
A novel binary matrix consisting of graphene oxide and caffeic acid for the analysis of scutellarin and its metabolites in mouse kidney by MALDI imaging†
Tao
Wang‡
a,
Hin Kiu
Lee‡
a,
Grace Gar Lee
Yue
b,
Arthur Chi Kong
Chung
*ab,
Clara Bik San
Lau
b and
Zongwei
Cai
 *a
*a
aDepartment of Chemistry and State Key Laboratory of Environmental and Biological Analysis, Hong Kong Baptist University, Kowloon Tong, Kowloon, Hong Kong SAR, P. R. China. E-mail: chungack@hkbu.edu.hk; zwcai@hkbu.edu.hk
bMedicine & Therapeutics and Institute of Chinese Medicine and State Key Laboratory of Research on Bioactivities and Clinical Applications of Medicinal Plants, The Chinese University of Hong Kong, Hong Kong, China
First published on 20th October 2020
Abstract
Although the in vivo metabolic pathways of scutellarin, a traditional Chinese medicine, have been investigated via different liquid chromatography techniques, studies on the distribution and location of scutellarin within organ tissue sections have not been reported. Matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) can generate in situ spatial distribution profiles for scutellarin and its metabolites in a kidney section. However, the direct detection of a small molecule (m/z < 600) using conventional matrices often results in ion suppression and matrix interferences. In this study, we demonstrated a novel methodology using MALDI-MSI for the in situ spatial localization of scutellarin and its metabolites in kidney tissues by applying a binary matrix of graphene oxide (GO) and caffeic acid (CA). The results indicated that the binary matrix (GO/CA) significantly improved the detection efficiency of scutellarin and its metabolites with relatively high sensitivity, selectivity and reproducibility on tissue sections. This methodology was successfully applied to map scutellarin and its metabolites with MALDI-MSI in mouse kidney tissues. Specifically, scutellarin and scutellarein were found to be located in the cortex and medulla regions of the kidney with relatively high abundance, whereas the remaining metabolites appeared in the cortex with low abundance. We believe that the novel imaging methodology may also be used for the studies of cancerous tissues and inform the development of the future therapies of kidney tumors.
Introduction
Scutellarin, a known flavonoid glycoside, is mainly extracted from the traditional Chinese botanic drug Erigeron breviscapus. Modern pharmacological studies have reported that scutellarin was widely useful in cardiovascular disease treatment, blood vessel dilation, cerebral blood flow increment, and platelet aggregation inhabitation.1–3 Given the potential benefits of scutellarin in health care, there is a growing interest in investigating its absorption, pharmacokinetics, metabolism, and excretion in the biological system. However, with most studies mainly targeted at biological samples such as blood and urine, little research attention has been paid to the presence of scutellarin and its metabolites in organ tissues. In order to observe the presence of scutellarin and its metabolites in organ tissues, it is important to visualize in situ spatial distribution of scutellarin for pharmaceutical development.One of the challenges of obtaining in situ distribution information for scutellarin and its metabolites is the choice of a suitable analytical technique. High-performance liquid chromatography coupled with mass spectrometry (HPLC/MS) is the most commonly used tool to study the concentration and biochemical changes of scutellarin.4–6 However, HPLC/MS fails to provide information on the spatial distribution of scutellarin and its metabolites because it analyzed only extracted samples from treated tissues, thus limiting its usage and application. Fluorescence microscopy can map indirectly the analyte distribution. However, it can only be used to detect the analytes being tracked by fluorescence; it cannot distinguish parent drug and its metabolites simultaneously.7–9 Matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) is a superior and powerful imaging technique for the visualization of biological tissues. The technique can in situ visualize the localization and distribution of a drug and its metabolites simultaneously.10–14 Also, with this technique, the accurate information of drug distribution for small molecules of drugs and their metabolites in target organs can be obtained with specificity and without the use of a label.10–14 Nowadays, the applications of MALDI-MSI in traditional chinese medicine (TCM) have gained currency in the pharmaceutical industry as a potential imaging technique.15–19 As an alternative and innovative technique, MALDI-MSI could be applied to visualize the localization and distribution of scutellarin and its metabolites.
