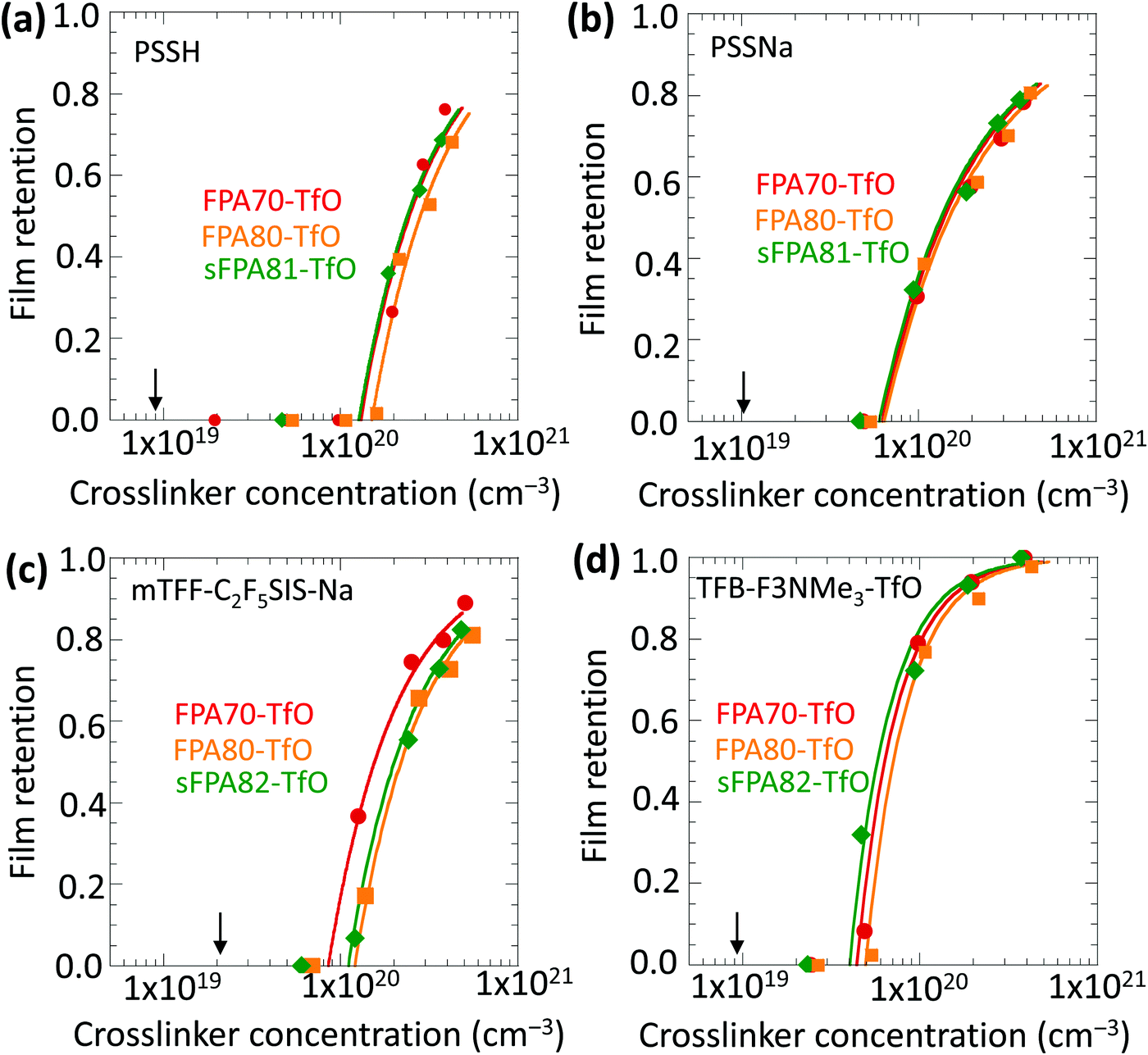 | ||
| Fig. 4 Plot of film retention against concentration of the crosslinkers for different polyelectrolytes. (a) PSSH (Mw 75k), (b) PSSNa (Mw 75k), (c) mTFF-C2F5SIS-Na (Mw 40k), and (d) TFB-F3NMe3-TfO (Mw 78k). The black arrow marks the theoretical gel point of the polymers, which is 9.6 × 1018 (PSSH), 1.0 × 1019 (PSSNa), 2.3 × 1019 (mTFF-C2F5SIS-Na), and 9.3 × 1018 cm−3 (TFB-F3NMe3-TfO). The curves are a guide to the eye, fitted to: η = 1 − (cc/c)n, where η is the film retention after photocrosslinking and solvent development, cc is the experimental gel point given by the onset of film retention plot, and c is the crosslinker concentration. n is a fitted characteristic exponent, which is n = 1.1 (PSSH), n = 0.82 (PSSNa), n = 1.15 (mTFF-C2F5SIS-Na), and n = 1.9 (TFB-F3NMe3-TfO). The starting film thickness is ca. 100 nm thick. Density of mTFF-C2F5SIS-Na is taken to be 1.55 g cm−3 and for the rest of the polyelectrolyte, it is 1.2 g cm−3. | ||
The crosslinker number concentration is computed from: 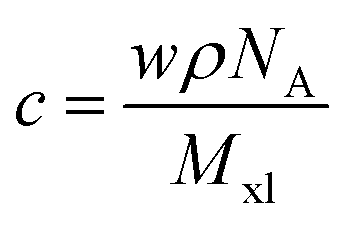 , where w is the weight ratio of the crosslinker to the polyelectrolyte matrix, ρ is the mass density of the matrix, NA is the Avogadro constant, and Mxl is the crosslinker molar mass. The film retention η is the ratio of the final film thickness to the initial film thickness, after photo exposure and solvent development. The gel point cc is the crosslinker concentration at the onset of the gelation, i.e., where an insoluble infinite-molecular-weight polymer fraction first emerges, indicated by the take-off in the film retention curve. The theoretical gel point is the crosslinker concentration given by:
