
Spatial proteomics for understanding the tissue microenvironment
Yiheng
Mao
ab,
Xi
Wang
bc,
Peiwu
Huang
b and
Ruijun
Tian
 *b
*b
aSchool of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin, 150001, China. E-mail: tianrj@sustech.edu.cn; Tel: +86-755-88018905
bDepartment of Chemistry, College of Science, Southern University of Science and Technology, Shenzhen 518055, China
cShenzhen People's Hospital, The First Affiliated Hospital of Southern University of Science and Technology, Shenzhen 518020, China
First published on 21st May 2021
Abstract
The human body comprises rich populations of cells, which are arranged into tissues and organs with diverse functionalities. These cells exhibit a broad spectrum of phenotypes and are often organized as a heterogeneous but sophisticatedly regulated ecosystem – tissue microenvironment, inside which every cell interacts with and is reciprocally influenced by its surroundings through its life span. Therefore, it is critical to comprehensively explore the cellular machinery and biological processes in the tissue microenvironment, which is best exemplified by the tumor microenvironment (TME). The past decade has seen increasing advances in the field of spatial proteomics, the main purpose of which is to characterize the abundance and spatial distribution of proteins and their post-translational modifications in the microenvironment of diseased tissues. Herein, we outline the achievements and remaining challenges of mass spectrometry-based tissue spatial proteomics. Exciting technology developments along with important biomedical applications of spatial proteomics are highlighted. In detail, we focus on high-quality resources built by scalpel macrodissection-based region-resolved proteomics, method development of sensitive sample preparation for laser microdissection-based spatial proteomics, and antibody recognition-based multiplexed tissue imaging. In the end, critical issues and potential future directions for spatial proteomics are also discussed.
1. Introduction
The human body comprises a massive number of cells, which are in turn arranged into tissues and organs with diverse functionalities for each compartment.1 Driven by the internal molecular features such as genetic codes, cells exhibit a broad spectrum of phenotypes and are often organized as a heterogeneous but harmonious ecosystem – tissue microenvironment inside the human body. Within the tissue microenvironment consisting of various cell types, subpopulations, and substructures, every cell interacts with and is reciprocally influenced by its surroundings through its entire life span. Therefore, it is critical to interrogate the tissue microenvironment for a comprehensive understanding of the cellular machinery and inner biological processes, for example, in the field of cancer biology.2–5 Generally, solid tumors are made up of neoplastic cells together with stromal parts including extracellular matrix (ECM), cancer-associated fibroblasts (CAFs), endothelial cells, immune cells, etc.6–9 Spatiotemporal heterogeneity within the tumor microenvironment (TME) has been proven to play a critical role in tumor initiation, progression, metastasis, therapeutic response, and drug resistance.10–13 Hence, systematic characterizing and accurate assessing this intricate network is considered as a prerequisite for deciphering the ecological and evolutionary nature of cancer as well as developing effective therapies.14,15 In recent years, efforts have been contributed to map the molecular features in TME with the help of a diversity of analytical and biological approaches. Among which, immunohistochemistry (IHC) staining of tissue sections has now become a routine technique in clinical diagnosis and biomedical research.16 IHC and other histochemical staining methods allow visual assessment of the tissue morphology, cell type distribution and expression level of endogenous molecules such as proteins. Nonetheless, the traditional IHC protocol is quantitatively imprecise and limited by a low throughput for measuring multiple targets in the same slide at the same time.17Under this circumstance, the concept of single-cell analysis has emerged for dissecting the cellular heterogeneity in single-cell resolution in the past decade.18,19 In order to make single-cell suspensions, tissue samples are usually disaggregated enzymatically. Afterwards, the cells are sorted based on antibody recognition or diluted using microfluidic chips following by single-cell analysis.20 Besides single cells, this procedure can also produce high-purity cell subpopulations, however, it will inevitably cause the loss of spatial information and important TME components such as ECM. In addition, whether results from single-cell analysis can well represent the cell states in the real TME is still controversial. Taking advantage of the advanced sequencing technologies with increased throughput and sensitivity in the past decade, it has become routinely available for performing in-depth transcriptome profiling at single-cell resolution. For example, the combination of single-cell sequencing technologies with traditional histological imaging has now enabled multiplexed and spatially resolved transcriptome analysis of the same tissue section by unbiased mRNA capture using spatially barcoded microarray of printed spots (55–100 μm in diameter) or densely packed beads (2–10 μm in diameter).21–25 These strategies, such as Spatial Transcriptomics technology,26 have now made it feasible to characterize the TME in higher dimension by measuring spatial patterns of gene expression in cryosections.27–30 Despite the extraordinary achievements of spatial transcriptomics in methodology developments and biomedical applications, it is worth noting that proteins rather than DNA or RNA control virtually all cellular processes and execute biological functions. Often, the genomic or transcriptomic datasets are used to predict the phenotype on the basis of the central dogma, however, poor correlations have been observed between mRNA copy number and protein abundance in several systematic and comparative studies.31–33
Mass spectrometry (MS)-based proteomics has now become a major tool to explore the structure and function of proteome owing to the great progress for the development of sample preparation and fractionation methods, high-performance instruments and data analysis algorithms.34–36 During the past decade, substantial efforts have been made for characterizing the proteome landscapes in different cell lines,37 body fluids,38 organs from healthy or diseased mammals.39–41 Typically, the tissue proteome is profiled by analyzing protein lysates extracted from bulk tissues which often contain massive number of cells. This streamlined workflow is very robust and can usually produce high-quality resources of thousands of proteins together with their post-translational modifications (PTMs) status.42 However, the homogenization process will inevitably result in an average effect by blurring the spatial and cell-type information.43 To this end, the past decade has seen increasing advances in the field of spatial proteomics, the purpose of which is to characterize the abundance and spatial distribution of proteins and their PTMs in tissue to describe and understand the TME in a systematic manner.44–48 In general, the analytical strategies of spatial proteomics for tissue can be divided into three categories: (1) physical dissection, (2) antibody recognition, and (3) mass spectrometry imaging (MSI). Among these strategies, the most prevalent and direct one is physical dissection by separating the heterogeneous organ or tissue sections into spatially resolved pieces by scalpel or pulsed laser with spatial resolutions ranging from millimeter to sub-micrometer based on the precision of dissecting tools. The dissection-based proteomic experiments can reach a depth of hundreds to thousands of proteins identified with their spatial information. Akin to conventional IHC staining, the second class of strategies depend on the recognition of targeted proteins on tissue sections by antibodies which should carry special reporters for downstream detection such as fluorescent groups for multiplexed imaging or metal-labeled complexes for mass cytometry. As for MSI, such as matrix-assisted laser desorption/ionization (MALDI)49,50 desorption electrospray ionization (DESI),51 and secondary ion MS(SIMS)-based technologies,52 have been widely used to unbiasedly measure the spatial localization and abundance of a variety of analytes mainly including peptides, metabolites, and drug molecules in tissue sections in a high-throughput manner. Taking advantage of the fast scan speed of these in situ analytical technologies, efforts have been made to apply MSI to characterize the TME mainly based on the spatial distribution of small molecules such as lipids.53 However, the proteome coverage and quantification precision of MSI is very limited due to its inherent technical shortages including ionization suppression, complicated procedure for proteomics sample preparation, and absence of efficient peptide separation. To this end, several strategies including on-tissue digestion,54,55 liquid extraction surface analysis (LESA)56,57 and utilization of ion mobility-based separation58 have been tested by coupling to MSI, but large-scale in situ peptide identification in tissue still remains challenging by far.
In this review, we outline the achievements and remaining challenges of MS-based tissue spatial proteomics. Exciting technology developments along with important biomedical applications of spatial proteomics are highlighted (Table 1). In addition, several novel technologies with great potential for spatial proteomics are also discussed. Since this is a technical review, the following parts are organized according to the analytical methods, including scalpel macrodissection, laser microdissection, and antibody recognition (Fig. 1). As for MSI, we only discuss it here as an annotation tool for laser microdissection-based spatial proteomics in section 3, because lots of comprehensive reviews introducing the latest progress of MSI and its application have been published with different highlights in the past decade.53,58–63
| Dissection methods | Representative works/technologies | Starting materials | Proteome coverage | MS instruments | Ref. |
|---|---|---|---|---|---|
| Scalpel macrodissection | Sharma et al. | Fresh mouse brain | >12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 proteins for eight regions and four cell types 000 proteins for eight regions and four cell types |
Q Exactive (DDA) | 64 |
| Doll et al. | Fresh human heart | >10000 proteins for sixteen regions and four cell types |
Q Exactive HF (DDA) | 67 | |
| Ni et al. | Fresh human gastric mucosa | >6000 proteins for seven regions | Orbitrap Fusion (DDA) | 71 | |
| Dyring-Andersen et al. | Fresh human skin | >10000 proteins for four layers and nine cell types |
Q Exactive HF-X (DDA) | 70 | |
| Laser microdissection | FFPE-FASP | H&E stained FFPE human colon tumor | >4000 proteins for cancer cells of 17.5 mm2 × 10 μm | LTQ-Velos (DDA) | 111 |
| LCM-SISPROT | H&E stained fresh-frozen human colon tumor | >5000 proteins for cancer cells of 5 mm2 × 10 μm | Orbitrap Fusion (DDA) | 83 | |
| FISGlyco | H&E stained fresh-frozen mouse brain | >1800 N-glycosites for isocortex of 15 mm2 × 20 μm | Q Exactive HF-X (DDA) | 128 | |
| IHC-SISPROT | IHC stained formalin-fixed human liver tumor | >6600 proteins for cancer cells of 5 mm2 × 12 μm | Q Exactive HF-X (DIA) | 130 | |
| TFE-based method | H&E stained FFPE human ovary tumor | >5000 proteins for cancer cells of 1.5 mm2 × 10 μm | Q Exactive HF-X (DIA) | 100 | |
| LCM-nanoPOTS | H&E stained fresh-frozen mouse brain | >1000 proteins for brain of 0.01 mm2 × 12 μm | Fusion Lumos (DDA) | 88 | |
| SP3-TMT | H&E stained FFPE human brain | >4000 proteins for substantia nigra of 1 mm2 × 10 μm | Orbitrap Fusion (TMT) | 90 | |
| Antibody recognition | MIBI | FFPE human breast tumor | 10 targets simultaneously at a resolution of 200 nm | NanoSIMS 50L MS | 171 |
| IMC | FFPE human breast tumor | 32 targets simultaneously at a resolution of 1 μm | Laser ablation-CyTOF | 172 |
 | ||
| Fig. 1 Scheme illustration of spatial proteomics technologies for understanding the tissue microenvironment. | ||
2. Scalpel macrodissection-based, region-resolved proteomics
In anatomy, organs and tissues in mammalian body can often be divided to different compartments. For instance, the human muscle is mostly composed of belly part and tendon part with distinct morphological characteristics and complementary functions during muscle movement. The initial attempts of spatial proteomics started with MS-based proteomic analysis of different anatomic regions dissected from organs using scalpel by clinical experts. Often, tens to hundreds of micrograms of starting material can be obtain from scalpel-based macrodissected tissues, therefore the experimental protocols used in this section is quite standard including tissue lysis, protein digestion, desalting, peptide fractionation, and liquid chromatography (LC)-MS analysis. Herein, we highlighted several recent works which provided rich proteomic resources of important organs with high data quality.Brain is the most important part in mammalian central nervous system and has a highly complex structural and functional organization. To understand the development and function of brain, several groups have profiled the global proteomes of major anatomic regions and cell types in human and mouse brain. In 2015, Sharma et al. performed an in-depth proteomic and transcriptomic analysis of eight major regions and four cell types isolated from adult mouse brain samples.64 Taking advantage of the match-between-runs (MBR) algorithm embedded in MaxQuant software,65 an average of ∼11000 proteins for each brain region and cell type were identified with high reproducibility. In the proof-of-concept analysis, a class of cell surface proteins were found over-expressed and then defined as cell-type-specific markers. This large-scale resource provided a successful example for the following efforts on the establishment of spatial- and cell-type-resolved brain proteomic landscapes. Two years later, the same group profiled the soluble and insoluble proteome of four brain regions in wild-type mouse and Huntington's disease (HD) mouse model at three different stages of disease progression to investigate the mechanisms of neurodegeneration from a spatiotemporal view. By using the raw files produced in their previous work64 as an additional spectral library to gain matching peptide identifications with MBR algorithm, over 8000 proteins were confidently quantified across different samples. Genetically engineered animal models are often considered unable to cover the entire proteome changes that occur in the human specimen, so several other works also profiled the spatial- and cell-type-resolved proteome in human brain samples. In 2017, Carlyle et al. conducted a comprehensive proteomic survey of seven regions in postnatal human brains of sixteen individuals at varied ages.32 About 5000 proteins in total were reliably quantified with MBR algorithm. Given that a more comprehensive proteomic atlas of human brain would promote the understanding of brain diseases such as neurodegeneration and dementia, the Coon group recently identified an average of ∼7000 proteins in nine anatomic regions in three aged human brains using high-pH reversed-phased (HpRP) fractionation and an Orbitrap Fusion Lumos MS.66
In addition to brain, heart is another life-control organ in human body, which can be dissected into four cavities, four valves with four major cell types. In 2017, the Mann group established a spatial- and cell-type-resolved proteome map of healthy human heart with three fresh samples from postmortem.67 Employing their high-sensitivity sample preparation and fractionation toolkit (inStageTip68 and loss-less nano-fractionator69), over 10000 protein groups were quantified from low-microgram protein lysates extracted from sixteen anatomic human heart regions and four cell types. By systematic comparison between these proteomes, regional differences in human heart and potential protein markers for atrial fibrillation were found. With the development of instrumentation, it has become much easier to obtain high-quality datasets. Recently, the same group generated the first spatial proteomic atlas of healthy human skin and over 10000 protein groups were quantified with location and cell-type information by a Q Exactive HF-X MS70 Due to the complex structures in the human skin, the authors employed curettage, biopsy punch, and fluorescence-activated cell sorting (FACS) to separate different tissue layers and cell types. By initial analysis of the atlas, an interesting spatial proteomic gradient phenomenon across different human skin layers was discovered and novel cell-type-specific structural and functional proteins were defined (Fig. 2).
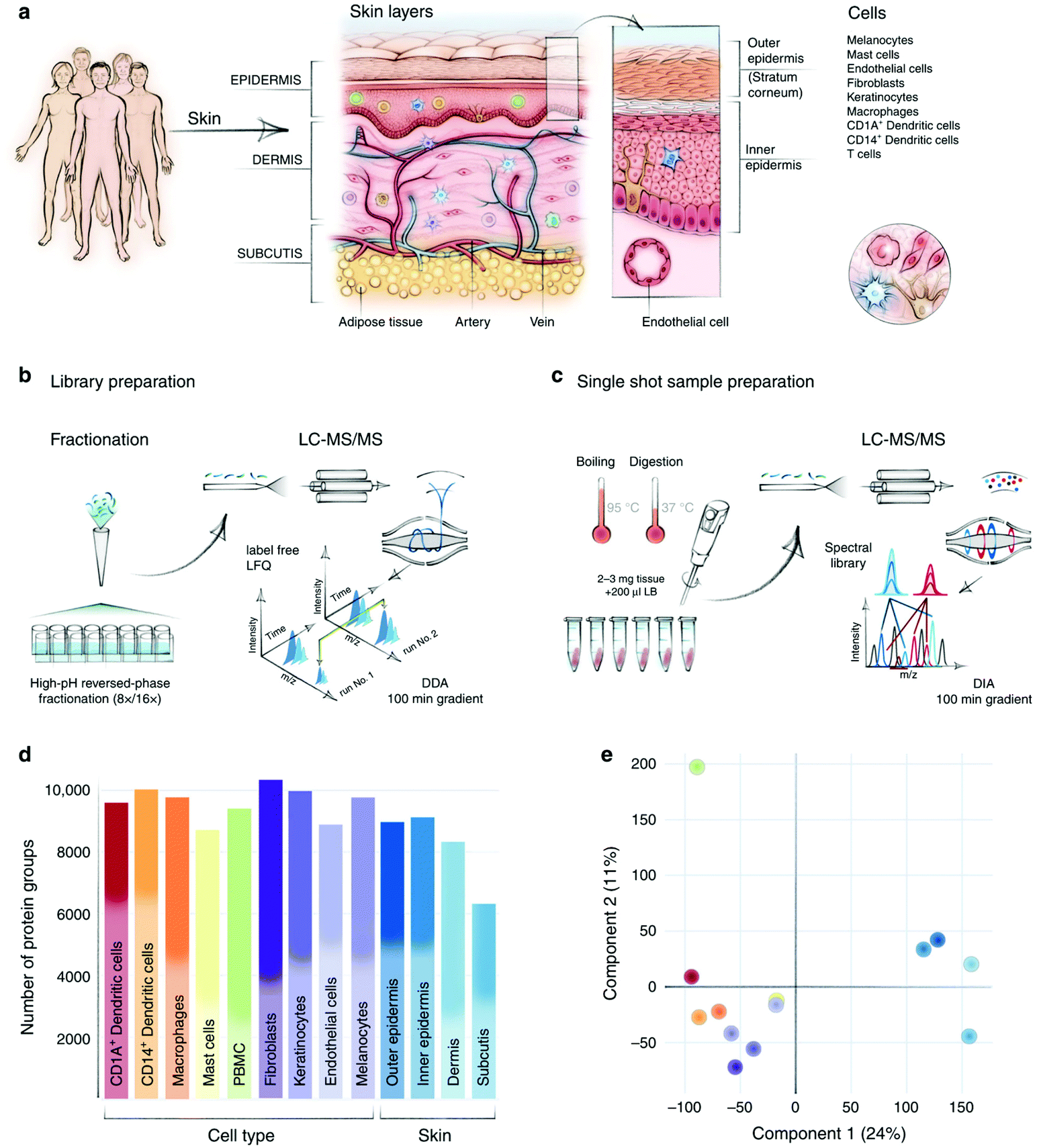 | ||
| Fig. 2 In-depth analysis of skin layers by macrodissection-based spatial proteomics. (a) Compartments and cell types in human skin. (b–c) Sample preparation and MS analysis workflow. (d–e) Protein identification number and PCA of different skin layers and cell types. Reproduced from ref. 70 with permission from the Springer Nature, copyright 2020. | ||
An important issue that attracts much attention is whether the molecular characteristics in anatomic-different tissue regions are distinct as well. In 2018, Ni et al. performed a global proteome profiling of mucosa samples from healthy human stomach.71 Anatomically, the human stomach mucosa can be divided into seven regions, but the generated regional-resolved proteomes in their study can only be classified into two clusters in the principal component analysis (PCA). This interesting result well demonstrated the complexity of the tissue microenvironment and the necessity for spatial proteomics technology with higher resolution. The precision of tissue dissecting tool determines the resolution of generated spatial proteome map. The scalpel-based macrodissection can achieve millimeter resolution by experienced hands,72 and several newly-introduced sampling technologies including hydrogel extraction54,55 and LESA56 can only reach sub-millimeter resolution with low proteome coverage. To this end, single-cell resolution dissecting tools are required to interrogate the cellular heterogeneity in tissue microenvironment.
3. Laser microdissection-based spatial proteomics
Laser microdissection was first described by Emmert-Buck et al.73 in 1996 as the laser capture microdissection (LCM) system following by the development of its different upgraded versions.74,75 At present, LMD has become a universal platform for cell enrichment at sub-micrometer resolution.76,77 LMD enables isolation and collection of regions of interest (ROIs) on tissue section in any shape by laser cutting with microscopic visualization.78 Then, the collected ROI pieces can be used for various downstream analysis including MS-based proteomics.79 Datasets derived from these samples can then be annotated with their original location, thus preserving spatial and cell type information.80 Although LMD has been successfully integrated into MS-based proteomics workflow, there are two major bottlenecks for the implementation of LMD-based spatial proteomics: (1) the lack of overall sensitivity in conventional MS-based proteomics workflow for low cell amounts, and (2) histopathological evaluation of tissue sections especially clinical samples usually requires guidance from experienced pathologists. Substantial efforts have been devoted to addressing these two technical problems, respectively. To increase the overall sensitivity, a variety of integrated sample preparation approaches have been developed and adapted to LMD-based spatial proteomics workflow. To improve the feasibility of histopathological evaluation, several automated and precise annotation methods have been introduced and combined with LMD-based cell isolation. The advance of these technologies has facilitated the in-depth proteome profiling of small-size tissue under convenient guidance. In this section, technical achievements and representative applications of LMD-based spatial proteomics are highlighted and discussed.3.1 Integrated sample preparation for higher sensitivity
Currently, the latest LC-MS/MS systems have achieved remarkable sensitivity for analyzing nanogram-level cell digests.81 Compared to the development of commercially available instrumentations and bioinformatic tools, proteomics sample preparation has largely lagged behind. For conventional proteomics workflow, multi-step manual operations are required and reactions often happen in large-volume solution. This protocol is very robust and works well with hundreds of micrograms of tissue lysates. However, when handling small amounts of sample such as LMD-collected tissues, considerable sample loss would lead to decreased proteome coverage with low reproducibility. For this reason, sample preparation technologies with miniaturized devices or integrated workflows have been developed to increase the overall sensitivity of LMD-based spatial proteomics.82 These technologies often differ in workflow according to tissue preservation methods.Fresh-frozen tissue is the ideal starting material for in-depth proteomic analysis because the modification, interaction, and structure of proteins can be well preserved once the tissue is immersed into liquid nitrogen and stored at −80 °C. For a balance of protein recovery and spatial resolution, fresh-frozen tissue for LMD is often cut into cryosections of 10–15 μm in thickness using a microtome to make theoretical single-cell layers. To prevent tissue from protein degradation, a quick fixation with ice cold organic solvents (e.g., acetone and methanol) is performed following by staining and dehydration. Since the protein components can be well preserved by the above procedures, the lysis buffer and sample preparation workflows designed for low-amount cell samples can also be used to process LMD-isolated cryosections with little modifications. However, the much tight and complex structure of these samples significantly increase the difficulty for extracting the protein components.
In 2018, our group combined LCM with the simple and integrated proteomics sample preparation technology (SISPROT) for processing rare clinical tissue samples.83 The SISPROT device is composed of ion exchange beads and C18 SPE disks in tandem within the same spintip. All the sample preparation steps including protein enrichment, reduction, alkylation, digestion, desalting, and high pH RP-based peptide fractionation can be carried out in one single tip within 2–3 hours.84 This unique design enables efficient processing of sub-microgram protein samples and removal of chemical dyes from hematoxylin and eosin (H&E) stained tissue slides (Fig. 3a). Four cell types in colon cancer tissue slice with 5 mm2 in area and 10 μm in thickness were collected by LCM and profiled by SISPROT-based proteomics with up to 5000 proteins identified. Furthermore, a proof-of-concept three-dimensional spatial and cell-type-resolved proteomic map of human colon tumor was generated.
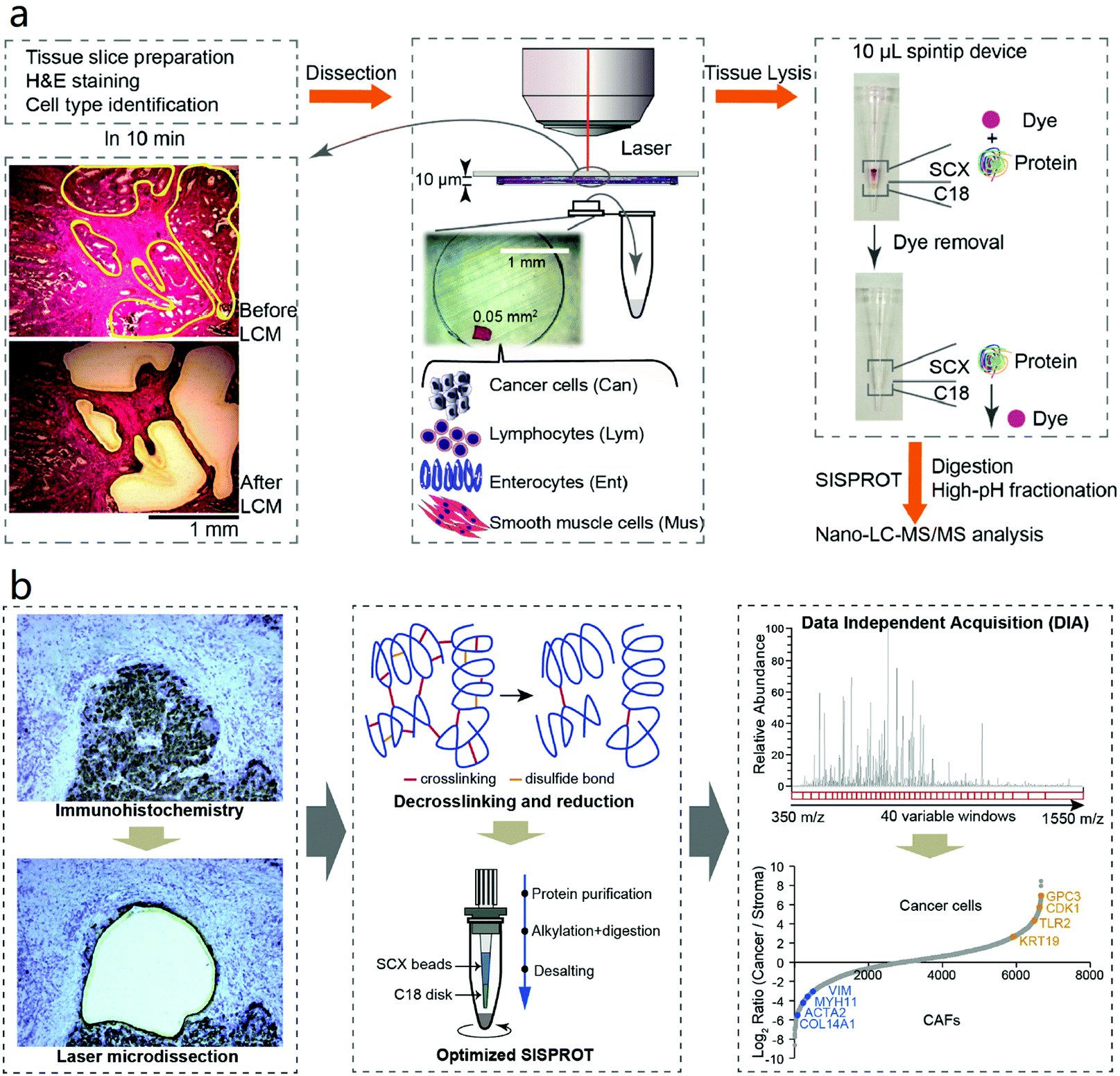 | ||
| Fig. 3 Workflows of (a) LCM-SISPROT and (b) IHC-SISPROT for spatial-resolution cell type proteome profiling of tumor microenvironment. Tissue sections were stained by H&E or IHC to discriminate different cell types which were further isolated by LCM and processed by SISPROT. Reproduced from ref. 83 and 130 with permission from the American Chemical Society, copyright 2018 and Elsevier, copyright 2020. | ||
Apart from developing novel devices, studies have been undertaken to optimize the performance of the conventional in-solution workflow. For example, Davis et al. recently tested different combinations of sample collection methods, lysis buffers, and digestion methods to maximize the proteome coverage with small-size tissue sample.85 In their optimized workflow, the LMD collection tube cap was pre-filled with trifluoroethanol (TFE) lysis buffer so that the tissue pieces can be directly lysed without the risk of sample loss during tube inverting and centrifugation. This in situ method showed good sensitivity for identifying 1500 protein groups from human brain tissue of 0.06 mm2 in area and 10 μm in thickness by using an Orbitrap Fusion Lumos MS.
The two examples above represent high sensitivity of sample preparation by manual operation on easy-to-use platforms while sophisticatedly designed microfluidic chip system, such as the nanodroplet processing in one pot for trace samples (nanoPOTS) platform, was also applied for processing LCM tissue samples.86 The original design of nanoPOTS has a glass chip fabricated with hydrophilic reaction vessels surrounded by a hydrophobic surface. Inside each vessel, several to hundreds of cells can be lysed and digested in nanodroplet before the peptides are concentrated and stored in a solid phase extraction (SPE) column for online desalting. For proof of concept, nanoPOTS was applied to profile the spatial proteomes of single human islet section and tomato tissue pieces.87 Later on, the nanoPOTS platform was further optimized by pre-populating dimethyl sulfoxide (DMSO) in the reaction vessels to improve tissue capture efficiency.88 Very recently, a combinatory strategy was reported by integrating LCM-nanoPOTS with an image processing software called Trelliscope which is capable to correlate the histological image with spatial proteomic datasets.89 In this report, “proteome imaging” was performed to investigate the tissue microenvironment in mouse uterus with ∼2000 protein groups identified from each “tissue voxel” of 0.01 mm2 in area and 12 μm in thickness on a modified Q Exactive Plus MS system (Fig. 4). Although the LCM-nanoPOTS system has shown extraordinary sensitivity, on the other hand, the nanowell design may limit its performance in terms of potential contamination by pre-populating organic solvent and sample loss by hydrophobic surface and off-line sample transfer to SPE column. Also, in-solution protocols are incapable of removing chemical dyes from H&E staining which have been proven detrimental to MS-based peptide identification.90 In addition to the approaches mentioned above, several other sample preparation technologies including immobilized enzyme reactor (IMER)91 and liquid handler-based processing platform92 have also been developed and applied for LMD-based spatial proteomics with fresh-frozen tissue sections in recent years.
 | ||
| Fig. 4 Workflow of the nanoPOTS imaging platform for high-throughput and spatially resolved proteomics. For proof of concept, “tissue voxels” of 0.01 mm2 in area were processed in nanodroplets and resulted proteome profiles were reconstructed as a protein map. Reproduced from ref. 89 with permission from the Springer Nature, copyright 2020. | ||
Despite the advantages of fresh-frozen tissue for MS-based proteomics, the preservation of frozen tissue or cryosection is less preferred for long-term low-temperature storage. On the other hand, poor rigidity of morphology is generally found in fresh-frozen tissue due to the formation of ice crystals during the freezing process.93 Therefore, formalin-fixed paraffin-embedded (FFPE) tissue specimens are more common as archives in clinical practice for more than a century.94 Clinical information, which is critical for linking proteomics data with corresponding clinical outcomes, is well documented for FFPE samples. Formalin (aqueous solution of formaldehyde) can form inter-molecular cross-links between proteins and intra-molecular chemical modifications with high reactivity and permeability, thus effectively preserve protein structure and tissue morphology in an ambient condition for decades.95 To make better use of the FFPE specimens in archives worldwide, numerous protocols have been developed to extract the protein contents in formalin-fixed tissues.96 Although the mechanism is not fully elucidated so far, protein fixation with formalin has been proven partially reversible in various extraction buffers (e.g., 6 M guanidine hydrochloride,97 300 mM Tris-HCl,98 1 M hydroxylamine hydrochloride,99 4% SDS,90 and 50% TFE100) and extraction conditions (e.g., heating,101 high pressure,102 and ultrasonication103). In an attempt to apply LMD-based spatial proteomics to FFPE tissue specimens, efficient protein extraction protocols in combination with integrated sample preparation technologies have been reported in recent years.
The combination of pressure cycling technology and sequential window acquisition of all theoretical fragment ion spectra-MS (PCT-SWATH) was first reported by the Aebersold group in 2015 attempting to convert tissue biopsy samples into “digital proteome maps” in an integrative and high-throughput fashion.104 Different from other protocols, the PCT method employs cycles between ultra-high and ambient pressure which enables in situ tissue homogenization, protein extraction, and tryptic digestion in about three hours.105 Since detergents are not required in the PCT workflow, the digested peptides can be directly injected for SWATH-MS analysis after C18 SPE desalting.106 In 2019, Zhu et al. integrated the acidic and alkaline hydrolysis steps into the PCT-SWATH workflow to process FFPE biopsies efficiently.102 Very recently, the FFPE-PCT protocol was well applied to profile the proteomic characteristics in different organs from COVID-19 patients.107 Because the PCT-SWATH method is designed for a few hundreds to thousands of micrograms of starting materials,108 further optimization might be adopted to facilitate its use in spatial proteomics by increasing the sensitivity.
Filter-aided sample preparation (FASP) is a widely-used proteomics sample preparation technology to remove low-molecular-weight detergent from tens of microgram of protein lysates and perform protein digestion on the filter.109,110 In order to increase the sensitivity for analyzing LMD-collected FFPE tissue samples, the FASP method was improved by adding 0.5% polyethylene glycol (PEG) in cell lysates as a carrier substance to reduce sample loss from nonspecific adsorption.111 In combination with SDS-based de-crosslinking/protein extraction and multi-enzymatic digestion strategy, as many as ∼10000 protein groups were surprisingly identified from 110 nL microdissected FFPE human colonic adenoma tissue sample using SAX-StageTip-based fractionation and a Q-Exactive MS.112 Later on, the FFPE-FASP protocol was applied to profile the proteomes of normal area, cancer cells, and metastases isolated from FFPE colon cancer tissue by LMD with about 8000–10000 protein groups quantified and the proteome remodeling phenomenon was investigated between different cell types.113,114 From the results above, the advantage for addition of PEG to reduce sample loss is obvious, however, PEG is also well-known as a detrimental contaminant in LC-MS analysis.
Single-pot, solid phase-enhanced sample-preparation (SP3) technology was developed by Hughes et al. in 2014 and became a popular proteomics sample clean-up approach due to its ease of use and good compatibility.115–117 The SP3 enables organic solvent precipitation-based physical capture of proteins and removal of interference components in lysis buffer using paramagnetic beads.118 In 2016, Hughes et al. reported a protocol termed SP3-Clinical Tissue Proteomics (SP3-CTP) by combining SDS-based decrosslinking/protein extraction, SP3 protein processing, tandem mass tags (TMT) labeling, HpRP fractionation, and MS3-based quantification.119 Buczak et al.120 and Griesser et al.90 further optimized and applied the SP3-CTP protocol to LMD-based spatial proteomics, respectively. In these two technical notes, similar performance was achieved with ∼5000 protein groups quantified from microgram-level starting materials by Orbitrap Fusion Lumos MS.
Although TMT-based isobaric labeling strategy is much preferred to increase the quantification precision and sensitivity in a lot of proteogenomic studies,121,122 potential drawbacks of TMT such as additional steps of sample preparation and inaccuracy (or ratio compression) caused by co-elution of peptides from different TMT channels123 have hindered its wider application in LMD-based spatial proteomics.124 To this end, several other MS data acquisition strategies such as data-independent acquisition (DIA) combined with integrated sample preparation technologies have been developed to increase the overall performance for analyzing LMD-collected FFPE tissue sample towards clinical needs. For example, the Mann group described a 96-well-based in-solution proteomics sample preparation protocol for performing DIA-based MS analysis of large FFPE tissue cohorts from histopathology glass slides or LMD-collected samples.100 Due to the large amount of FFPE specimen storage worldwide, high-throughput sample processing platforms with single-shot DIA analysis provide a good opportunity for broader applications of LMD-based spatial proteomics.
3.2 In-depth spatial PTM profiling in tissue section
In addition to the expression level of the whole proteome, it is ideal to measure the protein PTM at the same time. Indeed, MS-based analysis of important PTMs such as phosphorylation, glycosylation, and ubiquitylation has now become a widely-used technique via robust PTM protein/peptide enrichment strategies.125 Typically, at least hundreds of micrograms of protein lysates are required to perform an in-depth PTM profiling by MS because of the low stoichiometry of major PTMs in normal cell lines or tissue samples. To this end, global profiling of PTM in tissue section faces great challenges of the overall sensitivity. Up to now, there are very few reports for LMD-based spatial and in-depth PTM profiling in tissue sections. Under this circumstance, we and others set out to develop novel MS-based PTM profiling strategies with higher sensitivity for limited starting material such as LMD-collected tissue pieces.Protein glycosylation mediates various cellular processes including cell-cell communications, immune response, cell adhesion and so on.126 It is attractive to investigate the spatial distribution of glycome in clinical tissue specimens. In 2017, Hinneburg et al. reported a sensitive and glycan isomer-selective method for simultaneous N- and O-glycome profiling from FFPE tissue sections by immobilizing glycoproteins on PVDF membrane, releasing the N-glycan and O-glycan sequentially, and analyzing with porous graphitized carbon LC-MS.127 For proof of concept, N- and O-glycan profiles were determined from 2000 cells isolated from FFPE human liver tissue sections by LMD and significant global glycome changes were observed in comparison between tumor and non-tumor areas.
One major challenge to enrich glycoproteins from limited starting material is low glycoprotein recovery due to multiple sample transfer steps in conventional methods. To address this challenge, our group developed a fully integrated spintip-based glycoproteomic approach (FISGlyco) which integrated the glycoproteomic sample preparation workflow including glycoprotein enrichment, digestion, deglycosylation, and desalting in a spintip device.128 Taking advantage of the C18 matrix as a “switchable control” by SDS,129 the FISGlyco method greatly reduced processing time and sample loss resulted from unspecific adsorption by performing all the steps in a single tip (Fig. 5). For sensitivity assessment, ∼600 N-glycosylation sites were quantified from as low as 5 μg of mouse brain protein lysates. Moreover, the FISGlyco method was applied for LMD-based spatial glycoproteomics profiling to establish the first spatial-resolved N-glycoproteome profiling of four mouse brain regions with over 1000 N-glycosites identified using fresh-frozen tissue sections. For future perspective, sensitive technologies to profile other PTMs such as phosphorylation can also be possible for dedicatedly optimizing specific protocols for LMD samples.
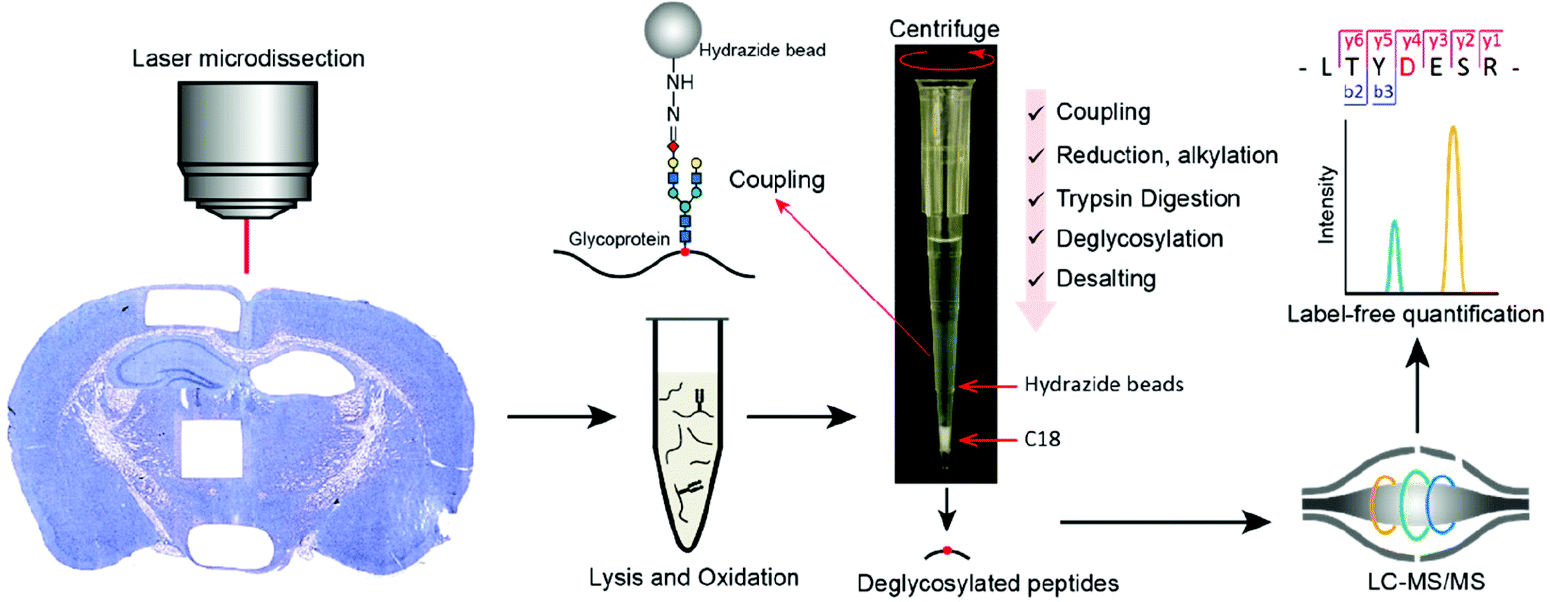 | ||
| Fig. 5 Workflow of the FISGlyco for spatially resolved identification and label-free quantification of N-linked glycoproteome in mouse brain. In FISGlyco, glycoprotein enrichment, digestion, deglycosylation, and desalting were integrated in the same spintip device. Reproduced from ref. 128 with permission from the American Chemical Society, copyright 2019. | ||
3.3 Annotation methods for LMD-based proteomics
For LMD-based proteomics, prior knowledge of molecular patterns or cell type distribution in tissue is mandatory to select ROI for downstream analysis. Often, it is evaluated using H&E by experienced pathologists. This strategy works well in region-resolved spatial proteomics studies mentioned above, whereas it is time-consuming and potentially imprecise for single-cell-resolved spatial analysis of large-area slides. Despite that IHC can provide more accurate spatial information about specific cell type by antibody recognition, most of the LMD-based spatial proteomics studies mentioned above used H&E-stained slides which is mainly because the high-abundance antibodies incubated on the tissue sections could interfere the proteomic analysis especially for low-input study. In order to make use of the easy-to-use IHC assay as an accurate guidance for LMD-based spatial proteomics, our group recently established the IHC-SISPROT approach for cell-type resolved proteome analysis of formalin-fixed tissue sample.130 Three major improvements were made by IHC-SISPROT compared to previous LCM-SISPROT technology: (1) IHC instead of H&E staining was used to define the sampling area for LMD to discriminate different cell types with higher accuracy by antibody recognition; (2) the SISPROT buffer was confirmed to enable efficient de-crosslinking and protein extraction by heating; and (3) superior sensitivity and reproducibility was achieved by using single-shot DIA analysis to alleviate the interference by antibodies. Using a Q Exactive HF-X MS, over 3500 protein groups were identified from formalin-fixed human liver cancer tissue section of 0.2 mm2 in area and 12 μm in thickness. Furthermore, cell type-specific ligands, receptors, and potential-novel cell–cell communication events were identified by analyzing liver cancer cells and CAFs enriched by LMD (Fig. 3b).In addition to IHC, two annotation methods including MALDI-imaging and artificial intelligence (AI)-image recognition have been also developed aiming to assist manual evaluation for higher precision and throughput in LMD-based spatial proteomics. MSI enables profiling the spatial distribution of a diversity of analytes directly in biological tissues in a label-free manner. Among MSI technologies with different ion sources, MALDI-MSI has become a widely-used tool to characterize the tissue morphology at molecular scale as a complementary dimension to histology.53 Using this feature, a MALDI-MSI-guided LMD-based spatial proteomics workflow was designed, in which MALDI-MS images of peptide distribution were used to defined and isolate the ROI by the LMD system. Recently, MALDI-MSI and LMD-based spatial proteomics were combined to study the FFPE human breast cancer tissue sections with a good correlation found between the datasets derived from two methods.131 In 2019, Dewez et al. systematically assessed the accuracy of co-registration of spatial annotation provided by MSI in the LMD system with error controlled less than 13 μm.132 In order to further improve the performance and robustness of MSI-guided LMD-based spatial proteomics, the same group recently explore the feasibility of performing the MSI and in-depth LC-MS analysis on the same timsTOF-fleX instrument equipped with a hybrid source.133 The MALDI source allowed rapid MSI scanning and the electron spray ionization (ESI) source enabled high-sensitivity MS/MS acquisitions. With the state-of-art MS platform, ∼2000 protein groups were quantified from tissue segments comprising 2000 cells isolated by LMD.
Artificial intelligence (AI) and machine learning tools have been widely used in digital pathology including cell type annotation in the last decade.134 In 2018, Brasko et al. established a computer-assisted microscopy isolation (CAMI) pipeline to recognize individual cells in tissue section and automatically guide LMD-based cell isolation by combining high-throughput microscopy, image analysis algorithms (to identify and segment cells), and machine learning software (to recognize cell phenotypes).135 The CAMI pipeline was then successfully applied to single-cell genome/transcriptome profiling. Very recently, the Mann group reported a “Deep Visual Proteomics (DVP)” strategy,136 which integrated a novel image analysis software termed BIAS with high-performance commercial instrumental platforms including Zeiss Axio Scan.Z1 microscope for high-resolution whole slide scanning, Leica LMD7 system for automated and precise single cell isolation, as well as Bruker timsTOF Pro in diaPASEF mode137 for high-throughput proteome profiling. For proof of concept, the DVP strategy was applied to investigate cellular heterogeneity in FFPE tissue sample of salivary gland carcinoma.
3.4 LMD-based proteomic applications
LMD has been employed in numerous studies since robust LMD systems and protocols were available. For example, Han et al. identified several cell-type-specific proteins and potential therapeutic targets in active multiple sclerosis lesions isolated by LMD and analyzed by MS-based proteomics in 2008.138 The Mann group developed a sensitive chromatographic system and identified ∼2400 protein groups from kidney glomeruli isolated by LMD in 2009.139 Valleix et al. profiled the proteome of amyloid fibrils extracted from cardiac and hepatic FFPE specimens by LMD in 2012.140 However, because of technical limitations, most of these early attempts of LMD-based spatial- or cell-type-resolved proteomics did not achieve a deep proteome coverage.With the advances of methodology and instrumentation for MS-based proteomics, we have seen an increasing number of high-quality biomedical studies employing LMD-based spatial proteomics for investigating the TME, especially in the recent five years. In 2016, Marakalala et al. analyzed the proteomes of five regions of granulomas sampling from human tuberculosis lung specimens by LMD in order to decipher the function and formation mechanism of granulomas.141 With a depth of ∼4400 protein groups identified in total, a spatially organized inflammatory signaling driven by functional proteins was found by spatial proteomic analysis and further validated by high-resolution MALDI-MSI and IHC. In 2018, Carnielli et al. identified differentially expressed proteins in six regions isolated from FFPE human oral squamous cell carcinoma tissues by LMD.142 Using machine learning-based bioinformatic analysis, potential prognostic signatures were proposed and further validated by IHC and SRM assay. In 2019, Eckert et al. isolated tumor and stromal compartments from in situ and metastatic lesions of human ovarian cancer samples by LMD.143 Using a sensitive and label-free workflow, ∼7000 protein groups were quantified from the four types of region. By comparative analysis, an extensive expression of N-methyltransferase (NNMT) and its related downstream proteins were found in the metastasis-associated stromal compartments. Moreover, NNMT was validated as a key regulator of CAFs in ovarian cancer. Apart from the aforesaid works, LMD was also applied to MS-based spatial proteomic studies of several other lethal diseases such as pancreatic ductal adenocarcinoma (PDAC),144,145 hepatocellular carcinoma (HCC),146 Alzheimer's disease (AD),147 Krabbe disease,148 and mitochondrial myopathy.149
4. Antibody recognition-based multiplexed tissue imaging
Owing to technical limitations, compromise exists between spatial resolution and identification depth for the development of spatial proteomics methodology at present (Fig. 1). In addition to abovementioned MSI-based spatial proteomics technology, several other modalities of multiplexed tissue imaging platforms with high sensitivity have also emerged in the past decade. All of these methods rely on available antibodies, which can specifically target proteins of interest in biological contexts. Essentially, these antibody recognition-based approaches are highly multiplexed versions of IHC staining. Aiming to ensure both the spatial resolution and multiplexed protein measurement, these approaches tend to fall into two classes of sequential or simultaneous analyses based on different instrumentations including immunofluorescence (IF) microscopy,150 Raman microscopy,151 mass cytometry,152 and NGS.153 In this section, we briefly introduce two representative platforms from each class.4.1 IF-based sequential tissue imaging
IF-based tissue imaging is a routine technique for in situ identification of single or several target proteins by chemical fluorophore-conjugated antibodies with high resolution and streamlined procedure.16 As an impressive example, Thul et al. recently established a “subcellular map of the human proteome” by applying 13993 antibodies to 22 human cell lines and created a huge resource of IF images showing the subcellular localization of 12003 proteins.154 Due to emission spectra overlap, conventional IF can only measure up to seven targets in one experiment simultaneously.155 To this end, a simple pipeline of iterative tissue staining, IF imaging, and antibody removal has been proposed to enhance the multiplexing capability of IF-based tissue imaging.156 Yet, several factors limit the performance of this pipeline including decay in antigenicity influenced by long hours of tissue processing in harsh conditions as well as high background resulting from incomplete fluorescence quenching/antibody removal. Aiming for the best fluorescent performance, different signal quenching methods have been proposed including photobleaching of fluorophore by strong excitation,156,157 chemical inactivation of fluorescent dyes/proteins by oxidation,158–160 using fluorescent antibody with cleavable linker,161,162 and stripping secondary antibody by optimized buffers.150,163 In another hand, several approaches for protecting the physicochemical properties of tissue section during serial staining were also developed such as system-wide control of interaction time and kinetics of chemicals (SWITCH) by multifunctional fixatives164 and stabilization under harsh conditions via intramolecular epoxide linkages to prevent degradation (SHIELD) by flexible poly-epoxides.165
Along with the efforts mentioned above, great progress contributed by IF-based multiplexed tissue imaging methods have been reported to characterize the TME in human samples. For a recent example, Gut et al. designed a 40-plex iterative indirect IF imaging (4i) workflow in an automated and high-throughput manner by using an optimized antibody stripping buffer.150 By measuring the localization and abundance of targeted proteins in cultured cells and human tissues, different biological processes including subcellular reorganization upon pharmacological perturbations were visualized and investigated.
4.2 Mass cytometry-based tissue imaging
Two mass cytometry-centric technologies were developed in 2014 and considered as the “next-generation IHC” for highly multiplexed tissue imaging.166 The platform of mass cytometry, also known as cytometry by time-of-flight (CyTOF), is based on an atomic MS which enables discrimination of element isotopes with high accuracy introduced by Bandura et al. in 2009.167 Notably, similar concept of indirect detection of biomolecules by ICP-MS was proposed by Zhang et al. early in 2001.168 In 2011, the Nolan group optimized this platform and applied it to single-cell analysis of human hematopoietic continuum by measuring 31 target proteins simultaneously.169 When performing a CyTOF experiment, the cell suspension is incubated with a cocktail of antibodies conjugated to chelated metal isotopes, and then injected to atomic MS for mass-to-charge measurement of metal ion which serves as an indirect proxy for the expression level of the targeted protein.170The CyTOF platform was developed as a MS-based highly multiplexed version of FACS, similarly, the same idea was soon introduced to tissue analysis. In 2014, the Nolan group and the Bodenmiller group reported two versions of mass cytometry-based tissue imaging platforms, named imaging mass cytometry (IMC) and multiplexed ion beam imaging (MIBI) with similar design and complementary features.171,172 These two platforms utilize laser or ion beam to rasterize the tissue section and generate a stream of particles which are analyzed by atomic MS in sequence, then the mass-to-charge datasets of all raster positions are re-constructed and visualized as two-dimensional images matching the tissue morphology and showing the localization and relative abundance of targeted proteins. In IMC, a UV laser ablation system is coupled to an inductively coupled plasma (ICP)-MS with pixel resolution of ∼1 μm. In MIBI, an ion beam system is coupled to a secondary ion (SI)-MS with pixel resolution of ∼200 nm.
In practice, 30–50 targeted proteins can be measured at the same time using commercialized platforms of IMC (http://www.fluidigm.com/applications/imaging-mass-cytometry). Mass cytometry-based tissue imaging should outperforms IF imaging in several ways, including lower background signal, better quantification performance, and higher robustness.152,173 Taking advantage of this powerful technology, tissue microenvironments of different human samples, including breast tumors,174–176 diseased pancreas,177,178 and squamous cell tumor179 have been characterized by IMC and analyzed by novel multiscale analysis algorithms.180,181 Recently, the Bodenmiller group described a single-cell pathology landscape of breast cancer by simultaneously quantifying 35 biomarkers using IMC with hundreds of highly multiplexed tissue images produced from breast tumor samples.176 On the basis of this high-quality resource, the TME of breast tumor including cellular phenotypes, organization, and heterogeneity were characterized and applied to predict the clinical outcomes (Fig. 6).
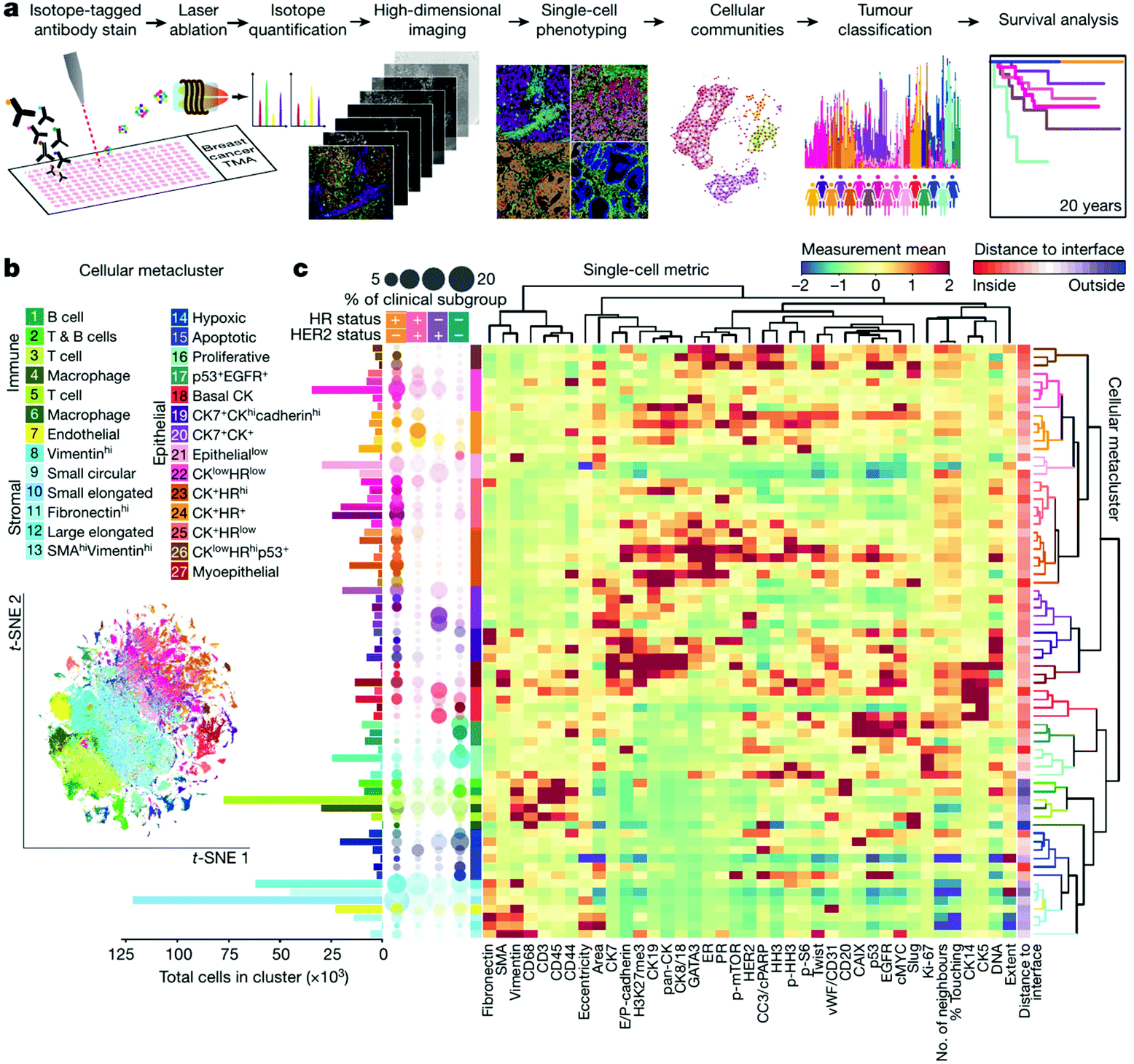 | ||
| Fig. 6 High-dimensional histopathology of breast cancer by imaging mass cytometry. (a) Workflow of acquisition and analysis of single-cell phenotypes in tissue sections by IMC. (b) t-Distributed stochastic neighbor embedding (t-SNE) map showing clusters of twenty-seven cell types identified. (c) Heat map showing the marker expression or distance to tumor-stroma interface of each cluster. In the left, the bar plot shows the cell counts of each cluster and the bubble plot shows the proportion of clusters in clinical subtypes. Reproduced from ref. 176 with permission from the Springer Nature, copyright 2020. | ||
5. Challenges and future perspective
In the ideal scenario, genome, epigenome, transcriptome, proteome, metabolome, and all the other important histological and cellular features could be measured simultaneously with subcellular resolution to achieve a complete characterization and understanding of the tissue microenvironment. Even though we are far away from this ultimate goal for now, substantial efforts have been made in the development of analytical methods and instrumentations. When it comes to spatial proteomics discussed in this review, we have seen great progress but also challenges especially in terms of methodologies. Herein, we highlight the remaining challenges as well as the potential future directions as follows.5.1 Importance of standard operating procedure (SOP)
The ischemia process starts in the tissue right after its removal from the human body. Absent from blood supply, changes will happen to molecular features such as protein phosphorylation within cells which can mask the actual status of TME in the tissue. More importantly, varied preservation protocols in different hospitals or laboratories could further influence the final results. For example, formaldehyde in aqueous solution diffuse into the bulk tissue at a rate of ∼1 mm per hour, which could lead to a crosslinking gradient from surface to core, and this phenomenon is more obvious for large specimens. Fresh-frozen is another solution, however, the ice crystal issue and varied cryosectioning protocol would also lead to technical variability for morphology and high-resolution spatial omics. Therefore, a meticulous SOP should be tested and finalized before any spatial proteomics experiments.5.2 Higher sensitivity for single-cell analysis
Underlying reasons for the slower pace of spatial proteomics compared to spatial genomics/transcriptomics include the complexity of proteome and lacking an approach to amplify proteins or peptides in vitro in a way like PCR. In spite of the emergence of other protein sequencing technologies such as the nanopore in recent years,182,183 mass spectrometry is still an indispensable and centric platform for global proteome profiling. Owing to the progress in sample preparation and instrumentation, single-cell proteomics has become possible on ultra-high sensitive platforms in several laboratories.19,184,185 For the next step, attention should be paid to increase the robustness and throughput of these platforms such as the nanoliter-scale oil-air-droplet (OAD) chip186 and extend their use to MS-based spatial proteomics. To this end, the LCM-nanoPOTS89 as well as the DVP platform136 have set good examples. For the next five years, we are looking forward to see comparable performance to spatial transcriptomics would be achieved by MS-based spatial proteomics.5.3 Opportunities for spatial multi-omics
Simultaneous measurement of mRNA and protein in the same tissue area has been achieved by several targeted approaches including DNA-PAINT,187 REAP-seq,188 CITE-seq,189 CODEX,190 Immuno-SABER,191 DBiT-seq,153 and Digital Spatial Profiling192 sharing similar designs by using DNA-barcoded or oligonucleotide-labeled antibodies to obtain both the abundance and localization information of certain epitope with high spatial resolution. For future perspective, we consider that more efforts should be put into developing new technologies for unbiased profiling the global proteome and PTM information in a small area of tissue section at the same time, because changes of PTM, such as phosphorylation and glycosylation, are more correlated to functional cellular events.193 Consequently, efficient and sensitive PTM protein/peptide enrichment technologies should be developed and integrated into current spatial proteomics workflow.5.4 New chemical biological technologies for spatial proteomics
Up to now, the realization of spatial resolution mostly relies on using physical dissection or antibody recognition. We believe that progress in other fields such as chemical biology also has the potential to promote MS-based spatial proteomics. For instance, the spatially targeted optical micro-proteomics (STOMP) technology was developed by Hadley et al.194 in 2015 and automatized recently.195 The STOMP utilized a photo-affinity chemical probe which covalently tags neighboring proteins in a diameter of 0.67 μm under two-photon excitation. This feature leads to the possibility to label and enrich the proteins within selected area of the tissue section. Inspired by this example, we are looking forward that other powerful chemical biological tools could also benefit the understanding of tissue microenvironment.196Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by funding from China State Key Basic Research Program Grants (2020YFE0202200, 2016YFA0501403, and 2016YFA0501404), National Natural Science Foundation of China (91953118), Guangdong Provincial Fund for Distinguished Young Scholars (2019B151502050), Guangdong Provincial Natural Science Grant (2016A030312016), Shenzhen Innovation of Science and Technology Commission (JCYJ20200109141212325 and JCYJ20170412154126026).References
- B. C. Hu, The human body at cellular resolution: the NIH Human Biomolecular Atlas Program, Nature, 2019, 574(7777), 187–192 CrossRef PubMed.
- O. Rozenblatt-Rosen, A. Regev, P. Oberdoerffer, T. Nawy, A. Hupalowska, J. E. Rood, O. Ashenberg, E. Cerami, R. J. Coffey, E. Demir, L. Ding, E. D. Esplin, J. M. Ford, J. Goecks, S. Ghosh, J. W. Gray, J. Guinney, S. E. Hanlon, S. K. Hughes, E. S. Hwang, C. A. Iacobuzio-Donahue, J. Jane-Valbuena, B. E. Johnson, K. S. Lau, T. Lively, S. A. Mazzilli, D. Pe’er, S. Santagata, A. K. Shalek, D. Schapiro, M. P. Snyder, P. K. Sorger, A. E. Spira, S. Srivastava, K. Tan, R. B. West, E. H. Williams and Human Tumor Atlas Network, The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution, Cell, 2020, 181(2), 236–249 CrossRef CAS PubMed.
- A. Marusyk, V. Almendro and K. Polyak, Intra-tumour heterogeneity: a looking glass for cancer?, Nat. Rev. Cancer, 2012, 12(5), 323–334 CrossRef CAS PubMed.
- S. H. Gohil, J. B. Iorgulescu, D. A. Braun, D. B. Keskin and K. J. Livak, Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy, Nat. Rev. Clin. Oncol., 2021, 18, 244–256 CrossRef PubMed.
- W. H. Fridman, F. Pages, C. Sautes-Fridman and J. Galon, The immune contexture in human tumours: impact on clinical outcome, Nat. Rev. Cancer, 2012, 12(4), 298–306 CrossRef CAS PubMed.
- N. McGranahan and C. Swanton, Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future, Cell, 2017, 168(4), 613–628 CrossRef CAS PubMed.
- D. F. Quail and J. A. Joyce, Microenvironmental regulation of tumor progression and metastasis, Nat. Med., 2013, 19(11), 1423–1437 CrossRef CAS PubMed.
- J. Hausser and U. Alon, Tumour heterogeneity and the evolutionary trade-offs of cancer, Nat. Rev. Cancer, 2020, 20(4), 247–257 CrossRef CAS PubMed.
- D. A. Lawson, K. Kessenbrock, R. T. Davis, N. Pervolarakis and Z. Werb, Tumour heterogeneity and metastasis at single-cell resolution, Nat. Cell Biol., 2018, 20(12), 1349–1360 CrossRef CAS PubMed.
- P. L. Bedard, A. R. Hansen, M. J. Ratain and L. L. Siu, Tumour heterogeneity in the clinic, Nature, 2013, 501(7467), 355–364 CrossRef CAS PubMed.
- I. Dagogo-Jack and A. T. Shaw, Tumour heterogeneity and resistance to cancer therapies, Nat. Rev. Clin. Oncol., 2018, 15(2), 81–94 CrossRef CAS PubMed.
- M. Binnewies, E. W. Roberts, K. Kersten, V. Chan, D. F. Fearon, M. Merad, L. M. Coussens, D. I. Gabrilovich, S. Ostrand-Rosenberg, C. C. Hedrick, R. H. Vonderheide, M. J. Pittet, R. K. Jain, W. Zou, T. K. Howcroft, E. C. Woodhouse, R. A. Weinberg and M. F. Krummel, Understanding the tumor immune microenvironment (TIME) for effective therapy, Nat. Med., 2018, 24(5), 541–550 CrossRef CAS PubMed.
- M. R. Junttila and F. J. de Sauvage, Influence of tumour micro-environment heterogeneity on therapeutic response, Nature, 2013, 501(7467), 346–354 CrossRef CAS PubMed.
- N. Zahir, R. Sun, D. Gallahan, R. A. Gatenby and C. Curtis, Characterizing the ecological and evolutionary dynamics of cancer, Nat. Genet., 2020, 52(8), 759–767 CrossRef CAS PubMed.
- W. C. Hahn, J. S. Bader, T. P. Braun, A. Califano, P. A. Clemons, B. J. Druker, A. J. Ewald, H. Fu, S. Jagu, C. J. Kemp, W. Kim, C. J. Kuo, M. McManus, G. B. Mills, X. Mo, N. Sahni, S. L. Schreiber, J. A. Talamas, P. Tamayo, J. W. Tyner, B. K. Wagner, W. A. Weiss, D. S. Gerhard and Cancer Target Discovery and Development Network, An expanded universe of cancer targets, Cell, 2021, 184(5), 1142–1155 CrossRef CAS PubMed.
- D. P. Bishop, N. Cole, T. Zhang, P. A. Doble and D. J. Hare, A guide to integrating immunohistochemistry and chemical imaging, Chem. Soc. Rev., 2018, 47(11), 3770–3787 RSC.
- L. Cohen and D. R. Walt, Highly Sensitive and Multiplexed Protein Measurements, Chem. Rev., 2019, 119(1), 293–321 CrossRef CAS PubMed.
- T. Stuart and R. Satija, Integrative single-cell analysis, Nat. Rev. Genet., 2019, 20(5), 257–272 CrossRef CAS PubMed.
- M. Labib and S. O. Kelley, Single-cell analysis targeting the proteome, Nat. Rev. Chem., 2020, 4(3), 143–158 CrossRef.
- J. Lamanna, E. Y. Scott, H. S. Edwards, M. D. Chamberlain, M. D. M. Dryden, J. Peng, B. Mair, A. Lee, C. Chan, A. A. Sklavounos, A. Heffernan, F. Abbas, C. Lam, M. E. Olson, J. Moffat and A. R. Wheeler, Digital microfluidic isolation of single cells for -Omics, Nat. Commun., 2020, 11(1), 5632 CrossRef CAS PubMed.
- C. Larsson, I. Grundberg, O. Soderberg and M. Nilsson, In situ detection and genotyping of individual mRNA molecules, Nat. Methods, 2010, 7(5), 395–397 CrossRef CAS PubMed.
- R. Ke, M. Mignardi, A. Pacureanu, J. Svedlund, J. Botling, C. Wahlby and M. Nilsson, In situ sequencing for RNA analysis in preserved tissue and cells, Nat. Methods, 2013, 10(9), 857–860 CrossRef CAS PubMed.
- J. H. Lee, E. R. Daugharthy, J. Scheiman, R. Kalhor, J. L. Yang, T. C. Ferrante, R. Terry, S. S. Jeanty, C. Li, R. Amamoto, D. T. Peters, B. M. Turczyk, A. H. Marblestone, S. A. Inverso, A. Bernard, P. Mali, X. Rios, J. Aach and G. M. Church, Highly multiplexed subcellular RNA sequencing in situ, Science, 2014, 343(6177), 1360–1363 CrossRef CAS PubMed.
- K. H. Chen, A. N. Boettiger, J. R. Moffitt, S. Wang and X. Zhuang, RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells, Science, 2015, 348(6233), aaa6090 CrossRef PubMed.
- R. Moncada, D. Barkley, F. Wagner, M. Chiodin, J. C. Devlin, M. Baron, C. H. Hajdu, D. M. Simeone and I. Yanai, Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas, Nat. Biotechnol., 2020, 38(3), 333–342 CrossRef CAS PubMed.
- P. L. Stahl, F. Salmen, S. Vickovic, A. Lundmark, J. F. Navarro, J. Magnusson, S. Giacomello, M. Asp, J. O. Westholm, M. Huss, A. Mollbrink, S. Linnarsson, S. Codeluppi, A. Borg, F. Ponten, P. I. Costea, P. Sahlen, J. Mulder, O. Bergmann, J. Lundeberg and J. Frisen, Visualization and analysis of gene expression in tissue sections by spatial transcriptomics, Science, 2016, 353(6294), 78–82 CrossRef CAS PubMed.
- N. Crosetto, M. Bienko and A. van Oudenaarden, Spatially resolved transcriptomics and beyond, Nat. Rev. Genet., 2015, 16(1), 57–66 CrossRef CAS PubMed.
- E. Lein, L. E. Borm and S. Linnarsson, The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing, Science, 2017, 358(6359), 64–69 CrossRef CAS PubMed.
- L. Larsson, J. Frisen and J. Lundeberg, Spatially resolved transcriptomics adds a new dimension to genomics, Nat. Methods, 2021, 18(1), 15–18 CrossRef CAS PubMed.
- X. Zhuang, Spatially resolved single-cell genomics and transcriptomics by imaging, Nat. Methods, 2021, 18(1), 18–22 CrossRef CAS PubMed.
- Y. Taniguchi, P. J. Choi, G. W. Li, H. Chen, M. Babu, J. Hearn, A. Emili and X. S. Xie, Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells, Science, 2010, 329(5991), 533–538 CrossRef CAS PubMed.
- B. C. Carlyle, R. R. Kitchen, J. E. Kanyo, E. Z. Voss, M. Pletikos, A. M. M. Sousa, T. T. Lam, M. B. Gerstein, N. Sestan and A. C. Nairn, A multiregional proteomic survey of the postnatal human brain, Nat. Neurosci., 2017, 20(12), 1787–1795 CrossRef CAS PubMed.
- J. Wang, Z. Ma, S. A. Carr, P. Mertins, H. Zhang, Z. Zhang, D. W. Chan, M. J. Ellis, R. R. Townsend, R. D. Smith, J. E. McDermott, X. Chen, A. G. Paulovich, E. S. Boja, M. Mesri, C. R. Kinsinger, H. Rodriguez, K. D. Rodland, D. C. Liebler and B. Zhang, Proteome Profiling Outperforms Transcriptome Profiling for Coexpression Based Gene Function Prediction, Mol. Cell. Proteomics, 2017, 16(1), 121–134 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Mass-spectrometric exploration of proteome structure and function, Nature, 2016, 537(7620), 347–355 CrossRef CAS PubMed.
- Y. Zhang, B. R. Fonslow, B. Shan, M. C. Baek and J. R. Yates 3rd, Protein analysis by shotgun/bottom-up proteomics, Chem. Rev., 2013, 113(4), 2343–2394 CrossRef CAS PubMed.
- M. Larance and A. I. Lamond, Multidimensional proteomics for cell biology, Nat. Rev. Mol. Cell Biol., 2015, 16(5), 269–280 CrossRef CAS PubMed.
- D. P. Nusinow, J. Szpyt, M. Ghandi, C. M. Rose, E. R. McDonald 3rd, M. Kalocsay, J. Jane-Valbuena, E. Gelfand, D. K. Schweppe, M. Jedrychowski, J. Golji, D. A. Porter, T. Rejtar, Y. K. Wang, G. V. Kryukov, F. Stegmeier, B. K. Erickson, L. A. Garraway, W. R. Sellers and S. P. Gygi, Quantitative Proteomics of the Cancer Cell Line Encyclopedia, Cell, 2020, 180(2), 387–402 CrossRef CAS PubMed.
- A. Hoshino, H. S. Kim, L. Bojmar, K. E. Gyan, M. Cioffi, J. Hernandez, C. P. Zambirinis, G. Rodrigues, H. Molina, S. Heissel, M. T. Mark, L. Steiner, A. Benito-Martin, S. Lucotti, A. Di Giannatale, K. Offer, M. Nakajima, C. Williams, L. Nogues, F. A. Pelissier Vatter, A. Hashimoto, A. E. Davies, D. Freitas, C. M. Kenific, Y. Ararso, W. Buehring, P. Lauritzen, Y. Ogitani, K. Sugiura, N. Takahashi, M. Aleckovic, K. A. Bailey, J. S. Jolissant, H. Wang, A. Harris, L. M. Schaeffer, G. Garcia-Santos, Z. Posner, V. P. Balachandran, Y. Khakoo, G. P. Raju, A. Scherz, I. Sagi, R. Scherz-Shouval, Y. Yarden, M. Oren, M. Malladi, M. Petriccione, K. C. De Braganca, M. Donzelli, C. Fischer, S. Vitolano, G. P. Wright, L. Ganshaw, M. Marrano, A. Ahmed, J. DeStefano, E. Danzer, M. H. A. Roehrl, N. J. Lacayo, T. C. Vincent, M. R. Weiser, M. S. Brady, P. A. Meyers, L. H. Wexler, S. R. Ambati, A. J. Chou, E. K. Slotkin, S. Modak, S. S. Roberts, E. M. Basu, D. Diolaiti, B. A. Krantz, F. Cardoso, A. L. Simpson, M. Berger, C. M. Rudin, D. M. Simeone, M. Jain, C. M. Ghajar, S. K. Batra, B. Z. Stanger, J. Bui, K. A. Brown, V. K. Rajasekhar, J. H. Healey, M. de Sousa, K. Kramer, S. Sheth, J. Baisch, V. Pascual, T. E. Heaton, M. P. La Quaglia, D. J. Pisapia, R. Schwartz, H. Zhang, Y. Liu, A. Shukla, L. Blavier, Y. A. DeClerck, M. LaBarge, M. J. Bissell, T. C. Caffrey, P. M. Grandgenett, M. A. Hollingsworth, J. Bromberg, B. Costa-Silva, H. Peinado, Y. Kang, B. A. Garcia, E. M. O'Reilly, D. Kelsen, T. M. Trippett, D. R. Jones, I. R. Matei, W. R. Jarnagin and D. Lyden, Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers, Cell, 2020, 182(4), 1044–1061 CrossRef CAS PubMed.
- J. B. Muller, P. E. Geyer, A. R. Colaco, P. V. Treit, M. T. Strauss, M. Oroshi, S. Doll, S. Virreira Winter, J. M. Bader, N. Kohler, F. Theis, A. Santos and M. Mann, The proteome landscape of the kingdoms of life, Nature, 2020, 582(7813), 592–596 CrossRef PubMed.
- L. Jiang, M. Wang, S. Lin, R. Jian, X. Li, J. Chan, G. Dong, H. Fang, A. E. Robinson, GTEx Consortium and M. P. Snyder, A Quantitative Proteome Map of the Human Body, Cell, 2020, 183(1), 269–283 CrossRef CAS PubMed.
- M. Uhlen, L. Fagerberg, B. M. Hallstrom, C. Lindskog, P. Oksvold, A. Mardinoglu, A. Sivertsson, C. Kampf, E. Sjostedt, A. Asplund, I. Olsson, K. Edlund, E. Lundberg, S. Navani, C. A. Szigyarto, J. Odeberg, D. Djureinovic, J. O. Takanen, S. Hober, T. Alm, P. H. Edqvist, H. Berling, H. Tegel, J. Mulder, J. Rockberg, P. Nilsson, J. M. Schwenk, M. Hamsten, K. von Feilitzen, M. Forsberg, L. Persson, F. Johansson, M. Zwahlen, G. von Heijne, J. Nielsen and F. Ponten, Tissue-based map of the human proteome, Science, 2015, 347(6220), 1260419 CrossRef PubMed.
- P. Mertins, L. C. Tang, K. Krug, D. J. Clark, M. A. Gritsenko, L. Chen, K. R. Clauser, T. R. Clauss, P. Shah, M. A. Gillette, V. A. Petyuk, S. N. Thomas, D. R. Mani, F. Mundt, R. J. Moore, Y. Hu, R. Zhao, M. Schnaubelt, H. Keshishian, M. E. Monroe, Z. Zhang, N. D. Udeshi, D. Mani, S. R. Davies, R. R. Townsend, D. W. Chan, R. D. Smith, H. Zhang, T. Liu and S. A. Carr, Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry, Nat. Protoc., 2018, 13(7), 1632–1661 CrossRef CAS PubMed.
- I. Dapic, L. Baljeu-Neuman, N. Uwugiaren, J. Kers, D. R. Goodlett and G. L. Corthals, Proteome analysis of tissues by mass spectrometry, Mass Spectrom. Rev., 2019, 38(4–5), 403–441 CrossRef CAS PubMed.
- L. Yang, J. George and J. Wang, Deep Profiling of Cellular Heterogeneity by Emerging Single-Cell Proteomic Technologies, Proteomics, 2020, 20(13), e1900226 CrossRef PubMed.
- M. Waas and T. Kislinger, Addressing Cellular Heterogeneity in Cancer through Precision Proteomics, J. Proteome Res., 2020, 19(9), 3607–3619 CrossRef CAS PubMed.
- F. von Eggeling and F. Hoffmann, Microdissection-An Essential Prerequisite for Spatial Cancer Omics, Proteomics, 2020, 20(17–18), e2000077 CrossRef PubMed.
- X. K. Lun and B. Bodenmiller, Profiling Cell Signaling Networks at Single-cell Resolution, Mol. Cell. Proteomics, 2020, 19(5), 744–756 CrossRef CAS PubMed.
- N. W. Bateman and T. P. Conrads, Recent advances and opportunities in proteomic analyses of tumour heterogeneity, J. Pathol., 2018, 244(5), 628–637 CrossRef PubMed.
- R. Casadonte and R. M. Caprioli, Proteomic analysis of formalin-fixed paraffin-embedded tissue by MALDI imaging mass spectrometry, Nat. Protoc., 2011, 6(11), 1695–1709 CrossRef CAS PubMed.
- R. Van de Plas, J. Yang, J. Spraggins and R. M. Caprioli, Image fusion of mass spectrometry and microscopy: a multimodality paradigm for molecular tissue mapping, Nat. Methods, 2015, 12(4), 366–372 CrossRef CAS PubMed.
- K. Y. Garza, C. L. Feider, D. R. Klein, J. A. Rosenberg, J. S. Brodbelt and L. S. Eberlin, Desorption Electrospray Ionization Mass Spectrometry Imaging of Proteins Directly from Biological Tissue Sections, Anal. Chem., 2018, 90(13), 7785–7789 CrossRef CAS PubMed.
- A. M. Kotowska, G. F. Trindade, P. M. Mendes, P. M. Williams, J. W. Aylott, A. G. Shard, M. R. Alexander and D. J. Scurr, Protein identification by 3D OrbiSIMS to facilitate in situ imaging and depth profiling, Nat. Commun., 2020, 11(1), 5832 CrossRef CAS PubMed.
- A. R. Buchberger, K. DeLaney, J. Johnson and L. Li, Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights, Anal. Chem., 2018, 90(1), 240–265 CrossRef CAS PubMed.
- G. A. Harris, J. J. Nicklay and R. M. Caprioli, Localized in situ hydrogel-mediated protein digestion and extraction technique for on-tissue analysis, Anal. Chem., 2013, 85(5), 2717–2723 CrossRef CAS PubMed.
- D. G. Rizzo, B. M. Prentice, J. L. Moore, J. L. Norris and R. M. Caprioli, Enhanced Spatially Resolved Proteomics Using On-Tissue Hydrogel-Mediated Protein Digestion, Anal. Chem., 2017, 89(5), 2948–2955 CrossRef CAS PubMed.
- D. J. Ryan, N. H. Patterson, N. E. Putnam, A. D. Wilde, A. Weiss, W. J. Perry, J. E. Cassat, E. P. Skaar, R. M. Caprioli and J. M. Spraggins, MicroLESA: Integrating Autofluorescence Microscopy, In Situ Micro-Digestions, and Liquid Extraction Surface Analysis for High Spatial Resolution Targeted Proteomic Studies, Anal. Chem., 2019, 91(12), 7578–7585 CrossRef CAS PubMed.
- R. L. Griffiths, E. C. Randall, A. M. Race, J. Bunch and H. J. Cooper, Raster-Mode Continuous-Flow Liquid Microjunction Mass Spectrometry Imaging of Proteins in Thin Tissue Sections, Anal. Chem., 2017, 89(11), 5683–5687 CrossRef CAS PubMed.
- E. K. Sisley, E. Illes-Toth and H. J. Cooper, In situ analysis of intact proteins by ion mobility mass spectrometry, Trends Anal. Chem., 2020, 124, 115534 CrossRef CAS.
- J. Y. Han, H. Permentier, R. Bischoff, G. Groothuis, A. Casini and P. Horvatovich, Imaging of protein distribution in tissues using mass spectrometry: An interdisciplinary challenge, Trends Anal. Chem., 2019, 112, 13–28 CrossRef CAS.
- J. J. Xue, Y. Bai and H. W. Liu, Recent advances in ambient mass spectrometry imaging, Trends Anal. Chem., 2019, 120, 115659 CrossRef CAS.
- J. L. Norris and R. M. Caprioli, Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research, Chem. Rev., 2013, 113(4), 2309–2342 CrossRef CAS PubMed.
- K. Schwamborn and R. M. Caprioli, Molecular imaging by mass spectrometry–looking beyond classical histology, Nat. Rev. Cancer, 2010, 10(9), 639–646 CrossRef CAS PubMed.
- B. Rocha, C. Ruiz-Romero and F. J. Blanco, Mass spectrometry imaging: a novel technology in rheumatology, Nat. Rev. Rheumatol., 2017, 13(1), 52–63 CrossRef CAS PubMed.
- K. Sharma, S. Schmitt, C. G. Bergner, S. Tyanova, N. Kannaiyan, N. Manrique-Hoyos, K. Kongi, L. Cantuti, U. K. Hanisch, M. A. Philips, M. J. Rossner, M. Mann and M. Simons, Cell type- and brain region-resolved mouse brain proteome, Nat. Neurosci., 2015, 18(12), 1819–1831 CrossRef CAS PubMed.
- S. Tyanova, T. Temu and J. Cox, The MaxQuant computational platform for mass spectrometry-based shotgun proteomics, Nat. Protoc., 2016, 11(12), 2301–2319 CrossRef CAS PubMed.
- J. McKetney, R. M. Runde, A. S. Hebert, S. Salamat, S. Roy and J. J. Coon, Proteomic Atlas of the Human Brain in Alzheimer's Disease, J. Proteome Res., 2019, 18(3), 1380–1391 CrossRef CAS PubMed.
- S. Doll, M. Dressen, P. E. Geyer, D. N. Itzhak, C. Braun, S. A. Doppler, F. Meier, M. A. Deutsch, H. Lahm, R. Lange, M. Krane and M. Mann, Region and cell-type resolved quantitative proteomic map of the human heart, Nat. Commun., 2017, 8(1), 1469 CrossRef PubMed.
- N. A. Kulak, G. Pichler, I. Paron, N. Nagaraj and M. Mann, Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells, Nat. Methods, 2014, 11(3), 319–324 CrossRef CAS PubMed.
- N. A. Kulak, P. E. Geyer and M. Mann, Loss-less Nano-fractionator for High Sensitivity, High Coverage Proteomics, Mol. Cell. Proteomics, 2017, 16(4), 694–705 CrossRef CAS PubMed.
- B. Dyring-Andersen, M. B. Lovendorf, F. Coscia, A. Santos, L. B. P. Moller, A. R. Colaco, L. Niu, M. Bzorek, S. Doll, J. L. Andersen, R. A. Clark, L. Skov, M. B. M. Teunissen and M. Mann, Spatially and cell-type resolved quantitative proteomic atlas of healthy human skin, Nat. Commun., 2020, 11(1), 5587 CrossRef CAS PubMed.
- X. Ni, Z. Tan, C. Ding, C. Zhang, L. Song, S. Yang, M. Liu, R. Jia, C. Zhao, L. Song, W. Liu, Q. Zhou, T. Gong, X. Li, Y. Tai, W. Zhu, T. Shi, Y. Wang, J. Xu, B. Zhen and J. Qin, A region-resolved mucosa proteome of the human stomach, Nat. Commun., 2019, 10(1), 39 CrossRef PubMed.
- N. Gebert, C. W. Cheng, J. M. Kirkpatrick, D. Di Fraia, J. Yun, P. Schadel, S. Pace, G. B. Garside, O. Werz, K. L. Rudolph, H. Jasper, O. H. Yilmaz and A. Ori, Region-Specific Proteome Changes of the Intestinal Epithelium during Aging and Dietary Restriction, Cell Rep., 2020, 31(4), 107565 CrossRef CAS PubMed.
- M. R. Emmert-Buck, R. F. Bonner, P. D. Smith, R. F. Chuaqui, Z. Zhuang, S. R. Goldstein, R. A. Weiss and L. A. Liotta, Laser capture microdissection, Science, 1996, 274(5289), 998–1001 CrossRef CAS PubMed.
- R. F. Bonner, M. Emmert-Buck, K. Cole, T. Pohida, R. Chuaqui, S. Goldstein and L. A. Liotta, Laser capture microdissection: molecular analysis of tissue, Science, 1997, 278(5342), 1481 CrossRef CAS PubMed , 1483.
- K. Schutze and G. Lahr, Identification of expressed genes by laser-mediated manipulation of single cells, Nat. Biotechnol., 1998, 16(8), 737–742 CrossRef CAS PubMed.
- S. Datta, L. Malhotra, R. Dickerson, S. Chaffee, C. K. Sen and S. Roy, Laser capture microdissection: Big data from small samples, Histol. Histopathol., 2015, 30(11), 1255–1269 CAS.
- C. Bevilacqua and B. Ducos, Laser microdissection: A powerful tool for genomics at cell level, Mol. Aspects Med., 2018, 59, 5–27 CrossRef CAS PubMed.
- V. Espina, J. D. Wulfkuhle, V. S. Calvert, A. VanMeter, W. Zhou, G. Coukos, D. H. Geho, E. F. Petricoin 3rd and L. A. Liotta, Laser-capture microdissection, Nat. Protoc., 2006, 1(2), 586–603 CrossRef CAS PubMed.
- R. Longuespee, R. Casadonte, K. Schwamborn, D. Reuss, D. Kazdal, K. Kriegsmann, A. von Deimling, W. Weichert, P. Schirmacher, J. Kriegsmann and M. Kriegsmann, Proteomics in Pathology, Proteomics, 2018, 18(2), 700361 CrossRef PubMed.
- T. De Marchi, R. B. Braakman, C. Stingl, M. M. van Duijn, M. Smid, J. A. Foekens, T. M. Luider, J. W. Martens and A. Umar, The advantage of laser-capture microdissection over whole tissue analysis in proteomic profiling studies, Proteomics, 2016, 16(10), 1474–1485 CrossRef CAS PubMed.
- F. Meier, A. D. Brunner, M. Frank, A. Ha, I. Bludau, E. Voytik, S. Kaspar-Schoenefeld, M. Lubeck, O. Raether, N. Bache, R. Aebersold, B. C. Collins, H. L. Rost and M. Mann, diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition, Nat. Methods, 2020, 17(12), 1229–1236 CrossRef CAS PubMed.
- X. T. Ye, J. Tang, Y. H. Mao, X. Lu, Y. Yang, W. D. Chen, X. Y. Zhang, R. L. Xu and R. J. Tian, Integrated proteomics sample preparation and fractionation: Method development and applications, Trends Anal. Chem., 2019, 120, 115667 CrossRef CAS.
- R. Xu, J. Tang, Q. Deng, W. He, X. Sun, L. Xia, Z. Cheng, L. He, S. You, J. Hu, Y. Fu, J. Zhu, Y. Chen, W. Gao, A. He, Z. Guo, L. Lin, H. Li, C. Hu and R. Tian, Spatial-Resolution Cell Type Proteome Profiling of Cancer Tissue by Fully Integrated Proteomics Technology, Anal. Chem., 2018, 90(9), 5879–5886 CrossRef CAS PubMed.
- W. Chen, S. Wang, S. Adhikari, Z. Deng, L. Wang, L. Chen, M. Ke, P. Yang and R. Tian, Simple and Integrated Spintip-Based Technology Applied for Deep Proteome Profiling, Anal. Chem., 2016, 88(9), 4864–4871 CrossRef CAS PubMed.
- S. Davis, C. Scott, O. Ansorge and R. Fischer, Development of a Sensitive, Scalable Method for Spatial, Cell-Type-Resolved Proteomics of the Human Brain, J. Proteome Res., 2019, 18(4), 1787–1795 CrossRef CAS PubMed.
- Y. Zhu, P. D. Piehowski, R. Zhao, J. Chen, Y. Shen, R. J. Moore, A. K. Shukla, V. A. Petyuk, M. Campbell-Thompson, C. E. Mathews, R. D. Smith, W. J. Qian and R. T. Kelly, Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells, Nat. Commun., 2018, 9(1), 882 CrossRef PubMed.
- Y. Liang, Y. Zhu, M. Dou, K. Xu, R. K. Chu, W. B. Chrisler, R. Zhao, K. K. Hixson and R. T. Kelly, Spatially Resolved Proteome Profiling of <200 Cells from Tomato Fruit Pericarp by Integrating Laser-Capture Microdissection with Nanodroplet Sample Preparation, Anal. Chem., 2018, 90(18), 11106–11114 CrossRef CAS PubMed.
- Y. Zhu, M. Dou, P. D. Piehowski, Y. Liang, F. Wang, R. K. Chu, W. B. Chrisler, J. N. Smith, K. C. Schwarz, Y. Shen, A. K. Shukla, R. J. Moore, R. D. Smith, W. J. Qian and R. T. Kelly, Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets, Mol. Cell. Proteomics, 2018, 17(9), 1864–1874 CrossRef CAS PubMed.
- P. D. Piehowski, Y. Zhu, L. M. Bramer, K. G. Stratton, R. Zhao, D. J. Orton, R. J. Moore, J. Yuan, H. D. Mitchell, Y. Gao, B. M. Webb-Robertson, S. K. Dey, R. T. Kelly and K. E. Burnum-Johnson, Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-mum spatial resolution, Nat. Commun., 2020, 11(1), 8 CrossRef CAS.
- E. Griesser, H. Wyatt, S. Ten Have, B. Stierstorfer, M. Lenter and A. I. Lamond, Quantitative Profiling of the Human Substantia Nigra Proteome from Laser-capture Microdissected FFPE Tissue, Mol. Cell. Proteomics, 2020, 19(5), 839–851 CrossRef PubMed.
- G. Clair, P. D. Piehowski, T. Nicola, J. A. Kitzmiller, E. L. Huang, E. M. Zink, R. L. Sontag, D. J. Orton, R. J. Moore, J. P. Carson, R. D. Smith, J. A. Whitsett, R. A. Corley, N. Ambalavanan and C. Ansong, Spatially-Resolved Proteomics: Rapid Quantitative Analysis of Laser Capture Microdissected Alveolar Tissue Samples, Sci. Rep., 2016, 6, 39223 CrossRef CAS.
- E. L. de Graaf, D. Pellegrini and L. A. McDonnell, Set of Novel Automated Quantitative Microproteomics Protocols for Small Sample Amounts and Its Application to Kidney Tissue Substructures, J. Proteome Res., 2016, 15(12), 4722–4730 CrossRef CAS.
- H. Meng, P. M. Janssen, R. W. Grange, L. Yang, A. H. Beggs, L. C. Swanson, S. A. Cossette, A. Frase, M. K. Childers, H. Granzier, E. Gussoni and M. W. Lawlor, Tissue triage and freezing for models of skeletal muscle disease, J. Visualized Exp., 2014,(89), 51586 Search PubMed.
- S. Magdeldin and T. Yamamoto, Toward deciphering proteomes of formalin-fixed paraffin-embedded (FFPE) tissues, Proteomics, 2012, 12(7), 1045–1058 CrossRef CAS PubMed.
- T. Tayri-Wilk, M. Slavin, J. Zamel, A. Blass, S. Cohen, A. Motzik, X. Sun, D. E. Shalev, O. Ram and N. Kalisman, Mass spectrometry reveals the chemistry of formaldehyde cross-linking in structured proteins, Nat. Commun., 2020, 11(1), 3128 CrossRef CAS.
- S. R. Shi, C. R. Taylor, C. B. Fowler and J. T. Mason, Complete solubilization of formalin-fixed, paraffin-embedded tissue may improve proteomic studies, Proteomics: Clin. Appl., 2013, 7(3–4), 264–272 CAS.
- X. Jiang, X. Jiang, S. Feng, R. Tian, M. Ye and H. Zou, Development of efficient protein extraction methods for shotgun proteome analysis of formalin-fixed tissues, J. Proteome Res., 2007, 6(3), 1038–1047 CrossRef CAS PubMed.
- Y. Kawashima, Y. Kodera, A. Singh, M. Matsumoto and H. Matsumoto, Efficient extraction of proteins from formalin-fixed paraffin-embedded tissues requires higher concentration of tris(hydroxymethyl)aminomethane, Clin. Proteomics, 2014, 11(1), 4 CrossRef.
- D. K. Lee, S. S. Rubakhin, I. Kusmartseva, C. Wasserfall, M. A. Atkinson and J. V. Sweedler, Removing Formaldehyde-Induced Peptidyl Crosslinks Enables Mass Spectrometry Imaging of Peptide Hormone Distributions from Formalin-Fixed Paraffin-Embedded Tissues, Angew. Chem., 2020, 59(50), 22584–22590 CrossRef CAS.
- F. Coscia, S. Doll, J. M. Bech, L. Schweizer, A. Mund, E. Lengyel, J. Lindebjerg, G. I. Madsen, J. M. Moreira and M. Mann, A streamlined mass spectrometry-based proteomics workflow for large-scale FFPE tissue analysis, J. Pathol., 2020, 251(1), 100–112 CrossRef CAS PubMed.
- R. Longuespee, D. Alberts, C. Pottier, N. Smargiasso, G. Mazzucchelli, D. Baiwir, M. Kriegsmann, M. Herfs, J. Kriegsmann, P. Delvenne and E. De Pauw, A laser microdissection-based workflow for FFPE tissue microproteomics: Important considerations for small sample processing, Methods, 2016, 104, 154–162 CrossRef CAS PubMed.
- Y. Zhu, T. Weiss, Q. Zhang, R. Sun, B. Wang, X. Yi, Z. Wu, H. Gao, X. Cai, G. Ruan, T. Zhu, C. Xu, S. Lou, X. Yu, L. Gillet, P. Blattmann, K. Saba, C. D. Fankhauser, M. B. Schmid, D. Rutishauser, J. Ljubicic, A. Christiansen, C. Fritz, N. J. Rupp, C. Poyet, E. Rushing, M. Weller, P. Roth, E. Haralambieva, S. Hofer, C. Chen, W. Jochum, X. Gao, X. Teng, L. Chen, Q. Zhong, P. J. Wild, R. Aebersold and T. Guo, High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification, Mol. Oncol., 2019, 13(11), 2305–2328 CrossRef CAS PubMed.
- D. M. Marchione, I. Ilieva, K. Devins, D. Sharpe, D. J. Pappin, B. A. Garcia, J. P. Wilson and J. B. Wojcik, HYPERsol: High-Quality Data from Archival FFPE Tissue for Clinical Proteomics, J. Proteome Res., 2020, 19(2), 973–983 CrossRef CAS PubMed.
- T. Guo, P. Kouvonen, C. C. Koh, L. C. Gillet, W. E. Wolski, H. L. Rost, G. Rosenberger, B. C. Collins, L. C. Blum, S. Gillessen, M. Joerger, W. Jochum and R. Aebersold, Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps, Nat. Med., 2015, 21(4), 407–413 CrossRef CAS PubMed.
- H. Gao, F. Zhang, S. Liang, Q. Zhang, M. Lyu, L. Qian, W. Liu, W. Ge, C. Chen, X. Yi, J. Zhu, C. Lu, P. Sun, K. Liu, Y. Zhu and T. Guo, Accelerated Lysis and Proteolytic Digestion of Biopsy-Level Fresh-Frozen and FFPE Tissue Samples Using Pressure Cycling Technology, J. Proteome Res., 2020, 19(5), 1982–1990 CrossRef CAS PubMed.
- S. Shao, T. Guo, V. Gross, A. Lazarev, C. C. Koh, S. Gillessen, M. Joerger, W. Jochum and R. Aebersold, Reproducible Tissue Homogenization and Protein Extraction for Quantitative Proteomics Using MicroPestle-Assisted Pressure-Cycling Technology, J. Proteome Res., 2016, 15(6), 1821–1829 CrossRef CAS PubMed.
- X. Nie, L. Qian, R. Sun, B. Huang, X. Dong, Q. Xiao, Q. Zhang, T. Lu, L. Yue, S. Chen, X. Li, Y. Sun, L. Li, L. Xu, Y. Li, M. Yang, Z. Xue, S. Liang, X. Ding, C. Yuan, L. Peng, W. Liu, X. Yi, M. Lyu, G. Xiao, X. Xu, W. Ge, J. He, J. Fan, J. Wu, M. Luo, X. Chang, H. Pan, X. Cai, J. Zhou, J. Yu, H. Gao, M. Xie, S. Wang, G. Ruan, H. Chen, H. Su, H. Mei, D. Luo, D. Zhao, F. Xu, Y. Li, Y. Zhu, J. Xia, Y. Hu and T. Guo, Multi-organ proteomic landscape of COVID-19 autopsies, Cell, 2021, 184(3), 775–791 CrossRef CAS PubMed.
- S. Shao, T. Guo, C. C. Koh, S. Gillessen, M. Joerger, W. Jochum and R. Aebersold, Minimal sample requirement for highly multiplexed protein quantification in cell lines and tissues by PCT-SWATH mass spectrometry, Proteomics, 2015, 15(21), 3711–3721 CrossRef CAS.
- J. R. Wisniewski, A. Zougman, N. Nagaraj and M. Mann, Universal sample preparation method for proteome analysis, Nat. Methods, 2009, 6(5), 359–362 CrossRef CAS PubMed.
- D. C. Liebler and A.-J. L. Ham, Spin filter–based sample preparation for shotgun proteomics, Nat. Methods, 2009, 6(11), 785–785 CrossRef CAS PubMed.
- J. R. Wisniewski, P. Ostasiewicz and M. Mann, High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers, J. Proteome Res., 2011, 10(7), 3040–3049 CrossRef CAS.
- J. R. Wisniewski, K. Dus and M. Mann, Proteomic workflow for analysis of archival formalin-fixed and paraffin-embedded clinical samples to a depth of 10 000 proteins, Proteomics: Clin. Appl., 2013, 7(3–4), 225–233 CAS.
- J. R. Wisniewski, P. Ostasiewicz, K. Dus, D. F. Zielinska, F. Gnad and M. Mann, Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma, Mol. Syst. Biol., 2012, 8, 611 CrossRef PubMed.
- J. R. Wisniewski, K. Dus-Szachniewicz, P. Ostasiewicz, P. Ziolkowski, D. Rakus and M. Mann, Absolute Proteome Analysis of Colorectal Mucosa, Adenoma, and Cancer Reveals Drastic Changes in Fatty Acid Metabolism and Plasma Membrane Transporters, J. Proteome Res., 2015, 14(9), 4005–4018 CrossRef CAS PubMed.
- C. S. Hughes, S. Foehr, D. A. Garfield, E. E. Furlong, L. M. Steinmetz and J. Krijgsveld, Ultrasensitive proteome analysis using paramagnetic bead technology, Mol. Syst. Biol., 2014, 10, 757 CrossRef PubMed.
- S. Moggridge, P. H. Sorensen, G. B. Morin and C. S. Hughes, Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics, J. Proteome Res., 2018, 17(4), 1730–1740 CrossRef CAS PubMed.
- C. S. Hughes, S. Moggridge, T. Muller, P. H. Sorensen, G. B. Morin and J. Krijgsveld, Single-pot, solid-phase-enhanced sample preparation for proteomics experiments, Nat. Protoc., 2019, 14(1), 68–85 CrossRef CAS PubMed.
- T. S. Batth, M. A. X. Tollenaere, P. Ruther, A. Gonzalez-Franquesa, B. S. Prabhakar, S. Bekker-Jensen, A. S. Deshmukh and J. V. Olsen, Protein Aggregation Capture on Microparticles Enables Multipurpose Proteomics Sample Preparation, Mol. Cell. Proteomics, 2019, 18(5), 1027–1035 CrossRef CAS PubMed.
- C. S. Hughes, M. K. McConechy, D. R. Cochrane, T. Nazeran, A. N. Karnezis, D. G. Huntsman and G. B. Morin, Quantitative Profiling of Single Formalin Fixed Tumour Sections: proteomics for translational research, Sci. Rep., 2016, 6, 34949 CrossRef CAS PubMed.
- K. Buczak, J. M. Kirkpatrick, F. Truckenmueller, D. Santinha, L. Ferreira, S. Roessler, S. Singer, M. Beck and A. Ori, Spatially resolved analysis of FFPE tissue proteomes by quantitative mass spectrometry, Nat. Protoc., 2020, 15(9), 2956–2979 CrossRef CAS PubMed.
- Y. J. Chen, T. I. Roumeliotis, Y. H. Chang, C. T. Chen, C. L. Han, M. H. Lin, H. W. Chen, G. C. Chang, Y. L. Chang, C. T. Wu, M. W. Lin, M. S. Hsieh, Y. T. Wang, Y. R. Chen, I. Jonassen, F. Z. Ghavidel, Z. S. Lin, K. T. Lin, C. W. Chen, P. Y. Sheu, C. T. Hung, K. C. Huang, H. C. Yang, P. Y. Lin, T. C. Yen, Y. W. Lin, J. H. Wang, L. Raghav, C. Y. Lin, Y. S. Chen, P. S. Wu, C. T. Lai, S. H. Weng, K. Y. Su, W. H. Chang, P. Y. Tsai, A. I. Robles, H. Rodriguez, Y. J. Hsiao, W. H. Chang, T. Y. Sung, J. S. Chen, S. L. Yu, J. S. Choudhary, H. Y. Chen, P. C. Yang and Y. J. Chen, Proteogenomics of Non-smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression, Cell, 2020, 182(1), 226–244 CrossRef CAS PubMed.
- Q. Gao, H. Zhu, L. Dong, W. Shi, R. Chen, Z. Song, C. Huang, J. Li, X. Dong, Y. Zhou, Q. Liu, L. Ma, X. Wang, J. Zhou, Y. Liu, E. Boja, A. I. Robles, W. Ma, P. Wang, Y. Li, L. Ding, B. Wen, B. Zhang, H. Rodriguez, D. Gao, H. Zhou and J. Fan, Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma, Cell, 2019, 179(2), 561–577 CrossRef CAS PubMed.
- L. Ting, R. Rad, S. P. Gygi and W. Haas, MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics, Nat. Methods, 2011, 8(11), 937–940 CrossRef CAS PubMed.
- A. Hogrebe, L. von Stechow, D. B. Bekker-Jensen, B. T. Weinert, C. D. Kelstrup and J. V. Olsen, Benchmarking common quantification strategies for large-scale phosphoproteomics, Nat. Commun., 2018, 9(1), 1045 CrossRef PubMed.
- P. Mertins, J. W. Qiao, J. Patel, N. D. Udeshi, K. R. Clauser, D. R. Mani, M. W. Burgess, M. A. Gillette, J. D. Jaffe and S. A. Carr, Integrated proteomic analysis of post-translational modifications by serial enrichment, Nat. Methods, 2013, 10(7), 634–637 CrossRef CAS PubMed.
- K. K. Palaniappan, C. R. Bertozzi and C. Glycoproteomics, Chem. Rev., 2016, 116(23), 14277–14306 CrossRef CAS PubMed.
- H. Hinneburg, P. Korac, F. Schirmeister, S. Gasparov, P. H. Seeberger, V. Zoldos and D. Kolarich, Unlocking Cancer Glycomes from Histopathological Formalin-fixed and Paraffin-embedded (FFPE) Tissue Microdissections, Mol. Cell. Proteomics, 2017, 16(4), 524–536 CrossRef CAS PubMed.
- P. Huang, H. Li, W. Gao, Z. Cai and R. Tian, A Fully Integrated Spintip-Based Approach for Sensitive and Quantitative Profiling of Region-Resolved in Vivo Brain Glycoproteome, Anal. Chem., 2019, 91(14), 9181–9189 CrossRef CAS PubMed.
- Y. Mao, P. Chen, M. Ke, X. Chen, S. Ji, W. Chen and R. Tian, Fully Integrated and Multiplexed Sample Preparation Technology for Sensitive Interactome Profiling, Anal. Chem., 2021, 93(5), 3026–3034 CrossRef CAS PubMed.
- P. Huang, Q. Kong, W. Gao, B. Chu, H. Li, Y. Mao, Z. Cai, R. Xu and R. Tian, Spatial proteome profiling by immunohistochemistry-based laser capture microdissection and data-independent acquisition proteomics, Anal. Chim. Acta, 2020, 1127, 140–148 CrossRef PubMed.
- D. Alberts, C. Pottier, N. Smargiasso, D. Baiwir, G. Mazzucchelli, P. Delvenne, M. Kriegsmann, D. Kazdal, A. Warth, E. De Pauw and R. Longuespee, MALDI Imaging-Guided Microproteomic Analyses of Heterogeneous Breast Tumors-A Pilot Study, Proteomics: Clin. Appl., 2018, 12(1), 1700062 Search PubMed.
- F. Dewez, M. Martin-Lorenzo, M. Herfs, D. Baiwir, G. Mazzucchelli, E. De Pauw, R. M. A. Heeren and B. Balluff, Precise co-registration of mass spectrometry imaging, histology, and laser microdissection-based omics, Anal. Bioanal. Chem., 2019, 411(22), 5647–5653 CrossRef CAS PubMed.
- F. Dewez, J. Oejten, C. Henkel, R. Hebeler, H. Neuweger, E. De Pauw, R. M. A. Heeren and B. Balluff, MS Imaging-Guided Microproteomics for Spatial Omics on a Single Instrument, Proteomics, 2020, 20(23), e1900369 CrossRef PubMed.
- K. Bera, K. A. Schalper, D. L. Rimm, V. Velcheti and A. Madabhushi, Artificial intelligence in digital pathology - new tools for diagnosis and precision oncology, Nat. Rev. Clin. Oncol., 2019, 16(11), 703–715 CrossRef PubMed.
- C. Brasko, K. Smith, C. Molnar, N. Farago, L. Hegedus, A. Balind, T. Balassa, A. Szkalisity, F. Sukosd, K. Kocsis, B. Balint, L. Paavolainen, M. Z. Enyedi, I. Nagy, L. G. Puskas, L. Haracska, G. Tamas and P. Horvath, Intelligent image-based in situ single-cell isolation, Nat. Commun., 2018, 9(1), 226 CrossRef PubMed.
- A. Mund, F. Coscia, R. Hollandi, F. Kovács, A. Kriston, A.-D. Brunner, M. Bzorek, S. Naimy, L. M. Rahbek Gjerdrum, B. Dyring-Andersen, J. Bulkescher, C. Lukas, C. Gnann, E. Lundberg, P. Horvath and M. Mann, AI-driven Deep Visual Proteomics defines cell identity and heterogeneity, bioRxiv, 2021.2001.2025.427969, 2021.
- F. Meier, A. D. Brunner, S. Koch, H. Koch, M. Lubeck, M. Krause, N. Goedecke, J. Decker, T. Kosinski, M. A. Park, N. Bache, O. Hoerning, J. Cox, O. Rather and M. Mann, Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer, Mol. Cell. Proteomics, 2018, 17(12), 2534–2545 CrossRef CAS PubMed.
- M. H. Han, S. I. Hwang, D. B. Roy, D. H. Lundgren, J. V. Price, S. S. Ousman, G. H. Fernald, B. Gerlitz, W. H. Robinson, S. E. Baranzini, B. W. Grinnell, C. S. Raine, R. A. Sobel, D. K. Han and L. Steinman, Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets, Nature, 2008, 451(7182), 1076–1081 CrossRef CAS PubMed.
- L. F. Waanders, K. Chwalek, M. Monetti, C. Kumar, E. Lammert and M. Mann, Quantitative proteomic analysis of single pancreatic islets, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(45), 18902–18907 CrossRef CAS PubMed.
- S. Valleix, J. D. Gillmore, F. Bridoux, P. P. Mangione, A. Dogan, B. Nedelec, M. Boimard, G. Touchard, J. M. Goujon, C. Lacombe, P. Lozeron, D. Adams, C. Lacroix, T. Maisonobe, V. Plante-Bordeneuve, J. A. Vrana, J. D. Theis, S. Giorgetti, R. Porcari, S. Ricagno, M. Bolognesi, M. Stoppini, M. Delpech, M. B. Pepys, P. N. Hawkins and V. Bellotti, Hereditary systemic amyloidosis due to Asp76Asn variant beta2-microglobulin, N. Engl. J. Med., 2012, 366(24), 2276–2283 CrossRef CAS PubMed.
- M. J. Marakalala, R. M. Raju, K. Sharma, Y. J. Zhang, E. A. Eugenin, B. Prideaux, I. B. Daudelin, P. Y. Chen, M. G. Booty, J. H. Kim, S. Y. Eum, L. E. Via, S. M. Behar, C. E. Barry 3rd, M. Mann, V. Dartois and E. J. Rubin, Inflammatory signaling in human tuberculosis granulomas is spatially organized, Nat. Med., 2016, 22(5), 531–538 CrossRef CAS PubMed.
- C. M. Carnielli, C. C. S. Macedo, T. De Rossi, D. C. Granato, C. Rivera, R. R. Domingues, B. A. Pauletti, S. Yokoo, H. Heberle, A. F. Busso-Lopes, N. K. Cervigne, I. Sawazaki-Calone, G. V. Meirelles, F. A. Marchi, G. P. Telles, R. Minghim, A. C. P. Ribeiro, T. B. Brandao, G. de Castro Jr., W. A. Gonzalez-Arriagada, A. Gomes, F. Penteado, A. R. Santos-Silva, M. A. Lopes, P. C. Rodrigues, E. Sundquist, T. Salo, S. D. da Silva, M. A. Alaoui-Jamali, E. Graner, J. W. Fox, R. D. Coletta and A. F. Paes Leme, Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer, Nat. Commun., 2018, 9(1), 3598 CrossRef PubMed.
- M. A. Eckert, F. Coscia, A. Chryplewicz, J. W. Chang, K. M. Hernandez, S. Pan, S. M. Tienda, D. A. Nahotko, G. Li, I. Blazenovic, R. R. Lastra, M. Curtis, S. D. Yamada, R. Perets, S. M. McGregor, J. Andrade, O. Fiehn, R. E. Moellering, M. Mann and E. Lengyel, Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts, Nature, 2019, 569(7758), 723–728 CrossRef CAS PubMed.
- Y. Hiroshima, R. Kasajima, Y. Kimura, D. Komura, S. Ishikawa, Y. Ichikawa, M. Bouvet, N. Yamamoto, T. Oshima, S. Morinaga, S. R. Singh, R. M. Hoffman, I. Endo and Y. Miyagi, Novel targets identified by integrated cancer-stromal interactome analysis of pancreatic adenocarcinoma, Cancer Lett., 2020, 469, 217–227 CrossRef CAS PubMed.
- T. Y. Le Large, G. Mantini, L. L. Meijer, T. V. Pham, N. Funel, N. C. van Grieken, B. Kok, J. Knol, H. W. van Laarhoven, S. R. Piersma, C. R. Jimenez, G. Kazemier, E. Giovannetti and M. F. Bijlsma, Microdissected pancreatic cancer proteomes reveal tumor heterogeneity and therapeutic targets, JCI Insight, 2020, 5(15), e138290 CrossRef PubMed.
- K. Buczak, A. Ori, J. M. Kirkpatrick, K. Holzer, D. Dauch, S. Roessler, V. Endris, F. Lasitschka, L. Parca, A. Schmidt, L. Zender, P. Schirmacher, J. Krijgsveld, S. Singer and M. Beck, Spatial Tissue Proteomics Quantifies Inter- and Intratumor Heterogeneity in Hepatocellular Carcinoma (HCC), Mol. Cell. Proteomics, 2018, 17(4), 810–825 CrossRef CAS PubMed.
- F. Xiong, W. Ge and C. Ma, Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer's disease, Alzheimer's Dementia, 2019, 15(3), 429–440 CrossRef PubMed.
- D. Pellegrini, A. Del Grosso, L. Angella, N. Giordano, M. Dilillo, I. Tonazzini, M. Caleo, M. Cecchini and L. A. McDonnell, Quantitative Microproteomics Based Characterization of the Central and Peripheral Nervous System of a Mouse Model of Krabbe Disease, Mol. Cell. Proteomics, 2019, 18(6), 1227–1241 CrossRef CAS PubMed.
- M. Murgia, J. Tan, P. E. Geyer, S. Doll, M. Mann and T. Klopstock, Proteomics of Cytochrome c Oxidase-Negative versus -Positive Muscle Fiber Sections in Mitochondrial Myopathy, Cell Rep., 2019, 29(12), 3825–3834 CrossRef CAS PubMed.
- G. Gut, M. D. Herrmann and L. Pelkmans, Multiplexed protein maps link subcellular organization to cellular states, Science, 2018, 361(6401), eaar7042 CrossRef PubMed.
- L. Wei, Z. Chen, L. Shi, R. Long, A. V. Anzalone, L. Zhang, F. Hu, R. Yuste, V. W. Cornish and W. Min, Super-multiplex vibrational imaging, Nature, 2017, 544(7651), 465–470 CrossRef CAS PubMed.
- B. Bodenmiller, Multiplexed Epitope-Based Tissue Imaging for Discovery and Healthcare Applications, Cell Syst., 2016, 2(4), 225–238 CrossRef CAS PubMed.
- Y. Liu, M. Yang, Y. Deng, G. Su, A. Enninful, C. C. Guo, T. Tebaldi, D. Zhang, D. Kim, Z. Bai, E. Norris, A. Pan, J. Li, Y. Xiao, S. Halene and R. Fan, High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue, Cell, 2020, 183(6), 1665–1681 CrossRef CAS PubMed.
- P. J. Thul, L. Akesson, M. Wiking, D. Mahdessian, A. Geladaki, H. Ait Blal, T. Alm, A. Asplund, L. Bjork, L. M. Breckels, A. Backstrom, F. Danielsson, L. Fagerberg, J. Fall, L. Gatto, C. Gnann, S. Hober, M. Hjelmare, F. Johansson, S. Lee, C. Lindskog, J. Mulder, C. M. Mulvey, P. Nilsson, P. Oksvold, J. Rockberg, R. Schutten, J. M. Schwenk, A. Sivertsson, E. Sjostedt, M. Skogs, C. Stadler, D. P. Sullivan, H. Tegel, C. Winsnes, C. Zhang, M. Zwahlen, A. Mardinoglu, F. Ponten, K. von Feilitzen, K. S. Lilley, M. Uhlen and E. Lundberg, A subcellular map of the human proteome, Science, 2017, 356(6340), eaal3321 CrossRef PubMed.
- K. M. Dean and A. E. Palmer, Advances in fluorescence labeling strategies for dynamic cellular imaging, Nat. Chem. Biol., 2014, 10(7), 512–523 CrossRef CAS PubMed.
- W. Schubert, B. Bonnekoh, A. J. Pommer, L. Philipsen, R. Bockelmann, Y. Malykh, H. Gollnick, M. Friedenberger, M. Bode and A. W. Dress, Analyzing proteome topology and function by automated multidimensional fluorescence microscopy, Nat. Biotechnol., 2006, 24(10), 1270–1278 CrossRef CAS PubMed.
- M. Friedenberger, M. Bode, A. Krusche and W. Schubert, Fluorescence detection of protein clusters in individual cells and tissue sections by using toponome imaging system: sample preparation and measuring procedures, Nat. Protoc., 2007, 2(9), 2285–2294 CrossRef CAS PubMed.
- M. J. Gerdes, C. J. Sevinsky, A. Sood, S. Adak, M. O. Bello, A. Bordwell, A. Can, A. Corwin, S. Dinn, R. J. Filkins, D. Hollman, V. Kamath, S. Kaanumalle, K. Kenny, M. Larsen, M. Lazare, Q. Li, C. Lowes, C. C. McCulloch, E. McDonough, M. C. Montalto, Z. Pang, J. Rittscher, A. Santamaria-Pang, B. D. Sarachan, M. L. Seel, A. Seppo, K. Shaikh, Y. Sui, J. Zhang and F. Ginty, Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(29), 11982–11987 CrossRef CAS PubMed.
- J. R. Lin, M. Fallahi-Sichani and P. K. Sorger, Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method, Nat. Commun., 2015, 6, 8390 CrossRef CAS PubMed.
- Z. Du, J. R. Lin, R. Rashid, Z. Maliga, S. Wang, J. C. Aster, B. Izar, P. K. Sorger and S. Santagata, Qualifying antibodies for image-based immune profiling and multiplexed tissue imaging, Nat. Protoc., 2019, 14(10), 2900–2930 CrossRef CAS PubMed.
- R. M. Schweller, J. Zimak, D. Y. Duose, A. A. Qutub, W. N. Hittelman and M. R. Diehl, Multiplexed in situ immunofluorescence using dynamic DNA complexes, Angew. Chem., Int. Ed., 2012, 51(37), 9292–9296 CrossRef CAS PubMed.
- M. Mondal, R. Liao, L. Xiao, T. Eno and J. Guo, Highly Multiplexed Single-Cell In Situ Protein Analysis with Cleavable Fluorescent Antibodies, Angew. Chem., Int. Ed., 2017, 56(10), 2636–2639 CrossRef CAS PubMed.
- T. Tsujikawa, S. Kumar, R. N. Borkar, V. Azimi, G. Thibault, Y. H. Chang, A. Balter, R. Kawashima, G. Choe, D. Sauer, E. El Rassi, D. R. Clayburgh, M. F. Kulesz-Martin, E. R. Lutz, L. Zheng, E. M. Jaffee, P. Leyshock, A. A. Margolin, M. Mori, J. W. Gray, P. W. Flint and L. M. Coussens, Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis, Cell Rep., 2017, 19(1), 203–217 CrossRef CAS PubMed.
- E. Murray, J. H. Cho, D. Goodwin, T. Ku, J. Swaney, S. Y. Kim, H. Choi, Y. G. Park, J. Y. Park, A. Hubbert, M. McCue, S. Vassallo, N. Bakh, M. P. Frosch, V. J. Wedeen, H. S. Seung and K. Chung, Simple, Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems, Cell, 2015, 163(6), 1500–1514 CrossRef CAS PubMed.
- Y. G. Park, C. H. Sohn, R. Chen, M. McCue, D. H. Yun, G. T. Drummond, T. Ku, N. B. Evans, H. C. Oak, W. Trieu, H. Choi, X. Jin, V. Lilascharoen, J. Wang, M. C. Truttmann, H. W. Qi, H. L. Ploegh, T. R. Golub, S. C. Chen, M. P. Frosch, H. J. Kulik, B. K. Lim and K. Chung, Protection of tissue physicochemical properties using polyfunctional crosslinkers, Nat. Biotechnol., 2018, 37(1), 73–83 CrossRef PubMed.
- D. L. Rimm, Next-gen immunohistochemistry, Nat. Methods, 2014, 11(4), 381–383 CrossRef CAS PubMed.
- D. R. Bandura, V. I. Baranov, O. I. Ornatsky, A. Antonov, R. Kinach, X. Lou, S. Pavlov, S. Vorobiev, J. E. Dick and S. D. Tanner, Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry, Anal. Chem., 2009, 81(16), 6813–6822 CrossRef CAS PubMed.
- C. Zhang, F. Wu, Y. Zhang, X. Wang and X. Zhang, A novel combination of immunoreaction and ICP-MS as a hyphenated technique for the determination of thyroid-stimulating hormone (TSH) in human serum, J. Anal. At. Spectrom., 2001, 16(12), 1393–1396 RSC.
- S. C. Bendall, E. F. Simonds, P. Qiu, A. D. Amir el, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum, Science, 2011, 332(6030), 687–696 CrossRef CAS PubMed.
- X. Lou, G. Zhang, I. Herrera, R. Kinach, O. Ornatsky, V. Baranov, M. Nitz and M. A. Winnik, Polymer-based elemental tags for sensitive bioassays, Angew. Chem., Int. Ed., 2007, 46(32), 6111–6114 CrossRef CAS PubMed.
- M. Angelo, S. C. Bendall, R. Finck, M. B. Hale, C. Hitzman, A. D. Borowsky, R. M. Levenson, J. B. Lowe, S. D. Liu, S. Zhao, Y. Natkunam and G. P. Nolan, Multiplexed ion beam imaging of human breast tumors, Nat. Med., 2014, 20(4), 436–442 CrossRef CAS PubMed.
- C. Giesen, H. A. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schuffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Gunther and B. Bodenmiller, Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry, Nat. Methods, 2014, 11(4), 417–422 CrossRef CAS PubMed.
- M. H. Spitzer and G. P. Nolan, Mass Cytometry: Single Cells, Many Features, Cell, 2016, 165(4), 780–791 CrossRef CAS PubMed.
- L. Keren, M. Bosse, D. Marquez, R. Angoshtari, S. Jain, S. Varma, S. R. Yang, A. Kurian, D. Van Valen, R. West, S. C. Bendall and M. Angelo, A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging, Cell, 2018, 174(6), 1373–1387 CrossRef CAS PubMed.
- D. Schulz, V. R. T. Zanotelli, J. R. Fischer, D. Schapiro, S. Engler, X. K. Lun, H. W. Jackson and B. Bodenmiller, Simultaneous Multiplexed Imaging of mRNA and Proteins with Subcellular Resolution in Breast Cancer Tissue Samples by Mass Cytometry, Cell Syst., 2018, 6(1), 25–36 CrossRef CAS PubMed.
- H. W. Jackson, J. R. Fischer, V. R. T. Zanotelli, H. R. Ali, R. Mechera, S. D. Soysal, H. Moch, S. Muenst, Z. Varga, W. P. Weber and B. Bodenmiller, The single-cell pathology landscape of breast cancer, Nature, 2020, 578(7796), 615–620 CrossRef CAS PubMed.
- N. Damond, S. Engler, V. R. T. Zanotelli, D. Schapiro, C. H. Wasserfall, I. Kusmartseva, H. S. Nick, F. Thorel, P. L. Herrera, M. A. Atkinson and B. Bodenmiller, A Map of Human Type 1 Diabetes Progression by Imaging Mass Cytometry, Cell Metab., 2019, 29(3), 755–768 CrossRef CAS PubMed.
- Y. J. Wang, D. Traum, J. Schug, L. Gao, C. Liu, H. Consortium, M. A. Atkinson, A. C. Powers, M. D. Feldman, A. Naji, K. M. Chang and K. H. Kaestner, Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes, Cell Metab., 2019, 29(3), 769–783 CrossRef CAS PubMed.
- A. L. Ji, A. J. Rubin, K. Thrane, S. Jiang, D. L. Reynolds, R. M. Meyers, M. G. Guo, B. M. George, A. Mollbrink, J. Bergenstrahle, L. Larsson, Y. Bai, B. Zhu, A. Bhaduri, J. M. Meyers, X. Rovira-Clave, S. T. Hollmig, S. Z. Aasi, G. P. Nolan, J. Lundeberg and P. A. Khavari, Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma, Cell, 2020, 182(2), 497–514 CrossRef CAS PubMed.
- D. Schapiro, H. W. Jackson, S. Raghuraman, J. R. Fischer, V. R. T. Zanotelli, D. Schulz, C. Giesen, R. Catena, Z. Varga and B. Bodenmiller, histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data, Nat. Methods, 2017, 14(9), 873–876 CrossRef CAS PubMed.
- S. Chevrier, H. L. Crowell, V. R. T. Zanotelli, S. Engler, M. D. Robinson and B. Bodenmiller, Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry, Cell Syst., 2018, 6(5), 612–620 CrossRef CAS PubMed.
- L. Restrepo-Perez, C. Joo and C. Dekker, Paving the way to single-molecule protein sequencing, Nat. Nanotechnol., 2018, 13(9), 786–796 CrossRef CAS PubMed.
- W. Timp and G. Timp, Beyond mass spectrometry, the next step in proteomics, Sci. Adv., 2020, 6(2), eaax8978 CrossRef CAS PubMed.
- A.-D. Brunner, M. Thielert, C. G. Vasilopoulou, C. Ammar, F. Coscia, A. Mund, O. B. Hoerning, N. Bache, A. Apalategui, M. Lubeck, S. Richter, D. S. Fischer, O. Raether, M. A. Park, F. Meier, F. J. Theis and M. Mann, Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation, bioRxiv, 2020.2012.2022.423933, 2021.
- R. T. Kelly, Single-cell Proteomics: Progress and Prospects, Mol. Cell. Proteomics, 2020, 19(11), 1739–1748 CrossRef CAS PubMed.
- Z. Y. Li, M. Huang, X. K. Wang, Y. Zhu, J. S. Li, C. C. L. Wong and Q. Fang, Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis, Anal. Chem., 2018, 90(8), 5430–5438 CrossRef CAS PubMed.
- R. Jungmann, M. S. Avendano, J. B. Woehrstein, M. Dai, W. M. Shih and P. Yin, Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT, Nat. Methods, 2014, 11(3), 313–318 CrossRef CAS PubMed.
- V. M. Peterson, K. X. Zhang, N. Kumar, J. Wong, L. Li, D. C. Wilson, R. Moore, T. K. McClanahan, S. Sadekova and J. A. Klappenbach, Multiplexed quantification of proteins and transcripts in single cells, Nat. Biotechnol., 2017, 35(10), 936–939 CrossRef CAS PubMed.
- M. Stoeckius, C. Hafemeister, W. Stephenson, B. Houck-Loomis, P. K. Chattopadhyay, H. Swerdlow, R. Satija and P. Smibert, Simultaneous epitope and transcriptome measurement in single cells, Nat. Methods, 2017, 14(9), 865–868 CrossRef CAS PubMed.
- Y. Goltsev, N. Samusik, J. Kennedy-Darling, S. Bhate, M. Hale, G. Vazquez, S. Black and G. P. Nolan, Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging, Cell, 2018, 174(4), 968–981 CrossRef CAS PubMed.
- S. K. Saka, Y. Wang, J. Y. Kishi, A. Zhu, Y. Zeng, W. Xie, K. Kirli, C. Yapp, M. Cicconet, B. J. Beliveau, S. W. Lapan, S. Yin, M. Lin, E. S. Boyden, P. S. Kaeser, G. Pihan, G. M. Church and P. Yin, Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues, Nat. Biotechnol., 2019, 37(9), 1080–1090 CrossRef CAS PubMed.
- C. R. Merritt, G. T. Ong, S. E. Church, K. Barker, P. Danaher, G. Geiss, M. Hoang, J. Jung, Y. Liang, J. McKay-Fleisch, K. Nguyen, Z. Norgaard, K. Sorg, I. Sprague, C. Warren, S. Warren, P. J. Webster, Z. Zhou, D. R. Zollinger, D. L. Dunaway, G. B. Mills and J. M. Beechem, Multiplex digital spatial profiling of proteins and RNA in fixed tissue, Nat. Biotechnol., 2020, 38(5), 586–599 CrossRef CAS PubMed.
- J. D. Scott and T. Pawson, Cell signaling in space and time: where proteins come together and when they're apart, Science, 2009, 326(5957), 1220–1224 CrossRef CAS PubMed.
- K. C. Hadley, R. Rakhit, H. Guo, Y. Sun, J. E. Jonkman, J. McLaurin, L. N. Hazrati, A. Emili and A. Chakrabartty, Determining composition of micron-scale protein deposits in neurodegenerative disease by spatially targeted optical microproteomics, eLife, 2015, 4, e09579 CrossRef PubMed.
- B. Yin, R. Mendez, X. Y. Zhao, R. Rakhit, K. L. Hsu and S. E. Ewald, Automated Spatially Targeted Optical Microproteomics (autoSTOMP) to Determine Protein Complexity of Subcellular Structures, Anal. Chem., 2020, 92(2), 2005–2010 CrossRef CAS PubMed.
- W. Qin, K. F. Cho, P. E. Cavanagh and A. Y. Ting, Deciphering molecular interactions by proximity labeling, Nat. Methods, 2021, 18(2), 133–143 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |