
Particulate kidney extracellular matrix: bioactivity and proteomic analysis of a novel scaffold from porcine origin†
Rita
Sobreiro-Almeida
 ab,
Maria Elena
Melica
c,
Laura
Lasagni
c,
Hugo
Osório
def,
Paola
Romagnani
cg and
Nuno M.
Neves
*ab
ab,
Maria Elena
Melica
c,
Laura
Lasagni
c,
Hugo
Osório
def,
Paola
Romagnani
cg and
Nuno M.
Neves
*ab
a3B's Research Group, I3Bs – Research Institute on Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark, Parque de Ciência e Tecnologia, Zona Industrial da Gandra, 4805-017 Barco, Guimarães, Portugal. E-mail: nuno@i3bs.uminho.pt
bICVS/3B's – PT Government Associate Laboratory, Braga, Guimarães, Portugal
cDepartment of Biomedical, Experimental and Clinical Medicine, University of Florence, Florence, Italy
di3S – Instituto de Investigação e Inovação em Saúde, Universidade do Porto, 4200-135 Porto, Portugal
eIpatimup – Institute of Molecular Pathology and Immunology of the University of Porto, 4200-135 Porto, Portugal
fFMUP – Faculty of Medicine of the University of Porto, 4200-319 Porto, Portugal
gNephrology and Dialysis Unit, Meyer Children's University Hospital, Florence, Italy
First published on 4th November 2020
Abstract
Decellularized matrices are attractive substrates, being able to retain growth factors and proteins present in the native tissue. Several biomaterials can be produced by processing these matrices. However, new substrates capable of being injected that reverse local kidney injuries are currently scarce. Herein, we hypothesized that the decellularized particulate kidney porcine ECM (pKECM) could support renal progenitor cell cultures for posterior implantation. Briefly, kidneys are cut into pieces, decellularized by immersion on detergent solutions, lyophilized and reduced into particles. Then, ECM particles are analyzed for nuclear material remaining by DNA quantification and histological examination, molecular conformation by FITR and structural morphology by SEM. Protein extraction is also optimized for posterior identification and quantification by mass spectrometry. The results obtained confirm the collagenous structure and composition of the ECM, the effective removal of nucleic material and the preservation of ECM proteins with great similarity to human kidneys. Human renal progenitor cells (hRPCs) are seeded in different ratios with pKECM, on 3D suspensions. The conducted assays for cell viability, proliferation and distribution over 7 days of culture suggest that these matrices as biocompatible and bioactive substrates for hRPCs. Also, by analyzing CD133 expression, an optimal ratio for specific phenotypic expression is revealed, demonstrating the potential of these substrates to modulate cellular behavior. The initial hypothesis of developing and characterizing a particulate ECM biomaterial as a consistent substrate for 3D cultures is successfully validated. The findings in this manuscript suggest these particles as valuable tools for regenerative nephrology by minimizing surgeries and locally reversing small injuries which can lead to chronic renal disfunction.
1. Introduction
The extracellular matrix (ECM) is known to directly influence cell adhesion, migration, proliferation and differentiation. It is a specific product of resident cells providing not only support, but also biochemical cues crucial for determining cell fate and survival.1,2 Thus, tissue engineers have been trying to mimic not only the morphology but also the composition of the ECM for promoting the repair and regeneration of tissues. Many studies use candidate biomaterials by using micro- and nanofibrillar fabrication techniques, such as electrospinning3–5 and/or supramolecular self-assembly.6–8 However, these materials are often composed of synthetic polymers, which fail to reproduce the intricate network of proteins, growth factors, glycosaminoglycans and cytokines, inherent to the natural ECM.9,10In the past few years, decellularized tissues have been extensively studied because they enable retaining proteins and other components present in the native tissue. This naturally occurring ECM has shown to be biocompatible, lacking immunogenic cell membrane proteins, and to promote site-specific remodeling of tissues, minimizing scar formation.11,12 Decellularized tissues are also advantageous because of the high degree of evolutionary conservation of many ECM components, which allows for the use of xenogeneic sources. These matrices are currently being studied as an alternative source for organ transplantation, where whole-organ decellularization, repopulation,13,14 and posterior in vivo implantation are achieved by using perfusion strategies.15,16 Despite the promising results obtained, a number of concerns remain to be addressed to translate this technique to clinical practice. It is also important to consider new solutions for initial-stage repetitive episodes of acute kidney injury (AKI), which can lead to irreversible renal damage and evolve into chronic kidney disease (CKD).17
Several authors already reported the use of the decellularized and processed kidney extracellular matrix to obtain several ECM-derived biomaterials, suitable for addressing early symptoms of kidney disease. Kidney ECM hydrogels from human origin have already been developed and characterized for their properties and ability to support kidney endothelial cell culture.18,19 Other authors also studied the growth and metabolism of kidney stem cells when seeded on hydrogels from three different regions of the kidney, revealing tissue and region-specificity.20 However, cells were seeded on top of the hydrogels and, therefore, in 2D. Another approach is to combine the kidney-derived ECM with other components (natural or synthetic) in order to enhance its limited mechanical properties. For instance, a small portion (10%) of porcine derived kidney ECM was combined with a synthetic polymer to fabricate a porous scaffold. This scaffold was implanted in a partially nephrectomized mice in vivo, demonstrating regeneration of renal glomerular tissue, demonstrating some inflammatory response.21 Other studies have also followed this rationale, developing alginate and gelatin-based hydrogels in combination with kidney matrices as new bioinks for 3D printing.22,23 Although hybrid biomaterials can provide diverse cellular responses, it is also important to study the composition-related response of the host tissues. Having a particulate form of ECM can also be of advantage because of their small size and shape, which allows for their implantation by non-invasive techniques, such as injections,24 enabling cell delivery on a specific defect and/or injury of any size or shape.25 This particulate ECM can also circumvent the problems of low cellular migration on hydrogels, allowing for more efficient cell spreading. Some studies were published in the past few years using particulate ECM as a cell carrier for regeneration of several tissues, such as cartilage,25 adipose tissue26 and bone.27
In this work, we hypothesized that decellularized porcine kidney ECM particles possess a similar composition to human kidneys, enabling the 3D culture of human-origin cells, without changing their phenotypic expression. To the best of our knowledge this is the first time that the porcine kidney matrix is fully characterized by proteomic analysis and also the first time that renal progenitor cells are cultured in 3D with a natural ECM. We believe that this substrate can be promising for cell delivery and, ultimately, promote kidney regeneration.
2. Results
Naturally occurring ECMs have been gaining interest in the past few years in the tissue engineering field. This matrix, in a particulate form, enables developing injectable advanced therapies. Herein, we aim at characterizing the ECM particulate morphology, composition and bioactivity when supplemented in cultures of specific renal progenitor cells.2.1. Evaluation of pKECM substrates
This section will report on the studies aiming at characterizing the particulate ECM structure and morphology. Its protein and DNA content are compared with those of native kidney tissue. Also, the effects of ethylene oxide sterilization on the thermal and conformational behavior were evaluated. Its protein composition was extensively characterized by mass spectrometry and further compared with human renal proteomic content.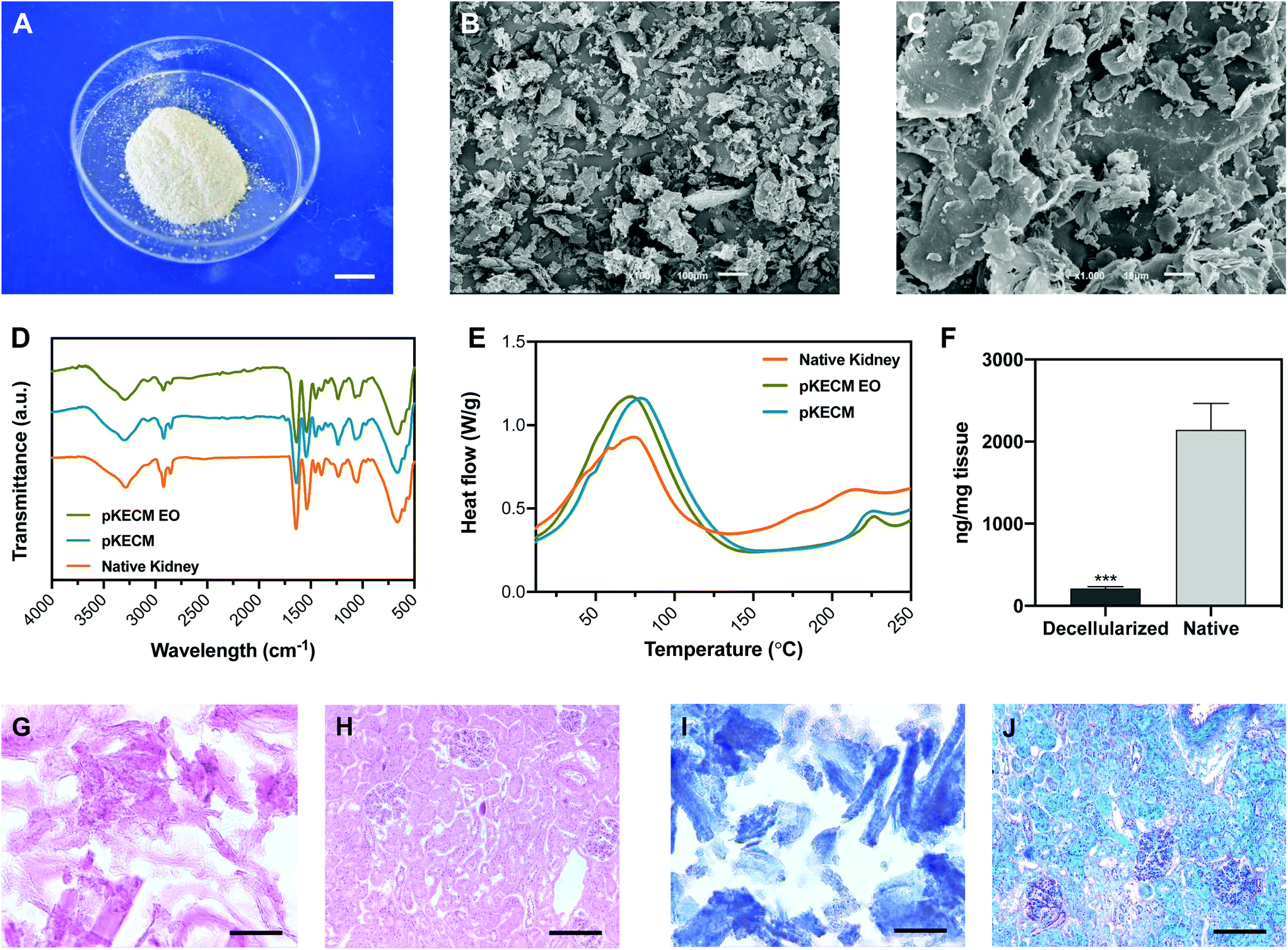 | ||
| Fig. 1 Particulate kidney matrix characterization. (A) Gross appearance of the powders obtained after decellularization, freeze-drying and milling process. Scale bar: 1 cm; (B–C) scanning electron microscopy micrographs of the powders. A broad distribution of sizes is presented, with collagen-characteristic sheet-like conformation. Scale bars: 100 and 10 μm. (D) Fourier transform infrared spectroscopy and (E) differential scanning calorimetry characterization of native kidney (orange), decellularized matrix (pKECM, blue) and decellularized matrix subjected to ethylene oxide sterilization (pKECM EO, green). The results suggest maintenance of molecular composition and thermal proprieties of the matrix after decellularization and after the ethylene oxide sterilization process. (F) DNA quantification of decellularized and native tissue demonstrating a ∼90% decrease on the dsDNA content (n = 3). ***p < 0.001 vs. native; (G–H) hematoxylin and eosin and (I–J) Masson's trichrome staining on cryosections of native tissue (H–J) and decellularized matrix (G and I). Nuclear staining is not present on decellularized matrix sections and collagenous staining is seen (blue). Scale bars: (G–I) 50 μm, (H–J) 200 μm. | ||
To assess the effect of decellularization and processing of the matrix on the molecular composition, ATR-FTIR analysis was performed to native, decellularized and sterile pKECM samples (Fig. 1D). The spectra, characteristic of collagenous components, present several bands and peaks that will be described herein. The band at 3284 cm−1 is characteristic of amide A for NH stretching. Also, the peaks at 2848 and 2916 cm−1 are characteristic of OH, NH2, and NH stretching vibration in the amide group. The peaks at 1631 and 1537 cm−1, respectively, are characteristic of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (amide I) stretching vibration and the bending and stretching vibration of NH and CN (amide II).31 The peak at 1232 cm−1 is relative to the NH plane bending from amide linkages (amide III), associated with the secondary conformation of collagen. The peak identified at 1450 cm−1 and the region in the range of 1417–1360 cm−1 corresponds to the stereochemistry of the pyrrolidine rings of proline and hydroxyproline.32 No significant difference was found between the spectra of native kidneys, pKECM and pKECM subjected to the EO sterilization process, suggesting that the molecular conformation of the ECM and collagen does not change by the decellularization and/or sterilization process.
O (amide I) stretching vibration and the bending and stretching vibration of NH and CN (amide II).31 The peak at 1232 cm−1 is relative to the NH plane bending from amide linkages (amide III), associated with the secondary conformation of collagen. The peak identified at 1450 cm−1 and the region in the range of 1417–1360 cm−1 corresponds to the stereochemistry of the pyrrolidine rings of proline and hydroxyproline.32 No significant difference was found between the spectra of native kidneys, pKECM and pKECM subjected to the EO sterilization process, suggesting that the molecular conformation of the ECM and collagen does not change by the decellularization and/or sterilization process.
Differential scanning calorimetry (DSC) was used to compare the thermal proprieties of pKECM substrates before and after the sterilization process to the thermal properties of native samples (Fig. 1E). In all samples the matrix demonstrates a broad transition temperature from approximately 15 °C to 140 °C, with one distinct endothermic peak. Enthalpy has a shift from 181 to 306 J g−1 when samples are decellularized. The melting temperature (Tm) peaks range from approximately 73 to 77 °C, which are within the range of collagen melting temperature. Additionally, another endothermic peak appears around 225 °C for both decellularized conditions and also for native samples, but to a minor extent.
To assess the effective removal of the cellular content, specimens were embedded in OCT and the obtained cryosections were stained with H&E and MT. Also, dsDNA quantification enables a quantitative measurement of the effectiveness of the decellularization process. H&E micrographs demonstrate no violet staining for the nucleus (Fig. 1G), representing a successful decellularization, in comparison with the native tissue (Fig. 1H). As eosin Y binds to positively charged amino-acid side chains such as lysine and arginine, we found that the pKECM stains positive for it, evidencing their protein content and structure in 2D. This staining is comparable to the native tissue (Fig. 1H), where the pink color can also be found surrounding the nucleus, evidencing the cytoplasm and ECM. Corroborating these results, MT staining for pKECM revealed no red staining for Weigert's iron hematoxylin (Fig. 1I), comparing with the native tissue (Fig. 1J). Double-stranded DNA quantification confirms an effective removal of >90% of the nucleic material when comparing with the native tissue, confirming an adequate protocol for kidney decellularization (Fig. 1F).