, where w is the weight ratio of the crosslinker to the polyelectrolyte matrix, ρ is the mass density of the matrix, NA is the Avogadro constant, and Mxl is the crosslinker molar mass. The film retention η is the ratio of the final film thickness to the initial film thickness, after photo exposure and solvent development. The gel point cc is the crosslinker concentration at the onset of the gelation, i.e., where an insoluble infinite-molecular-weight polymer fraction first emerges, indicated by the take-off in the film retention curve. The theoretical gel point is the crosslinker concentration given by: 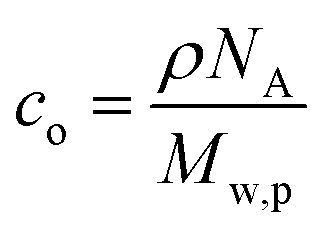 , where Mw,p is the weight-average molar mass of the polyelectrolyte.27 This corresponds to the situation of an average of one crosslink per polymer chain, where an infinite-molecular-weight fraction should appear. This is marked with an arrow in the film retention plots. The experimental gel point, however, occurs at a higher crosslinker concentration, i.e. cc ≥ cc,o. The photo-crosslinking efficiency ξ is then given by the ratio of co to cc, i.e.
, where Mw,p is the weight-average molar mass of the polyelectrolyte.27 This corresponds to the situation of an average of one crosslink per polymer chain, where an infinite-molecular-weight fraction should appear. This is marked with an arrow in the film retention plots. The experimental gel point, however, occurs at a higher crosslinker concentration, i.e. cc ≥ cc,o. The photo-crosslinking efficiency ξ is then given by the ratio of co to cc, i.e.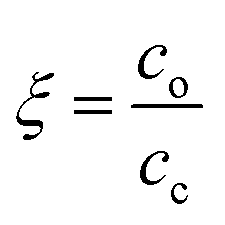 . The ξ values are tabulated in Table 1.
. The ξ values are tabulated in Table 1.
| Photocrosslinker | Photo-crosslinking efficiency ξ | |||
|---|---|---|---|---|
| PSSH (Mw 75k) | PSSNa (Mw 75k) | mTFF-C2F5SIS-Na (Mw 40k) | TFB-F3NMe3-TfO (Mw 78k) | |
| FPA70-TfO | 0.074 | 0.16 | 0.27 | 0.20 |
| FPA80-TfO | 0.064 | 0.16 | 0.19 | 0.18 |
| sFPA82-TfO | 0.076 | 0.16 | 0.20 | 0.23 |
For the same polymer, the ξ values for all three crosslinkers are more or less similar, indicating that conjugation extension and steric substitution do not degrade the photo crosslinking efficiency. For PSSH, the average ξ value is 0.07 ± 0.005; for PSSNa, it is 0.16. The lower ξ in the acidic polyelectrolyte matrix may be related to the rapid protonation of the singlet phenyl nitrene, which then undergoes nucleophilic degradation, interfering with singlet insertion.28–30 The average ξ value of mTFF-C2F5SIS-Na is 0.22 ± 0.05, 35% higher than that of PSSNa, despite the fact that they are both polyanions with the same counter-ion. We attribute this to a better dispersion of the crosslinkers across the smaller ion clusters present in the conjugated polyelectrolyte, because of its lower ion volume fraction. A stronger aggregation of crosslinkers in PSSNa would lead to quenching of the desired crosslinking reaction by nitrene ‘self-reaction’ with azide, or ionic nucleophilic degradation. Finally, the average ξ value for the conjugated polycation TFB-F3NMe3-TfO is 0.20 ± 0.03, similar to that of mTFF-C2F5SIS-Na. The ion volume fractions of both polymers are similar. Their limited ξ is attributed to the tethered ion pairs in these conjugated polyelectrolytes aggregated into relatively large multiplets and clusters in the spaces between the polymer backbone. Hence, when ionic crosslinkers are added, they are often either embedded inside or located on the surface of the large ion cluster. As a result, photogenerated nitrene is occluded from the polymer matrix, which suppress photo-crosslinking efficiency. For poly(acrylic acid), where ion clusters are not present, and the polymer is not strongly acidic, the FPA70 crosslinker appears to be fully molecularly dispersed, and ξ can reach 1.24
Conclusion
In conclusion, we have developed the first FPA-based both UV and i-line photo-crosslinkers, FPA80 and sFPA82, suitable for generic photo-crosslinking of polyelectrolytes. Their photo-crosslinking efficiencies are not inferior to the original deep UV variants. These crosslinkers are easy to use, and give relatively efficient photo-crosslinking. Their absorption bands extend throughout the entire UV range (100–400 nm), allowing photo exposure in principle at these wavelengths where the polyelectrolyte matrix absorbs least strongly, limited by a commercially-available lamp.Methods
Materials
PSSH (Mw 75k) and PSSNa (Mw 75k) are obtained from Sigma-Aldrich and are used as received. Poly(9,9-bis(3-(pentafluoroethanesulfonylimidosulfonyl)propyl)fluorene-2,7-diyl-alt-1,4-phenylene-(m-trifluoromethylphenylimino)-1,4-phenylene) sodium salt (mTFF-C2F5SIS-Na) of Mw 40k is synthesized in-house following Tang et al.22,31 The molecular weights are determined by gel permeation chromatography with N,N-dimethylformamide and 0.1 M lithium bromide as an eluent. Poly(9,9-bis(3′-tetramethylamino)fluorene-2,7-diyl-1,4-phenylene-N-(p-sec-butylphenyl)-imino-1,4-phenylene) (TFB-F3NMe3-TfO) of Mw 78k is synthesized following Tang et al.32 Its molecular weight is determined by gel permeation chromatography with chloroform and 0.25% tetraethylammonium as an eluent.Synthesis of photo-crosslinkers with an ionic linker
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (2:1) afforded FPA70-TfO as a white solid in 60% yield (450 mg). 1H NMR (400 MHz, CD3OD): δ 2.08–2.15 (m, 4H), 3.15 (s, 6H), 3.40–3.44 (m, 4H), 3.50 (t, J = 6.7 Hz, 4H); 19F NMR (377 MHz, CD3OD): δ −153.41 to −153.32 (m, 4F), −144.80 to −144.70 (m, 4F), −80.14 (s, 3F).
:methanol (2:1) afforded FPA70-TfO as a white solid in 60% yield (450 mg). 1H NMR (400 MHz, CD3OD): δ 2.08–2.15 (m, 4H), 3.15 (s, 6H), 3.40–3.44 (m, 4H), 3.50 (t, J = 6.7 Hz, 4H); 19F NMR (377 MHz, CD3OD): δ −153.41 to −153.32 (m, 4F), −144.80 to −144.70 (m, 4F), −80.14 (s, 3F).
UV-Vis spectroscopy
Crosslinkers are dissolved in acetonitrile at a concentration of 0.01 mg mL−1. Their UV-Vis absorption spectra are then measured using an Aligent Cary 60 UV-Vis spectrophotometer with a quartz cuvette of 1 cm path length.Preparation of crosslinker in polyelectrolyte solution
25 mg mL−1 stock solutions of crosslinkers are prepared in acetonitrile. Selected 20 mg mL−1 stock solutions of polyelectrolytes are also prepared: PSSNa and PSSH in methanol/water (1:1 v/v); mTFF-C2F5SIS-Na and TFB-F3NMe3-TfO in acetonitrile. Solutions of desired wt/wt% of crosslinker in polyelectrolyte are then prepared from these stock solutions, and spin-cast onto desired substrates.
Photolysis test
200 nm-thick 10 wt/wt% crosslinkers in PSSNa film on an intrinsic Si wafer (1 cm × 1 cm) are prepared and annealed (110 °C, 5 min) in a N2 glove-box (pH2O < 1 ppm). The film is mounted into a vacuum chamber for spectral collection using a Nicolet 8700 FTIR spectrophotometer from Thermo Scientific. This first spectrum is taken as the reference 0s exposure data. Subsequently, the film is exposed to 254 nm UV-light in a N2 glove-box and the spectrum is collected from the spectrophotometer again. The spectra are collected at increasing exposure times until there is no visible νas(N3) intensity. A photolysis test with 365 nm i-line light exposure is also performed. The 254 nm and 365 nm light fluxes are 0.74 mW cm−2 and 3.76 mW cm−2 respectively.Thermal stability
200 nm-thick 10 wt/wt% crosslinkers in PSSH film on an intrinsic Si wafer (1 cm × 1 cm) are prepared in a N2 environment inside a glove-box (pH2O < 1 ppm). The film is mounted into a vacuum chamber for spectral collection using a Nicolet 8700 FTIR spectrophotometer from Thermo Scientific. Subsequently, the film is heated at 100 °C (5 min) in a N2 glove-box and the spectrum is collected from the spectrophotometer again. The spectra are also collected upon higher temperature treatment at 120 °C, 130 °C, 140 °C, 150 °C and 180 °C, with 5 min duration for each temperature.Photo-crosslinking efficiency
About 50–80 nm-thick polyelectrolyte films with different crosslinker loadings on Si native wafer (1 cm × 1 cm) were prepared, annealed (110 °C, 5 min), and photo-crosslinked (UV 254 nm, 5 min) in a N2 glove-box (pH2O < 1 ppm). To determine film retention, their initial and remaining film thickness after development with acetronitrile (30 s) are measured by using a profiler (KLA-Tencor AlphaStep D-600). Film retention is then calculated as the ratio of the film thickness after solvent development to the initial film thickness. Film retention η is plotted against different crosslinking loading c to obtain the gel dose curve. The experimental data are then fitted with the equation: η = 1 − (cc/c)n, where n is a characteristic exponent. The concentration at which film retention exhibits an abrupt increase is the gel point, cc.Author contributions
L. L. C. planned the experiments. Z. J. synthesized the photo-crosslinkers. Q. J. S. syntheized the conjugated polyelectrolytes. D. W. Y. T. and Z. J. performed characterizations including UV-Vis, FTIR, conductivity and device measurements. L. L. C. and R. Q. P. supervised the work. L. L. C., Z. J. and D. W. Y. T. wrote the manuscript. The authors declare no competing financial interests.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Research Foundation, Prime Minister's Office, Singapore under its Competitive Research Programme (CRP Award No. NRF CRP 11-2012-03: R-144-000-339-281, R-143-000-608-281). We thank P. K. H. Ho for the discussions.References
- R. Q. Png, P. J. Chia, J. C. Tang, B. Liu, S. Sivaramakrishnan, M. Zhou, S. H. Khong, H. S. O. Chan, J. H. Burroughes, L. L. Chua, R. H. Friend and P. K. H. Ho, Nat. Mater., 2010, 9, 152–158 CrossRef CAS PubMed.
- H. J. Kim, A. R. Han, C. H. Cho, H. Kang, H. H. Cho, M. Y. Lee, J. M. J. Frechet, J. H. Oh and B. J. Kim, Chem. Mater., 2012, 24, 215–221 CrossRef CAS.
- G. Wantz, L. Derue, O. Dautel, A. Rivaton, P. Hudhomme and C. Dagron-Lartigau, Polym. Int., 2014, 1346–1361 CrossRef CAS.
- J. W. Rumer and I. McCulloch, Mater. Today, 2015, 18, 425–435 CrossRef CAS.
- N. Aizawa, Y. J. Pu, H. Sasabe and J. Kido, Org. Electron., 2013, 14, 1614–1620 CrossRef CAS.
- E. Leyva, D. Munoz and M. S. Platz, J. Org. Chem., 1989, 54, 5938–5945 CrossRef CAS.
- J. F. W. Keana and S. X. Cai, J. Org. Chem., 1990, 55, 3640–3647 CrossRef CAS.
- R. Poe, K. Schnapp, M. J. T. Young, J. Grayzar and M. S. Platz, J. Am. Chem. Soc., 1992, 114, 5054–5067 CrossRef CAS.
