
Carbon aerogel-based solid-phase microextraction coating for the analysis of organophosphorus pesticides†
Piia
Jõul
 *,
Merike
Vaher
and
Maria
Kuhtinskaja
*,
Merike
Vaher
and
Maria
Kuhtinskaja
Department of Chemistry and Biotechnology, Tallinn University of Technology, Akadeemia Tee 15, 12618 Tallinn, Estonia. E-mail: piia.joul@gmail.com
First published on 20th November 2020
Abstract
The current study is focused on the in situ synthesis of a carbon aerogel (CA)-based solid-phase microextraction (SPME) fiber coating on stainless steel wire and evaluation of the suitability of CAs as SPME coating materials for the analysis of selected organophosphorus pesticides (OPPs) contained in environmental samples. A CA-based coating was obtained by pyrolyzing organic aerogels, which were prepared by the sol–gel polymerization of formaldehyde and 5-methylresorcinol, an oil shale processing by-product. The results demonstrated, for the first time, the in situ synthesis of a CA-based SPME fiber coating on stainless steel wire and its suitability for the extraction and preconcentration of six OPPs. Main parameters affecting the extraction efficiency were investigated and optimized. The direct immersion (DI)-SPME procedure combined with gas chromatography-mass spectrometry (GC-MS) for the simultaneous analysis of selected OPPs was successfully applied to the efficient and sensitive determination of analytes of interest in environmental matrices of honey and natural water samples. The developed CA-coated SPME fiber showed good linearity (R2 = 0.981–0.994), low detection limits (0.11–0.83 μg L−1) and satisfactory single fiber and fiber-to-fiber reproducibilities (8.8–12.3%, n = 5 and 11.4–17.2%, n = 3). The performance of the CA-coating was compared with that of commercially available SPME fiber coatings.
1. Introduction
Solid-phase microextraction (SPME) is an extraction and preconcentration technique, which was introduced by Arthur and Pawliszyn1 to address the need for a rapid sample preparation method.2 In this microextraction technique, a small amount of extracting phase on a small diameter fiber is exposed to the sample and after a defined period of time the analytes are partitioned into the stationary phase, which combines the processes of sampling, isolation and enrichment in a single step. Then the coated fiber is removed from the sample and the analytes are desorbed either by thermally directly into a gas chromatograph or using a desorption solvent.1,6The main goal of sample pretreatment is to eliminate interfering compounds from the complex matrix by using a minimum number of steps, to give a reproducible methodology.2 Being an easy-to-use method for analyzing compounds in complicated matrices and due to the miniaturized design of SPME fibers, it precisely serves the purpose. It enables the analysis of wine volatiles, in vivo analysis of pollutants, on-site analysis of soil and water samples and food analysis; in addition, it makes in vitro and in vivo metabolomic studies and pharmaceutical and biomedical analyses possible, and even allows for analyses without separation techniques, for example direct coupling of SPME to MS (mass spectrometry).3 Moreover, this versatile and nonexhaustive sample preparation technique has been gaining wide interest, because in comparison to other sample extraction methods, including traditional solid-phase extraction (SPE), it is rather inexpensive due to the reduced consumption of high-purity solvents. As a result, the need for solvent deposal is diminished and implementation of Green Chemistry principles in analytical practice is supported.3–5
Affecting the efficiency, sensitivity and selectivity of extraction, the use of an appropriate SPME fiber coating for a particular application is highly important.3 There are several kinds of commercial SPME fibers available, including polydimethylsiloxane (PDMS), polyacrylate (PA), polyethyleneglycol, divinylbenzene (DVB), DVB-PDMS, Carbopack Z-PDMS, DVB-CAR-PDMS, Carboxen (CAR)-PDMS and DVB-CAR-PDMS coated fibers.7 However, despite their successful utilization in various applications, commercial SPME fibers have also some limitations in relation to operation temperature, selectivity, robustness, carryover, swelling in solvents, and poor affordability. Considering this the development of alternative SPME coating materials presents a very active area of research.3,4 Graphene, porous carbon, molecularly imprinted polymers, metal–organic frameworks, polymers, metal or metal oxide nanoparticles and ionic liquids are only a few examples of new coating materials, which have been already applied for the extraction of different sample matrices and compounds.4,8,9 Among these materials, porous carbons have attracted a lot of attention. High surface area, physicochemical stability and density as well as well-developed pore structure make them suitable materials to be used as SPME fiber coatings, providing excellent sorption and enrichment capability.8,10,11
Carbon aerogels (CAs) are one of those porous carbon materials, which have great potential for use as sorbents in different sample preparation techniques, including SPME to achieve high selectivity, sensitivity and throughput of analysis of various compounds contained in complex matrices.12,13 Aerogels are porous nanostructured materials whose production process involves three main stages: sol–gel preparation, aging and drying. CAs can be produced by an additional stage of organic aerogel production process, carbonization or pyrolysis.12,14 Due to their well-developed porous structure and advantageous properties, CAs can be used in various applications, for example, preparation of biosensors,15 storage of hydrogen,16 preparation of catalyst supports,17 electrodes and supercapacitors,18,19 adsorbents,20–22etc. In the last few decades, interest in using CAs as sorbent materials in sample preparation techniques, especially SPE,23,24 has been increasing.
CAs as SPE sorbents have been successfully applied for the simultaneous extraction of sulfur mustard degradation products from environmental water samples.25,26 A CA-based SPE sorbent was obtained by pyrolyzing organic aerogels, which were prepared by the sol–gel polymerization of formaldehyde (FA) and 5-methylresorcinol (MR), an oil shale processing by-product.12 Dong et al.23 used CAs for the analysis of plant growth regulators by employing micro-SPE and magnetic SPE techniques, referred to as environmentally friendly methods. However, there have been but a few studies on the use of CAs as coating materials of SPME fibers.
In addition to using a suitable SPME coating material, the choice of a suitable coating support and method is similarly important. The most common supports are metal or fused silica ones, while dip coating, glue method, sol–gel and deposition outweigh other coating methods for carbon-based materials.8
Zhu et al.27 were one of the first researchers to use CAs as SPME coating materials. The investigators compared the performance of two kinds of SPME fibers coated with porous carbon materials, namely CAs and wormhole-like mesoporous carbons, with that of a commercial PDMS fiber for the headspace solid-phase microextraction (HS-SPME) of four non-polar compounds and five polar compounds from water samples. Six types of fibers were prepared by three different coating methods using stainless steel wire as a supporting core. The results indicated the efficiency of CA-based fibers to be higher than those of other fibers in the extraction of both non-polar and polar analytes.27 Zheng et al.10 coated stainless steel wire with powdered CA by using the glue method and obtained a suitable fiber for the HS-SPME of hydrophobic analytes with one or two benzene rings from water samples. Compared to other fibers such as commercial PDMS and PDMS/DVB, as well as powdered polymer aerogel-coated fibers, the enrichment factors of target analytes obtained by HS-SPME-GC-FID using powdered CA-coated fibers were higher. By that, factors influencing the enrichment effect most included π–π interactions, van der Waals forces and hydrophobic interactions, as well as surface area and microporosity.10
Organophosphorus pesticides (OPPs) are widely used as insecticides in agriculture, but quite often, this use is related to poisoning cases. The abuse or erroneous application of pesticides, including highly persistent, bioaccumulative and biocidal OPPs, causes harm to the environment and contaminates foodstuff.28 Thus, the development of a selective and sensitive method for the determination of OPPs in environmental water samples and foodstuff is highly required. The most widely applied analytical instruments for the determination of OPPs are gas chromatography (GC) and high-performance liquid chromatography (HPLC) coupled with various detectors, but the complex and diversity of environmental sample matrices make the direct analysis of pesticides very difficult, requiring, an appropriate sample pretreatment technique.28,29 Honey is one of such matrices and OPPs in honey may originate from contaminated plants where bees collect nectar to make honey, or from pest- and disease-controlled beehives.30 SPME is considered a suitable sample pretreatment technique for the extraction of OPPs from environmental samples, including honey and natural water.29,31 For this purpose, the method uses both commercial32–37 and lab-made SPME fibers.38–42 However, to the best of the authors' knowledge, CAs have not been applied in SPME as fiber coating materials to analyze OPPs in complex sample matrices.
Hence, the current study is focused on the in situ synthesis of a CA-based SPME fiber coating on stainless steel wire, as well as evaluation of the applicability of coated fibers for the analysis of selected OPPs in environmental matrices, such as honey and natural water samples. The direct immersion solid-phase microextraction (DI-SPME) procedure coupled with gas chromatography-mass spectrometry (GC-MS) is optimized for the analysis of six OPPs: heptenophos, paraoxon-ethyl, tetrachlorvinphos, chlorfenvinphos, parathion and malathion. These OPPs were chosen for this study as model compounds based on their structural and thermal stability as well as volatility for the thermal desorption procedure. Moreover, these OPPs are the typical pesticides the residues of which are monitored in food/feed items and in the environment. The performance of novel SPME fibers is compared with that of the most common commercial SPME fibers.
2. Experimental
2.1 Reagents and materials
Organophosphorus pesticides heptenophos, paraoxon-ethyl, tetrachlorvinphos, chlorfenvinphos, parathion and malathion, and chemical compounds sodium chloride (NaCl) and sodium hydroxide (NaOH) were purchased from Sigma-Aldrich (Germany). Pure OPP standards were refrigerated in their original packages at 4 °C. Methanol and acetonitrile (HPLC grade ≥ 99.9%) were purchased from Sigma-Aldrich (Germany). All the chemicals were of analytical grade and were used as received. Deionized water from a Milli-Q water purification system (Millipore S. A. Molsheim, France) was used throughout the study.For the comparative analysis of SPME fibers with 65 μm PDMS/DVB, 50/30 μm DVB/CAR/PDMS, 85 μm CAR/PDMS and 85 μm PA coatings were purchased from Supelco (Bellefonte, PA, USA).
MR with a reported purity of >99% was provided by AS VKG (Estonia). The FA solution (37 wt% in H2O) and sodium carbonate (Na2CO3) powder (≥99.0%) were purchased from Sigma-Aldrich (Germany). Stainless steel wire 200 μm thick and epoxy glue were purchased from a local hardware store. Tubings with ID of 395 μm were purchased from IDEX Health & Science LLC (USA).
2.2 Sample preparation
The stock solutions of OPP standards were prepared with MeOH, then stored in the dark and refrigerated at −20 °C. The working solutions were prepared daily by appropriate dilution of the stock solutions and stored at 4 °C.Natural water samples were taken from the Pirita River and the Tallinn Bay, Baltic Sea (Estonia), spiked and analyzed directly by DI-SPME-GC-MS. Honey samples were obtained from local beekeepers. In order to study the honey matrix effect and the recovery of the DI-SPME-GC-MS analysis procedure, the samples were spiked with a certain amount of each pesticide standard to the 0.5 mL aliquot of the honey–water mixture (1 mL of the sample was dissolved in 1 mL of MilliQ water), mixed at least for 30 min and then analyzed following the above DI-SPME-GC-MS procedure. Before spiking the samples were checked for any kind of target analytes. The concentrations of OPPs for the spiked samples, as well as optimization and calibration standards were carefully selected to ensure adequate sensitivity to all the analytes.
2.3 Preparation of carbon aerogel-based solid-phase microextraction fibers
The CA-based coating was in situ synthesized on stainless steel wire. Before the coating procedure, 200 μm stainless steel wire was cut to pieces, each 1.5 cm in length. The wire pieces were immersed in acetone for 30 min, then in 1 M NaOH for 30 min and finally washed with MilliQ water for 30 min. Thereafter they were taken out and dried at room temperature.After that the stainless steel wire pieces were inserted into empty 1.7 cm tubings (ID 0.395 mm) and filled with the initial mixture, a fresh water solution of MR, Na2CO3 and FA (molar ratios MR/Na2CO3 = 90, water/MR = 45 and MR/FA = 0.5) prepared according to the literature.12 Then both ends of the tubings were closed with Parafilm and the filled tubings were kept overnight at room temperature for proper gelation. After gelation, both ends of the tubings were opened and the filled tubings were placed in acetone. Acetone was changed every 24 hours in the course of four days. To obtain organic aerogel-coated metal wires, the gel inside the tubes had to be dried with supercritical CO2. Thus, at first, acetone in the gel pores was replaced by liquid CO2 (120 bar, 25 °C for 1 h) and then subjected to supercritical drying (100 bar, 45 °C for 1.5 h). For this purpose a supercritical extraction system including a 100 mL double-clamp autoclave (NWA Analytische Meßgeräte GmbH, Germany) was used. After the supercritical drying the wires coated with organic aerogel were removed from the tubings.
In the next step, the CA-coated wires were fabricated by pyrolysing organic aerogel-coated wires. The pyrolysis was carried out in a N2 atmosphere by using an MTF 12/38/400 pyrolysis oven (Carbolite, England). The carbonization temperature program of the oven was as follows. The initial oven temperature was 25 °C, then the temperature was increased to 300 °C at a rate of 10 °C min−1 (held for 10 min), thereafter increased to 550 °C at 10 °C min−1 (held for 10 min) and finally increased to 900 °C at 10 °C min−1 (held for 1 h). After pyrolysis, the furnace was allowed to cool down to room temperature in a N2 atmosphere. The prepared coated SPME fiber was fixed with epoxy glue to a commercial fiber assembly, after removing the original core from the inner tube (Fig. S1 and S2, ESI†).
2.4 DI-SPME procedure
The CA-based fiber assembly was attached to the manual SPME holder and conditioned in the injection port of the gas chromatograph at 300 °C for 30 min prior to use. Commercial fibers were conditioned according to the manufacturer's instructions. The CA-coated SPME fibers were preconditioned using a MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MilliQ water solution (1:1, v/v) for 5 min. For the DI-SPME procedure the coated fiber protected in the septum piercing needle was inserted into the sample vial, then pushed out from the needle and immersed into the sample solution (0.5 mL) for 20 min at room temperature. Both procedures, preconditioning and sorption, were carried out using an ELMI DOS-20M Digital Orbital Shaker (agitation 200 rpm). Before desorption, the fiber was rinsed with MilliQ water for 5 s to remove possible interfering matrix components. After that the fiber was withdrawn from the needle and inserted into the GC injection port where the analytes were thermally desorbed for the GC-MS analysis.
:MilliQ water solution (1:1, v/v) for 5 min. For the DI-SPME procedure the coated fiber protected in the septum piercing needle was inserted into the sample vial, then pushed out from the needle and immersed into the sample solution (0.5 mL) for 20 min at room temperature. Both procedures, preconditioning and sorption, were carried out using an ELMI DOS-20M Digital Orbital Shaker (agitation 200 rpm). Before desorption, the fiber was rinsed with MilliQ water for 5 s to remove possible interfering matrix components. After that the fiber was withdrawn from the needle and inserted into the GC injection port where the analytes were thermally desorbed for the GC-MS analysis.
2.5 GC-MS analysis
Chromatographic separations were performed on an Agilent Technologies 7890A GC system equipped with an ultra inert splitless liner (Agilent Technologies, type 5190-2293) to eliminate matrix-induced chromatographic response enhancement. The gas chromatograph was coupled to an Agilent 5975C mass spectrometer with an electron ionization source and a quadrupole mass analyzer. Instrument control was carried out with MassHunter Acquisition software. The separation of pesticides from the samples was performed on a ZB-5MSi capillary column (30 m × ID 0.25 mm, film thickness 0.25 μm, Agilent Technologies). Helium (6.0 purity) was used as a carrier gas at a constant flow rate of 1.3 mL min−1. Sample injection was performed in splitless mode at 275 °C for 2.0 min (an optimized value for SPME thermal desorption). The oven temperature programming was performed as follows. The initial temperature, 60 °C, was held for 1 min, then increased to 180 °C at a rate of 25 °C min−1, held for 2 min and finally increased to 250 °C at a rate of 15 °C min−1 and held for 2.5 min. The total analysis time was 15 min starting from fiber introduction into the injector block.The analyte ionization was performed in electron ionization mode using the electron energy of 70 eV. The temperatures of the interface, ion source and mass analyzer were set at 280, 230 and 150 °C, respectively. All analytes were monitored using selected ion monitoring (SIM) mode. Chromatographic peaks were identified by measuring the retention times of individual OPP standards and confirmed by the fragmentation pattern of molecules and database results (NIST17). Quantitative analysis was carried out in SIM mode using a specific quantifying ion for each target analyte (Table 1). Data treatment was performed with Agilent MassHunter Workstation software.
| Pesticide | Quantitation ion, m/z | Linearity, μg L−1 | R 2 | LOD, μg L−1 | LOQ, μg L−1 | RSD single fiber (n = 5), % | RSD fiber-to-fiber (n = 3), % |
|---|---|---|---|---|---|---|---|
| Heptenophos | 215.0 | 0.50–62.7 | 0.981 | 0.15 | 0.50 | 9.3 | 11.4 |
| Paraoxon-ethyl | 275.0 | 2.8–68.8 | 0.985 | 0.83 | 2.8 | 10.3 | 17.2 |
| Malathion | 173.0 | 0.66–165.2 | 0.990 | 0.20 | 0.66 | 8.8 | 16.5 |
| Parathion | 291.0 | 0.58–145.6 | 0.994 | 0.18 | 0.58 | 10.1 | 14.9 |
| Chlorfenvinphos | 323.0 | 0.42–103.8 | 0.990 | 0.12 | 0.42 | 12.3 | 14.8 |
| Tetrachlorvinphos | 331.0 | 0.37–91.5 | 0.983 | 0.11 | 0.37 | 9.6 | 17.1 |
3. Results and discussion
3.1 Characterization of the coating material
The surface morphology of the CA-coated SPME fiber was examined with a high resolution scanning electron microscope (HR-SEM Zeiss Merlin). The SEM image of the surface of the obtained carbon coated fiber is shown in Fig. 1. As can be seen, the fiber surface looks like a solid shell with evenly distributed pores. The morphology of the cross-section of the obtained fiber was similar to that of the fiber surface. | ||
| Fig. 1 SEM image of the surface of the CA-coated SPME fiber. | ||
The nitrogen adsorption–desorption isotherms of CA were recorded using a Quantachrome Autosorb iQ-C instrument. The results of the pore size distribution analysis, which was based on the density functional theory (DFT), showed that the fiber coating contained mainly micropores, diameter < 2 nm according to IUPAC (Fig. S4 and S5, ESI†). The thickness of the coating was about 20 μm, which varied between different fibers insignificantly. The specific surface area obtained by Brunauer–Emmett–Teller (BET) analysis was 501.157 m2 g−1, giving evidence of the formation of a large adsorption area.
It is known that carbon-based sorbent/analyte interactions involve more than one adsorption mechanism.10,43 During the pyrolysis of organic aerogels most of the hydrophilic functional groups, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, O–H and C–H, are eliminated under the influence of high temperature, as a result of which a relatively highly hydrophobic material is formed. The X-ray photoelectron spectroscopy (XPS, Kratos AXIS Ultra DLD) analysis of the obtained coating material indicated the dominance of carbon atoms in the sp2 (66.8%) and sp3 (12.90%) hybridization as well as a minor presence of C–O (8.52%), CO (4.05%) and –O– (2.08%) groups (Fig. S3 and Table S1, ESI†). Taking into account the nonpolar and hydrophobic nature of both interacting sides, we assume that hydrophobic interactions play one of the most important roles in the sorption process. Moreover, due to the existence of conjugated systems in the analytes under study and sp2 carbon, π–π interactions (π-stacking as well as π donor–acceptor interactions) may be considered as another strong supramolecular force responsible for the mechanism of sorption. In addition, a considerable amount of C–O, CO and other oxygen functionalities on the fiber surface allows the occurrence of dipole–dipole interactions between the phosphorus atom of pesticides and the electronegative oxygen of the sorbent.
O, C–O, O–H and C–H, are eliminated under the influence of high temperature, as a result of which a relatively highly hydrophobic material is formed. The X-ray photoelectron spectroscopy (XPS, Kratos AXIS Ultra DLD) analysis of the obtained coating material indicated the dominance of carbon atoms in the sp2 (66.8%) and sp3 (12.90%) hybridization as well as a minor presence of C–O (8.52%), CO (4.05%) and –O– (2.08%) groups (Fig. S3 and Table S1, ESI†). Taking into account the nonpolar and hydrophobic nature of both interacting sides, we assume that hydrophobic interactions play one of the most important roles in the sorption process. Moreover, due to the existence of conjugated systems in the analytes under study and sp2 carbon, π–π interactions (π-stacking as well as π donor–acceptor interactions) may be considered as another strong supramolecular force responsible for the mechanism of sorption. In addition, a considerable amount of C–O, CO and other oxygen functionalities on the fiber surface allows the occurrence of dipole–dipole interactions between the phosphorus atom of pesticides and the electronegative oxygen of the sorbent.
3.2 Optimization of extraction conditions
To achieve the highest extraction efficiency of the lab-made CA-based fiber, optimization of SPME conditions such as extraction time, desorption time, desorption temperature and salt concentration was necessary. All the experiments were performed in triplicate with 0.5 mL water samples spiked with target OPPs at a final concentration of 2.5 mg L−1 heptenophos, 2.8 mg L−1 paraoxon-ethyl, 3.3 mg L−1 malathion, 2.9 mg L−1 parathion, 2.6 mg L−1 chlorfenvinphos, and 3.7 mg L−1 tetrachlorvinphos. | ||
| Fig. 2 The effect of different factors on SPME efficiency. (a) Extraction time, (b) desorption temperature, (c) desorption time, (d) NaCl concentration. The concentration of OPPs in 0.5 mL of working solution, 2.5 mg L−1 heptenophos, 2.7 mg L−1 paraoxon-ethyl, 3.7 mg L−1 tetrachlorvinphos, 2.6 mg L−1 chlorfenvinphos, 2.9 mg L−1 parathion and 3.3 mg L−1 malathion. | ||
The desorption time at optimal temperature should be as short as possible to decrease the analysis time. However, a very short time may lead to a carryover effect and may not enable a complete transfer of pesticides by the carrier gas to the separation column. The desorption time of 0.5–3 min was used to investigate the desorption of analytes from the CA-coated SPME fiber to the gas phase (Fig. 2c). The experimental results demonstrated that the efficiency of pesticide extraction was the highest, at a temperature of 275 °C during 2.0 min with their complete desorption. Moreover, no carry over effect of pesticides was observed, which allowed the fiber to be used repeatedly.
3.3 Performance of DI-SPME-GC-MS under optimal extraction conditions
Based on the results of experiments, the extraction time of 20 min, desorption temperature of 275 °C, desorption time of 2.0 min proved to be the most optimal for DI-SPME. The effect of the preconcentration of the CA-coated fiber on pesticides extraction under optimized conditions was investigated using a solution of target OPPs (1.3 mg L−1 heptenophos, 1.4 mg L−1 paraoxon-ethyl, 1.7 mg L−1 malathion, 1.5 mg L−1 parathion, 1.3 mg L−1 chlorfenvinphos, and 1.8 mg L−1 tetrachlorvinphos). Fig. 3 compares two extracted ion chromatograms obtained by DI-SPME-GC-MS of water samples spiked with OPPs at the final concentration of 1.3 mg L−1 heptenophos (1), 1.4 mg L−1 paraoxon-ethyl (2), 1.7 mg L−1 malathion (3), 1.5 mg L−1 parathion (4), 1.3 mg L−1 chlorfenvinphos-exists in two isometric forms (5), and 1.8 mg L−1 tetrachlorvinphos (6) (colored) and injecting the test solution, the same concentration of target OPPs as in the spiked water sample, in acetonitrile, into GC-MS without the DI-SPME procedure (black). | ||
| Fig. 3 Extracted ion chromatograms of DI-SPME-GC-MS of water sample spiked with OPPs at the final concentration of 1.3 mg L−1 heptenophos (1), 1.4 mg L−1 paraoxon-ethyl (2), 1.7 mg L−1 malathion (3), 1.5 mg L−1 parathion (4), 1.3 mg L−1 chlorfenvinphos exists in two isometric forms (5), and 1.8 mg L−1 tetrachlorvinphos (6) (colored) under optimum extraction conditions and of GC-MS of the same concentration of target OPPs, as in the spiked water sample, in acetonitrile (black). | ||
In the latter case, the recommended maximum volume of solution to be injected was 2 μL to avoid the carryover effect. As seen from Fig. 3, the coating material has a significant binding affinity for all target analytes, demonstrating its ability towards preconcentration of analytes and improvement of the method sensitivity.
3.4 Evaluation of the performance of DI-SPME-GC-MS
The analytical performance of the DI-SPME-GC-MS method using the CA-coated fiber was evaluated under optimized extraction conditions and validated using MilliQ water samples spiked with target pesticide standards. For this purpose, the chromatographic peak area of each pesticide was plotted against the corresponding concentration. The linearity, coefficients of determination (R2), limits of detection (LODs), limits of quantification (LOQs) and repeatability as relative standard deviation (RSD) of single fiber and fiber-to-fiber were determined. As revealed by Table 1, for all pesticides the linearity R2 ranged from 0.981 to 0.994. LODs based on the signal-to-noise ratio of three (S/N = 3) for the analytes varied from 0.11 to 0.83 μg L−1, and LOQs (S/N = 10) of the method were in the range of 0.37–2.8 μg L−1. Under the optimized conditions of DI-SPME-GC-MS, RSDs varied from 8.8 to 12.3%, while all the replicate experiments were performed using a single fiber (n = 5). The repeatability for the three CA-coated SPME fibers was in the range of 11.4–17.2%. The thermal and solvent stabilities of the coating were determined as well. The experiments showed that the same fiber could be used at least in 80 sorption/desorption cycles with no significant decrease in extraction efficiency. Nevertheless, the fragile coating material is prone to damage and requires caution in handling. The obtained results show that the CA-based fiber coating provides satisfactory accuracy and precision of extraction and is suitable to be used for sample pretreatment and further GC-MS analysis of target OPPs.3.5 Functionality of the CA-based coating on real samples
To evaluate the applicability of the CA-based fiber coating for the analysis of selected OPPs in actual environmental samples, the novel DI-SPME-GC-MS analysis method was applied to the determination of six OPPs in three different samples. Two natural water samples were collected from the Pirita River and the Tallinn Bay, Baltic Sea (Estonia) and their recoveries were measured by spiking the samples with the initial concentrations of OPPs, as listed in Table 2. The recoveries recorded were in the range of 81.8–96.1% (n = 3, RSD < 14%). Another promising application of the developed method, which was tested to demonstrate the fiber coating applicability, was the analysis of OPPs in a more complex sample matrix, honey. In which case the recovery was 82.5–88.1%, n = 3, and RSD < 13%. The recoveries of the spiked natural water and honey samples are given in Table 2. All the blank samples subjected to analysis showed no traces of target OPPs. The results of analysis of real samples demonstrated that the CA-coated fiber was suitable for this kind of extraction and preconcentration. No such matrix effect in the determination OPPs in natural water and honey samples was observed. However, further matrix-matched calibration experiments will be required to estimate the applicability of the novel CA-based DI-SPME-GC-MS method to the analysis of other complex food and environmental samples.| Pesticide | Spiked concentration, μg L−1 | Recovery, % | ||
|---|---|---|---|---|
| River water | Seawater | Honey | ||
| Heptenophos | 2.5 | 82.3 | 96.1 | 82.5 |
| Paraoxon-ethyl | 13.8 | 85.8 | 91.8 | 86.6 |
| Malathion | 6.6 | 83.6 | 85.1 | 84.9 |
| Parathion | 1.5 | 85.4 | 83.9 | 87.0 |
| Chlorfenvinphos | 20.8 | 89.8 | 81.8 | 88.1 |
| Tetrachlorvinphos | 1.8 | 88.6 | 92.9 | 85.7 |
3.6 Comparison of CA-coated and commercial fibers
The extraction performance of CA-coated fibers in relation to six OPPs was compared with that of four commercial fibers (65 μm PDMS/DVB, 50/30 μm DVB/CAR/PDMS, 85 μm CAR/PDMS, 85 μm PA) under the optimized extraction conditions. The normalized peak areas obtained using the CA-coated and commercial fibers are shown in Fig. 4. The figure displays that the ability of CA-coated fibers to extract most target OPPs was similar to or even higher than that of commercial fibers. However, in the case of parathion and malathion, the extraction performance of CA-coated fibers was significantly higher compared to commercial ones. At the same time, commercial CAR-coated fibers exhibited a higher capacity to extract one of the pesticides, heptenophos. The high binding affinity of CA-coated SPME fibers for target OPPs may be due to the high surface area and porosity of CA, as well as strong π–π interactions and other sorption mechanisms typical of carbon-based materials.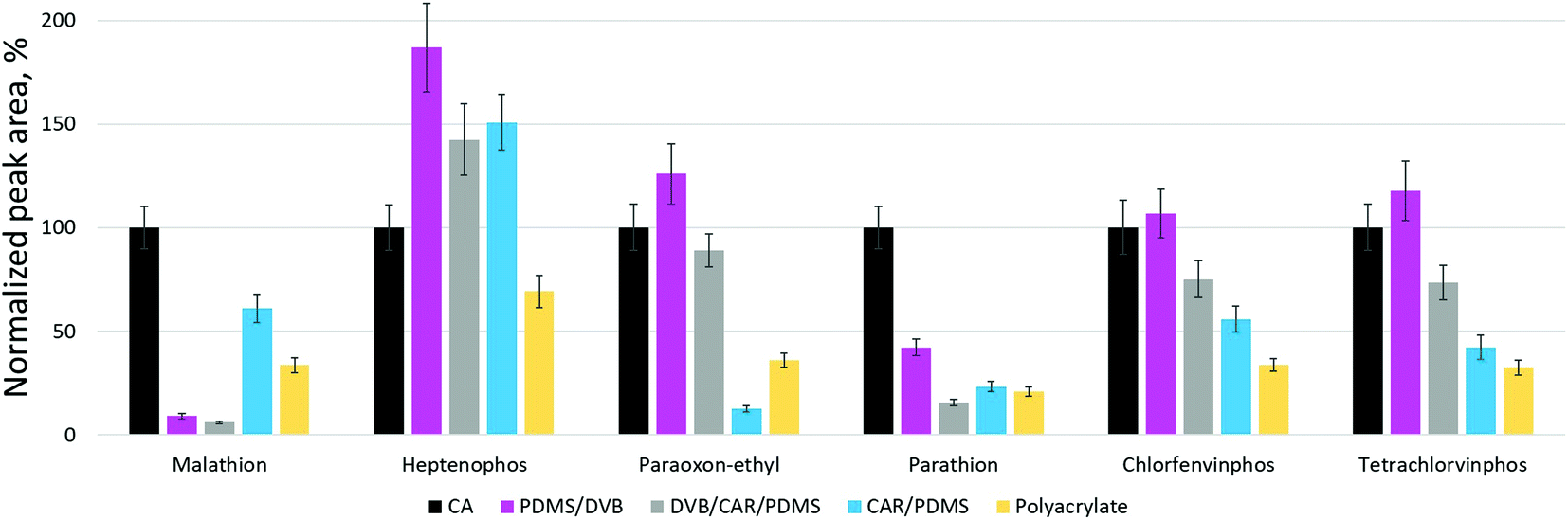 | ||
| Fig. 4 Extraction of OPPs from water samples using different SPME fibers. | ||
4. Conclusions
The results obtained verify the suitability of wire-supported CAs as SPME fiber coatings for the extraction and preconcentration of six OPPs. The DI-SPME-GC-MS method was successfully applied to the simultaneous determination of analytes of interest in complex environmental matrices such as honey and natural water samples. The extraction efficiency of CA-based SPME fibers was comparable to that of commercial ones, being in some cases even higher. The current study gives evidence of the high analytical potential of this type of coating procedure and sorbents for microextraction techniques and micro total analysis systems of samples of interest. Further studies of CA-based sorbents in microextraction techniques are in progress, including the improvements of coating durability and some additional aspects affecting the extraction process and applicability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Estonian Research Council (Institutional Research Fund No. 33-20), the European Regional Development Fund and the Graduate School of Functional Materials and Technologies, University of Tartu, receiving funding from the European Regional Development Fund. The authors would like to thank Dr Olga Volobujeva for providing SEM images, Dr Mati Danilson for performing XPS analysis and Dr Heidi Lees for carrying out N2 adsorption analysis.References
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- J. Pawliszyn, Handbook of Solid Phase Microextraction, Elsevier, Sydney, 2012 Search PubMed.
- N. Reyes-Garcés, E. Gionfriddo, G. A. Gómez-Ríos, M. N. Alam, E. Boyacl, B. Bojko, V. Singh, J. Grandy and J. Pawliszyn, Anal. Chem., 2018, 90, 302–360 CrossRef.
- F. A. Hansen and S. Pedersen-Bjergaard, Anal. Chem., 2020, 92, 2–15 CrossRef CAS.
- S. Armenta, S. Garrigues, F. A. Esteve-Turrillas and M. de la Guardia, Trends Anal. Chem., 2019, 116, 248–253 CrossRef CAS.
- H. Lord and J. Pawliszyn, J. Chromatogr. A, 2000, 885, 153–193 CrossRef CAS.
- E. Y. Zeng, Persistent Organic Pollutants (POPs): Analytical Techniques, Environmental Fate and Biological Effects, Elsevier, Amsterdam, 2015 Search PubMed.
- J. Zheng, J. Huang, Q. Yang, C. Ni, X. Xie, Y. Shi, J. Sun, F. Zhu and G. Ouyang, Trends Anal. Chem., 2018, 108, 135–153 CrossRef CAS.
- Á. I. Fresco-Cala, B. López-Lorente, M. L. Soriano, R. Lucena and S. Cárdenas, in Handbook of Smart Materials in Analytical Chemistry, ed. M. de la Guardia and F. A. Esteve-Turrillas, John Wiley & Sons, Chichester, 2019, pp. 273–308 Search PubMed.
- J. Zheng, J. Huang, F. Xu, F. Zhu, D. Wu and G. Ouyang, Nanoscale, 2017, 9, 5545–5550 RSC.
- O. J. Portillo-Castillo, R. Castro-Riós, A. Chávez-Montes, A. González-Horta, N. Cavazos-Rocha, N. H. W. De Torres and M. Garza-Tapia, Rev. Anal. Chem., 2018, 37, 1–22 Search PubMed.
- F. Pérez-Caballero, A.-L. Peikolainen, M. Uibu, R. Kuusik, O. Volobujeva and M. Koel, Microporous Mesoporous Mater., 2008, 108, 230–236 CrossRef.
- V. Jalili, A. Barkhordari and M. Heidari, Microchem. J., 2019, 147, 948–954 CrossRef CAS.
- R. W. Pekala, J. Mater. Sci., 1989, 24, 3221–3227 CrossRef CAS.
- S. Dong, N. Li, G. Suo and T. Huang, Anal. Chem., 2013, 85, 11739–11746 CrossRef CAS.
- H. Kabbour, T. F. Baumann, J. H. Satcher, A. Saulnier and C. C. Ahn, Chem. Mater., 2006, 18, 6085–6087 CrossRef CAS.
- X. Hou, J. Zhao, J. Liu, Y. Han, Y. Pei and J. Ren, New J. Chem., 2019, 43, 9430–9438 RSC.
- R. Liu, L. Wan, S. Liu, L. Pan, D. Wu and D. Zhao, Adv. Funct. Mater., 2015, 25, 526–533 CrossRef CAS.
- A. N. Malkova, N. A. Sipyagina, I. O. Gozhikova, Y. A. Dobrovolsky, D. V. Konev, A. E. Baranchikov, O. S. Ivanova, A. E. Ukshe and S. A. Lermontov, Molecules, 2019, 24, 1–13 CrossRef.
- S. Abolhasani, A. Ahmadpour, T. Rohani Bastami and A. Yaqubzadeh, J. Mol. Liq., 2019, 281, 261–268 CrossRef CAS.
- F. Carrasco-Marín, D. Fairén-Jiménez and C. Moreno-Castilla, Carbon, 2009, 47, 463–469 CrossRef.
- A. K. Meena, G. K. Mishra, P. K. Rai, C. Rajagopal and P. N. Nagar, J. Hazard. Mater., 2005, 122, 161–170 CrossRef CAS.
- S. Dong, G. Huang, M. Su and T. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 22256–22263 CrossRef CAS.
- J.-H. Lee and S.-J. Park, Carbon, 2020, 163, 1–18 CrossRef CAS.
- P. Jõul, H. Lees, M. Vaher, E.-G. Kobrin, M. Kaljurand and M. Kuhtinskaja, Electrophoresis, 2015, 36, 1202–1207 CrossRef.
- P. Jõul, M. Vaher and M. Kuhtinskaja, Chemosphere, 2018, 198, 460–468 CrossRef.
- F. Zhu, J. Guo, F. Zeng, R. Fu, D. Wu, T. Luan, Y. Tong, T. Lu and G. Ouyang, J. Chromatogr. A, 2010, 1217, 7848–7854 CrossRef CAS.
- S. Osamu and K. Watanabe, Drugs and Poisons in Humans, Springer, Berlin, 2005 Search PubMed.
- J. Chen, C. Duan and Y. Guan, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 1216–1225 CrossRef CAS.
- B. Albero, C. Sánchez-Brunete and J. L. Tadeo, J. Agric. Food Chem., 2004, 52, 5828–5835 CrossRef CAS.
- P. A. Souza Tette, L. R. Guidi, M. B. De Abreu Glória and C. Fernandes, Talanta, 2016, 149, 124–141 CrossRef CAS.
- M. Shamsipur, N. Yazdanfar and M. Ghambarian, Food Chem., 2016, 204, 289–297 CrossRef CAS.
- N. Campillo, R. Peñalver, N. Aguinaga and M. Hernández-Córdoba, Anal. Chim. Acta, 2006, 562, 9–15 CrossRef CAS.
- C. Blasco, G. Font and Y. Pico, Food Addit. Contam., 2007, 25, 59–69 CrossRef.
- C. Blasco, M. Fernández, Y. Picó and G. Font, J. Chromatogr. A, 2004, 1030, 77–85 CrossRef CAS.
- Y. Jabali, M. Millet and M. El-Hoz, Microchem. J., 2019, 147, 83–92 CrossRef CAS.
- X. Li, P. Gan, R. Peng, C. Huang and H. Yu, J. Chromatogr. Sci., 2010, 48, 183–187 CAS.
- S. Zali, F. Jalali, A. Es-haghi and M. Shamsipur, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 1002, 387–393 CrossRef CAS.
- J. Yu, C. Wu and J. Xing, J. Chromatogr. A, 2004, 1036, 101–111 CrossRef CAS.
- F. Wei, Y. He, X. Qu, Z. Xu, S. Zheng, D. Zhu and H. Fu, Anal. Chim. Acta, 2019, 1078, 70–77 CrossRef CAS.
- W. A. Wan Ibrahim, K. V. Veloo and M. M. Sanagi, J. Chromatogr. A, 2012, 1229, 55–62 CrossRef CAS.
- J. Hu, C. Qian, Y. Zhang, Y. Tian and Y. Duan, Anal. Methods, 2020, 12, 3954–3963 RSC.
- F. Ghaemi, A. Amiri and R. Yunus, Trends Anal. Chem., 2014, 59, 133–143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM image of the surface of the uncoated stainless steel wire and CA-coated SPME fiber; the image of the prepared coated SPME fibers fixed by epoxy glue to a recycled commercial SPME fiber assembly; XPS C1s curve fitting, the quantification data, N2 adsorption and desorption isotherms and DFT pore-size distribution of the CA-based SPME coating material. See DOI: 10.1039/d0ay02002h |
| This journal is © The Royal Society of Chemistry 2021 |