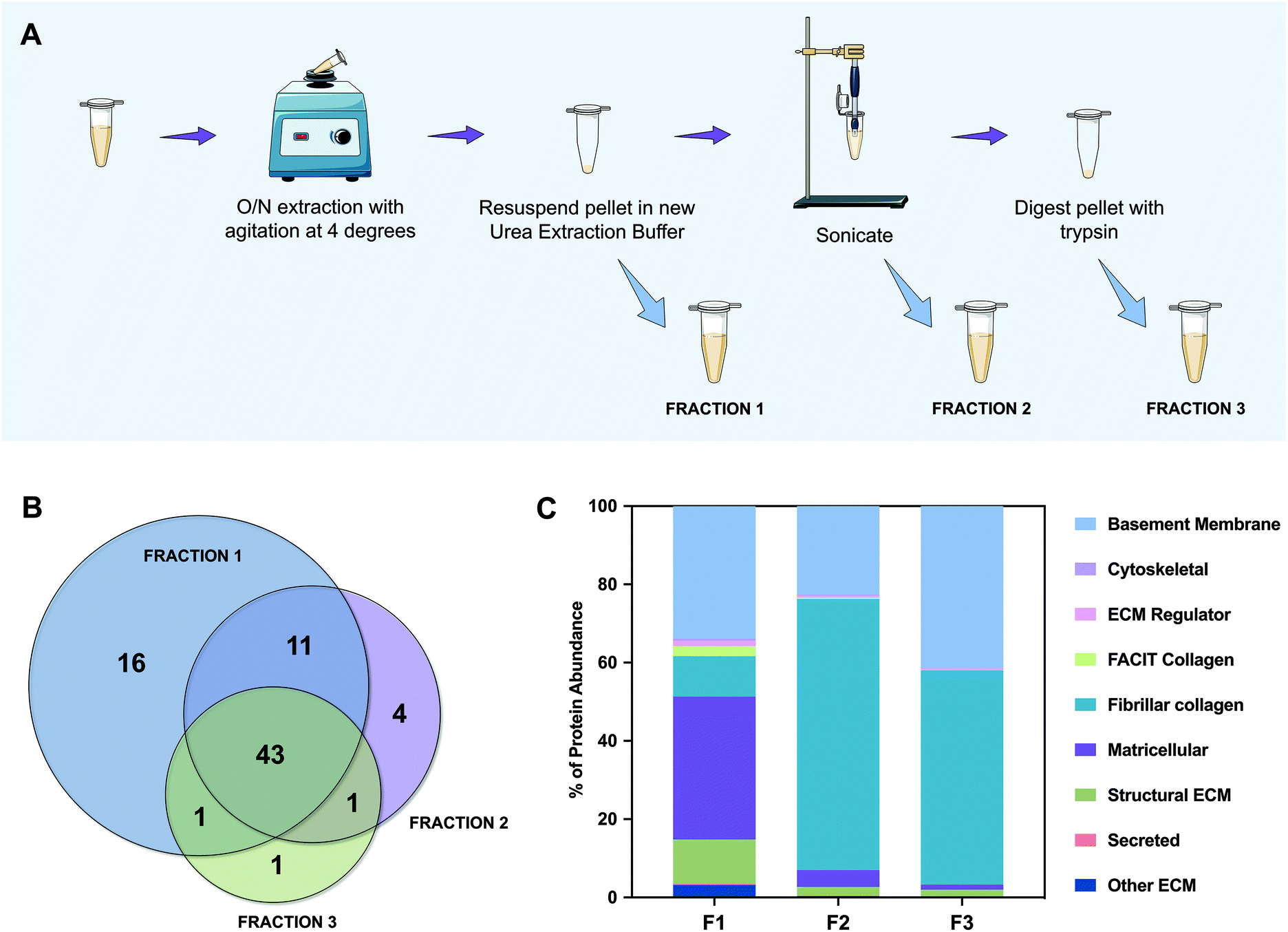 | ||
| Fig. 2 Schematic representation of the methodology and the unique profile of the ECM fractions. (A) Fraction 1 was obtained by homogenization with urea extraction buffer, fraction 2 was obtained after sonication of the remaining pellet and fraction 3 was obtained after tryptic digestion of the fraction 2 pellet; (B) 43 proteins are common to the three fractions being most proteins identified on fraction 1 (F1), while only 6 new proteins were found in fraction 2 and 3 (F2 and F3). (C) F2 and F3 are mainly composed of basement membrane proteins and fibrillar collagen, which lead us to conclude that F1 abundances do not correspond to the total protein isolated, and that several steps of protein homogenization are required to extract the total amount of protein. | ||
To better categorize the proteins that are present in each fraction, we performed gene ontology analysis of the characterized ECM proteins. This categorization was not completely automatic because the UniProt database for porcine (Sus scrofa) is less informative than that of the human (Homo sapiens). Therefore, and because some proteins were still “uncharacterized”, the majority were confirmed and classified manually. The most abundant of them is collagen type IV, which is herein named “COL4*” because the NC1 domain could not be identified. We have noticed that although we were able to extract the majority of proteins in F1 (59 out of 77), they did not represent the abundance expected from a naturally sourced biological kidney matrix. So, we graphed all of the identified proteins as a percentage of the total protein abundance by gene ontology classification (Fig. 2C). From this analysis we could observe that fibrillar collagen only constitutes ∼10% of the proteins identified in F1, while matricellular proteins account for ∼37% of whole protein abundance. Furthermore, by analyzing F2 and F3, ∼69% and ∼55% of their content is composed of fibrillar collagen, and ∼23% and ∼41% is composed of basement membrane proteins, respectively. Accordingly, the amount of proteins representing ECM regulators, FACIT collagens, and secreted and matricellular proteins decreases radically from F1 to F3. Indeed, in F3, only ∼4% of the proteins correspond to these kinds of proteins, the remaining being ∼96% basement membrane and fibrillar collagen proteins. These results support our initial hypothesis; the chaotropic insoluble pellet, which is typically discarded in the proteomic analysis of this kind, is mainly composed of fibrillar collagen and basement membrane proteins, essential components of the ECM. Encouragingly, cytoskeletal components represent less than 1% of the total amount of proteins, which confirms once more a successful protocol of decellularization.
A more detailed description of the identified proteins is shown in Table S1.† The percentages of each protein present on each analyzed kidney sample (K1, K2 and K3) and their coefficient of variation (CV) were calculated. We could notice that high variances (greater than 1) were mainly detected on ECM regulator family of proteins, rather than in the most abundant components of the matrix. This feature is expectable due to the decellularization process, in which those proteins are more easily removed. In proteins such as collagen type IV, collagen type I, collagen type VI, perlecan and nidogens, CV calculated values were lower than 0.5.
Absolute quantitative abundances of the 30 more expressed ECM proteins were graphed in a heat map for each separate sample that was analyzed by MS (Fig. S1†), where green represents the lowest abundances, red represents the higher abundances and black denotes the middle-term. We managed to extract the majority of proteins in F1, a consistent result within the 3 replicas. However, as previously noticed in Fig. 2C, the extraction of fibrillar (collagens type I and III) and basement membrane collagens (collagen type IV) revealed higher abundances in F2 and F3 extractions. In contrast, matricellular collagens (collagen type VI) show a decrease in abundance from F1 to F3. The same phenomena occurred for all of the remaining proteins identified. We could not notice the presence of nidogen-1 in F3, which was the only protein in this set which could not be detected in the 3 fractions. The overall profile denotes that chaotrope protein extraction may give us a reasonable idea of the group of proteins that constitute the matrix, but cannot give us a rigorous quantification, which was only possible when separating the fractions and quantifying separately.
To have a stronger analysis of our protein profile, we have performed pie chart analysis for total ECM components of the kidney considering the three independent tissue replicas (Fig. 3). Percentages were obtained by dividing the individual abundance of each identified protein by the total ECM abundance content. Fibrillar collagens, basement membrane and matricellular proteins were further analyzed, and a sub-pie chart is presented for each one of these categories, given that together they constitute more than 92% of the total protein content. As expected, the main component of basement membranes is collagen type IV (70%). Matricellular protein analysis revealed that collagen type VI composes 76% of the total amount of identified proteins and collagen type I composes 92% of fibrillar collagen existent in porcine kidneys.
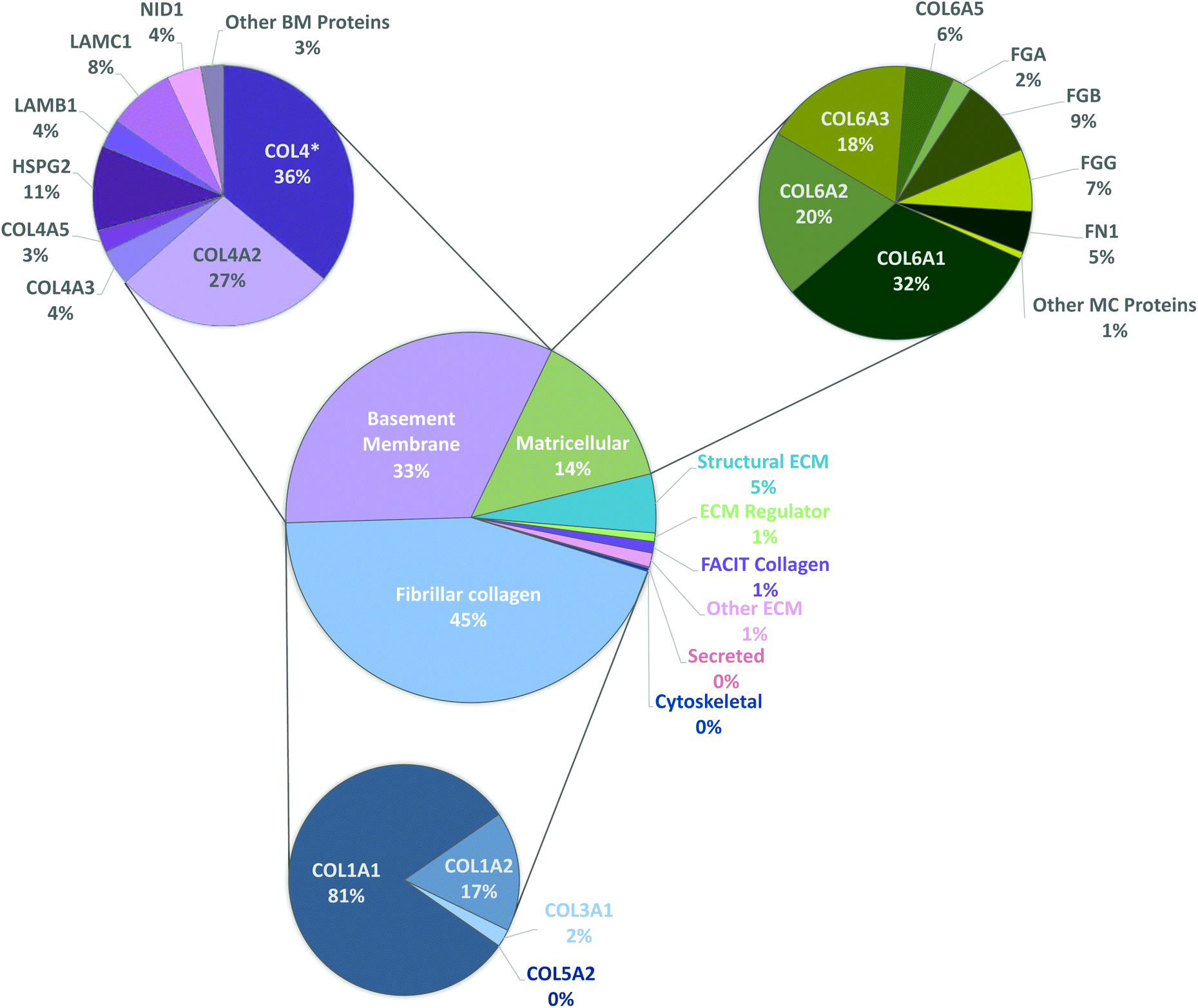 | ||
| Fig. 3 Quantitative assessment of the proteomic profile of pKECM by LC-MS/MS. A sub graph is demonstrated for the most abundant ECM categories: fibrillar collagens (blue), basement membrane proteins (purple/pink) and matricellular proteins (green). Quantitative proteomics demonstrate that collagen type IV comprises nearly 70% of basement membranes; collagen type I comprises 92% of total fibrillar collagen content and collagen type VI is 76% of total matricellular proteins. Proteins are identified with the respective gene name. This proteomic analysis enlightens the three major collagenous components of the porcine kidney ECM. | ||
2.2. 3D culture of renal progenitor cells and pKECM
This section includes the evaluation of the bioactivity of the particulate ECM to stimulate specific renal progenitor cells in a 3D culture. Cytocompatibility and cell proliferation were evaluated, as well as the capacity of the ECM to guide phenotypic changes.To assess pKECM cytotoxicity, we first evaluated cell viability when in contact with pKECM conditioned medium. Fig. 4B and D show cells after 2 and 24 h of conditioned culture, respectively. No morphological differences are observed between these cells and cells subjected to regular culture medium (Fig. 4A and C), giving first insights into the biocompatibility of this matrix. Next, the matrix was directly cultured with particulate ECM for 7 days and live/dead staining was performed with Calcein AM and PI for 1, 2 and 4 mg of pKECM (Fig. 4E, F and G, respectively). No significant red staining is visible, indicative of the biocompatibility of this substrate, regardless of the quantity used for the assays. However, a higher quantity of cells can be seen on 1 and 2 mg of matrix compared with 4 mg. Also, an alteration on morphology is present, being that cells cultured on 4 mg of matrix are bigger and rounder, suggesting lack of adhesion points to the pKECM. On the other hand, cells cultured with lower quantities of the matrix demonstrate even distribution and a higher amount of green staining, also suggesting higher cell viability and proliferation. Quantification of fluorescence intensity was also performed, corroborating the previous findings (Fig. 4H). Although not significant, PI staining is only visible on cells cultured on 1 mg of pKECM. This can be explained due to the high proliferation and higher cell confluence present on these constructs, which can lead to cell death. As the quantity of matrix increases, the cell number diminishes and, as a consequence, the number of live and dead cells also decrease, leading to no dead cells quantified on 4 mg of pKECM.
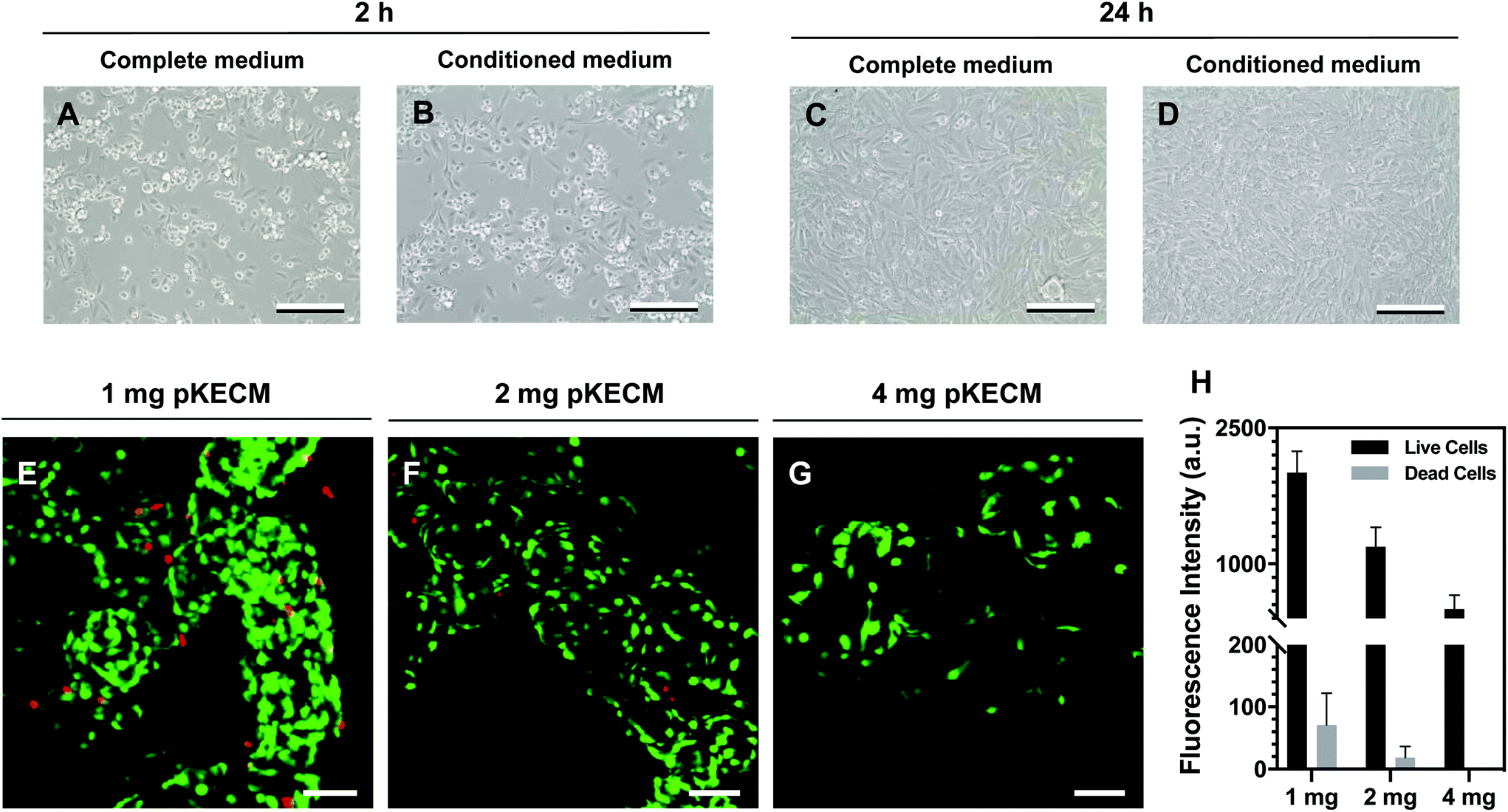 | ||
| Fig. 4 Cytocompatibility of human renal progenitor cells. (A–D) Optical microscopy images of hRPCs after 2 and 24 h of seeding in complete medium (EGM-MV) or conditioned medium (EGM-MV prior exposed to pKECM during 24 h). Conditioned medium cultures (B and D) demonstrate no morphological changes on the hRPCs compared with complete medium cultures (A and C). Scale bar: 200 μm; qualitative results for viability were obtained by immunofluorescence micrographs of live/dead assay for hRPCs after a 7-day culture time with (E) 1 mg, (F) 2 mg and (G) 4 mg of matrix. Cells were incubated with Calcein AM (stains green for live cells) and PI (stains red for dead cells). Scale bar: 100 μm; (H) quantification of fluorescence intensity of live and dead cells was performed on ImageJ, corroborating live/dead stainings (n = 3). | ||
As a corroborative assay, hRPCs metabolic activity was quantitatively assessed overtime using alamarBlue on hRPCs seeded on pKECM substrates (1, 2 and 4 mg) and on tissue culture plastic (0 mg) (Fig. 5A). We noticed that hRPCs are viable in all timepoints when in contact with pKECM, revealing no cytotoxicity. We can additionally notice a common tendency for a peak increase in intermediate culture times and then a decrease of the metabolic activity after 7 days of culture. We hypothesized this event to be a result of achieving cell confluence after 4 days, reducing cellular metabolic activity. We can also observe that after 2 days of culture, cells were shown more metabolically active when cultured on intermediate amounts of the matrix (2 mg), which was also observed at day 4, being statistically different from all the other conditions. At day 7, cells are shown more metabolically active when cultured on a higher amount of matrix, which is also attributed to the lack of cell confluence on these substrates.
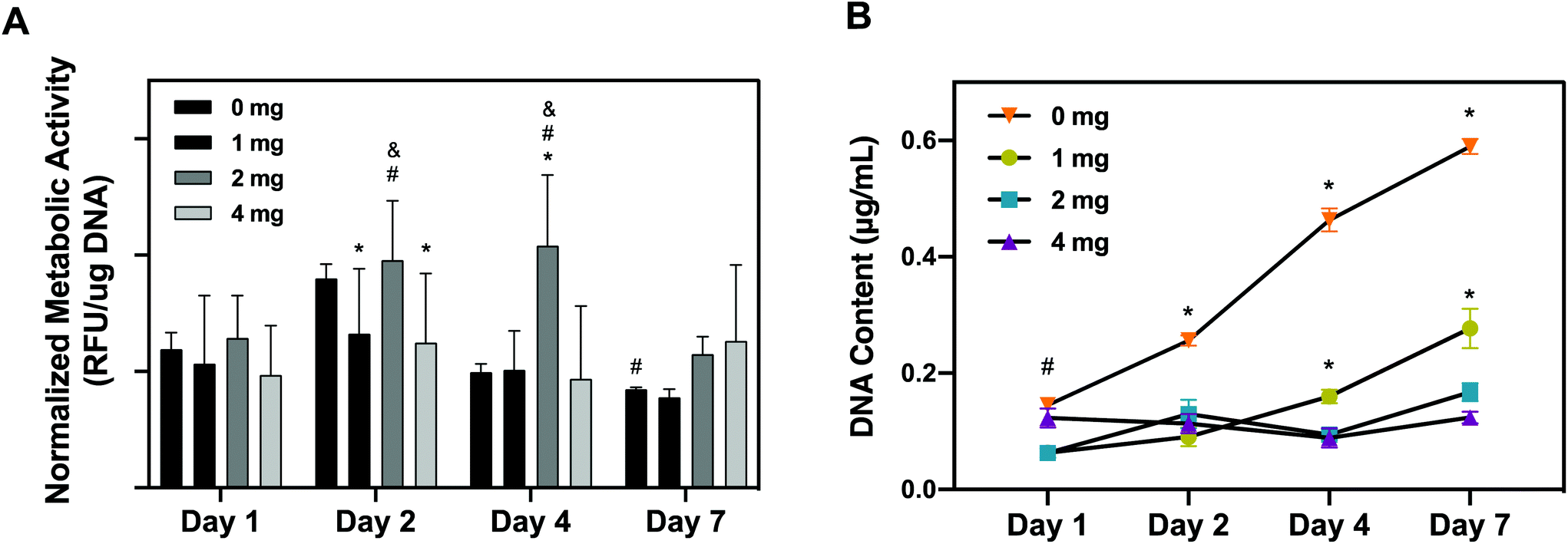 | ||
| Fig. 5 Quantitative analysis of viability and proliferation of hRPCs cultured on pKECM. (A) Viability assay demonstrated by alamarBlue fluorescence over 7 days at different timepoints. Control condition (0 mg) represents hRPCs cultured on tissue culture plastic; RFU = relative fluorescence units; (*) statistically different from 0 mg; (&) statistically different from 1 mg; (#) statistically different from 4 mg (n = 9); (B) proliferation assay demonstrated by quantification of DNA over time. (*) statistically different from all conditions; (#) statistically different from 1 and 2 mg conditions; (n = 9). | ||
Cellular proliferative capacity was inferred from the quantification of double-stranded DNA present on hRPCs (Fig. 5B). After 24 h, RPCs cultured on 0 and 4 mg substrates present the higher proliferation capacity. However, throughout the timepoints, cells seem to not be proliferating on the 4 mg condition, the concentration of dsDNA being maintained after 7 days of culture on the substrates. Nevertheless, there is a tendency for the increase of DNA content from day 1 to day 7 on the cells cultured on lower amounts of pKECM (1 and 2 mg). This is a good indicator of cell preference towards lower quantities of the particulate matrix. It is also important to notice a tendency from day 4 to day 7 of higher proliferation capacity of the cells when cultured on 1 mg instead of 2 mg of pKECM. This can be again explained by the lower quantity of the substrate, which enables a higher surface area for cells to attach and proliferate. Instead, in 2 mg of matrix, although cells can proliferate in between the ECM particles, this effect becomes reduced as the amount of matrix increases. This can also be corroborated by the lower DNA content demonstrated on 4 mg of pKECM. The increasing values observed for the 0 mg condition translate the proliferating capacity of these cells on tissue culture plastic, although it cannot be directly compared with our 3D suspension conditions.
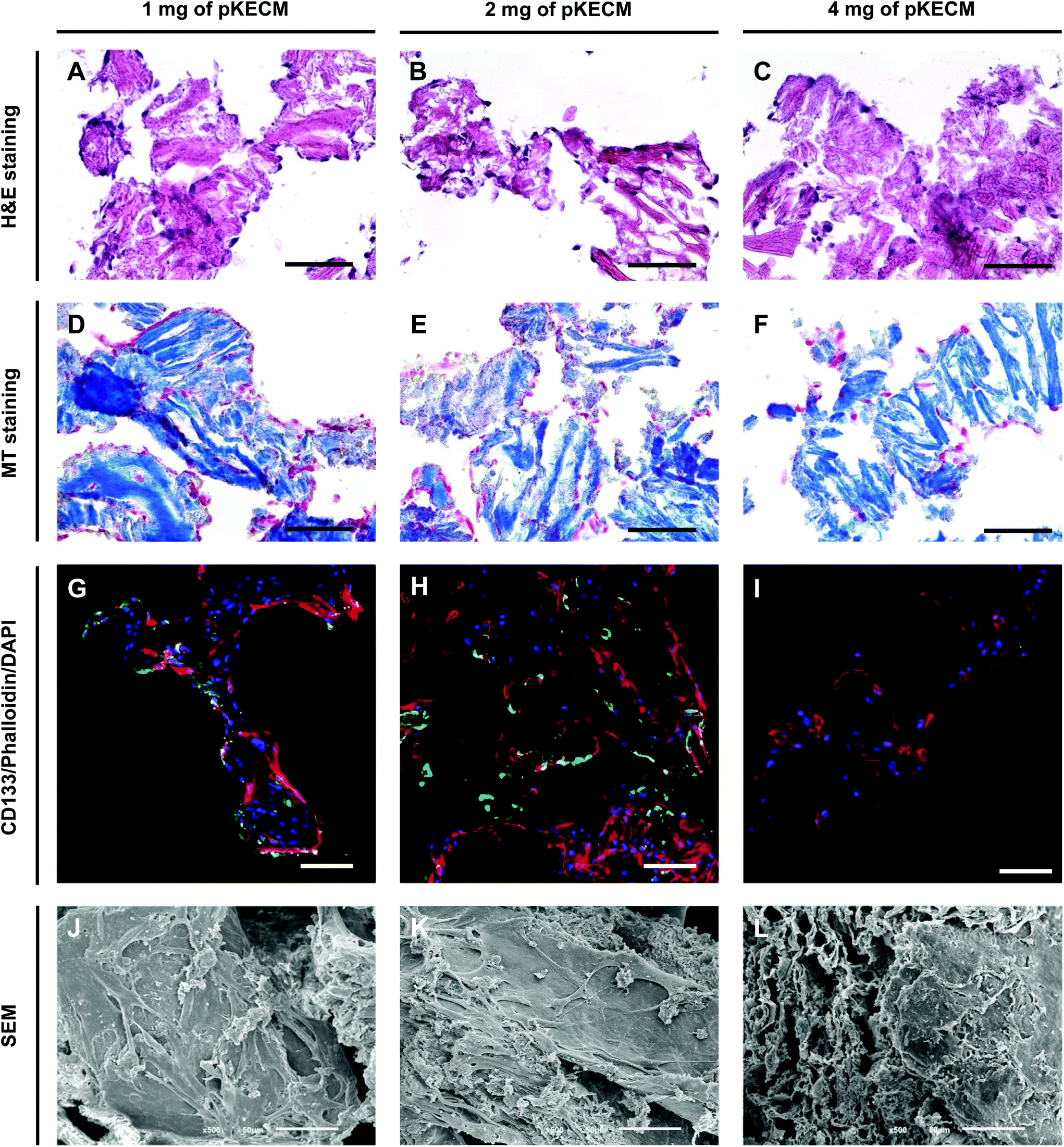 | ||
| Fig. 6 Spatial distribution of hRPCs cultured on pKECM substrates. Immunohistochemistry on matrix cryosections for hematoxylin and eosin (A–C) and Masson's trichrome (D–F) demonstrate good cellular coverage around the powders mainly on 1 and 2 mg of pKECM after 7 days of culture. Scale bars: 100 μm. (G–I) Immunofluorescence of CD133 (green), phalloidin for cytoskeleton (red) and DAPI for nuclei (blue) demonstrate greater fluorescence of the stem marker on 2 mg of pKECM. Scale bar: 100 μm. (J–L) Scanning electron microscopy images of hRPCs cultured on the matrix after being fixed and dehydrated. After 7 days, the overall cell coverage of the substrate was found on 1 and 2 mg of pKECM. Scale bar: 50 μm. | ||
3. Discussion
This work was focused on developing a particulate kidney matrix to act as a new advanced therapy for kidney diseases. We performed structural, morphological and proteomic characterization and as a second aim, we optimized a strategy to culture cells of human origin in vitro with the matrix. We suggest that decellularized kidney particles retain the biological efficacy needed to maintain the stemness of renal progenitor cells. The injection of cells alone has a number of shortcomings. Without a substrate to attach, a lot of cells are lost in the process or migrate out of the intended site of action and frequently there is insufficient cell engraftment to obtain clinically relevant efficacy. Therefore, we propose a synergistic effect of using pKECM as a substrate in which hRPCs could be delivered, helping in the renal repair after injury.To ensure that the cells can interact, attach and proliferate in the particulate matrix, it is important to evaluate the structure and composition of the ECM. SEM micrographs (Fig. 1B and C), allow detecting two distinct surface ultrastructures within the particles: smooth surfaces, demonstrating the sheet conformation of basement membranes and fibrous surfaces, which can be assigned to the interstitial matrix.24 We also noticed the resemblance of the matrix to those of other previously published studies.30 Using ATR-FTIR we could not detect any alterations in molecular conformation between native and decellularized samples (Fig. 1D), indicating the preservation of the structure. Moreover, we can infer from the FTIR spectrum that the integrity of the triple helix was conserved by the ratio of 1235 cm−1 (amide III) and 1450 cm−1 (pyrrolidine ring) bands, which is close to 1 in both samples.34 By DSC, we could confirm that decellularization and sterilization did not change the thermal properties of the native tissue. Additionally, the broad transition temperature range presented in the DSC thermogram, indicates the specific distribution of the collagenous population in the particulate matrix.31 The difference observed between melting peaks (4 °C range) is attributed to variability between samples, since they were not performed on the same kidney sample. The enthalpy values also differ between native and decellularized samples. To the best of our knowledge, this is the first report on the thermal behavior between native and decellularized samples of kidney tissue. We suggest that this difference is due to the natural plasticizer effect of the cells, which reduces the forces of attraction between ECM fibers, thereby reducing the enthalpy needed for denaturation, as previously reported.35 We also investigated the remaining cellular and nuclear materials by DNA quantification and hematoxylin staining of pKECM frozen sections. We demonstrated that >90% of the nucleic material was removed after powder digestion (Fig. 1F). Additionally, no hematoxylin staining for DNA was found on IHC stainings (Fig. 1G and I). By comparing our findings with previously published results, we were able to notice structural, morphological and thermal proprieties very similar to other decellularized matrices.30,36 Thus, we were able to validate our developed particulate matrix in terms of particle size, collagenous content, thermal properties and immunogenic content.
The proteomic content of our matrices was investigated by nanoLC-MS/MS. Herein, we employed for the first time an ECM targeted homogenization method and proteomic analysis to define the molecular components of porcine-derived renal matrices at a quantitative level. We demonstrate the presence of essential ECM proteins in the pellets that arise from protein homogenization, which are typically discarded. We also considered the biological variability existing between kidney donors, and thus we analyzed three kidneys representative of different animals which were independently subjected to the decellularization process. The variability between ECM components of each kidney presented on Table S1† can be attributed to donor-to-donor variability, since it is dependent on donor age, species of origin and also the history of stimulus/insults that each kidney experienced.37 By applying this methodology, we were able to identify proteins which are typically lost during decellularization procedures, such as some glycoproteins, proteoglycans and other secreted factors. These proteins are extremely important on stabilizing elastin and collagen networks being therefore essential to maintain the basement membrane stability and integrity.38 We were also able to retain the majority of renal basement proteins, such as collagen type IV and laminins, which are important because of their abundant binding sites for both endothelial and epithelial cells and their capacity to regulate cellular migration and proliferation.39 We have also noticed the preservation of fibrillar collagens, which provide tensile strength to the organs, including the kidneys.
Until now, our understanding of the matrix composition of renal decellularized matrices relied on a previous mass spectrometry study.18 However, this study does not describe donor-to-donor variability and no gene ontology classification was performed. Other authors also performed a fractioned proteomic analysis in lung and myocardial tissues.40,41 Unlike those reports, we did not use toxic digestion agents and we managed to extract the majority of proteins in F1, a consistent result within the 3 replicas (Fig. S1†). The results presented on Fig. S1† suggest that subpopulations of proteins are able to solubilize according with their location in the matrix as well as their interactions with other matrix proteins, making some of them insoluble in the commonly used buffer solutions.42
Comparing our results with a recently published study by Louzao-Martinez et al.,43 where the human kidney proteome was analyzed, we can detect proteomic similarities between porcine and human kidneys, where 49 proteins were common to both proteomes (Fig. S3†). However, we also could notice that a lot of essential proteins are not present in the human kidney proteomic analysis, such as alpha-3, 4, 5 and 6 chains of collagen type IV, alpha-1 and 2 chains of collagen type V and even apha-1 laminin. Collagen type IV and laminins being the main components of human kidney basement membranes,44 and collagen type V a fibrillar collagen that comprises the majority of the interstitial ECM,45 these should have been detected and identified in the human proteomic analysis. As aforementioned, we noticed that fibrillar and more crosslinked collagens could only be extracted in its total abundance in the fractions F2 and F3, with a more severe method of homogenization. We hypothesize that the different extraction protocols may be responsible for the differences found when comparing human and porcine kidney proteomics. Therefore, an accurate comparison cannot be performed on this study between porcine and human kidney ECM. We have additionally noticed that we are lacking the identification of some renal ECM proteins on porcine kidneys, such as laminin alpha-4 and 5 and small proteoglycans such as biglycan and decorin. Instead, we identified other 5 sub-domains of laminin and the core proteins of heparan sulfate, lumican, perlecan, collagen type XVIII and agrin (Table S1†).45
As a second aim, we investigated the bioactivity of the particulate ECM by evaluating the growth, proliferation, morphology and phenotypic changes of hRPCs. These cells represent a subset of multipotent progenitors present in the Bowman's capsule. They can be purified from cultured kidney fragments, revealing self-renewal potential and a high cloning efficiency.46 When injected into SCID mice with acute renal failure, hRPCs have demonstrated to ameliorate the morphologic and functional kidney damage.33 Our results demonstrate high biocompatibility and mitogenic activity on a 3D culture with ECM particles (Fig. 4 and 5). We also noticed the presence of cell–cell and cell–substrate interactions when cultured on 1 and 2 mg of matrix (Fig. 6), indicative of good cellular environment adaptation over 7 days of in vitro culture. The IHC micrographs are the result of a 10 μm section of the whole 3D culture, being not representative of the cell–cell interactions but on how cells are distributed. We could notice the cells surrounding the matrix, creating an evenly distributed monolayer when cultured on 1 and 2 mg of matrix. In contrast, when augmenting the quantity of matrix to 4 mg, but with the same cell concentration, the cells lose their cell-to-cell interactions. Consequently, very few cells or no cells can be spotted on IHC micrographs. However, they can be found when performing a z-stack on IF micrographs. Regarding CD133 expression (Fig. 6G–I), it was interesting to spot a change on the morphology and size of the cells with the increasing content of the matrix. This change consequently caused the loss of CD133 expression on 4 mg of pKECM. Additionally, very little CD133 expression is found on cells cultured on 1 mg of matrix, suggesting that cell–cell and cell–matrix interactions led the guidance of phenotypic expression, where a mild proliferation capacity together with a mild cell–cell interaction lead to maintenance of stemness by using 2 mg of matrix. A 3D reconstruction of cellular organization around the matrix can be seen in Fig. S4.† Although these results are promising and consistent with each other in terms of cell distribution, morphology and interaction with the substrate, the stem nature of hRPCs is only confirmed by immunofluorescence. In the future, we intend to do a follow-up of the cell markers CD133 and CD24 by flow cytometry, which will enable for a stronger evaluation of cellular phenotype. Moreover, in a future in vivo work, we intend not only to perform injectability tests but also functional analysis assays in order to evaluate the full potential of our therapy.
These findings suggest an optimal ratio for the maintenance cell phenotype and an optimal cell–cell interaction. The particulate matrix has shown to be highly supportive for a 3D progenitor cell culture, suggesting that a scaled-up culture of these cells with pKECM could be achieved in vitro for posterior patient engraftment.
4. Conclusions
This work highlights the potential of ECM-based biomaterials of porcine origin for kidney advanced therapies. We successfully confirmed our initial hypothesis of developing and characterizing a particulate extracellular matrix biomaterial as a consistent substrate for 3D cultures of adherent renal progenitor cells. We herein show a similar protein composition with human kidneys. Indeed, the matrix demonstrated being an excellent substrate for supporting renal progenitor cell attachment, proliferation and growth. Moreover, it was shown to modulate the morphology and phenotypic expression of renal progenitor cells. To the best of our knowledge, this the first report investigating particulate kidney ECM as a promising 3D substrate for cell cultures. Our results suggest that this particulate ECM together with reparative progenitor cells may become a powerful tool to address renal pathologies in its early stages.5. Materials and methods
Expanded methods for morphological, structural, histological and biochemical analysis of the particulate kidney ECM as well as the nano-liquid chromatography–tandem mass spectrometry (nanoLC-MS/MS) methodology for characterization and relative quantification of protein composition can be found in ESI Experimental section.† Detailed information about the isolation and characterization of human renal progenitor cells (hRPC) is also provided.5.1. Processing and characterization of kidney ECM derived powders (pKECM)
 | ||
| Scheme 1 Sequence of procedures used in the preparation of pKECM. (A) Porcine kidneys were obtained in a local slaughterhouse and cut into sagittal sections before being washed in distilled water. (B) Sections were immersed in detergent solutions of 1% SDS and 1% Triton X-100 and placed on an orbital shaker for 7 days. Decellularization was achieved by frequent renewal of the solutions. (C) The decellularized tissue was freeze-dried. (D) pKECM was obtained using a cryogenic mill. Scale bar: 1 cm. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 rpm and the supernatant, referred to as Fraction 1, was removed and saved. The pellet was once again homogenized and centrifuged. The resultant supernatant was joined to the soluble Fraction 1 (F1). Urea extraction buffer was again added to the resultant pellet and vortexed. The mixture was sonicated at 40% amplitude while being refrigerated. After centrifuging, the supernatant was saved as Fraction 2 (F2). The remaining pellet was digested with trypsin as described in section 2.2.5 to yield Fraction 3 (F3).
700 rpm and the supernatant, referred to as Fraction 1, was removed and saved. The pellet was once again homogenized and centrifuged. The resultant supernatant was joined to the soluble Fraction 1 (F1). Urea extraction buffer was again added to the resultant pellet and vortexed. The mixture was sonicated at 40% amplitude while being refrigerated. After centrifuging, the supernatant was saved as Fraction 2 (F2). The remaining pellet was digested with trypsin as described in section 2.2.5 to yield Fraction 3 (F3).
5.2. In vitro renal progenitor cell culture studies on pKECM
To evaluate hRPCs proliferation on the different cell/matrix ratios, dsDNA quantification was performed. After the viability assay, constructs were rinsed in DPBS to release the alamarBlue dye. After, specimens were immersed in Milli-Q water and cells were lysed by thermal and osmotic shock. The resulting supernatant was used for DNA quantification with the Quanti-iT Picogreen dsDNA assay kit (Thermo Fisher). The fluorescence of each specimen was measured using a microplate reader (excitation: 485 nm; emission: 528 nm). The DNA concentration for each sample was calculated using a standard curve.
For the histological analysis of the distribution of cells on the matrix after 7 days, constructs were fixed in 10% neutral buffered formalin, and embedded in OCT compound for cryosectioning as previously described. H&E and MT stainings were performed on the constructs. Staining intensity and cell distribution were observed under light microscopy (DM750, Leica).
The stemness potential of hRPCs cultured in pKECM was evaluated by immunocytochemistry after 7 days of culture. Briefly, constructs were fixed with 10% neutral buffered formalin for 1 h at 4 °C and then washed in PBS. Cells were further incubated with antibody against the CD133 marker for hRPCs (Miltenyi Biotec; 1:2.5 diluted in 3% BSA/PBS) for 1 h at RT. Samples were washed with PBS and incubated with the secondary antibody Alexa Fluor 488 (Alfagene; 1:500 diluted in PBS) at RT in the dark for 1 h. DAPI (Sigma Aldrich, 1:500) and Alexa Fluor 546 Phalloidin (Alfagene; 1:200) were incubated along with the secondary antibody. After several rinses in PBS, constructs were analyzed under a Leica SP5 AOBS confocal microscope. Cells cultured in conventional polystyrene plates without pKECM were used as control. Images were recorded digitally and further processed using LASX software (Leica).
5.3. Statistical analysis
Statistical analyses were performed with GraphPad Prism 8 (GraphPad Software, California, USA). Data were expressed as means ± standard deviation of experiments with at least three independent assays. Differences between groups were analyzed by unpaired t test or two-way analysis of variance (ANOVA) in the case of experiments conducted over time, using Tukey test for post hoc assessments of the differences between samples. Statistical significance was defined as p < 0.05.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the European Regional Development Fund (ERDF) on the project FROnTHERA (NORTE-01-0145-FEDER-000023) and the FCT PhD Grant on the Doctoral Program on Advanced Therapies for Health (PATH) (PD/BD/128102/2016). Mass spectrometry and proteomics were performed at the Proteomics i3S Scientific Platform with the assistance of Hugo Osório. This work was supported by the Portuguese Mass Spectrometry Network, integrated in the National Roadmap of Research Infrastructures of Strategic Relevance (ROTEIRO/0028/2013; LISBOA-01-0145-FEDER-022125).References
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS.
- D. Philp, S. S. Chen, W. Fitzgerald, J. Orenstein, L. Margolis and H. K. Kleinman, Stem Cells, 2005, 23, 288–296 CrossRef.
- E.-R. Kenawy, J. M. Layman, J. R. Watkins, G. L. Bowlin, J. A. Matthews, D. G. Simpson and G. E. Wnek, Biomaterials, 2003, 24, 907–913 CrossRef CAS.
- M. Alves da Silva, A. Martins, A. R. Costa-Pinto, N. Monteiro, S. Faria, R. L. Reis and N. M. Neves, Biotechnol. J., 2017, 12, 1700073 CrossRef.
- A. Martins, R. L. Reis and N. M. Neves, Int. Mater. Rev., 2008, 53, 257–274 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS.
- G. Jiménez, S. Venkateswaran, E. López-Ruiz, M. Perán, S. Pernagallo, J. J. Díaz-Monchón, R. F. Canadas, C. Antich, J. M. Oliveira, A. Callanan, R. Walllace, R. L. Reis, E. Montañez, E. Carrillo, M. Bradley and J. A. Marchal, Acta Biomater., 2019, 90, 146–156 CrossRef.
- A. Carretero, D. Soares da Costa, R. L. Reis and I. Pashkuleva, J. Mater. Chem. B, 2017, 5, 3103–3106 RSC.
- F. J. O'Brien, B. A. Harley, I. V. Yannas and L. J. Gibson, Biomaterials, 2005, 26, 433–441 CrossRef.
- G. Orlando, C. Booth, Z. Wang, G. Totonelli, C. L. Ross, E. Moran, M. Salvatori, P. Maghsoudlou, M. Turmaine, G. Delario, Y. Al-Shraideh, U. Farooq, A. C. Farney, J. Rogers, S. S. Iskandar, A. Burns, F. C. Marini, P. De Coppi, R. J. Stratta and S. Soker, Biomaterials, 2013, 34, 5915–5925 CrossRef CAS.
- Y. L. Yu, Y. K. Shao, Y. Q. Ding, K. Z. Lin, B. Chen, H. Z. Zhang, L. N. Zhao, Z. B. Wang, J. S. Zhang, M. L. Tang and J. Mei, Biomaterials, 2014, 35, 6822–6828 CrossRef CAS.
- H. C. Ott, T. S. Matthiesen, S.-K. Goh, L. D. Black, S. M. Kren, T. I. Netoff and D. A. Taylor, Nat. Med., 2008, 14, 213–221 CrossRef CAS.
- M. Abolbashari, S. M. Agcaoili, M. K. Lee, I. K. Ko, T. Aboushwareb, J. D. Jackson, J. J. Yoo and A. Atala, Acta Biomater., 2016, 29, 52–61 CrossRef CAS.
- I. K. Ko, M. Abolbashari, J. Huling, C. Kim, S.-H. Mirmalek-Sani, M. Moradi, G. Orlando, J. D. Jackson, T. Aboushwareb, S. Soker, J. J. Yoo and A. Atala, Technology, 2014, 02, 243–253 CrossRef.
- J. J. Song, J. P. Guyette, S. E. Gilpin, G. Gonzalez, J. P. Vacanti and H. C. Ott, Nat. Med., 2013, 19, 646–651 CrossRef CAS.
- E. A. J. Hoste, J. A. Kellum, N. M. Selby, A. Zarbock, P. M. Palevsky, S. M. Bagshaw, S. L. Goldstein, J. Cerdá and L. S. Chawla, Nat. Rev. Nephrol., 2018, 14, 607–625 CrossRef CAS.
- R. J. Nagao, J. Xu, P. Luo, J. Xue, Y. Wang, S. Kotha, W. Zeng, X. Fu, J. Himmelfarb and Y. Zheng, Tissue Eng., Part A, 2016, 22, 1140–1150 CrossRef CAS.
- J. Su, S. C. Satchell, R. N. Shah and J. A. Wertheim, J. Biomed. Mater. Res., Part A, 2018, 106A(9), 2448–2462 CrossRef.
- J. D. O'Neill, D. O. Freytes, A. J. Anandappa, J. A. Oliver and G. V. Vunjak-Novakovic, Biomaterials, 2013, 34, 9830–9841 CrossRef.
- E. Lih, W. Park, K. W. Park, S. Y. Chun, H. Kim, Y. K. Joung, T. G. Kwon, J. A. Hubbell and D. K. Han, ACS Cent. Sci., 2019, 5, 458–467 CrossRef CAS.
- N. K. Singh, W. Han, S. A. Nam, J. W. Kim, J. Y. Kim, Y. K. Kim and D. W. Cho, Biomaterials, 2020, 232, 119734 CrossRef CAS.
- M. Ali, P. R. Anil Kumar, J. J. Yoo, F. Zahran, A. Atala and S. J. Lee, Adv. Healthcare Mater., 2019, 8(7), e1800992 CrossRef.
- T. W. Gilbert, D. B. Stolz, F. Biancaniello, A. Simmons-Byrd and S. F. Badylak, Biomaterials, 2005, 26, 1431–1435 CrossRef CAS.
- H. Yin, Y. Wang, Z. Sun, X. Sun, Y. Xu, P. Li, H. Meng, X. Yu, B. Xiao, T. Fan, Y. Wang, W. Xu, A. Wang, Q. Guo, J. Peng and S. Lu, Acta Biomater., 2016, 33, 96–109 CrossRef CAS.
- J. S. Choi, H.-J. Yang, B. S. Kim, J. D. Kim, J. Y. Kim, B. Yoo, K. Park, H. Y. Lee and Y. W. Cho, J. Controlled Release, 2009, 139, 2–7 CrossRef CAS.
- L. Penolazzi, S. Mazzitelli, R. Vecchiatini, E. Torreggiani, E. Lambertini, S. Johnson, S. F. Badylak, R. Piva and C. Nastruzzi, J. Cell. Physiol., 2012, 227, 857–866 CrossRef CAS.
- R. Sobreiro-Almeida, D. R. Fonseca and N. M. Neves, Mater. Sci. Eng., C, 2019, 103, 109866 CrossRef CAS.
- R. Sobreiro-Almeida, M. Elena Melica, L. Lasagni, P. Romagnani and N. M. Neves, Acta Physiol., 2020, 230(1), e13491 CAS.
- S. Zahiri, E. Masaeli, E. Poorazizi and M. H. Nasr-Esfahani, J. Biomed. Mater. Res., Part A, 2018, 106, 2463–2471 CrossRef CAS.
- M. C. Bottino, M. V. Jose, V. Thomas, D. R. Dean and G. M. Janowski, Dent. Mater., 2009, 25, 1109–1115 CrossRef CAS.
- Z. Movasaghi, S. Rehman and D. I. ur Rehman, Appl. Spectrosc. Rev., 2008, 43, 134–179 CrossRef CAS.
- C. Sagrinati, G. S. Netti, B. Mazzinghi, E. Lazzeri, F. Liotta, F. Frosali, E. Ronconi, C. Meini, M. Gacci, R. Squecco, M. Carini, L. Gesualdo, F. Francini, E. Maggi, F. Annunziato, L. Lasagni, M. Serio, S. Romagnani and P. Romagnani, J. Am. Soc. Nephrol., 2006, 17, 2443–2456 CrossRef CAS.
- T. Riaz, R. Zeeshan, F. Zarif, K. Ilyas, N. Muhammad, S. Z. Safi, A. Rahim, S. A. A. Rizvi and I. U. Rehman, Appl. Spectrosc. Rev., 2018, 53, 703–746 CrossRef CAS.
- G. Chi Ting Au-Yeung, U. Sarig, H. Sarig, H. Bogireddi, T. Bronshtein, L. Baruch, A. Spizzichino, J. Bortman, B. Y. C. Freddy, M. Machluf and S. S. Venkatraman, Biomater. Sci., 2017, 5(6), 1183–1194 RSC.
- S. Baiguera, C. Del Gaudio, E. Lucatelli, E. Kuevda, M. Boieri, B. Mazzanti, A. Bianco and P. Macchiarini, Biomaterials, 2014, 35, 1205–1214 CrossRef CAS.
- J. L. Dziki and S. F. Badylak, Regener. Eng. Transl. Med., 2019, 5, 414–419 CrossRef.
- J. R. Couchman, D. R. Abrahamson and K. J. McCarthy, Kidney Int., 1993, 43(1), 79–84 CrossRef CAS.
- J. D. Cameron, A. P. N. Skubitz and L. T. Furcht, Invest. Ophthalmol. Visual Sci., 1991, 32(10), 2766–2773 CAS.
- R. C. Hill, E. A. Calle, M. Dzieciatkowska, L. E. Niklason and K. C. Hansen, Mol. Cell. Proteomics, 2015, 14, 961–973 CrossRef CAS.
- T. D. Johnson, R. C. Hill, M. Dzieciatkowska, V. Nigam, A. Behfar, K. L. Christman and K. C. Hansen, Proteomics: Clin. Appl., 2016, 10, 75–83 CAS.
- D. R. Eyre, M. A. Paz and P. M. Gallop, Annu. Rev. Biochem., 1984, 53, 717–748 CrossRef CAS.
- L. Louzao-Martinez, C. G. M. van Dijk, Y. J. Xu, A. Korn, N. J. Bekker, R. Brouwhuis, M. N. Nicese, J. A. A. Demmers, M.-J. T. H. Goumans, R. Masereeuw, D. J. Duncker, M. C. Verhaar and C. Cheng, Matrix Biol. Plus, 2019, 4, 100011 CrossRef.
- P. N. Furness, J. Clin. Pathol., 1996, 49, 355–359 CrossRef CAS.
- F. Genovese, A. A. Manresa, D. J. Leeming, M. A. Karsdal and P. Boor, Fibrog. Tissue Repair, 2014, 7, 4 CrossRef.
- M. L. Angelotti, E. Ronconi, L. Ballerini, A. Peired, B. Mazzinghi, C. Sagrinati, E. Parente, M. Gacci, M. Carini, M. Rotondi, A. B. Fogo, E. Lazzeri, L. Lasagni and P. Romagnani, Stem Cells, 2012, 30, 1714–1725 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01272f |
| This journal is © The Royal Society of Chemistry 2021 |