- K. A. Schnapp, R. Poe, E. Leyva, N. Soundararajan and M. S. Platz, Bioconjugate Chem., 1993, 4, 172–177 CrossRef CAS PubMed.
- A. Marcinek, M. S. Platz, S. Y. Chan, R. Floresca, K. Rajagopalan, M. Golinski and D. Watt, J. Phys. Chem., 1994, 98, 412–419 CrossRef CAS.
- G. B. Schuster and M. S. Platz, Advances in Photochemistry, John Wiley & Sons, 1992, vol. 17, pp. 69–143 Search PubMed.
- M. Avadanei, J. Appl. Polym. Sci., 2015, 134, 44694 Search PubMed.
- E. P. Kyba and R. A. Abramovitch, J. Am. Chem. Soc., 1980, 102, 735–740 CrossRef CAS.
- N. Gritsan and M. Platz, Organic Azides: Syntheses and Applications, John Wiley & Sons, Ltd, 2010 Search PubMed.
- A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, Inc., 2002 Search PubMed.
- L. Derue, O. Dautel, A. Tournebize, M. Drees, H. Pan, S. Berthumeyrie, B. Pavageau, E. Cloutet, S. Chambon, L. Hirsch, A. Rivaton, P. Hudhomme, A. Facchetti and G. Wantz, Adv. Mater., 2014, 26, 5831–5838 CrossRef CAS PubMed.
- J. W. Rumer, R. S. Ashraf, N. D. Eisenmenger, Z. Huang, I. Meager, C. B. Nielsen, B. C. Schroeder, M. L. Chabinyc and I. McCulloch, Adv. Energy Mater., 2015, 5, 1401426 CrossRef.
- D. Landerer, C. Sprau, D. Baumann, P. Pingel, T. Leonhard, D. Zimmermann, C. L. Chochos, H. Krüger, S. Janietz and A. Colsmann, Sol. RRL, 2019, 3, 1800266 CrossRef.
- H. Mehenni, V. Pourcelle, J. F. Gohy and J. Marchand-Brynaert, Aust. J. Chem., 2012, 193–201 CrossRef CAS.
- M. S. Platz, Acc. Chem. Res., 1995, 28, 487–492 CrossRef CAS.
- R. Q. Png, P. J. Chia, S. Sivaramakrishnan, L. Y. Wong, M. Zhou, L. L. Chua and P. K. H. Ho, Appl. Phys. Lett., 2007, 91, 013511 CrossRef.
- C. G. Tang, M. C. Y. Ang, K. K. Choo, V. Keerthi, J. K. Tan, N. S. Mazlan, T. Kugler, J. H. Burroughes, R. Q. Png, L. L. Chua and P. K. H. Ho, Nature, 2016, 539, 536 CrossRef CAS PubMed.
- F. J. Touwslager, N. P. Willard and D. M. de Leeuw, Appl. Phys. Lett., 2002, 81, 4556–4558 CrossRef CAS.
- S. H. Khong, S. Sivaramakrishnan, R. Q. Png, L. Y. Wong, P. J. Chia, L. L. Chua and P. K. H. Ho, Adv. Funct. Mater., 2007, 17, 2490–2499 CrossRef CAS.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, Inc., 3rd edn, 1990 Search PubMed.
- L. K. Dyall and J. E. Kemp, Aust. J. Chem., 1966, 20, 1395–1402 CrossRef.
- R. F. T. Stepto, Polymer Networks: Principles of Their Formulation, Structure and Properties, Blackie Academic & Professional, London, UK, 1998 Search PubMed.
- J. Michalak, H. B. Zhai and M. S. Platz, J. Phys. Chem., 1996, 100, 14028–14036 CrossRef CAS.
- R. A. McClelland, T. A. Gadosy and D. Ren, Can. J. Chem., 1998, 76, 1327–1337 CrossRef CAS.
- R. A. McClelland, M. J. Kahley, P. A. Davidse and G. Hadzialic, J. Am. Chem. Soc., 1996, 118, 4794–4803 CrossRef CAS.
- M. C. Y. Ang, C. G. Tang, Q. M. Koh, C. Zhao, Q. J. Seah, Y. Wang, M. Callsen, Y. P. Feng, R. Q. Png and L. L. Chua, Mater. Horiz., revised manuscript submitted Search PubMed.
- C. G. Tang, M. Nur Syafiqah, Q. M. Koh, C. Zhao, Z. Jamal, Q. J. Seah, M. J. Cass, M. J. Humphries, I. Grizzi, J. H. Burroughes, R. Q. Png, L. L. Chua and P. K. H. Ho, Nature, 2019, 573, 519–525 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tc04060a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |