
An optomicrofluidic device for the detection and isolation of drop-encapsulated target cells in single-cell format†
R.
Gaikwad
and
A. K.
Sen
 *
*
Department of Mechanical Engineering, Indian Institute of Technology Madras, Chennai-600036, India. E-mail: ashis@iitm.ac.in
First published on 12th October 2020
Abstract
Single-cell analysis has emerged as a powerful method for genomics, transcriptomics, proteomics, and metabolomics characterisation at the individual cell level. Here, we demonstrate a technique for the detection and selective isolation of target cells encapsulated in microdroplets in single-cell format. A sample containing a mixed population of cells with fluorescently labelled target cells can be focused using a sheath fluid to direct cells in single file toward a droplet junction, wherein the cells are encapsulated inside droplets. The droplets containing the cells migrate toward the centre of the channel owing to non-inertial lift force. The cells present in the droplets are studied and characterised based on forward scatter (FSC), side scatter (SSC), and fluorescence (FL) signals. The FL signals from the target cells can be used to activate a selective isolation module based on electro-coalescence, using suitable electronics and a program to sort droplets containing the target cells in single-cell format from droplets containing background cells. We demonstrated the detection and isolation of target cells (cancer cells: HeLa and DU145) from mixed populations of cells, peripheral blood mononuclear cells (PBMC) + cervical cancer cells (HeLa) and PBMC + human prostate cancer cells (DU145), at a concentration range of 104–106 ml−1 at 300 cells per s. The performance of the device is characterised in terms of sorting efficiency (>97%), enrichment (>1800×), purity (>98%), and recovery (>95%). The sorted target cells were found to be viable (>95% viability) and showed good proliferation when cultured, showing the potential of the proposed sorting technique for downstream analysis.
1. Introduction
In recent years, single-cell analysis has received immense attention mainly due to its ability to overcome the limitations of conventional cell analysis methods, which are only capable of providing average information about a population of cells.1–3 The average information from the analysis of a population of cells fails to present detailed biological characteristics relating to individual cells. The detection, isolation, and analysis of biological cells in single-cell format could provide answers to critical questions relating to cancer cells4 and other rare cells,5 stem cell biology,5 and immunology,5 and could facilitate the development of new drugs and therapies.5,6 Rare cells, such as cancer cells, fetal cells, and stem cells in the blood, hold significant opportunities for disease diagnostics and prognostics.7 Thus, it is important to detect and isolate rare cells for downstream analysis.Single-cell analysis techniques involve the separation and compartmentalisation of cells of interest from large cell populations in single-cell format. The cells of interest are isolated either based on physical properties, such as size,8 density,9 and deformability,10 or depending on biological properties, such as cell adhesion, via protein expression.5 There are several conventional techniques available for the selective isolation of target cells, which include immunomagnetic separation,11 filtration,12 centrifugation,13 and flow cytometry.7 Flow cytometry is a widely used technique for single-cell analysis due to its improved accuracy and higher sensitivity compared to other techniques. It is a vital technique for various clinical applications, such as immunology,14 cancer biology,15 and haematology.16,17
Flow cytometers based on optical,17 magnetic,19 and electrical (impedance)20 techniques have been developed. However, optofluidics-based flow cytometry is the most widely used technique due to its unique advantages in terms of speed (throughput), sensitivity, and specificity.17,18 In opto-flow cytometry, cells present in a sample are focused two-dimensionally, typically at the centre of a capillary or channel, using a sheath fluid so that the cells flow in single-file with an adequate distance between them to prevent cell–cell interactions. The focused cells are made to pass through an optical interrogation window, created by a laser beam intersecting the flow path, giving rise to scattering signals. The forward scatter (FSC) signal represents the size of the cells, whereas the side scatter (SSC) signal represents the internal structure of the cells.21 If the cells are labelled with fluorescent molecules, the fluorescent (FL) signals emitted by the cells can be used for the detection of specific types of cells.15
A microfabricated fluorescence-activated cell sorter (FACS) analyses a small volume of fluid to detect and sort the biological cells present in the sample fluid.22 However, conventional FACS systems are very expensive and thus are available only in centralised research facilities and major healthcare centres.23 Similarly, due to their complexity, regular maintenance and skilled expertise are required to operate the machine, analyse data, and make reports. Besides, skilled technicians are needed to fix any functional failures and for troubleshooting. These factors add considerable cost to the maintenance of the machines and thus increase the cost per test during diagnosis. In the last few years, owing to advancements in microfluidics technology, research focus has shifted towards developing cost-effective and portable micro-FACS devices.
However, one of the main hindrances in the development of micro-FACS devices is that complicated techniques are used for two-dimensional focusing and controlling the distance between biological cells in an optical window in a microchannel.23 Various active24–26 and passive27–31 techniques exist for focusing particles inside microchannels, such as multilayer sheath flow, inertial microfluidics, acoustophoresis, magnetophoresis, and elasto-inertial microfluidics. In multilayer sheath flow, hydrodynamic forces govern particle focusing.32 In the case of one-dimensional flow focusing, the sample fluid is focused using a co-flowing sheath fluid on both sides of the central sample stream (in the horizontal plane).33 Here, since the particles are not focused in the vertical plane, two particles present inside the sample fluid can have different velocities and hence arrive at the detection zone at the same time; this can generate erroneous results. On the contrary, in two-dimensional focusing, the sample fluid is focused using a sheath fluid in both the horizontal and vertical planes.34 However, here, the requirements for complicated fabrication procedures and multiple inlets are challenges that need to be addressed. Inertial and viscoelastic flow focusing techniques exclude the use of a sheath fluid.35 Inertial focusing requires longer channels for focusing; this can be addressed by using spiral channels, but it further introduces an additional force known as Dean drag.36 Viscoelastic focusing demands a viscoelastic fluid as the medium for suspending particles and uses viscoelastic forces to focus the particles.37 Various active techniques, such as acoustic, magnetic, optical, and DEP methods, incorporate external stimuli to focus the particles inside microchannels; however, such techniques require additional equipment, thus leading to complicated setups.38 In the proposed technique, the cells are focused in single file using a sheath fluid and are subsequently encapsulated inside droplets at a flow focusing junction. The droplets encapsulating the cells self-align toward the centre of the channel due to non-inertial lift force and move into the detection window in single file, thus overcoming the challenges stated above.
Another critical challenge in the development of micro-FACS systems is the isolation of target cells after detection, preferably in single-cell format for downstream single-cell analysis. In the literature, various techniques have been reported to achieve the isolation of target cells, such as hydrodynamic,39 dielectrophoresis,40 optical,41 and piezoelectric22 methods. However, such techniques either require high voltages or high shear forces, thus affecting cell viability and properties, offering low throughput, and/or requiring complicated instrumentation; thus, they are not amenable to the development of a microfluidic sorter. Also, none of these techniques allow for the extraction and isolation of the target cells in single-cell format. Here, we report a method wherein, upon the detection of the droplet-encapsulated target cells, selective electro-coalescence is used to isolate droplets containing target cells for downstream analysis. In the literature, electric fields have been employed for the coalescence of droplets for microparticle extraction and droplet sorting.42–44 The coalescence of droplets in an emulsion along the direction of flow has been explored.45 The coalescence of aqueous droplets parallel to an aqueous phase stream in a direction normal to the flow direction has also been investigated.43,46 However, the latter device requires very high voltage (∼thousands of volts) and a large electric field (107 V m−1); therefore, it is not suitable for biological applications due to cell viability issues.43 In the proposed method, the aqueous droplets containing cells are in microscopic contact with an aqueous-oil co-flow interface in the electric field region, thus requiring a significantly lower voltage and smaller electric field for coalescence. Our approach thus enables the rapid extraction of the target cells in single-cell format without causing cell damage.
Here, we demonstrate the optical detection and electro-coalescence-based isolation of target cells encapsulated in microdroplets in single-cell format for downstream analysis. A sample containing a mixture of target and background cells is first focused into a single-file stream using a sheath fluid, and individual cells are encapsulated inside droplets. Further, the droplet-encapsulated cells are analysed via measuring the FSC, SSC, and FL signals. Upon the detection of a target cell, an electro-coalescence-based selective isolation module is activated using suitable software and electronics so that the droplets containing the target cells are sorted from those containing the background cells. The performance of the system is evaluated in terms of sorting efficiency, enrichment, purity, and recovery. The viability of the target cells was verified using trypan blue assays, and cells could be cultured, indicating that the isolated target cells could be used for downstream analysis.
2. Experiments
2.1 Device geometry and fabrication
Fig. 1a shows a schematic diagram of the experimental device, which consists of three working modules with a depth of 130 μm throughout. First is the particle focusing and encapsulation module (Fig. 1b), which has a channel width of 70 μm in the focusing region and a throat width of 25 μm in the droplet generation region. Second is the optical detection module (Fig. 1c), which consists of four grooves that contain optical fibres that are placed at a distance of 100 μm from the channel walls. Groove I, which is perpendicular to the fluid channel, is used as an excitation fibre (incident beam); groove II is placed at an angle of 45° to groove I and is used for fluorescence (FL) detection; and groove III is placed at an angle of 135° to groove I and is used for the detection of the side scattered (SSC) signal. The forward scattered (FSC) signal fibre (groove IV) is placed at a 5° angle to the incident beam to avoid the direct exposure of the optical detector to the incident beam. The grooves each have a width and depth of 130 μm and are designed to hold standard single-mode (10 μm ID and 125 μm OD) and multimode (62.5 μm ID and 125 μm OD) optical fibres in place. Third is the electro-coalescence (EC) module (Fig. 1e), which consists of a pair of electrodes placed at the bottom of the channel to execute EC. The electrodes have a width of 50 μm and are placed with a gap of 50 μm between them. Device fabrication was carried out in two steps. First, standard photolithography was used to fabricate the silicon master, which was further used to fabricate the PDMS microchannel layer via soft lithography. The details of the master fabrication and soft lithography processes are discussed elsewhere.47 Second, the electrode layer was fabricated via the deposition of silver onto a glass slide using thermal evaporation and then electrode patterning was carried out using photolithography and etching. The detailed process for the fabrication of the electrodes is explained elsewhere.48 | ||
| Fig. 1 (a) Schematic diagrams of the experimental device showing the three different modules used for the detection and isolation of cells, (b) the particle focusing and encapsulation module, (c) the optical detection module, and (d) the electro-coalescence module. | ||
2.2 Working principle
In the particle focusing and encapsulation (PFE) module, particles/cells present in the sample are focused using an aqueous sheath fluid so that particles/cells move in single file when approaching the droplet generation junction. The width of the focused stream (wf) and the distance between the particles/cells (g2) can be controlled via adjusting the sheath-to-sample flow-rate ratio (r = Qsh/Qs).49 During droplet generation, particles/cells get encapsulated inside droplets. The flow rates (discrete Qd and continuous Qc) are adjusted to establish stable droplet generation/encapsulation.50 Biocompatible oil is used as the continuous phase. In the focusing region, an adequate distance between adjacent particles/cells is maintained so that each droplet contains no more than one cell. The cells can be labelled with fluorescent antibodies that are specific to the target cells (T) or with suitable fluorescent dyes. As the droplets encapsulating the particles/cells pass through the laser beam, FSC, SSC, and FL signals (Fig. 1d) are measured. The droplet generation frequency is controlled via adjusting the flow rate ratio (r) so that the droplets cross the laser beam in single file. Once a target cell is detected, an optoelectronic circuit sends a signal to the electrodes to selectively isolate the target droplet from background droplets (B) (Fig. 1d).To maintain stable droplets, a surfactant is added to the continuous phase (oil). The flow rate of the aqueous co-flow stream (Qa) is adjusted to ensure that the width of the continuous phase in the electro-coalescence region is less than the droplet size so that the droplets are in microscopic contact with the oil-aqueous interface. In the absence of an electric field, surfactant molecules covering the drop-oil and water stream-oil interfaces exhibit tail-tail repulsion (known as disjoining pressure) and therefore prevent the coalescence of water drops with the water stream. Upon the application of an electric field, there is a reorientation of the surfactant molecules, allowing the aqueous phases across the thin oil-layer (aqueous drops and stream) to merge or electro-coalesce with each other.51 In our case, we only need the voltage/electric field to overcome the disjoining pressure due to the surfactant molecules. However, in electro-coalescence systems reported in the literature,44,51 typically, electro-coalescence is a two-step process: first, the aqueous drops are brought into contact with other aqueous drops or an aqueous interface due to coulombic force acting on the drops; and second, the electric field/voltage overcomes the disjoining pressure, leading to electro-coalescence. The first step requires a considerably higher voltage, which is not required in the present case; therefore, in our case, electro-coalescence can be achieved at a lower voltage and with a smaller electric field.
Two sorting schemes are employed (Fig. 1d). In the positive sorting scheme, under default conditions, the electrodes are kept energised and background droplets continuously coalesce with the aqueous stream; thus, background particles/cells are extracted into the aqueous stream. Upon the detection of a target droplet and the arrival of the particular droplet at the electrodes, the electrodes are de-energised; thus, the target droplet does not coalesce with the aqueous stream and is directed toward outlet 1. In the negative sorting scheme, under default conditions, the electrodes are de-energised and the background droplets are collected at outlet 1. Upon the detection of a target drop, the electrodes are energised, the target drop coalesces with the aqueous stream, and the target particle/cell is extracted into the aqueous stream and collected at outlet 2.
2.3 Experimental setup: fluidics and optoelectronics
A schematic diagram and a photograph of the experimental setup are depicted in Fig. 2a and b, respectively. Syringe pumps (neMESYS Pump, Cetoni, Germany) are used for infusing liquid samples. Polyethylene tubing is used to establish a fluidic connection between the syringe pumps and device inlets. The optics setup consists of an excitation laser source and optical detectors. The excitation laser source is a 532 nm continuous wave laser (Wave Form Systems, Inc., USA). The beam generated from the laser is coupled with a standard single-mode optical fibre (10/125 μm), which is inserted into the excitation fibre groove in the device to focus the laser beam inside the channel. When the droplets pass through the incident beam, different optical signals are generated, and these are collected with the help of multimode optical fibres (62.5/125 μm) placed inside the collection grooves. To minimise reflection losses, the fibre grooves are filled with an index matching fluid (Fiber Instruments Sales Inc., USA) before inserting the optical fibres. For the detection of FSC, SSC, and FL signals, a fibre-coupled photodiode (PD) (Thorlabs Inc. USA), an avalanche photodiode (APD) (Hamamaatsu Photonics, Japan), and a single photon counting module (SPCM) (Thorlabs Inc. USA), respectively, are used. The FSC signal is collected in the forward direction of the incident beam, hence the signal strength is adequate for detection with a PD. The SSC signal is weaker than the FSC signal, hence it requires a highly sensitive APD detector to detect the signal. The FL signal is weaker compared to the FSC and SSC signals, and thus a SPCM, which has higher sensitivity compared to an APD, is used. Further, the weak FL signal suggests that a particular range of light wavelengths should be allowed into the detector. Hence, a bandpass filter (ET575/50m, Chroma Technology Corp. USA) is mounted onto the SPCM detector for coupling the SPCM with the optical fibre. Additionally, a microscope system (Axiovert A1, Carl Zeiss, Germany) coupled with a high-speed camera (Photron Inc., USA) and a fluorescence lamp is also used to trace the droplets. A data acquisition (DAQ) system (NI USB-6251, National Instruments, USA) is used to acquire data using the Labview program. The APD and PD modules are connected to the analog input pins of the DAQ system and data acquisition is done at a sampling rate of a thousand samples per second. In the feedback system, a high voltage amplifier (Amp-Line Corp., USA) is used to amplify the signal (sinusoidal waveform, 0–5 V, 15 kHz) coming from the function generator, and the amplified signal (0–25 V) is further amplified using a transformer. A maximum AC voltage of 60 V is used in our experiments, although our setup is capable of delivering AC voltages in the range of 25 V to 150 V. The output of the transformer is applied to the electrodes through a solid-state relay (capacity: input, 4–32 V DC; output, 240 V AC), which acts as a switch to modulate the signal applied to the electrodes and is controlled by the Arduino board (Uno R3) via the Labview platform. The acquisition and post-processing of optical signals are carried out using computer programs developed on the Labview platform, and the details are presented in section S1.† The signal to the relay for the deactivation of the electrode during positive sorting or its activation during negative sorting is triggered ∼3 ms before a target cell arrives at the electrodes (i.e., at t = ts), taking into account the response time of the electronics. Time evolution plots of the detection and electro-coalescence-based sorting of target cells from background cells in positive and negative sorting modes are presented in Fig. 2c and d, respectively. | ||
| Fig. 2 (a) A schematic diagram of the experimental setup, (b) a photograph of the experimental setup, and the time evolution of the detection and electro-coalescence-based sorting of target cells from background cells in (c) positive and (d) negative sorting modes. | ||
2.4 Materials and methods
DI water (resistivity: 18.2 MΩ cm, DI purification system, ELGA, UK) is used for suspending microparticles, and this suspension is then used as the discrete aqueous phase for generating droplets. Non-fluorescent microbeads (size: 10 μm) and fluorescent microbeads (size: 15 μm) (microParticles GmbH, Germany) are suspended in a mixture of DI water and glycerol. To avoid the sedimentation and agglomeration of particles, 23% (wt/wt) glycerol (Sigma Aldrich Bangalore, India) and 0.5% (wt/wt) Tween 80 (Sigma Aldrich Bangalore, India) are added to the deionised (DI) water. The resulting suspension was sonicated for 5 min in an ultrasonic cleaner (Maxsell, India). Mineral oil (Sigma Aldrich, Bangalore, India) is used as the continuous phase. A mixture of DI water with 23% (wt/wt) glycerol is used as the sheath fluid to focus the microparticles. Mineral oil was filtered using PTFE filters (0.2 μm) to avoid the presence of any dust particles, which may cause channel clogging. To improve droplet stability, 5% (wt/wt) Span 85 (Sigma Aldrich, Bangalore, India) is added to the mineral oil. Peripheral blood mononuclear cells (PBMCs) are isolated from whole blood samples using a density gradient method.9 The experiments were performed after obtaining ethical clearance from our institute. Cancer cell lines (CTCs; HeLa and DU145) were procured from the National Centre for Cell Science (NCCS, Pune, India) and cultured using a standard protocol.9 The cultured cells are tagged with either anti-EpCAM-FITC antibodies or rhodamine 6G dye for separate experiments. For tagging cells with dye, the cells were labelled with rhodamine 6G (R6G) (TCI Chemicals (India) Pvt. Ltd) and then suspended in 100 μl of phosphate buffered saline (PBS). 5 μl of rhodamine 6G was added to the cell suspension and this was incubated at a temperature of 37 °C for 1 h. After incubation, cells were washed thrice with 1.0 ml of PBS under centrifugation (1500 rpm for 10 min) before use. For antibody tagging, the cells were labelled with anti-EpCAM-FITC antibodies (Sigma Aldrich, Bangalore, India). Cell pellets with a maximum concentration of 106 were suspended in 100 μL of PBS. 20 μL of reagent was added to the cell suspension and the mixture was then incubated on ice in the dark for 30 min. After incubation, the cells were washed thrice with 1.0 ml of PBS under centrifugation at 1500 rpm for 10 min before use. The concentrations of cells (PBMCs + CTCs) used in our experiments range from 108 to 1010 per mL with 103 to 104 CTCs per mL.3. Results and discussion
3.1 Particle focusing and drop encapsulation
The focusing of particles (15 μm, 3.6 × 104 per ml) in an aqueous sample at a flow rate of 1 μl min−1 using a sheath fluid (a mixture of DI water and glycerol, 23% wt/wt) at a flow rate of 7 μl min−1 in the focusing region is presented in Fig. S1a in section S2 (see Video S1†). The variation of the width of the focused stream, wf, with the ratio of the sheath to sample flow rates, r = (Qsh/Qs), is shown in Fig. S1b.† As observed, wf decreases with an increase in r. wf is correlated with r as follows: wf = 34.8r−0.6 (R2 = 0.98). For the complete focusing of particles at the sidewall, wf should be equal to or smaller than the particle/cell size. In the case of samples containing differently sized particles or cells, the r value required for the focusing of the smallest particle/cell needs to be used to ensure the focusing of particles of all sizes. The spacing between two adjacent particles in the focused region, g2, was studied upon varying the sample concentration, cs, which affects the average spacing between particles before focusing, g1, and the results are presented in Fig. S1c.† As observed, at a fixed value of cs, the spacing ratio, G = (g2/g1), linearly increases with an increase in r. If r remains fixed, g2 decreases with cs.49 Based on g2 and the velocity of the particles, Up, the frequency of the arrival of particles at the droplet generation junction could be calculated, and it was found to be a function of the particle concentration, cs, and the total flow rate.At the flow-focusing junction, three different regimes, namely squeezing, dripping, and jetting, can be used for generating droplets52 (Fig. S2a in section S2†). The squeezing regime was observed at a very low capillary number, Ca < 0.02, whereas the dripping regime was observed for Ca values in the range of 0.02 < Ca < 0.1. The jetting regime is observed for much higher Ca values >0.1. Of the above regimes, the dripping regime is the most stable and results in a homogenous size distribution; hence, it was used in our experiments. A comparison of the sizes of particle/cell-encapsulating and empty droplets at a continuous to discrete phase flow rate ratio of rf is shown in Fig. S2b.† As observed, the droplet diameter decreases with an increase53 in rf, as dd = 281rf−0.6. Thus, while operating in the dripping regime, the sizes of the particle/cell-encapsulating droplets and the empty droplets are the same when the ratio of the particle size to droplet diameter is less than 0.6. The distance between the droplets can be controlled via varying the continuous to droplet phase flow rate ratio, as shown in Fig. S2c.† The distance between droplets should be greater than the width of the optical detection zone (∼100 μm) so that droplets move in single file through the detection zone. It is observed that the droplet separation distance increases (or the frequency decreases) with an increase in the flow rate ratio, following the relationship sd = 23rf0.5.
The frequency of the arrival of particles should match the frequency of droplet generation to ensure that each droplet encapsulates only one particle. If the frequency of the arrival of particles is higher than the droplet generation frequency, some of the droplets will encapsulate more than one particle (see Fig. S2d†). On the other hand, if the frequency of the arrival of particles is lower than the droplet generation frequency, some of the droplets will not contain any particles, resulting in empty droplets (see Fig. S2d†). The encapsulation rate of single cells in droplets is studied at a droplet frequency of 300 drops per s, and the results are presented in Fig. S2e.† It is observed that the single-cell encapsulation rate increases with an increase in cell concentration (in the concentration range of 104–106), which can be attributed to better matching between the droplet generation rate and the arrival rate of particles at the droplet generation junction at higher particle concentrations. With a particle concentration of 106, we could achieve a single-cell encapsulation efficiency of 85%. Also, it is ensured that the minimum spacing between adjacent droplets (120 μm) is greater than the width of the optical window (100 μm).
The droplets encapsulating particles/cells migrate to the centre of the channel due to non-inertial lift force; thus, the proposed technique eliminates the need for complicated 2D particle/cell focusing. Simulation results (using the level set method in COMSOL, see the simulation model details in section S3†) show that an aqueous droplet in an oil medium is positioned at the centre of the channel cross-section, indicating the self-focusing of droplets (see Fig. S3†).
3.2 The optical detection of particles/cells
The self-focused droplets pass through the optical detection zone in single file, generating different optical signals. The signals are detected by different optical detectors, acquired with the help of NI-DAQ and Labview, and processed using Python programs, as mentioned above. The FSC, SSC, and FL signals from droplets with a size of 68 μm encapsulating fluorescent (R6G-tagged and anti-EpCAM-FITC-tagged) and non-fluorescent HeLa cells with an average size of 20 μm and from empty droplets of the same size are shown in Fig. 3. The cells labelled with R6G and anti-Ep-CAM-FITC have excitation wavelengths of 530 nm and 488 nm, and emission wavelengths of 560 nm and 520 nm, respectively. The FSC, SSC, and FL signals from the droplets (size: 60 μm) encapsulating fluorescent and non-fluorescent microbeads with an average size of 15 μm and from empty droplets of the same size are shown in Fig. S4.† When a droplet passes through the optical detection zone it obstructs the laser beam, which results in a sudden reduction in the FSC signal. The height and width of the FSC signal will depend on the droplet size and residence time (i.e., the time spent by a droplet in the detection zone), respectively. The sizes of the droplets containing FL- and non-FL cells are equal, and they travel at the same velocity, hence their FSC signals are identical. However, as particle-encapsulating droplets obstruct more light compared to empty droplets, the heights of the FSC signals from particle-encapsulating droplets are higher compared to those from empty droplets. The SSC signal will depend on the internal structure of the droplet and it generally occurs because of the reflection and refraction of light at an interface where there is a change in the refractive index. The presence of solid particles inside droplets results in a solid–liquid interface inside the drop, thus resulting in higher reflection. Therefore, the SSC signals from particle-encapsulating droplets are higher than those from empty droplets. The tagged cells show the same FSC and SSC signals as untagged cells. The FL signals from cells tagged with anti-EpCAM-FITC antibodies are much lower compared to those from cells tagged with rhodamine 6G. The intensities of the FL signals collected off-chip from cells tagged with anti-EpCAM-FITC antibodies and rhodamine 6G are compared in Fig. S5,† which further confirms this. In the present case, the tagged cells are encapsulated inside aqueous droplets that are present in an oil medium, so the FL signals will be attenuated. Therefore, we proceed with rhodamine 6G tagging in subsequent experiments.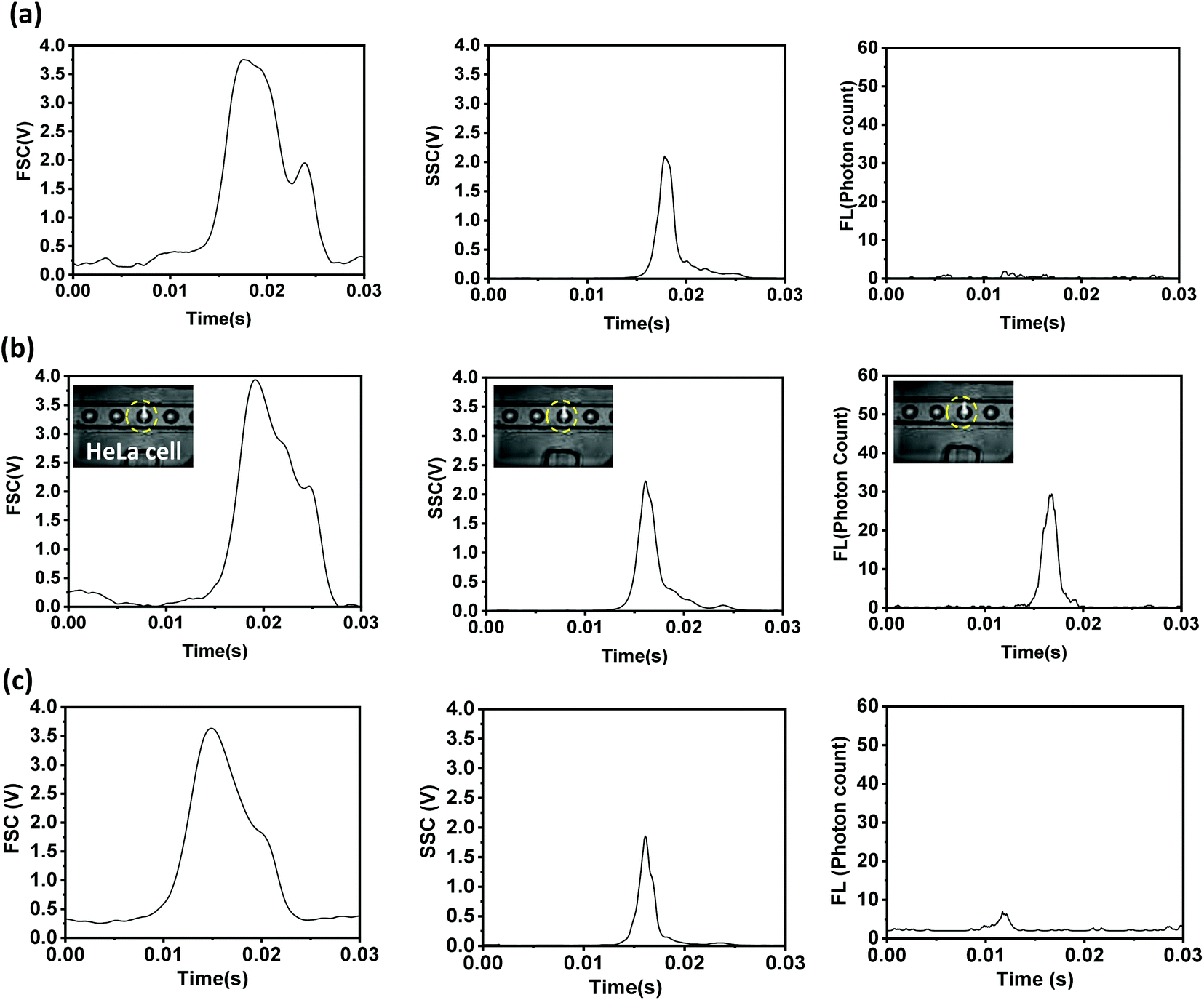 | ||
| Fig. 3 The FSC, SSC, and FL signals collected from 68 μm droplets encapsulating (a) non-fluorescent, and fluorescent (b) R6G-tagged and (c) anti-Ep-CAM-FITC-tagged HeLa cells. | ||
The variations in FSC signals with droplet size and flow rate are depicted in Fig. S6,† which shows that the height of the FSC signal increases with an increase in droplet size due to increased forward light scattering, whereas the width of the FSC signal decreases with an increase in droplet velocity due to a decrease in the residence time. It is observed that there are no significant changes to the SSC signal when the particle size is varied while keeping the droplet size fixed (Fig. S6†). The variations of the FL signal with the ratio of droplet diameter to particle size are shown in Fig. S6;† it is revealed that as the ratio of the droplet size to the particle size increases, the FL intensity decreases. This may be attributed to the fact that as the drop size increases relative to the particle size, higher attenuation of the FL signal occurs inside the droplet itself before further attenuation occurs as the emission tries to arrive at the detector interface. Thus, a smaller ratio of drop-to-particle diameter is suggested for achieving a good signal-to-noise ratio and good sensitivity.
Next, mixtures of fluorescent and non-fluorescent polystyrene microbeads with sizes of 15 μm and 10 μm, respectively, and mixtures of fluorescent-labelled (rhodamine 6G) cancer cells (HeLa and DU 145) and peripheral blood mononuclear cells (PBMCs) were analysed based on the FSC and FL signals in scatter plots (Fig. 4). The areas under the FSC or FL signal versus time plots are used to obtain the scatter plots. The bead and cell samples are prepared via following the protocol described above. Fig. 4a shows the scatter plot from the polystyrene microbeads, showing two separate clusters for the fluorescent and non-fluorescent microbeads. The FL signals (mean values) from the fluorescent microbeads are much higher (∼103 times) than the signals from the non-fluorescent microbeads. However, the FL signals from the non-fluorescent microbeads have non-zero values (although small), which can be attributed to background fluorescence signals (noise). The FSC signals (mean values) obtained from the 15 μm fluorescent microbeads are higher than those obtained from the 10 μm non-fluorescent microbeads, since FSC is proportional to particle size. Fig. 4b and c show scatter plots from mixtures of PBMCs + DU145 and PBMCs + HeLa cell samples, respectively. The sizes of the PBMCs are in the range of 9 to 20 μm (average: 10.5 μm), the sizes of the HeLa cells are in the range of 15 to 24 μm (average: 20 μm), and the sizes of the DU145 cells are in the range of 12 to 16 μm (average: 15 μm). Since the average sizes of HeLa and DU145 cells are larger than those of the PBMCs, the FSC signals, which are proportional to the cell size, are higher for the cancer cells compared to the PBMCs. Since the cancer cells are fluorescently labelled, the FL signals (mean values) from the cancer cells are much higher (∼102 times) than the FL signals from the PBMCs. The non-zero FL signals from the PBMCs can be attributed to background fluorescence (noise) from the biological cells.
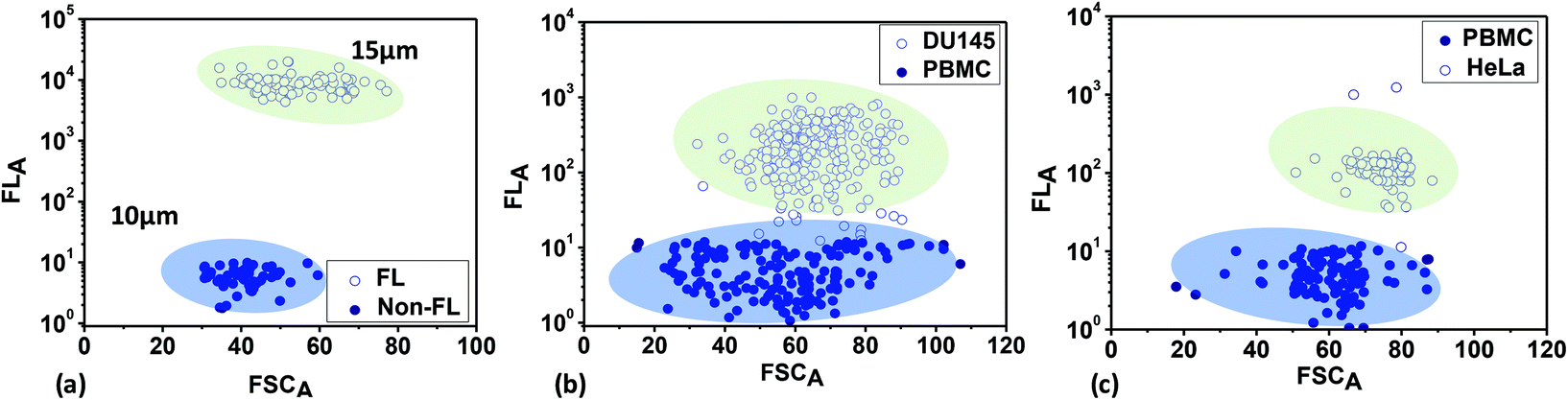 | ||
| Fig. 4 Scatter plots obtained from optical detection experiments with (a) fluorescent and non-fluorescent beads of different sizes, (b) PBMCs and DU145 cells, and (c) PBMCs and HeLa cells. | ||
3.3 Coalescence of drops with an aqueous co-flow stream
The coalescence of droplets with an aqueous co-flow stream is studied. Fig. S7a† shows the time evolution of the coalescence of droplets containing 15 μm microbeads and empty droplets. The particles/cells are encapsulated in 65 μm droplets in an oil stream. The flow rates of the oil stream and aqueous stream are maintained at 2 μl min−1 and 120 μl min−1, respectively, so that a stable interface is maintained, with the width of the oil stream being 62 μm. Since the width of the oil stream is smaller than the droplet size, there is microscopic contact between the droplets and the interface, which enables electro-coalescence at a lower voltage, as explained above. The variations in the width of the oil stream with the oil-to-aqueous stream flow rate ratio are depicted in Fig. S7b.†Via applying 60 V to the electrodes, which offer an electric field of 1.2 × 106 V m−1, adequate to overcome the disjoining pressure,43 the droplets coalesce with the aqueous stream and the particles/cells present inside the droplets are extracted into the aqueous stream. The effects of the size and velocity of the droplets on the electric field and voltage required for coalescence were studied (see Fig. S7c and S7d† and their insets). As the droplet size increases, the electric field required for coalescence reduces; this may be because of larger non-inertial lift force at a higher diameter, which requires a smaller electrical force and, hence, a smaller voltage to overcome the disjoining pressure. Similarly, as the velocity of the droplets decreases, the required electric field decreases, which is because at a smaller droplet velocity the residence time is higher and thus there is more time available for the electrical stress to overcome the disjoining pressure, even with a smaller electric field.3.4 Micro-FACS: the integrated optical detection and isolation of target droplets
The microfluidic fluorescence-activated sorting (micro-FACS) of droplets encapsulating target microbeads and cells was demonstrated via integrating the optical detection and isolation modules.44Fig. 5 shows the time evolution of the optical detection and isolation of droplets containing 15 μm FL microbeads from 10 μm non-FL microbeads. The beads were encapsulated inside 65 μm droplets with a distance between droplets of ∼100 μm, and the droplet speed was maintained at ∼50 mm s−1. Therefore, the droplets arrive at the electro-coalescence region at a rate of ∼500 drops per s. However, the response time of the electronics was found to be 3 ms, i.e., a signal needed to be maintained (low or high) for a duration of at least 3 ms, which is the minimum time required to write a particular signal on the Arduino board, activate the relay, and energise the electrodes. Therefore, the proposed micro-FACS system can sort droplets at a maximum rate of 300 drops per s. | ||
| Fig. 5 Experimental images showing the time evolution of target droplets (T) that contain FL microbeads and background droplets (B) that contain non-FL microbeads in the detection and electro-coalescence regions, and the ON/OFF control of the electrode voltage: (a) positive sorting and (b) negative sorting. | ||
First, a positive sorting technique is used with droplets containing 15 μm FL microbeads as target droplets (T) and 10 μm non-FL microbeads (and empty droplets) as background droplets (B) (Fig. 5a). Under default conditions, electrodes are energised at 60 V, so the background droplets continuously coalesce with the aqueous stream. In the present design, the distance between the detection and electro-coalescence zones is 8 mm. A droplet containing a target particle is detected at t = 0, and this arrives at the electro-coalescence zone at t = (L/ud) ms = 160 ms. Since the response time of the electronics is 3 ms, the ‘low’ signal is activated at ts = [(L/ud) − 3] ms = 157 ms to de-energize the electrodes (Fig. 2c). The target droplets containing 15 μm FL microbeads do not coalesce with the aqueous stream and collect at outlet 1 (see Video S2†). Experimental results demonstrating the negative sorting of 15 μm FL microbeads from 10 μm non-FL microbeads are presented in Fig. 5b (see Video S3†). The proposed technique can also be used for the coalescence of droplets in a specific sequence, for example, the positive/negative sorting of every alternate droplet, two consecutive droplets, and four consecutive droplets containing target particles, which is depicted in Fig. S8 (see Videos S4–S6†). The results clearly show the capabilities of the present system in terms of selective isolation accuracy, as long as the droplet frequency is ≤300 drops per s.
Next, experiments are performed using the micro-FACS system to separate target droplets encapsulating cancer cells (HeLa and DU145) from background cells (encapsulating PBMCs or empty drops). In both cases, the background droplets continuously coalesce. Once target droplets containing the cells are detected, the signal is turned OFF 3 ms before these target droplets arrive at the electrodes (i.e., at ts = 157 ms). Thus, the target droplets containing HeLa and DU145 cells do not coalesce and are collected at outlet 1. Experimental images showing the sorting of HeLa cells from PBMCs using the positive sorting technique are shown in Fig. 6a (see Video S7†). Experimental results demonstrating the negative sorting of HeLa cells from PBMCs are presented in Fig. 6b (see Video S8†).
 | ||
| Fig. 6 Experimental images showing the time evolution of target droplets (T) that contain FL cells (HeLa tagged with R6G) and background (B) droplets that contain PBMCs (non-tagged) in the detection and electro-coalescence regions, and the ON/OFF control of the electrode voltage: (a) positive sorting and (b) negative sorting. | ||
The sorting performances54 of the technique are studied for beads and cells separately via estimating various parameters, such as enrichment, efficiency, recovery, and purity. Enrichment is defined as the ratio of the number of target cells/particles to the number of background cells/particles at the respective outlets to the ratio of the same at the inlet, as follows:
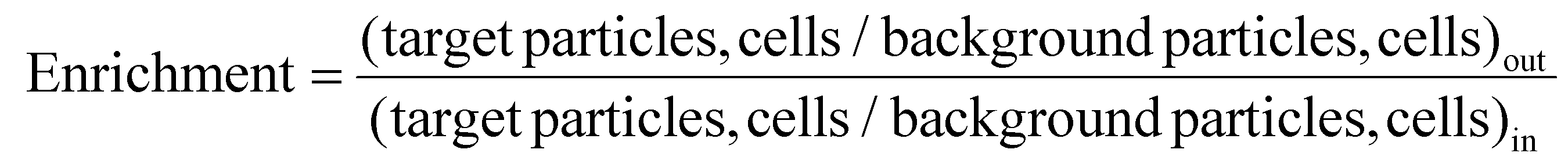 | (1) |
Efficiency is defined as the ratio of the number of target cells/particles at a particular outlet (ntrue) to the sum of the number of target cells/particles at the same outlet and the number of target cells/particle at the other outlet (nfalse), as follows:
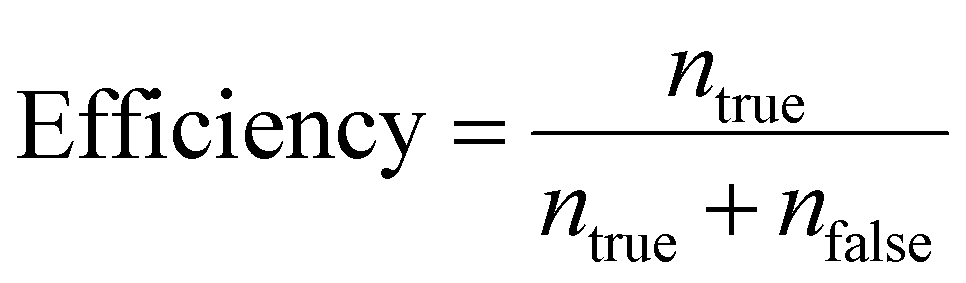 | (2) |
Recovery is defined as the ratio of the number of target cells/particles at the target outlet to the number of target cells/particles at the inlet, as follows:
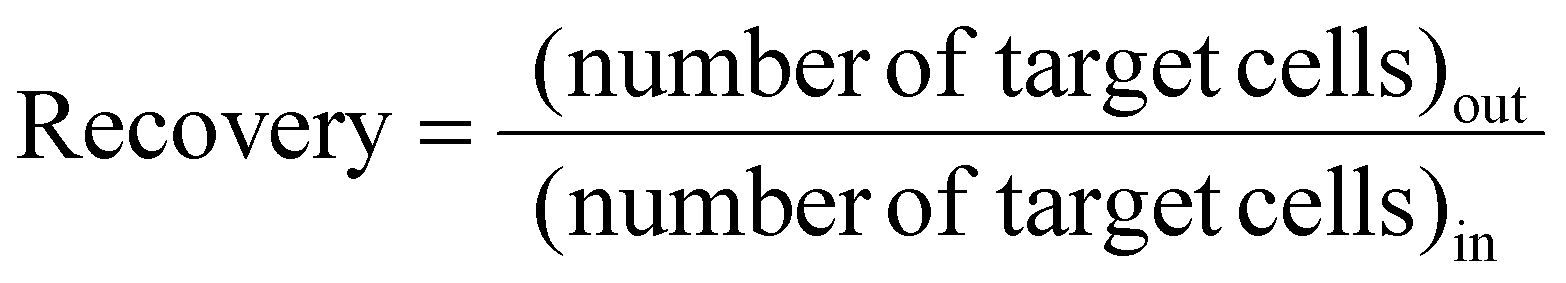 | (3) |
Purity is defined as the ratio of the number of target cells/particles at a particular outlet (ntrue) to the sum of the number of target cells/particles and number of background cells/particles (ncon) at the same outlet, as follows:
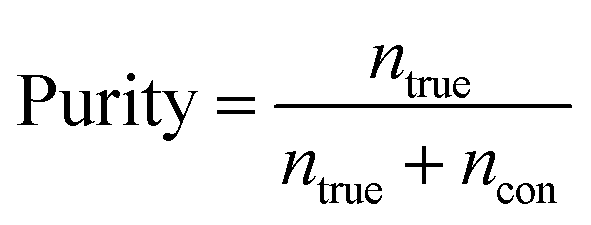 | (4) |
The initial concentrations of beads and different cells (PBMCs, HeLa, and DU145) are estimated before infusing samples at the device inlet. The concentrations at the respective outlets are estimated after the sorting of particles and cells. In positive sorting, target particles/cells are collected at outlet 1 and background particles are collected at outlet 2, while in negative sorting, target particles/cells are collected at outlet 2 and background particles are collected at outlet 1. The counting of cells and particles is carried out using fluorescence microscopy with a Neubauer cytometer chamber. Table S1† and Table 1 delineate the performance parameters from positive and negative sorting experiments, respectively. For all sorting experiments, the concentration of target particles and cells is maintained much lower (∼102 times) than the concentration of non-target or background particles and cells. In the bead sorting experiments, it is observed that the recovery is low (∼90%) compared to cells (∼98%), and the low recovery is due to difficulties in collecting the fluorescent beads at the outlet (beads stick to the tubing and channel outlet). A comparative study between positive and negative sorting shows that positive sorting results in better recovery compared to negative sorting. For both positive and negative sorting, the system shows very good efficiency (∼98%), purity (∼98%), and enrichment (∼1000×). The performance of the proposed device and method, i.e., enrichment, purity, efficiency, etc., may be affected when using real samples for the isolation of CTCs, due to their extremely low abundance. In that case, multi-stage sorting with detection and electro-coalescence modules in series or the serial processing of cell samples through a single-stage detection and electro-coalescence module (via re-feeding the cells collected at the outlets back into the device) may be adopted as possible solutions to maintain the performance of the device. The isolation of CTCs at smaller concentrations is the future scope of this work.
| Initial Conc. | Enrichment | Efficiency (%) | Recovery (%) | Purity (%) | |
|---|---|---|---|---|---|
| Fluorescent | 3.3 × 104 | 2900× | 98.71 | 88.18 | 98.98 |
| Non-fluorescent | 106 | ||||
| HeLa | 2.4 × 103 | 1800× | 98.19 | 97.5 | 98.56 |
| PBMC | 6.4 × 105 | ||||
| DU145 | 2.9 × 104 | 2300× | 97.54 | 95.17 | 98.58 |
| PBMC | 106 | ||||
Cell viability tests were performed using the sorted cells obtained from positive and negative sorting and unsorted cells using trypan blue staining assays (see the procedure outlined in section S6†). Fig. 7a(i–iii) shows representative trypan blue staining images (40×) of HeLa cells sorted using positive and negative sorting and unsorted HeLa cells. The counting data detailing the numbers of live and dead cells in each of the three different cases, i.e., the unsorted case and the sorted cases with positive and negative sorting techniques (with two trials for each case), and the % cell viability values are tabulated in section S6.† We observe that positive sorting results in ∼97% cell viability, whereas negative sorting results in ∼95% cell viability. The viability of unsorted cells was used as a control, and 99% of unsorted cells were found to be viable, which indicates that the actual cell viability due to the sorting technique alone is higher than what is stated above.
 | ||
| Fig. 7 (a) Trypan blue staining test images (40×) for cells obtained via (i) positive sorting and (ii) negative sorting, and (iii) unsorted cells (stain enters into dead cells, making them appear blue). (b) Images from the cell proliferation study: (i) an image of HeLa cells captured at the outlet; (ii–vi) images of cells during cell growth after 0 h, 24 h, 48 h, 72 h, and 96 h, respectively. (c) The variations of the normalized cell concentrations of sorted and unsorted cells plotted with time (in h). | ||
Further, we performed cell proliferation studies to demonstrate the suitability of the sorted cells (Fig. 7b(i)) for downstream assays. The sorted HeLa cells collected at the device outlet were washed twice with PBS, and the cell pellet was resuspended in 4 ml of DMEM (i.e., culture medium). The concentration of the collected cells was measured, which we refer to as the 0th h concentration. Further, the cell suspension was divided into four different parts, and the cells were seeded separately into four different wells of a 12-well cell culture plate. The cells were maintained at 37 °C with 5% CO2 throughout. Cell proliferation was studied via counting the cells at different time points, 0 h, 24 h, 48 h, 72 h, and 96 h, using a Neubauer hemocytometer and an inverted microscope (see above) with a 10× objective. At each time point, the cell count was performed five times to calculate the mean cell count and standard deviation. After counting the cells after 24 h, the cell media from one of the wells was discarded and the cells were cleaned with 0.5 ml of PBS. After cleaning, 0.5 ml of 2× trypsin was added into the well and the cells were incubated for 5 min at 37 °C for cell detachment. Once the cells were detached from the chamber surface, 1.0 ml of pre-warmed DMEM was added and the mixture was pipetted thoroughly. The suspension was then taken for determining the cell concentration using a hemocytometer. The above steps were repeated after 48 h, 72 h, and 96 h. Microscope images of the cells at each time point are shown in Fig. 7b(ii–vi), which show that the cells are intact and cell growth is maintained. To further characterise cell proliferation, we compared the growth curves of the sorted and unsorted cells. In both cases, the normalized cell growth on the y-axis is obtained via normalising the cell concentration at a particular time using the corresponding initial cell concentration. The results show that the cell growth curves are identical and match within 4%, as shown in Fig. 7c. The normalised cell concentration correlates with time following the relationship c = K0.05t (with R2 = 0.98 and 0.97), where K = 1.52 and 1.54 for sorted and unsorted cells, respectively. Graphs showing the actual cell concentrations are shown in Fig. S9;† the differences between the growth curves are attributed to differences in the starting concentrations of sorted and unsorted cells before culturing and the non-linear cell growth rate. However, the close match between the normalized curves in Fig. 7c shows that the growth rates are identical and proves the non-invasiveness of the sorting system.
4. Conclusions
We demonstrated the optical detection and selective isolation of target particles and cells encapsulated in microdroplets in single-cell format. The encapsulation of particles/cells inside droplets facilitated the auto-focusing of the cells in the optical interrogation window, thus eliminating the need for complicated two-dimensional focusing. Droplets were generated using a stable dripping regime to achieve uniform droplets, and the distance between droplets was adjusted to be larger than the width of the optical window via adjusting the discrete to continuous phase flow rate ratio. The particles and cells encapsulated in droplets were characterised based on FSC, SSC, and FL signals. The variation of the FL signal with the ratio of the drop-to-particle size showed that the FL intensity decreases with an increase in the droplet-to-particle size ratio. The continuous phase to aqueous stream width was adjusted via varying the ratio of the flow rates so that the droplets were in microscopic contact with the oil-aqueous interface. The electric field/voltage required for the coalescence of aqueous droplets with the aqueous stream with respect to droplet size and velocity showed the requirement for a higher voltage at a higher droplet velocity or smaller droplet size. The sorting of 15 μm FL microbeads from 10 μm non-FL microbeads and of HeLa and DU145 cancer cells from PBMCs was demonstrated. The sorting performance was characterised in terms of sorting efficiency, enrichment, purity, and recovery, and it was shown that cancer cells (∼104 ml−1) could be successfully sorted from PBMCs (104–106 ml−1) with very good efficiency (>97%), enrichment (>1800×), purity (>98%), and recovery (>95%) at a throughput of 300 droplets per s. It was found that the sorted cells remained viable (>95% viability) and showed good proliferation when cultured, which further confirms the potential application of the proposed technique to downstream analysis. We note that the FL signals from droplet-encapsulated cells tagged with anti-EpCAM-FITC antibodies, which are specific to CTCs, were found to be very low due to relatively weaker signals from the antibodies compared to from R6G. Anti-EpCAM-FITC antibodies bound to quantum dots,55 which are capable of providing signals that are significantly enhanced several-fold compared to antibody-tagging alone and can address the specificity of the detection system, can be employed, and this is the future scope of the present work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank SERB, DST, India (EMR/2014/001151), and I.I.T. Madras (MEE/15-16/843/RFTP/ASHS) for providing the financial support that enabled this work. We also acknowledge CNNP, IIT Madras, for supporting device fabrication.References
- C. Petchakup, K. H. H. Li and H. W. Hou, Advances in single cell impedance cytometry for biomedical applications, Micromachines, 2017, 8, 20 CrossRef.
- C. E. Sims and N. L. Allbritton, Analysis of single mammalian cells on-chip, Lab Chip, 2007, 7, 423–440 RSC.
- S. J. Altschuler and L. F. Wu, Cellular Heterogeneity: Do Differences Make a Difference?, Cell, 2010, 141, 559–563 CrossRef CAS.
- R. A. Harouaka, M. Nisic and S.-Y. Zheng, Circulating Tumor Cell Enrichment Based on Physical Properties, J. Lab. Autom., 2013, 18, 455–468 CrossRef CAS.
- Y. Chen, P. Li, P. H. Huang, Y. Xie, J. D. Mai, L. Wang, N. T. Nguyen and T. J. Huang, Rare cell isolation and analysis in microfluidics, Lab Chip, 2014, 14, 626–645 RSC.
- L. M. Fu, R. J. Yang, C. H. Lin, Y. J. Pan and G. B. Lee, Electrokinetically driven micro flow cytometers with integrated fiber optics for on-line cell/particle detection, Anal. Chim. Acta, 2004, 507, 163–169 CrossRef CAS.
- M. Antfolk and T. Laurell, Continuous flow microfluidic separation and processing of rare cells and bioparticles found in blood ? A review, Anal. Chim. Acta, 2017, 965, 9–35 CrossRef CAS.
- P. Sajeesh and A. K. Sen, Particle separation and sorting in microfluidic devices: A review, Microfluid. Nanofluid., 2014, 17, 1–52 CrossRef.
- S. Karthick, P. Pradeep, P. Kanchana and A. K. Sen, Acoustic impedance based size-independent isolation of circulating tumour cells View Article Online from blood using acoustophoresis, Lab Chip, 2018, 444, 45–78 Search PubMed.
- A. Raj, M. Dixit, M. Doble and A. K. Sen, A combined experimental and theoretical approach towards mechanophenotyping of biological cells using a constricted microchannel †, Lab Chip, 2017, 17, 3704–3716 RSC.
- A. Ulmer, O. Schmidt-Kittler, J. Fischer, U. Ellwanger, G. Rassner, G. Riethmüller, G. Fierlbeck and C. A. Klein, Immunomagnetic enrichment, genomic characterisation, and prognostic impact of circulating melanoma cells, Clin. Cancer Res., 2004, 10, 531–537 CrossRef CAS.
- S. T. Wortham, G. A. Ortolano and B. Wenz, A brief history of blood filtration: clot screens, microaggregate removal, and leukocyte reduction, Transfus. Med. Rev., 2003, 17, 216–222 CrossRef.
- R. Mulliken, The Separation of Liquid Mixtures by Centrifuging, J. Am. Chem. Soc., 1922, 44, 1729–1730 CrossRef CAS.
- M. G. Lee, S. Choi and J.-K. Park, Three-dimensional hydrodynamic focusing with a single sheath flow in a single-layer microfluidic device, Lab Chip, 2009, 9, 3155 RSC.
- J. Guo, X. Liu, K. Kang, Y. Ai, Z. Wang and Y. Kang, A Compact Optofluidic Cytometer for Detection and Enumeration of Tumor Cells, J. Lightwave Technol., 2015, 33, 3433–3438 CAS.
- D. Heikali and D. Di Carlo, A Niche for Microfluidics in Portable Hematology Analyzers, J. Assoc. Lab. Autom., 2010, 15, 319–328 CrossRef.
- Y. Zhao, Q. Li, X. Hu and Y. Lo, Microfluidic cytometers with integrated on-chip optical systems for red blood cell and platelet counting, Biomicrofluidics, 2016, 10, 064119 CrossRef.
- R. S. Gaikwad and A. K. Sen, The Microflow Cytometer. in Environmental, Chemical and Medical Sensors, Energy, Environment, and Sustainability, ed. S. Bhattacharya, A. Agarwal, N. Chanda, A. Pandey and A. Sen, Springer, Singapore, 2018, pp. 371–387 Search PubMed.
- M. Reisbeck, M. J. Helou, L. Richter, B. Kappes, O. Friedrich and O. Hayden, Magnetic fingerprints of rolling cells for quantitative flow cytometry in whole blood, Sci. Rep., 2016, 6, 32838 CrossRef CAS.
- K. C. Cheung, M. D. Berardino, G. Schade-Kampmann, M. Hebeisen, A. Pierzchalski, J. Bocsi, A. Mittag and A. Tárnok, Microfluidic impedance-based flow cytometry, Cytometry, Part A, 2010, 77, 648–666 CrossRef.
- P. Rosendahl, K. Plak, A. Jacobi, M. Kraeter, N. Toepfner, O. Otto, C. Herold, M. Winzi, M. Herbig, Y. Ge, S. Girardo, K. Wagner, B. Baum and J. Guck, Real-time fluorescence and deformability cytometry, Nat. Methods, 2018, 15, 355–358 CrossRef CAS.
- C. H. Chen, S. H. Cho, F. Tsai, A. Erten and Y. H. Lo, Microfluidic cell sorter with integrated piezoelectric actuator, Biomed. Microdevices, 2009, 11, 1223–1231 CrossRef CAS.
- S. H. Cho, J. M. Godin, C.-H. Chen, W. Qiao, H. Lee and Y.-H. Lo, Review Article: Recent advancements in optofluidic flow cytometer, Biomicrofluidics, 2010, 4, 043001–043023 CrossRef.
- O. Jakobsson, C. Grenvall, M. Nordin, M. Evander and T. Laurell, Correction: Acoustic actuated fluorescence activated sorting of microparticles, Lab Chip, 2015, 15, 4625–4625 RSC.
- G. R. Goddard, C. K. Sanders, J. C. Martin, G. Kaduchak and S. W. Graves, Analytical performance of an ultrasonic particle focusing flow cytometer, Anal. Chem., 2007, 79, 8740–8746 CrossRef CAS.
- M. Tenje, A. Fornell, M. Ohlin and J. Nilsson, Particle Manipulation Methods in Droplet Microfluidics, Anal. Chem., 2018, 90, 1434–1443 CrossRef CAS.
- C. Simonnet and A. Groisman, High-Throughput and High-Resolution Flow Cytometry in Molded Microfluidic Devices, Anal. Chem., 2006, 78, 5653–5663 CrossRef CAS.
- A. A. S. Bhagat, S. S. Kuntaegowdanahalli, N. Kaval, C. J. Seliskar and I. Papautsky, Inertial microfluidics for sheath-less high-throughput flow cytometry, Biomed. Microdevices, 2010, 12, 187–195 CrossRef.
- S. C. Hur, H. T. K. Tse and D. Di Carlo, Sheathless inertial cell ordering for extreme throughput flow cytometry, Lab Chip, 2010, 10, 274–280 RSC.
- S. Etcheverry, A. Faridi, H. Ramachandraiah, T. Kumar, W. Margulis, F. Laurell and A. Russom, High performance micro-flow cytometer based on optical fibres, Sci. Rep., 2017, 7, 5628 CrossRef CAS.
- K. W. Seo, H. J. Byeon, H. K. Huh and S. J. Lee, Particle migration and single-line particle focusing in microscale pipe flow of viscoelastic fluids, RSC Adv., 2014, 4, 3512–3520 RSC.
- G.-B. Lee, C.-C. Chang, S.-B. Huang and R.-J. Yang, The hydrodynamic focusing effect inside rectangular microchannels, J. Micromech. Microeng., 2006, 16, 1024–1032 CrossRef.
- T. E. Winkler, H. Ben-Yoav and R. Ghodssi, Hydrodynamic focusing for microfluidic impedance cytometry: a system integration study, Microfluid. Nanofluid., 2016, 20, 1–14 CrossRef CAS.
- Y. Zhao, Q. Li and X. Hu, Universally applicable three-dimensional hydrodynamic focusing in a single-layer channel for single cell analysis, Anal. Methods, 2018, 10, 3489–3497 RSC.
- A. J. Chung, D. R. Gossett and D. Di Carlo, Three dimensional, sheathless, and high-throughput microparticle inertial focusing through geometry-induced secondary flows, Small, 2013, 9, 685–690 CrossRef CAS.
- Q. Zhao, D. Yuan, S. Yan, J. Zhang, H. Du, G. Alici and W. Li, Flow rate-insensitive microparticle separation and filtration using a microchannel with arc-shaped groove arrays, Microfluid. Nanofluid., 2017, 21, 1–11 CrossRef CAS.
- M. Asghari, M. Serhatlioglu, B. Ortaç, M. E. Solmaz and C. Elbuken, Sheathless microflow cytometry using viscoelastic fluids, Sci. Rep., 2017, 7, 1–14 CrossRef CAS.
- M. Frankowski, J. Theisen, A. Kummrow, P. Simon, H. Ragusch, N. Bock, M. Schmidt and J. Neukammer, Microflow cytometers with integrated hydrodynamic focusing., Sensors, 2013, 13, 4674–4693 CrossRef.
- L. Wu, P. Chen, Y. Dong, X. Feng and B. F. Liu, Encapsulation of single cells on a microfluidic device integrating droplet generation with fluorescence-activated droplet sorting, Biomed. Microdevices, 2013, 15, 553–560 CrossRef CAS.
- S. Hasan, D. Geissler, K. Wink, A. Hagen, J. J. Heiland and D. Belder, Fluorescence lifetime-activated droplet sorting in microfluidic chip systems, Lab Chip, 2019, 19, 403–409 RSC.
- M. M. Wang, E. Tu, D. E. Raymond, J. M. Yang, H. Zhang, N. Hagen, B. Dees, E. M. Mercer, A. H. Forster, I. Kariv, P. J. Marchand and W. F. Butler, Microfluidic sorting of mammalian cells by optical force switching., Nat. Biotechnol., 2005, 23, 83–87 CrossRef CAS.
- L. M. Fidalgo, G. Whyte, D. Bratton, C. F. Kaminski, C. Abell and W. T. S. Huck, From microdroplets to microfluidics: Selective emulsion separation in microfluidic devices, Angew. Chem., Int. Ed., 2008, 47, 2042–2045 CrossRef CAS.
- A. Srivastava, S. Karthick, K. S. Jayaprakash and A. K. Sen, Droplet Demulsification Using Ultralow Voltage-Based Electrocoalescence, Langmuir, 2018, 34, 1520–1527 CrossRef CAS.
- V. Chokkalingam, Y. Ma, J. Thiele, W. Schalk, J. Tel and W. T. S. Huck, An electro-coalescence chip for effective emulsion breaking in droplet microfluidics, Lab Chip, 2014, 14, 2398–2402 RSC.
- A. R. Thiam, N. Bremond and J. Bibette, Breaking of an emulsion under an ac electric field, Phys. Rev. Lett., 2009, 102, 1–4 CrossRef.
- C. Frey, K. Göpfrich, S. Pashapour, I. Platzman and J. P. Spatz, Electrocoalescence of Water-in-Oil Droplets with a Continuous Aqueous Phase: Implementation of Controlled Content Release, ACS Omega, 2020, 5, 7529–7536 CrossRef CAS.
- P. Sajeesh, M. Doble and A. K. Sen, Hydrodynamic resistance and mobility of deformable objects in microfluidic channels, Biomicrofluidics, 2014, 8, 54112 CrossRef CAS.
- A. Srivastava, S. Karthick, K. S. Jayaprakash and A. K. Sen, Droplet Demulsification Using Ultralow Voltage-Based Electrocoalescence, Langmuir, 2018, 34, 1520–1527 CrossRef CAS.
- P. Sajeesh, S. Manasi, M. Doble and A. K. Sen, Microfluidic device with focusing and spacing control for resistance based sorting of droplets and cells, Lab Chip, 2015, 15, 3738–3748 RSC.
- D. J. Collins, A. Neild, A. deMello, A. Q. Liu and Y. Ai, The Poisson distribution and beyond: Methods for microfluidic droplet production and single cell encapsulation, Lab Chip, 2015, 15, 3439–3459 RSC.
- Z. Liu, S. T. Chan, H. A. Faizi, R. C. Roberts and H. C. Shum, Droplet-based electro-coalescence for probing threshold disjoining pressure, Lab Chip, 2015, 15, 2018–2024 RSC.
- K. S. Jayaprakash and A. K. Sen, Droplet encapsulation of particles in different regimes and sorting of particle-encapsulating-droplets from empty droplets, Biomicrofluidics, 2019, 13, 034108 CrossRef CAS.
- J. M. Chen, M.-C. Kuo and C.-P. Liu, Control of Droplet Generation in Flow-Focusing Microfluidic Device with a Converging-Diverging Nozzle-Shaped Section, Jpn. J. Appl. Phys., 2011, 50, 107301 CrossRef.
- S. Hazra, K. S. Jayaprakash, K. Pandian, A. Raj, S. K. Mitra and A. K. Sen, Non-inertial lift induced migration for label-free sorting of cells in a co-flowing aqueous two-phase system, Analyst, 2019, 144, 2574–2583 RSC.
- H. Min, S.-M. Jo and H.-S. Kim, Efficient Capture and Simple Quantification of Circulating Tumor Cells Using Quantum Dots and Magnetic Beads, Small, 2015, 11, 2536–2542 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data relating to the acquisition and post-processing of optical signals, particle focusing and drop encapsulation, numerical simulations showing the droplet position inside the microchannel, the optical detection of particles/cells, and the optical detection of particles/cells. See DOI: 10.1039/d0an00160k |
| This journal is © The Royal Society of Chemistry 2021 |