MALDI-MSI has been successfully applied for small molecules under the complex biological system, but the target analyte signals could be easily suppressed due to the matrix interferences from matrix-related clusters.20 Several strategies have been developed to overcome the disadvantages. The “binary combination matrix” approach has been adopted to improve the overall analyte detection efficiency and to reduce background noise. For instance, some studies revealed that a binary matrix mixture of 2,5-dihydroxybenzoic acid (DHB) and sinapinic acid (SA) improves the signal sensitivity for carbohydrates and glycoproteins.21 Moreover, the binary mixture of α-cyano-4-hydroxycinnamic acid (CHCA) and 9-aminoacridine (9-AA) can effectively reduce noise signal in low-molecular-weight regions.22 Graphene oxide (GO) has recently received considerable attention as one of the binary matrices owing to its unique chemical and electronic properties, including low matrix background interference, analyte sensitivity enhancement and good reproducibility.23–28 As previously reported, Zhang et al. has utilized GO directly as a matrix for scutellarin MALDI-MS detection.29 However, Zhang et al.'s application limits the detection on only MALDI-target plate, with no information provided on biological tissue samples. Considering the idea of this binary matrix and the results reported by Zhang et al. about the utilization of GO as a matrix for scutellarin detection, it is highly promising to combine GO with another matrices for the analysis of scutellarin and its metabolites by MALDI-MSI on animal tissues.
In this study, we developed a novel methodology for the localization and distribution of scutellarin and its metabolites in biological kidney tissue using the MALDI-MSI technique. We presented a new binary matrix mixture of GO and CA, which could effectively enhance the sensitivity and selectivity in detecting and locating scutellarin and its metabolites. The new binary matrix was successfully applied for the in vivo characterization and spatial distribution mapping of scutellarin and its metabolites in tissue sections of kidneys after intraperitoneal (ip) injection.
Results and discussion
Evaluation of the single matrix for the MALDI MS analysis
Typically, matrix type and deposition have a strong influence on the quality of the MALDI-MS measurement, particularly for the detection of lower-abundance species. Thus, a preliminary matrix optimization experiment was performed on blank tissue sections. Seven commonly used matrices, namely 2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB), 2,4,6-trihydroxyacetophenone (THAP), caffeic acid (CA), ferulic acid (FA), trans-bis(4-hydroxyphenyl)methane (BPF), α-cyano-4-hydroxycinnamic acid (CHCA) and 2,5-dihydroxybenzoic acid (DHB), were examined with scutellarin standard by MALDI-MS using the dried-droplet technique under identical conditions. The preparation of matrix solutions are listed in Table S1.† As shown in Fig. S1,† the predominant ion [M + H]+ at m/z 463.1 clearly observed in most suitable matrices except BPF and DCTB. BPF exhibited lower abundance in the positive ion mass spectrum, and DCTB severely produced background interference and the analyte signals could rarely be distinguished from background noise. However, in particular, FA and THAP had the best performances with an S/N ratio around 1.5–8 folds greater than other matrices for the scutellarin measurement, whereas a lower analyte signal intensity was detected with CHCA, DHB and CA matrices. Furthermore, a low-abundance sodium adduct [M + Na]+ ion at m/z 485.1 and a potassium adduct [M + K]+ ion at m/z 501.0 were also identified throughout the whole mass range for the FA, CHCA, THAP, DHB and CA matrices. The above-mentioned results indicate that FA, CHCA, THAP, DHB and CA matrices are sensitive towards the standard scutellarin analyte, and will be chosen for the analysis of scutellarin metabolites in the real biological system.The chosen matrices were further tested on the scutellarin-treated tissue section directly. First, scutellarin metabolites were identified and characterized on a kidney extract via ESI-MS/MS (Fig. S2 and S3†). All of the ESI† results were in accordance with the previous studies based on the characterization of scutellarin and its metabolites in mice.30–32 As shown in Fig. S4,† almost no metabolite ions could be observed with FA or DHB matrix by MALDI-MS, which might be explained by the structure and functional groups of the matrix. The predominate parent ion (M0, scutellarin) peak was observed when using the CA matrix, the metabolites’ intensities were significantly weaker. In addition, when using THAP as the matrix, only the scutellarein metabolite (M1, scutellarein) signal was detected, the parent ion M0 was totally lost. CHCA was capable of detecting both M0 and M1, simultaneously; however, the other two metabolite (M2, 6-O-methyl-scutellarin; M3, 6-O-methyl-scutellarein) signals were absent. Within all identified metabolites (M1–M3) by MS/MS, only M1 metabolite generated an excellent signal in positive mode via MALDI-MS. This may be because scutellarein is the primary metabolite species of scutellarin. All of these results suggested that the current single matrix could not extract and identify all metabolites from the complex tissue sample with an appropriate response.
Optimization of the binary matrix for the MALDI MS analysis
To increase the overall detection efficiency and improve the uniformity of sample-matrix co-crystallization, graphene oxide (GO) was used as the matrix in the experiment since previous studies reported that the detection efficiency for scutellarin could be significantly improved when using GO as the matrix on the MALDI-target plate.29 However, our experimental results showed that there was no significant improvement in the detection of scutellarin and its metabolites when GO was applied as a single matrix on biological tissue sections (Fig. S5†). It can drastically reduce the background noise after m/z 250. However, there were nearly no other signals of scutellarin and its metabolites in the spectrum. GO, alone, was not capable of detecting scutellarin and its metabolites. It is important to note that the binary matrix could promote detection sensitivity and reduce the background noise of small molecular drugs and their metabolites. Therefore, the effectiveness of binary matrices including GO and traditional organic matrix was evaluated. As displayed in Fig. 1A, the quality of the mass spectra was improved. The higher peaks intensities and higher S/N ratio (>3) with the binary matrix (GO/CA) were obtained in comparison with that of the single matrix CA in positive mode (Table S2†). In addition, metabolite (M2, M3) ion intensities were obviously enhanced with high abundance signals and excellent resolution in the mass spectra. It can be explained by the unique structure and properties of GO. Due to the large delocalized π-electron conjugated system of GO, matrix and analyte molecules can be easily adsorbed on the surface of GO. Together with the exclusive electron mobility of GO, the energy transfer process between matrices and analytes can be strengthened. In addition, the hydroxyl group from GO may act as a Lewis acid for protonation, leading to a higher degree of ionization on the general MS signal response. The GO/CHCA matrices observed that the intensity of the M1 signal was noticeably enhanced and the S/N ratio was significantly increased. Nevertheless, M0 and M2 signals were suppressed and had very low intensity and resolution. GO/THAP did not provide any good quality mass spectra (Fig. 1C). This may be because this binary matrix system was not stable in vacuum. Therefore, the binary matrix was not used for this study. Based on the above-mentioned results, the binary matrix (GO/CA) significantly improved the scutellarin and its metabolites detection efficiency with relatively high sensitivity and selectivity on tissue sections. Finally, we adapted GO/CA as our target matrix for further investigation.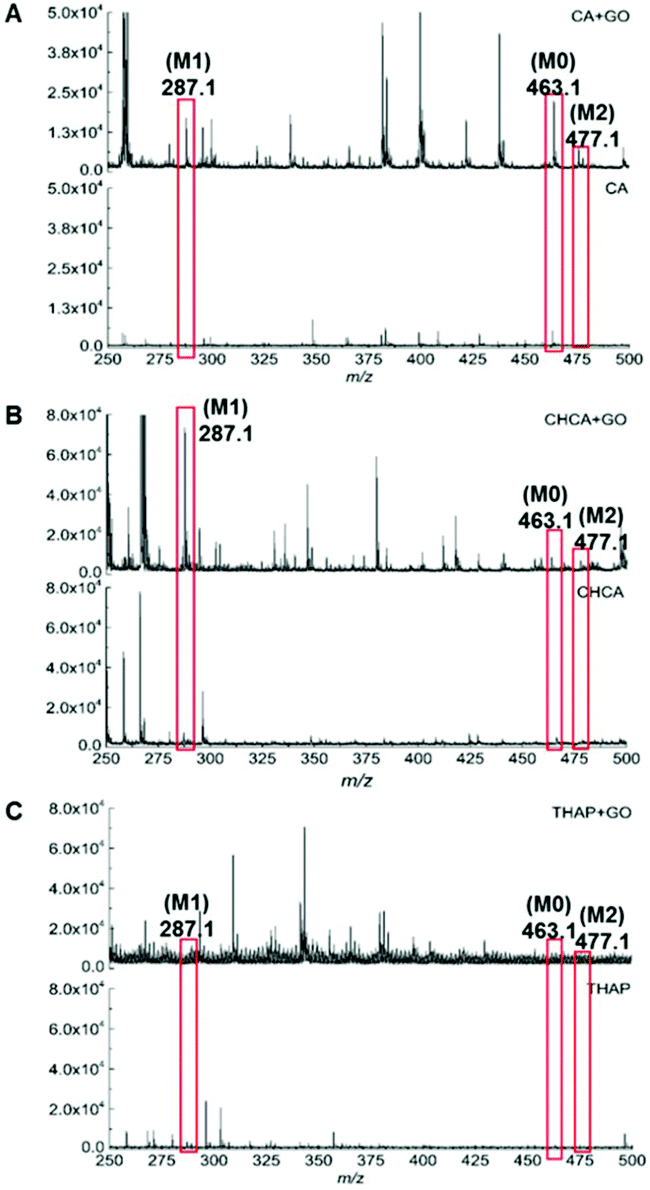 | ||
| Fig. 1 The MALDI-MS spectrum showing the difference between matrix added with GO and without GO: (A) GO/CA, (B) GO/CHCA, (C) GO/THAP. | ||
To optimize the performance of the GO/CA binary matrix for imaging analysis, GO and CA combinations of different ratios were subsequently tested on a kidney section. When the ratio of GO/CA was set as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, only M2 peak was clearly detected, while all other metabolite peaks were overlapped with the background signal in the mass spectra (Fig. 2A). In addition, M1 can also be identified besides M2 and generated a prominent peak in positive mode when the ratio was adjusted to 1:1 (Fig. 2B). This result of the ionization ability enhancement may be due to the π–π conjugated system of GO and energy transfer. Notably, when a GO/CA combination of 8:2 ratio was loaded, all species (M0–M3) peaks had an excellent intensity and resolution that was higher than all the ratios previously tested (Fig. 2C). However, when the proportion of GO was increased to 90%, all ion signals significantly suppressed (Fig. 2D). This result may be caused by the structure of CA, serving as a Lewis acid that can offer protons for analyte ionization. All of these findings and explanations were basically consistent with those existing GO matrix-based MALDI-MS studies. In short, our binary matrix has successfully achieved optimal efficiency in MALDI-MS ionization as a novel matrix. Considering its superior performance in detecting metabolites relative to other matrix combinations suggested by the experimental result, GO/CA (8:2) should be an ideal choice as our target matrix for scutellarin and its metabolite imaging analysis.
:9, only M2 peak was clearly detected, while all other metabolite peaks were overlapped with the background signal in the mass spectra (Fig. 2A). In addition, M1 can also be identified besides M2 and generated a prominent peak in positive mode when the ratio was adjusted to 1:1 (Fig. 2B). This result of the ionization ability enhancement may be due to the π–π conjugated system of GO and energy transfer. Notably, when a GO/CA combination of 8:2 ratio was loaded, all species (M0–M3) peaks had an excellent intensity and resolution that was higher than all the ratios previously tested (Fig. 2C). However, when the proportion of GO was increased to 90%, all ion signals significantly suppressed (Fig. 2D). This result may be caused by the structure of CA, serving as a Lewis acid that can offer protons for analyte ionization. All of these findings and explanations were basically consistent with those existing GO matrix-based MALDI-MS studies. In short, our binary matrix has successfully achieved optimal efficiency in MALDI-MS ionization as a novel matrix. Considering its superior performance in detecting metabolites relative to other matrix combinations suggested by the experimental result, GO/CA (8:2) should be an ideal choice as our target matrix for scutellarin and its metabolite imaging analysis.
 | ||
| Fig. 2 Binary matrix of four different ratios applied on the scutellarin-treated sample tissue: (A) GO:CA (1:9 v/v), (B) GO:CA (1:1 v/v), (C) GO:CA (8:2 v/v), (D) GO:CA (9:1 v/v). The large spectra displayed the metabolites ranged between m/z 455–480, meanwhile the insets show expanded spectra for metabolites within the m/z 280–305. Background (BG). | ||
In situ imaging of scutellarin and its metabolites
Targeted MALDI-MSI is a useful technique for mapping the distribution and localization of scutellarin and its metabolites with label-free. Because of the strong matrix interferences, the technique for small-molecule metabolite imaging is quite limited. The various sizes of organic matrix crystals and analyte extraction also affect the development of imaging in terms of spatial resolutions. In our study, all metabolites and parent compound cannot be detected simultaneously with single matrix. Only one metabolite and parent compound can be visualized in the spectrum using the CA matrix. However, our binary matrix with outstanding advantages was therefore used to address the issues mentioned above. All metabolites and the parent compound could be observed. The results of our research are consistent with Wu's findings.25 Prior to the MSI analysis, our binary matrix solution was deposited on the scutellarin-treated tissue sections by an auto-sprayer to achieve homogeneous crystallization with a high coverage density (Fig. S6†). In this case, GO has demonstrated its monolayer structure and good dispersion ability to ensure the formation of a uniform distribution domain while overcoming the limitation of analyte delocalization through sample-matrix co-crystallization. To profile the possible scutellarin and its metabolites, the kidney sample after a single ip injection was analyzed with MALDI-MSI. A 60 min administration model was taken as an illustrated example since it generally detected the metabolites with the highest response rates from our pre-experiment (Fig. S7†). The result indicates that scutellarin was absorbed quickly in the bloodstream and underwent extensive biotransformation to its different metabolites in a very short time in vivo. The spatial localization of scutellarin and its metabolites in kidney sections from 60 min is visualized in Fig. 3. MSI results clearly indicate that the imaging from scutellarin and its metabolites with the binary matrix are all visualized in the Experimental section with respect to control sections (60 min). The results of the H&E staining show the distinguishable structure of the mouse kidney. With the aid of the histopathology image, it was found that both scutellarin M0 and metabolite M1 were mainly distributed in the pelvis, medulla and cortex regions. Moreover, the M0 was also present in the medulla and cortex regions at a relatively lower abundance than that at other sites. From imaging results, the relative accumulation of M1 found in kidney was greater than M0 in general. This may be because most of the M0 were rapidly transformed into metabolites M1 in kidney. It is notable that metabolite M2 and M3 ions tended to localize predominantly at the cortex part, were nearly absent in the pelvis. For each analyte, the signal intensity of each characteristic peak in the mass spectrum was shown in different colors. For reproducibility assessment, repeated MSI analysis was conducted with the binary matrix in three different experimental tissue sections, respectively. As can be seen in Fig. S8,† comparable images were obtained for each respective scutellarin and its metabolites with similar intensity and distribution, confirming the consistency and repeatability of our results. All of the results indicate that the localization and distribution of scutellarin and its metabolites can be visualized simultaneously with our binary matrix in the same tissue section, and such distribution of scutellarin and scutellarein through uptake by renal cells may be useful for further kidney clinical applications. Taking everything into account, this study not only pioneers a new field for the applications of the GO binary matrix system, but also provides a new technique for the highly efficient facial analysis of low-molecular-weight compounds in areas including drug metabolism research and phenolic compound analysis.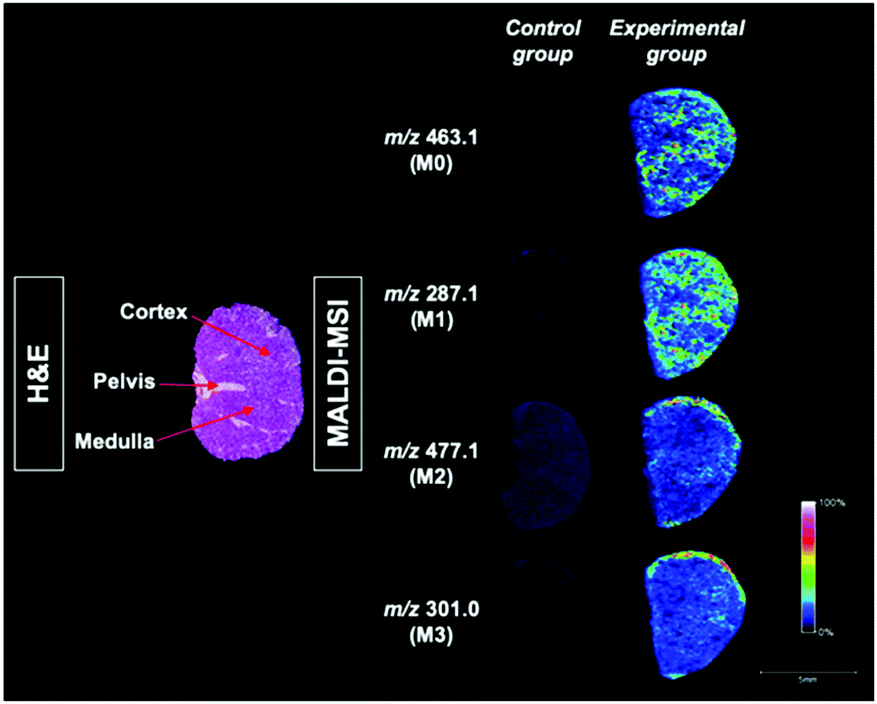 | ||
| Fig. 3 In situ visualized scutellarin and its metabolites in mouse kidney tissue after the ip injection with respective to the control tissue by MALDI-MSI. Also, the H&E staining tissue sections labelled with the cortex, medulla and corticomedullary junction in dotted lines. | ||
Conclusions
Despite extensive investigation on the characterization, metabolism and pharmacokinetics of scutellarin, the spatial distribution of scutellarin and its metabolites have received little research attention. The present study developed a novel MALDI-MSI-based assay for qualitatively analyzing the spatial distribution of scutellarin and its metabolites in kidney tissue sections. In the initial stage, our newly developed GO/CA binary matrix has successfully demonstrated its feasibility as an alternative matrix for the MALDI-MS analysis of biological tissue samples with improved ionization efficiency and homogeneous crystallization. Although GO could not function alone on tissue sections, when mixed with CA, a significant improvement in sensitivity and selectivity was observed compared to other conventional matrices. Besides, the optimized binary matrix was successfully applied to the study of the spatial distribution of scutellarin and its metabolites after the ip administration via MALDI-MSI. A significant difference on spatial visualization and intensity was found for different metabolites. Scutellarin and scutellarein were both located in the cortex and medulla regions of the kidney with a relatively high concentration, while the remaining metabolites appeared in the cortex region with rather low abundance. Due to the high absorption rate observed after drug injection, the accumulation of the drug to reach effective levels inside the kidney highlighted its potential for the prevention and treatment of kidney tumour.Experimental
Materials and reagents
Standard scutellarin (≥98%), was purchased from Xiya Chemical Technology Co., Ltd (Chengdu, China). For matrices, ferulic acid (FA, ≥99%), α-cyano-4-hydroxycinnamic acid (CHCA), caffeic acid (CA, ≥99%), bis(4-hydroxyphenyl)methane (BPF, ≥98%), 2,5-dihydroxybenzoic acid (DHB, ≥98%) and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB, ≥99%), 2,4,6-trihydroxyacetophenone (THAP, ≥99.5%) were bought from Sigma-Aldrich (St Louis, MO, USA), while graphene oxide (GO, ≥97%) powder was obtained from XFNANO Materials Tech Co., Ltd (Nanjing, China). For solvents, HPLC grade acetonitrile (ACN), methanol (MeOH) and trifluoroacetic acid (TFA, 99%) were purchased from VWR International company (Radnor, Pennsylvania). HPLC grade dichloromethane (DCM) was obtained from Duksan Pure Chemicals Co. Ltd (Republic of Korea). Distilled water was purified by a Milli-Q system (Millipore, Billerica, MA, USA).Sample preparation for MALDI-MSI
Male C57BL/6J mice (20 ± 2 g body weight) were obtained from the Chinese University of Hong Kong (CUHK) and were housed in sterile, individually ventilated cages (IVC) under controlled conditions (22 ± 2 °C temperature, 12 h light/dark cycle in each day) before using. All animal experiments were performed in accordance with the Guidelines of Care and Use of Animals for Experimental Purposes, and approved by the ethics committee at Hong Kong Baptist University. For the MALDI-MSI analysis of the scutellarin and its metabolites, scutellarin-treated mice were administered with scutellarin solution (10 mg mL−1 in PBS) via the ip injection at a dose of 100 mg kg−1 each, whereas the control mice were injected with the same volume of PBS solution in the same manner. Kidney tissues were collected at different time intervals after drug administration. Kidney samples were then snap-frozen under liquid nitrogen and stored under −80 °C until further use. Before the MALDI-MSI analysis, kidney sample tissues were cut into sections (12 μm thick) using a CyroStar Nx70 cryostat (Thermal Fisher Scientific Inc., Germany). The samples were thaw mounted on Indium-tin oxide (ITO)-coated conductive glass slides, and then dried in a vacuum desiccator until further use. All the tissue sections were sprayed with the binary matrix solution by an automatic matrix sprayer (ImagePrep, Bruker Daltonics, Billerica, MA) prior to the MALDI-MSI analysis.MS/MS procedure for scutellarin and its metabolites analysis
Scutellarin and its metabolites in a sample kidney were measured via direct infusion mass spectrometry using multiple reaction monitoring (MRM) and positive/negative switching. All samples were conducted by Dionex Ultimate 3000 (Thermo Scientific) linked to a triple quadrupole mass spectrometer, TSQ-Quantiva (Thermo Scientific, USA) equipped with an ESI† interface. The software Thermo Xcalibur (version 4.1) was employed for raw data extraction. The capillary voltage was set to ±3.5 kV and the cone voltage was ±70 V, the source and desolvation temperatures were 110 °C and 320 °C, respectively, the desolvation and nebuliser gas flow rates were 600 L h−1 and 75 L h−1, respectively. The collision energy was set at 35–45 eV.Solid-phase extraction (SPE) procedure
Sample kidney tissues after the ip injection were snap-frozen and stored under −80 °C. Approximately 10–15 mg for each tissue sample was placed into a 1.5 mL screw cap tube (RINO TUBES) and filled with 500 μL of MeOH. Tissue homogenization was operated with Bullet Blender X24 (Next Advance, Inc., NY, USA) with 1 × 2 min cycles by a bead beater with MeOH. The homogenize solutions were vortexed and centrifuged at 13000 rpm for 15 min at 4 °C. Supernatants were collected and evaporated to dryness under N2 gas at 40 °C. Following a simple SPE procedure was conducted before injection. The residue was reconstituted with 100 μL MeOH. The solution was then mixed with 400 μL of 0.1% phosphoric acid (H3PO4). The mixture was then transferred to a C8-SPE column, which was preconditioned with 1 ml of MeOH, 1 ml of water and 1 ml of 0.1% H3PO4. After loading the sample, the column was washed with 1 ml of 0.1% H3PO4. The analytes were eluted out with 1 mL of MeOH, and the eluate was evaporated under N2 gas. The residue was reconstituted with 60 μL of MeOH:0.1% formic acid (1:1, v/v), and then was subjected to the MS system.
MALDI-MSI and histological analysis
All matrix-coated sections were analyzed by RapifleX MALDI Tissuetyper (Bruker Doltonics, Germany) equipped with a laser emitting at 50–60% as the excitation source. Positive ion mass spectra were acquired with a 20 kV source voltage, frequency 10000 Hz, and 170 ns delayed extraction time, summed with 1000 laser shots over a 100 × 100 μm raster, in the m/z range of 100–1000. Ion images were filtered within tolerance ±0.2 Da. For instrumental calibration, external standards (SA, CHCA, DHB, Bradykinin (1–7) and Angiotensin II) were employed in positive ionization mode to minimize the mass measurement error. MALDI-TOF spectrometry is controlled by FlexControl 3.0. All images were then processed and visualized using Fleximaging 3.0 (Bruker Daltonics, Germany), which was applied for all spectrum analyses and exports. For the histological analysis, sections (12 μm) of mouse kidneys were stained with hematoxylin and eosin (H&E) staining. Histological changes of the kidney were acquired by a Leica DM 2500 microscope (Leica, Wetzlar, Germany) with microphotographs taken with x10 magnification.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (21577115 and 21477101); the Research Grant Council of Hong Kong (GRF 463612, 14104314, 12300114); Hong Kong Health and Medical Research Fund (HMRF/03144376); and HKASO research grant 2015-16. We thank Dr Simon Wang (Language Centre, HKBU) on improving the manuscript writing.References
- S. Chledzik, J. Strawa, K. Matuszek and J. Nazaruk, Am. J. Chin. Med., 2018, 46, 319–337 CrossRef CAS.
- L. P. Wang and Q. Ma, Pharmacol. Ther., 2018, 190, 105–127 CrossRef CAS.
- L. J. Xu, R. C. Chen, X. Y. Ma, Y. Zhu, G. B. Sun and X. B. Sun, Phytomedicine, 2020, 68, 153169–153170 CrossRef CAS.
- Q. F. Liu, Y. Shi, Y. Wang, J. Lu, W. J. Cong, G. A. Luo and Y. M. Wang, Talanta, 2009, 80, 84–91 CrossRef CAS.
- C. Y. Gao, X. Y. Chen and D. F. Zhong, Drug Metab. Dispos., 2011, 39, 2034–2044 CrossRef CAS.
- H. Tang, N. G. Li, Y. P. Tang, Q. P. Shi, J. M. Guo, W. Zhang, M. Z. Shen and J. A. Duan, Anal. Methods, 2014, 6, 4667–4673 RSC.
- T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545–580 CrossRef CAS.
- M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS.
- J. R. Lindner and J. Link, Circ. Cardiovasc. Imaging, 2018, 11, e005355 Search PubMed.
- T. Greer, R. Srurm and L. J. Li, J. Proteomics, 2011, 74, 2617–2631 CrossRef CAS.
- D. S. Cornett, S. L. Frappier and R. M. Caprioli, Anal. Chem., 2008, 80, 5648–5653 CrossRef CAS.
- L. Lamont, G. B. Eijkel, E. A. Jones, B. Flinders, S. R. Ellis, T. P. Siegel, R. M. A. Heeren and R. J. Vreeken, Anal. Chem., 2018, 90, 13229–13235 CrossRef CAS.
- E. Davoli, M. Zucchetti, C. Matteo, P. Ubezio, M. D'Incalci and L. Morosi, Mass Spectrom. Rev., 2020, 1–14 Search PubMed.
- L. R. Olsen, S. H. Hansen and C. Janfelt, Anal. Bioanal. Chem., 2015, 407, 2149–2158 CrossRef CAS.
- N. Bjarnholt, B. Li, J. D'Alvise and C. Janfelt, Nat. Prod. Rep., 2014, 31, 818–837 RSC.
- D. Sturtevant, Y. J. Lee and K. D. Chapman, Curr. Opin. Biotechnol., 2016, 37, 53–60 CrossRef CAS.
- P. M. Kumara, R. U. Shaanker and T. Pradeep, Phytochemistry, 2019, 159, 20–29 CrossRef.
- N. Araujo, L. M. Souza, F. E. Pinto, C. J. Macrino, C. M. Almeida, R. Mohana-Borges and W. Ramão, Anal. Methods, 2019, 11, 1757–1764 RSC.
- J. E. Spraker, G. T. Luu and L. M. Sanchez, Nat. Prod. Rep., 2020, 37, 150–162 RSC.
- D. S. Cornett, S. L. Frappier and R. M. Caprioli, Anal. Chem., 2008, 80, 5648–5653 CrossRef CAS.
- M. Lastovickova, J. Chmelik and J. Bobalova, Int. J. Mass Spectrom., 2009, 281, 82–88 CrossRef CAS.
- Z. Guo and L. He, Anal. Bioanal. Chem., 2007, 387, 1939–1944 CrossRef CAS.
- C. E. Rodríguez, J. Palacios, I. Fajardo, J. L. Urdiales, X. L. Guével, J. Lozano and F. S. Jiménez, J. Am. Soc. Mass Spectrom., 2016, 27, 366–369 CrossRef.
- Z. P. Zhu, J. J. Shen, Y. F. Xu, H. M. Guo, D. Kang, T. J. Yu, H. Wang, W. S. Xu, G. J. Wang and Y. Liang, J. Mass Spectrom., 2019, 54, 684–692 CrossRef CAS.
- H. N. Abdelhamid and H. F. Wu, Analyst, 2015, 5, 1555–1565 RSC.
- K. Liang, H. Y. Gao, Y. J. Gu, S. J. Yang, J. L. Zhang, J. J. Li, Y. L. Wang, Y. J. Wang and Y. Li, Anal. Chim. Acta, 2018, 1035, 108–118 CrossRef CAS.
- D. Zhou, S. Guo, M. Zhang, Y. J. Liu, T. J. Chen and Z. L. Li, Anal. Chim. Acta, 2017, 962, 52–59 CrossRef CAS.
- L. S. Cai, L. F. Sheng, M. C. Xia, Z. P. Li, S. C. Zhang, X. R. Zhang and H. Y. Chen, J. Am. Soc. Mass Spectrom., 2017, 28, 399–408 CrossRef CAS.
- Y. Liu, J. Y. Liu, P. Yin, M. X. Gao, C. H. Deng and X. M. Zhang, J. Mass Spectrom., 2011, 46, 804–815 CrossRef CAS.
- J. Xu, M. Zhao, D. W. Qian, E. X. Shang, S. Jiang, J. M. Guo, J. A. Duan and L. Du, Anal. Methods, 2014, 6, 2314–2319 RSC.
- X. Y. Chen, L. Cui, X. T. Duan, B. Ma and D. F. Zhong, Drug Metab. Dispos., 2006, 34, 1345–1352 CrossRef CAS.
- Q. F. Liu, Y. Shi, Y. Wang, J. Lu, W. J. Cong, G. A. Luo and Y. M. Wang, Talanta, 2009, 80, 84–91 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: MS/MS spectra, MALDI-MS spectra, optical microscope image, time profiles, MSI reproducibility and preparation of matrix solutions. See DOI: 10.1039/d0an01539c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |