
Revisiting gene delivery to the brain: silencing and editing
João
Conniot†
ab,
Sepehr
Talebian†
cd,
Susana
Simões†
 e,
Lino
Ferreira
*ef and
João
Conde
*ab
e,
Lino
Ferreira
*ef and
João
Conde
*ab
aNOVA Medical School, Faculdade de Ciências Médicas, Universidade Nova de Lisboa, 1169-056 Lisboa, Portugal. E-mail: joao.conde@nms.unl.pt
bCentre for Toxicogenomics and Human Health (ToxOmics), Genetics, Oncology and Human Toxicology, NOVA Medical School, Faculdade de Ciências Médicas, Universidade Nova de Lisboa, 1169-056 Lisboa, Portugal
cIntelligent Polymer Research Institute, ARC Centre of Excellence for Electromaterials Science, AIIM Facility, University of Wollongong, NSW 2522, Australia
dIllawarra Health and Medical Research Institute, University of Wollongong, Wollongong, NSW 2522, Australia
eCNC – Center for Neuroscience and Cell Biology, University of Coimbra, 3004-517 Coimbra, Portugal. E-mail: lino@uc-biotech.pt
fFaculty of Medicine, University of Coimbra, 3030-789 Coimbra, Portugal
First published on 24th November 2020
Abstract
Neurodegenerative disorders, ischemic brain diseases, and brain tumors are debilitating diseases that severely impact a person's life and could possibly lead to their demise if left untreated. Many of these diseases do not respond to small molecule therapeutics and have no effective long-term therapy. Gene therapy offers the promise of treatment or even a cure for both genetic and acquired brain diseases, mediated by either silencing or editing disease-specific genes. Indeed, in the last 5 years, significant progress has been made in the delivery of non-coding RNAs as well as gene-editing formulations to the brain. Unfortunately, the delivery is a major limiting factor for the success of gene therapies. Both viral and non-viral vectors have been used to deliver genetic information into a target cell, but they have limitations. Viral vectors provide excellent transduction efficiency but are associated with toxic effects and have limited packaging capacity; however, non-viral vectors are less toxic and show a high packaging capacity at the price of low transfection efficiency. Herein, we review the progress made in the field of brain gene therapy, particularly in the design of non-toxic and trackable non-viral vectors, capable of controlled release of genes in response to internal/external triggers, and in the delivery of formulations for gene editing. The application of these systems in the context of various brain diseases in pre-clinical and clinical tests will be discussed. Such promising approaches could potentially pave the way for clinical realization of brain gene therapies.
1. Introduction
The concept of gene therapy relies on the delivery of recombinant nucleic acids (DNA or RNA) to add, replace, repair, or even remove a gene sequence. The delivery of nucleic acids induces a change in the cell phenotype, which, in case of disease, means slowing down the disease progression or leads to the cure. Although the idea may seem extremely simple, gene regulation is highly complex, and many diseases result from a pattern of multiple altered genes, like some neurodegenerative diseases, such as Alzheimer's and Parkinson's diseases. Gene therapy can also be used to target cancer cells, by introducing a gene that will induce a phenotype more susceptible to complementary therapeutic strategies or is directly lethal. Even though the translation of gene therapy started in the early 1990s, the lack of knowledge concerning gene vectors, target cells, and, in particular, the diseases, has led to disappointing outcomes.1,2 In the beginning of the 2000s, the development of new vectors and gene-editing tools, combined with a deeper understanding of the target cells, renewed interest, and new trials evidenced sustained genetic modifications and clinical benefits in some.3–5RNA interference (RNAi), by which disease genes can be specifically silenced, has great potential to become a successful therapeutic strategy for neurological diseases.6 Small interfering RNAs (siRNAs) and microRNAs (miRNAs) have attracted considerable attention due to their role in gene regulation, which makes them likely targets for drug discovery and development.7 Therapeutic approaches based on siRNA involve the introduction of a synthetic siRNA into the target cells to elicit RNA interference (RNAi), thereby inhibiting the expression of a specific messenger RNA (mRNA) to produce a gene-silencing effect. By contrast, miRNA-based therapeutics comprise two approaches: miRNA inhibition and miRNA replacement.8 miRNA inhibition resembles antisense therapy, with synthetic single-stranded RNAs acting as miRNA antagonists (also known as antagomirs or anti-miRs) to inhibit the action of the endogenous miRNAs. On the other hand, in the replacement approach, synthetic miRNAs (also known as miRNA mimics) are used to mimic the function of the endogenous miRNAs, which leads to mRNA degradation/inhibition and produces a gene-silencing effect.9 In any case, the use of RNAi is limited to targets for which gene knockdown is beneficial. Also, RNAi often cannot fully repress gene expression and is therefore unlikely to provide a benefit for diseases in which complete ablation of gene function is necessary for therapy.10 Consequently, genome-editing technologies based on programmable nucleases, such as zinc finger nucleases (ZFNs),11 transcription activator-like effector nucleases (TALENs),12,13 and the clustered regularly interspaced short palindromic repeat (CRISPR)-associated nuclease Cas9,14 are opening up the possibility of therapeutic genome editing in diseased cells and tissues, resulting in the removal or correction of deleterious mutations or the insertion of protective mutations.
Growing interest in gene therapy has been motivated by the fact that, in theory, gene-based approaches may provide, with a single treatment, a sustained production of endogenous proteins by the target cells, which obviates the need for repeated administration of protein-based therapies. However, genetic manipulation of the CNS presents many challenges. The specific characteristics of most neurons, which are terminally differentiated, require specific delivery strategies, such as vectors that persist in postmitotic cells. In terms of gene therapy, which has shown promising results for the treatment of several diseases of the CNS, such as lysosomal storage diseases,15 Parkinson's disease,16 Alzheimer's disease,17 and brain tumors,18 gene delivery after systemic administration remains one of the main challenges, largely due to the blood–brain barrier (BBB).19 Before we achieve widespread application of gene delivery to the brain and the nervous system, specifically in the clinic, further efforts must be made to fine-tune targeted strategies and guarantee their safety and efficacy.
Herein, we review the most recent advances in brain gene delivery focusing on silencing and editing. Although recent studies have reviewed aspects of brain gene therapies,20,21 they were limited in integrating both silencing and editing strategies and cover non-viral delivery systems. Initially, we will highlight gene-delivery strategies to the brain using viral vectors (e.g. adenovirus, lentivirus, herpes simplex virus) and non-viral vectors (e.g. cationic liposomes, cationic polymers, organic and inorganic nanomaterials). Then, the importance of silencing gene therapies to treat brain diseases such as cancer and neurodegenerative diseases will be discussed. Finally, we will cover brain gene-editing strategies focusing essentially on the CRISPR/Cas system.
2. Gene-delivery strategies to the brain
Many chronic neurologic diseases do not respond to small-molecule therapeutics and have no effective long-term therapy.22 Gene therapy offers the promise of an effective cure for both genetic and acquired brain disease, yet progress in clinical trials has been slow, and a major limiting factor is delivery of the gene to brain.23 This is primarily due to complexity of the brain, side effects, and the impermeable blood-brain barrier (BBB).24 To this end, various vectors (vehicles) were used to move genetic information into a target cell, which mainly included viral and non-viral vectors (see Fig. 1).25,26 Each type of vector imposes its own set of advantages and disadvantages, and hence in the following section we will provide a comprehensive description of each vector type accompanied by relevant examples where they were used for gene delivery to brain. | ||
| Fig. 1 Schematic representation of viral (left) and non-viral (right) vector for gene delivery to the brain. | ||
2.1. Viral vectors
Ideal virus-based vectors for most gene-therapy applications harness the viral infection pathway but avoid the subsequent expression of viral genes that leads to replication and toxicity.27 In particular, the viral particles encapsulate a modified genome carrying a therapeutic gene cassette in place of the viral genome.28 Features of the ideal viral vector are: (i) specific tropism for highly efficient transduction of the target tissue and minimal transduction of ‘off-target’ cells, tissues, or organs; (ii) capacity to express the transgene for a period of time and at a level so as to have maximal therapeutic impact while eliciting a minimal host immune response; (iii) minimal side effects such as vector-related pathologies and/or host immune response.26 The spectrum of viral vectors used in basic research applications includes viruses with simple capsid virions—such as recombinant adenovirus and adeno-associated virus—and others such as retrovirus/lentivirus, and herpes virus, in which the capsid is surrounded by a lipid bilayer envelope.29 The difference between each class of viral vectors is their packaging capacity, transduction capability, integration into the host genome, duration of transgene expression, and induced pathogenesis, to name just a few.30 It should be noted that vectors derived from some retroviruses, such as Moloney murine leukaemia virus (MoMLV), have limited applications as vectors for the central nervous system (CNS) owing to their inability to deliver genes to non-dividing cells. Retroviral vectors have successfully been applied to modify non-neuronal cells ex vivo prior to transplantation in the nervous system.31 However, adeno-associated virus, lentivirus, and herpes virus are all suitable vectors to efficiently transduce neurons of the CNS.26 Nonetheless, because of the properties of binding and entry proteins on the virus capsid or envelope, some cells are intrinsically more susceptible to infection with certain vectors. Also, depending on the viral type, it can interact with different target cell receptors, which correspondingly mediates a different mechanism of endocytosis.32 Overall, viral vector delivery modalities can be grouped into those that attempt to achieve widespread gene delivery throughout the brain (global) and those that target specific cell populations within the brain (focal). Accordingly, focal delivery is often achieved through local administration where vectors are either injected directly into the brain parenchyma, ventricles, or vasculature, or indirectly injected via intrathecal or intranasal administration, with different types of vectors and modes of injection designed to hit selected targets.29 Local administration of these viral vectors implies that focal delivery modalities are capable of bypassing BBB, and they are also less susceptible to neutralization by circulating antibodies.33 Conversely, methods for global delivery include injection into the carotid artery, with promotion of entry across the blood–brain barrier through localized disruption of BBB (using magnetic resonance-guided focused ultrasound and IV administered microbubbles) or receptor-mediated transport via transcytosis (using the transferrin receptor, LDL receptor-related proteins, or insulin receptor), or using a viral vector that naturally crosses the BBB (archetypally, AAV9).34 The type of brain disease can determine the suitable method of viral delivery. For instance, focal delivery is preferred for treatment of Parkinson's disease by targeting focal areas of disease pathogenesis, whereas global delivery is more appropriate for widespread CNS diseases, such as brain tumors and lysosomal storage diseases.35 Currently, viral vector-mediated gene therapy for disorders of the central nervous system is focused on life-threatening or severely debilitating diseases, owing in part to unknown risk factors associated with virus vectors. Neurological conditions where these vectors might prove to be effective include stroke, spinal cord injury, neurodegenerative diseases, lysosomal storage diseases, brain tumours, and pain.29 In the following section, we will review the most common viral vectors used for gene delivery to the brain.Nevertheless, adeno-associated virus (AAV) and recombinant AAV vectors are commonly used to deliver various genes to brain mainly due to their favourable attributes such as capability to infect both dividing and non-dividing cells, as well as their minimal immunogenicity.49 Along similar lines, transgenes that encode therapeutic proteins,50–55 microRNAs (miRNAs),56–60 antibodies,61–63 and CRISPR-Cas9 guide RNA for gene editing64–68 have been successfully delivered to the CNS with AAVs in mice and other species (Fig. 2A). In addition, multiple capsids have been used across species to successfully target a variety of tissues and cell types within the CNS, including neurons, astrocytes, oligodendrocytes, and glioma cells.45,69–74
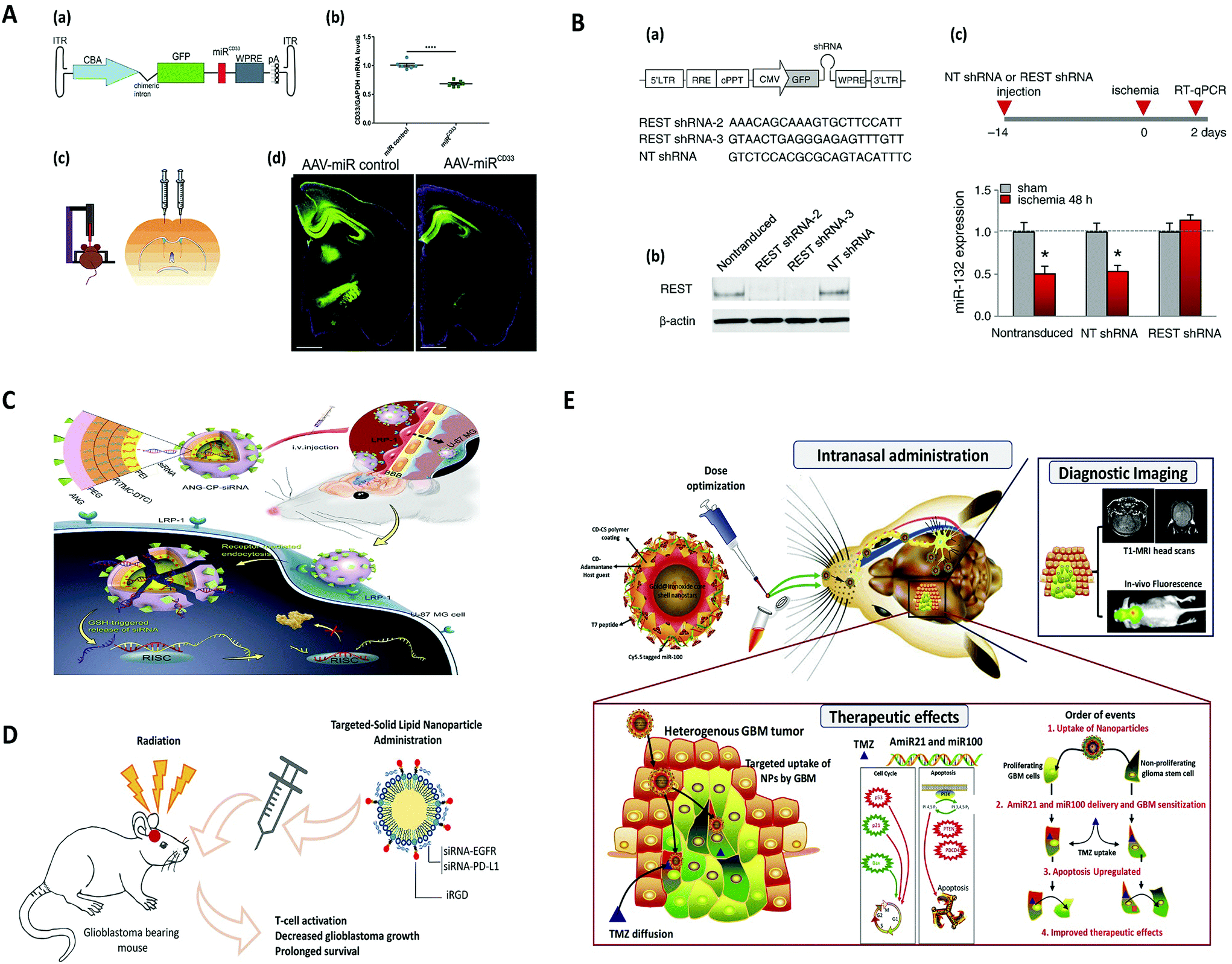 | ||
| Fig. 2 Viral and non-viral delivery for gene silencing in brain, (A) AAV-mediated SOD1 silencing by overexpression of miRNA against human SOD1 coding sequence, to prevent motoneuron degeneration caused by SOD1 mutation. Reproduced with permission.188 Copyright 2015, Wiley. (B) Lentivirus-mediated miRNA-guided neuron tag (“mAGNET”) to restrict transgene expression to cortical inhibitory (GABA+) neurons in the mouse neocortex (GABA mAGNET). Reproduced with permission.189 Copyright 2018, Elsevier. (C) RNAi therapy for human glioblastoma in vivo using siRNA-loaded nontoxic brain-targeting chimaeric polymersomes (ANG-CP-siRNA). Reproduced with permission.190 Copyright 2018, Elsevier. (D) A cyclic peptide iRGD (CCRGDKGPDC)-conjugated solid lipid nanoparticle (SLN) to deliver small interfering RNAs (siRNAs) against both epidermal growth factor receptor (EGFR) and PD-L1 for combined targeted and immunotherapy against glioblastoma. Reproduced with permission.138 Copyright 2019, American Chemical Society. (E) Targeted delivery of theranostic polyfunctional gold–iron oxide nanoparticle (polyGION) surface loaded with therapeutic miRNAs (miR-100 and antimiR-21) to glioblastoma in mice. Reproduced with permission.177 Copyright 2019, Elsevier. | ||
Lentiviral vectors can deliver 8 kb of sequence, and that means they can accommodate larger transgenes than AAV vectors.26 Lentiviral vectors have the advantages of high-efficiency infection of dividing and non-dividing cells, long-term stable expression of a transgene, and low immunogenicity.29 Conversely, this powerful tool has the potential to cause oncogenic, infectious, and other transformative changes to infected cells. LVV-infected cells can become cancerous through activation of oncogenes or inactivation of tumor-suppressor genes.84 One way to address this is application of the self-inactivating (SIN) vector in which the viral enhancer and promoter sequences have been deleted.85,86 Transgenes that encode therapeutic microRNAs (miRNAs),87–93 siRNAs,94–98 and shRNA99–103 have been successfully delivered to the CNS with LVVs in mice (Fig. 2B). LVVs have been demonstrated to transduce most cell types within the CNS in vivo, including neurons, astrocytes, adult neuronal stem cells, oligodendrocytes, and glioma cells.79 Efficient therapeutic effects of LVV-mediated transgene expression have been documented in animal models of lysosomal storage diseases (LSDs), Alzheimer's disease (AD), ALS, Huntington's disease (HD), and Parkinson's disease (PD), among others.79
HSV-1 is a neurotropic virus that can spread through the nervous system and establish a latent infection in neurons. Although sensory neurons are the natural reservoir of latency, gene-transfer experiments have shown that HSV-1 can also establish a latent infection in other neurons within the brain, as well as motor neurons.109 Generally, transgenes that encode therapeutic proteins110–113 and antibodies18,114–116 have been successfully delivered to the CNS with HSVs. Efficient therapeutic effects of HSV-mediated transgene expression have been documented in animal models of Parkinson's disease and brain tumors.117
2.2. Non-viral vectors
Although viral vectors have substantially advanced the field of gene therapy, several limitations are associated with viral vectors, including carcinogenesis, immunogenicity, broad tropism, limited DNA packaging capacity, and difficulty of vector production.118 To this end, non-viral gene therapy has the potential to address many of these limitations, particularly with respect to safety, as it is less immunogenic and non-integrating into the host genome.25 Non-viral vectors also have the potential to deliver larger genetic payloads and are typically easier to synthesize than viral vector.119 Nevertheless, few of these vectors have so far been developed clinically, owing to their low delivery efficiency relative to viral vectors.118 So far, a variety of non-viral vectors were used for gene delivery, including cationic lipids and liposomes, cationic polymers, polypeptides, and organic/non-organic nanoparticles.19,25,120–122 In the context of gene delivery, the ideal non-viral vector must target a specific population of cells to deliver its payload of condensed DNA or RNA to mediate an appropriate intracellular delivery of sufficient therapeutic to achieve a functional outcome.25 However, depending on the mode of administration (being systemic or local), the non-viral vectors can face different barriers.Accordingly, systemically delivered non-viral vectors need to prevent degradation by serum endonucleases and evade immune detection (which could be achieved by chemical modifications of nucleic acids and encapsulation of vectors). They also need to avoid renal clearance from the blood and prevent nonspecific interactions, using polyethylene glycol (PEG), or through specific characteristics of particles.118 Furthermore, for successful gene delivery to CNS, non-viral vectors must also pass the BBB. Accordingly, the structural and physicochemical properties of vectors including molecular size, charge, hydrogen-bonding potential, and lipophilicity will determine which is the passage mechanism across the BBB.123 Accordingly, the following transport mechanisms have been proposed: (i) vectors open tight junctions between endothelial cells or induce local toxic effects, which leads to a localized permeabilization of the BBB allowing the penetration of the cargo conjugated with the vectors (this could also be achieved by osmotic disruption, ultrasound disruption, or magnetic disruption, though they could be detrimental to integrity of the BBB124); (ii) vectors pass through endothelial cell by transcytosis (via absorptive-, transporter-, or receptor-mediated transcytosis125); or (iii) a combination of several of the mechanisms described previously.19 Moreover, after passage through the BBB, the vectors must selectively target diseased cells while minimizing the distribution into normal brain cells.126 Also, following internalization, vectors are challenged with a new set of intracellular obstacles (including endolysosomal escape, transport through the cytoplasm, nuclear localization, and unpacking) that must be overcome for successful gene delivery.119 Nevertheless, in order to achieve effective concentrations in the brain, the vectors must be administered in doses that are associated with adverse effects in peripheral organ.127
Consequently, local delivery of non-viral vectors was suggested as an alternative approach to bypass some of the obstacles associated with their systemic delivery. Hence, intraparenchymal (directly into the diseased brain regions facilitated by convection-enhanced delivery or inclusion into biodegradable polymeric implants), intraventricular (into the cerebrospinal fluid produced in the ventricles of the brain), intrathecal (into the intrathecal space surrounding the spinal cord), and intranasal administrations were practiced as local delivery approaches for delivery genes via non-viral vectors to CNS,128–130 albeit local delivery approaches are prone to limitations such as local trauma to the brain neuropil that leads to inflammation and toxicity, as well as limited diffusion from the injection site (for the intraparenchymal route), and an ultimate low ratio of successfully delivered vectors to the brain (for intrathecal and intranasal routes).131
In the following section, we will review the most commonly used non-viral vectors for gene delivery to the brain.
3 Gene therapy: replacement and silencing
3.1 Gene therapy in inherited diseases
Several innate genetic defects have a deleterious effect on the brain, such as lysosomal storage diseases (LSD). In LSD, the genetic defects lead to lysosomal dysfunction, which results in accumulation of uncleaved lipids, glycoproteins, and/or glycosaminoglycans. As a consequence, cell morphology can be altered by high oxidative stress, marked neuroinflammation, and impaired tissue and organ functions.191,192 Current available therapies for LSD are based on enzyme replacement or hematopoietic stem cell transplantation, which are extremely expensive and have unclear long-term effectiveness.193 These metabolic inherited diseases are promising candidates for brain gene therapy, because they generally result from a single gene defect, and the non-functional mutated gene can be replaced and the normal phenotype restored. Gene therapy may offer a more affordable, complete treatment to all the conditions in these diseases. Some pre-clinical studies and clinical trials have already shown promising results in different diseases, such as Gaucher disease,194 mucopolysaccharidosis,195,196 and the Niemann-pick type C1 disease,197,198 by replacement gene therapy.3.2 Gene therapy in acquired diseases
Even though the genetic pattern in acquired diseases is much more complex than in single gene inborn defects, gene therapy may also constitute a relevant approach in neurodegenerative conditions, such as Parkinson's (PD) and Alzheimer's (AD) diseases, or in brain tumors.Gene-therapy strategies for PD rely on the transfer of genes encoding neurotransmitter-synthesizing or metabolic enzymes to improve the function of the impaired synthesis. For instance, several approaches have reported the successful transfer cDNA of tyrosine hydroxylase (TH), the rate-limiting enzyme in the synthesis of DA to the striatum. Other genes, which encode complementary enzymes of DA synthesis, such as the aromatic amino acid decarboxylase (AADC)200,201 and the GTP cyclohydrolase (GTPCH)202 have also been delivered and have been shown to maximize DA production in the striatum. Silencing of the SNCA (alpha synuclein) gene with siRNA and shRNA has also shown promising results both in vitro and in vivo, with a reduction in SNCA in the brain and improvement in motor dysfunction.203,204 On the other hand, gene therapy can also be used to deliver genes of trophic factors and protective proteins to slow down or block the neurodegenerative process. Some examples of molecules that limit the progression of PD are anti-apoptotic, antioxidants, and trophic peptide factors, such as the glial-derived growth factor (GDNF).205–208 Although this protein does not cross the BBB after systemic administration, gene therapy has enabled their delivery to the striatum and substantia nigra, showing promising results.208,209
The current available therapies for AD are based on anticholinesterase inhibitors and NMDA antagonists, to improve cognitive function and behavior. However, the efficacy of these drugs has been very limited.212 Gene-therapy strategies so far have been focused on reducing Tau phosphorylation and Aβ plaques. The most obvious target for gene-therapy silencing is the amyloid precursor protein (APP), whose proteolysis leads to increased levels of Aβ protein. However, this silencing strategy has been avoided due to the role of APP in neuronal development and differentiation.213 Enzymes such as β-secretase, which are responsible for cleaving APP, leading to accumulation of Aβ protein, have been used as an alternative target. In vivo delivery of siRNA to silence β-secretase resulted in less accumulation of beta-amyloid (Aβ) in the hippocampus, as well as improvements in cognition and memory in mice.153 Selective gene silencing of glycogen synthase kinase 3 beta (GSK3β), an enzyme involved in Tau phosphorylation, induced memory preservation and synaptic plasticity in mice.214 In clinical trials, ex vivo gene delivery of nerve growth factor (NGF) improved the rate of cognitive decline.215,216
| Delivery method | Cargo | Administration route | Target | Disease | Ref. |
|---|---|---|---|---|---|
| AAV: adeno-associated virus; antimir: antisense oligonucleotides for miRNA inhibition; BACE1: beta-site amyloid precursor protein cleaving enzyme 1; MRP1: multidrug resistance-associated protein 1; NP: nanoparticles; PEG-PDMAEMA: PEGylated poly(2-(N,N-dimethylamino) ethyl methacrylate); PEI: poly(ethyleneimine); PLA: poly (lactic acid); STAT3: signal transducer and activator of transcription 3. | |||||
| Viral | |||||
| rAAV1/2 | siRNA | Interventricular injection | Glycogen synthase kinase-3 | Alzheimer's disease | 231 |
| Lentivirus | shRNA | Intracranial | Aquaporin-4 | Glial scar after traumatic brain injury | 232 |
| AAV5 | antimiR | Intrathecal | Huntingtin | Huntington's disease | 233 |
| Lentivirus | shRNA | Intracranial | α-synuclein | Parkinson's disease | 99 |
| Lentivirus | siRNA | Intracranial | BACE1 | Alzheimer's disease | 96 |
| Non-viral | |||||
| Gold NP | siRNA | Intravenous | Bcl12l2 | Recurrent glioblastoma multiforme or gliosarcoma | Clinical trials-NCT03020017 |
| Gold–iron oxide NP | antimiR | Intranasal | miR-21 | Glioblastoma | 177 |
| Exosomes | siRNA | Intravenous | BACE1 | Alzheimer's disease | 234 |
| PEI-capped porous silicon NP | siRNA | Intravenous | MRP1 | Glioblastoma | 235 |
| PEI complexes | siRNA | Intravenous | Claudin-5 | Cerebral oedema after traumatic brain injury | 236 |
| Poly(L)-lysines dendrigraft | siRNA | Intravenous | Caspase-3 | Parkinson's disease | 152 |
| PEG-PDMAEMA nanolexes | siRNA | Intravenous | BACE1 | Alzheimer's disease | 153 |
| PLA-hyperbranched polyglycerol NP | antimiR | Intratumoral | miR-21 | Glioblastoma | 237 |
| PEI nanoplexes | siRNA | Intranasal | Beclin-1 | HIV-infection brain reservoirs | 238 |
| Other | |||||
| Hydrophobic modification of siRNA | siRNA | Intrastriatal injection | Huntingtin | Huntington's disease | 239 |
| — | antimiR | Intravenous | miR-146a | Epilepsy | 240 |
| Aptamer-siRNA chimera | siRNA | Intraperitoneal | STAT3 | Glioblastoma | 241 |
4. Brain gene editing
Gene editing has emerged in recent years as a tool in the neurosciences, specifically to investigate the role of genes in normal brain–behaviour relationships, and as a potential therapeutic strategy for some neurodegenerative disorders. These tools rely on nucleases that are able to specifically recognize and induce double-strand DNA breaks (DSBs) at target locations in the genome. Genome editing tools based on site-specific DNA nucleases can include zinc finger nucleases (ZFNs),11,242 transcription activator-like effector nucleases (TALENs),243,244 and the Cas effector proteins of clustered regularly interspaced short palindromic repeat (CRISPR) systems such as Cas9.245,246 A major difference between the three genome editing tools is based on the fact that ZFNs and TALENs require alterations in the protein domains to target a specific gene site, whereas in the case of Cas proteins, the targeting is guided by a specific guide RNA sequence able to recognize a DNA target sequence by complementarity. Independently of the gene-editing process, after the induction of DSBs, the cell activates endogenous DNA repair pathways to fix the damage. There are two main types of repair processes, non-homologous end-joining (NHEJ) and homology directed repair (HDR).247–249 NHEJ is frequently associated with the presence of small insertions and deletions (indels) at the break site that can result in gene knockout. However, repair of DSBs by HDR involves the copying of DNA from a homologous donor template resulting in accurate correction of the DSB.249From a chronological point of view, the first endonucleases for gene editing were zinc finger nucleases (ZFNs).242 These enzymes are based on zinc finger proteins, a family of naturally occurring transcription factors, fused on an endonuclease FokI. Zinc finger domains can recognize a trinucleotide DNA sequence, whereas a set of linked zinc finger domains can recognize longer DNA sequences, providing desired on-target specificity. FokI endonuclease works as a dimer, which means that the double-strand DNA cleavage occurs only at sites of binding of two ZFNs to the opposite DNA strands.242 The system is based on two ZFNs engineered to recognize different closely located nucleotide sequences within the target site and requires the simultaneous recognition and binding of both ZFNs, which naturally limits off-target effects. The potential of ZFNs as a brain gene-editing tool has been demonstrated in Huntington's disease (HD).250,251 HD is a dominantly inherited neurodegenerative disorder caused by expanded CAG repeats in the huntingtin (HTT) gene. The gene editing was demonstrated in vivo, at gene and functional levels, in an HD mouse model after stereotaxic administration in the striatum of AAV1250 or AAV6251 particles encoding the ZFN. Importantly, the long-term expression of ZFNs in central neural system neurons induced significant gene repression activity for at least 6 months251,252 and did not induce measurable inflammation or neurodegeneration.253
TALENs were the second endonucleases to be evaluated in gene editing.243,254,255 A single TALE motif recognizes one nucleotide, and an array of TALEs can associate with a longer sequence. The activity of each TALE domain is restricted only to one nucleotide and does not affect the binding specificity of neighboring TALEs, making the engineering of TALENs much easier than ZFNs. Similar to ZFNs, TALE motifs are linked with FokI endonuclease, which requires dimerization for the cleavage to occur. This means that the binding of two different TALENs at opposite strands in close vicinity to the target DNA is needed. The gene editing of potential of TALENs was demonstrated in genes associated with autism (CACNA1C)255 and dementias (NTF3),244,256 among others. TALENs have been used in zebrafish257 and mouse258 models, but their potential as an in vivo brain editing system remains elusive.
The discovery of CRISPR/Cas9 gene editing system in 2012 changed our paradigm of gene editing and transformed the neurosciences research area.245 This system is composed of an enzyme (Cas) and a guide RNA sequence that binds to a specific target sequence of DNA. The RNA complexes with the Cas9 enzyme, resulting in the formation of the Cas9 ribonucleoprotein (RNP). The most frequent enzymes are Cas9 from the bacteria strains Streptococcus pyogenes (SpCas9)245 and Staphylococcus aureus (SaCas9).259 The guide RNA (gRNA) is composed of a dual RNA sequence crRNA (CRISPR RNA) with a target for genomic DNA and tracrRNA (trans-activating CRSPR RNA) that binds to the enzyme. These two RNAs can also be combined into a single hybrid guide RNA (sgRNA).245,260 Under the guidance of gRNA, Cas9 induces precise DSBs at target locations in the genome.14 CRISPR/Cas9 system is relatively easy to prepare because only the guide RNA is different for each application, very efficient and cost-saving in comparison with other gene-editing tools. Importantly, besides the conventional genome editing of Cas9, the enzyme can act as a programmable DNA-binding module to control transcription. In this case, Cas9 nuclease is inactivated (dead Cas9) via amino acid substitutions,260 and the resulting enzyme (dead Cas9) can be fused with transcriptional repressors such as KRAB261–263 or transcriptional activators such as VP64.262,264,265 The demonstration of the gene-editing potential of the CRISPR/Cas9 system has been demonstrated in the context of HD,266–269 Alzheimer's disease (AD),270,271 fragile X syndrome,272 Parkinson's disease,273,274 schizophrenia,275 and neural stem cell gene repression,276 among others, both in vitro and in vivo. The in vivo potential of the CRISPR/Cas9 system for brain applications has been confirmed in mice,266,268–270,272,276 rats277,278 and monkeys279via intracerebral266,268–272,275,280 and intrathecal275 administrations. Neurons,270,272,277,281–283 microglia,272 neural stem/progenitor cells,276,283 and astrocytes272 in animal models at embryonic,280,284 fetal, and adult270,277,284 stages have been edited. Brain cells at different locations including cerebral cortex,284 hippocampus,270,272,283,284 striatum,272,283,284 thalamus,284 hypothalamus,284 cerebellum,284 midbrain,284 and spinal cord284 have been edited, thus demonstrating the versatility of the gene-editing platform. Importantly, most of the gene-editing studies in the brain rely on the NHEJ282 repair mechanism with a success as high as 60%.277 The selection of NHEJ rather than a HDR repair mechanism is justified by the fact that neurons are post-mitotic cells, and thus the NHEJ repair mechanism has a high efficacy. The HDR-mediated gene modification of neurons has been demonstrated in mouse adult brain albeit with limited success (as high as ∼30% of the targeted neurons).284
4.1. Brain delivery of the CRISPR/Cas system
Despite the broad range of applications of the CRISPR/Cas9 system in the context of the brain, the efficient delivery of the CRISPR/Cas9 remains a key limiting factor. The CRISPR/Cas9 can be delivered in three different forms, including (i) DNA, (ii) messenger RNA (Cas9 mRNA), or (iii) Cas9 ribonucleoprotein (RNP) complex (Cas9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :gRNA complex).285 The strategies used for brain delivery of the CRISPR/Cas9 system (Table 2) can be grouped into physical delivery, viral vectors, and non-viral vectors (Fig. 3).
:gRNA complex).285 The strategies used for brain delivery of the CRISPR/Cas9 system (Table 2) can be grouped into physical delivery, viral vectors, and non-viral vectors (Fig. 3).
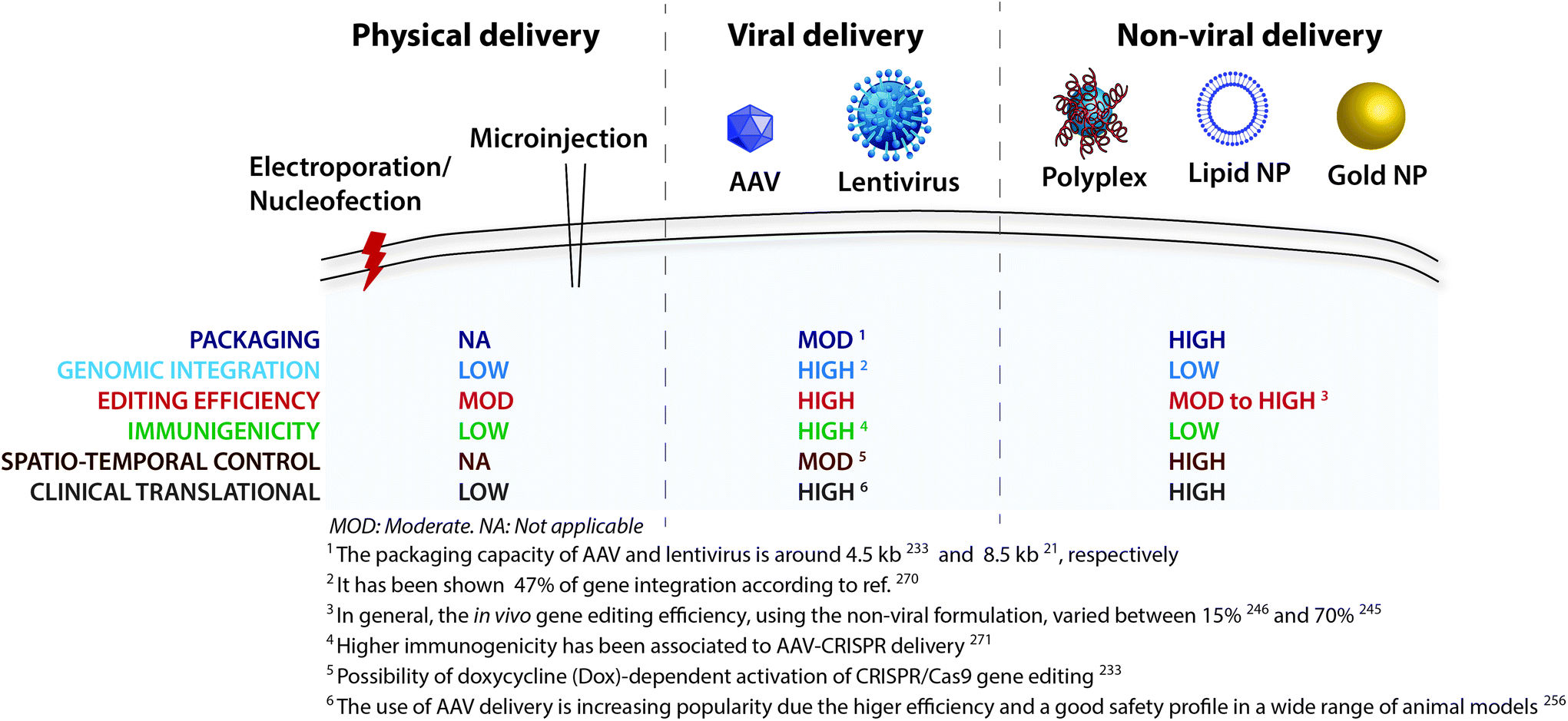 | ||
| Fig. 3 CRISPR/Cas9 delivery strategies. A variety of physical, viral and nonviral methods have been derived to achieve successful delivery across the cell membrane. | ||
| Delivery method | Cargo | Repair mechanism | Target | Efficiency | Brain disease target | Ref. |
|---|---|---|---|---|---|---|
| AAV: adeno-associated virus; NHEJ: nonhomologous end-joining; HDR: homology-directed repair (HDR); HIFI: homology-independent targeted integration; CRISPRi: CRISPR interference; SPH: SunTag-p65-HSF1; HD: Huntington's disease; SB: sleeping beauty transposon; NanoMEDIC: nanomembrane-derived extracellular vesicles for the delivery of macromolecular cargo; DMD: Duchenne muscular dystrophy; NA: not applicable. | ||||||
| Viral | ||||||
| AAV1/2 | AAV-U6-sgRNA-Cre vector | NHEJ | NeuN locus/Rbfox3 gene | High with an indel formation near the predicted cleavage site | Brain cancer mutations | 292 |
| AAV1/2 | Dual vector system: AAV-SpCas9 and AAV-SpGuide | NHEJ | Mecp2, Dnmt1, Dnmt3a and Dnmt3b genes | High | Neurodevelopmental disorders | 282 |
| AAV9 and AAV2g9 | Vectors packaging two guide RNAs (gRNAs) | NHEJ | MIR137 gene | Significant increase in mutant allele frequency | Schizophrenia | 300 |
| AAV serotypes 8 and 9 (HIFI) | Multiple vector system: AAV-Cas9, AAV-sgRNA and AAV-HDR-donor | HDR | Tubb3 gene | Low, partial recovery of vision | Retinitis pigmentosa and Hutchinson–Gilford progeria syndrome | 281 and 294 |
| MERTK gene | ||||||
| rAAV2/1 | Multiple vector system: rAAV2/1-SpCas9 and rAAV-hU6sgRNA | NHEJ | Mutant huntingtin gene (mHTT) | High knock out levels | Huntington's disease | 269 and 308 |
| AAV | AAV-SaCas9-sgRNA | NHEJ | CREB gene | Lower efficiency in vivo | Bipolar disorders | 293 |
| AAV1 and AAV9 | AAV-Cas9-gRNA | NHEJ | Mutant human APP gene (APPsw) | Limited gene-disruption efficiency in vivo | Alzheimer's disease | 270 |
| AAV8 | AAV-sgRNA | Upregulate gene expression | Neurogenic transcription factors: Ascl1, Neurog2 and Neurod1 | High | Parkinson's disease | 264 |
| AAV1 | AAV1-SaCas9-sgRNA | NHEJ | Huntingtin (HTT) gene | Moderate increases lifespan (∼15%) and improves certain motor deficits in these same mice | Huntington's disease | 309 |
| AAV-SB-CRISPR | AAV-SB-sgRNA | NHEJ | Pdia3 and Mgat5 gene | High | Glioblastoma | 310 |
| AAV1 and AAV9 | Dual vector system: AAV-SaCas9 and AAV-sgRNA | NHEJ | cbp gene (CREB-binding protein) | High | Rubinstein–Taybi syndrome | 278 |
| CRISPRi lentivirus | All-in-one lentiviral system (dCas9-KRAB and two or three sgRNAs) | NHEJ | Syt1 gene in the dentate gyrus (DG) | Very high | Neurodevelopmental disorder | 263 |
| lentiCRISPR | Lentiviral backbone plasmid (lentiCRISPR) encoding Cas9 and gRNA | Deletion | GRIN1 gene | high | Neurodevelopmental disorder | 291 |
| Physical | ||||||
| In utero electroporation | Cas9-gRNA plasmid | Knockout | Grin1 gene | High | Neurodevelopmental disorder | 311 |
| In utero electroporation | Cas9-sgRNA plasmid | Deletion | Nf1, Trp53 and Pten genes | High | Brain tumour | 289 |
| In-utero electroporation | Cas9-sgRNA plasmid | Knockout | Satb2 gene | High | Neurodevelopmental disorder | 312 |
| Nucleofection | Cas9-sgRNA plasmid and donor-plasmid | HDR | IL2RG, HBB, CCR5 genes | Very low in vivo correction | Krabbe disease | 286 |
| Nucleofection and Stereotaxic infusion of complexes | Cas9 ribonucleoprotein (RNP) complexes | loxP-stop cassette | High | NA | 283 | |
| Non-viral | ||||||
| Bio-reduceable lipids nanoparticles | Cas9 RNP | NHEJ | EGFP gene Cre-recombination | Highly protein delivery but minimal diffusion | NA | 92 |
| CRISPR-gold nanoparticles | Cas9 RNP | Knockout | loxP-stop cassette and mGluR5 gene | High | Fragile X syndrome | 272 |
| Magneto-electric nanoparticles | Cas9 RNP | Knockout | HIV-1-LTR gene | Significantly reduced latent infection in HIV cells | HIV-1 infection | 307 |
| Colloidal gold nanoparticles | Cas9 RNP and donor ssDNA | HDR | CCR5 receptor | Highest levels of HDR in CD34 + cells | Infectious diseases | 306 |
| No-editing was observed in brain | ||||||
| NanoMEDIC (extracellular vesicles) | spCas9-sgRNA plasmids | Deletion | SAMHD1 gene and DMD (Dystrophin) gene | High efficiently edit the SAMHD1 gene in neurons | Duchenne muscular dystrophy | 305 |
AAVs are the most used vectors for in vivo delivery of the CRISPR/Cas9 system. Smaller Cas9 orthologs, such as spCas9 (4.2 kb) and saCas9 (3.2 kb), are the most attractive ones because they can be packaged in a single AAV vector for brain gene editing.21,259 The following AAV particles have been used for in vivo studies: AAV1,266,270,277,282,284,292 AAV2,282,293 AAV2g9,275 AAV8,264 and AAV9.275,281,284 These viral vectors have been used for the transfection of brain cells in dentate gyrus,282 prefrontal cortex,292 primary visual cortex,282 olfactory bulb,275,284 striatum,266,275,284 hippocampus,270,275,284 and hypothalamus284 (Fig. 4A and B). According to some results, AAV2g9 seems to have a higher tropism to neurons than other brain cells, whereas AAV9 can transfect both neuronal and glial cells275 (Fig. 4C). In most studies, the viral particles were administered in the brain by intracerebral administration,266,275,282,292 and the introduction of knockout mutations through NHEJ266,269,275,282,292 and HDR.280,281,284 HDR-based targeted gene knock-in strategies have limitations because HDR is mainly active in dividing cells; however, some methodologies have been developed to allow DNA knock-in in both dividing and non-dividing cells in vitro and in the mouse brain.281,294 Importantly, studies have shown that AAVs can be used for the in vivo delivery of CRISPR/Cas9 to delete one or multiple genes in the brain.282 Due to the limited capacity of AAVs to package a large amount of genetic information, vectors containing different information have been used. For example, for the brain Mecp2 knockout, two AAVs have been used, one to express Cas9 and one to express the sgRNA and GFP.282 These viruses were mixed and injected into the hippocampus where they resulted in ∼80% co-transduction rate and up to 70% reduction in Mecp2-positive cells in the dentate gyrus. Similarly, a dual AAV-mediated strategy was successfully used to knock out the brain mutant huntingtin (HTT) gene in an HD mouse model268,269 as well as the APPswe mutation found in Tg2576 mice, an AD mouse model.270
 | ||
| Fig. 4 Viral and non-viral delivery in gene-editing efficiency. (A) AAV-HDR for the delivery of a sgRNA and HDR donor template to insert the human influenza hemagglutinin (HA) tag in the embryonic brain. HA tag sequence is fused either to the N terminus of endogenous β-actin (which expresses ubiquitously in most of cell types) or to CaMKIIa (which expresses exclusively in neurons). Confocal microscopic images of whole sagittal brain sections of Cas9 mice showing immunoreactivities for NeuN (magenta) and the HA tag (green) fused to the N terminus of endogenous β-actin (top) and CaMKIIa (bottom). Reproduced with permission.66 Copyright 2017, Elsevier. (B) Genome-editing efficiency in distinct cell types regulated by AAV-HDR administration in E10 mouse embryonic. Adapted from ref. 66. (C) Gene-editing efficiency in multiple brain cells types depends on the CRISPR delivery system. In the case of viral delivery, the AAV2g9 seems to have a higher tropism to transfect neurons than other brain cells, whereas AAV9 can transfect preferentially astrocytes, neuronal and glial cells.66 Non-viral delivery shows higher gene editing in astrocytes and microglia (more proliferative cells), but they also can target neuro stem cells and neurons, according to ref. 272 and 283. | ||
The viral delivery of the CRISPR/Cas9 system in brain cells is, in general, very efficient after intracerebral injection of the viral particles (even when two viral particles are needed to deliver the complete gene-editing formulation); however, the strategy has limitations in case the viral particles are administered by a less invasive route such as the intravenous route. Indeed, in most studies reported so far, the brain gene editing mediated by the CRISPR/Cas9 system occurred after administration of viral particles by stereotaxic injection; however, this allows the transduction of a few cubic millimetres of brain tissue, and the invasiveness of the procedure makes this delivery strategy less likely to be translated to the clinic. AAVs have been engineered for the efficient delivery of genes to the central nervous system after intravenous administration;295 however, the systemic administration of the virus still results in off-targets in the liver, kidney, and heart. Another limitation of the viral particles is linked to their immunogenicity and integration in the genome of the edited cell. A recent study has shown that up to 47% of AAVs were integrated in the genome of cells edited by the CRISPR/Cas9 system.296 In addition, the limited gene editing in some brain regions as well as the limited efficiency of HDR repair processes may require more than one administration of the gene-editing formulation. Unfortunately, the immunogenicity associated with AAVs297 makes this possibility unlikely. Another issue that deserves further attention is the control of the lifespan of Cas9 transgene expression in brain cells in order to reduce its immunogenicity and off-target editing. To reduce the potential toxicity of Cas9, several studies have used self-destructing versions of Cas9, which removed Cas9 expression via a Cas9-specific gRNA or homologous recombination.298 Yet, the complexity of multiple genetic modifications may be a problem in protein activity and can potentially induce neurotoxicity. As an alternative, Cas9 has been fused with cell-cycle regulators to restrict its expression in specific cell-cycle phases.299
The principles used for the preparation of the non-viral formulations took into consideration the selection of the CRISPR/Cas system components and strategies to facilitate the cellular internalization of the formulation (Fig. 5). First, apart from two studies,289,301 Cas proteins have been used as gene-editing enzymes. In general, the complexation of the sgRNA to Cas proteins has been made before the preparation of the nanoparticle. Second, the formulations have been prepared with components that enhanced (i) cellular internalization, (ii) endolysosomal compartment escape, and (iii) nuclear localization. To increase cellular uptake, the CRISPR/Cas9 system components were encapsulated or complexed with polycationic agents such as poly(ethyleneimine) (PEI),289 lipofectamine,283 or a cationic endosomal disruptive polymer PAsp (DET).272,304 To increase nuclear targeting, the Cas9 protein has been modified with four copies of SV40-NLS,283 whereas bio-reducible lipid nanoparticles have been used to increase endolysosomal escape.92 Third, some formulations, particularly those for HDR repair processes,272 have used the core of the nanoparticle to immobilize the repair template by hybridization with complementary oligonucleotides immobilized in the surface of the nanoparticle.
 | ||
| Fig. 5 CRISPR–Cas9 ribonucleoprotein complex challenges related to improving gene-editing efficiency (left) as well as tools and biomaterials for designing more efficient delivery systems in the future (right). | ||
In general, non-viral formulations compare favourably to viral formulations concerning the absence of its integration in the cellular genome, packaging capacity, and control in spatio-temporal release of the CRISPR/Cas cargo (Fig. 3). An important safety aspect related to brain gene editing is related to the immune response to the Cas9 RNP. Some studies indicate that Cas9RNP NPs delivered in the mouse brain did not activate microglia, the mainstay of the immune cells of the central nervous system.283 Therefore, the formulations seem to be safe; however, further studies are necessary to investigate this issue in greater detail (Fig. 5). In addition, although some formulations tested so far in the context of the brain showed a higher preference for neurons than for astrocytes,283 the control of gene editing in specific brain cell populations remains to be demonstrated. Another issue that remains to be investigated is whether the non-viral formulations can be administered by the intravenous route and accumulate in the brain. This strategy could simplify enormously the application of gene-editing formulations for brain applications. Recently, a magnetically guided non-invasive delivery of nanoparticles containing CRISPR/Cas9 vector was demonstrated that can cross the blood–brain barrier (BBB) to inhibit latent HIV-1 infection in microglial cells.307
Presently, brain gene-editing approaches have not reached the clinical trial stage. However, gene-editing approaches outside the brain are now in clinical trials. For example, a recent study has demonstrated the safety and feasibility of gene-editing human T lymphocytes in cancer patients in order to improve their antitumor activity.324 In coming years, clinical trials in brain gene editing are expected. CRISPR/Cas9-mediated genome editing will provide important advances in novel therapeutic targets or in multiple targets. Non-viral systems have great potential for packaging and delivering large genetic payloads and reducing the risk of insertional errors that can occur with viral gene delivery. Gene-therapy formulations for the silencing of specific brain genes may also find applications in the context of certain neurodegenerative diseases. For example, Patisiran, a lipid-siRNA formulation developed by Alnylam® Pharmaceuticals (https://www.alnylam.com/), has been approved in the US and Europe to target transthyretin-mediated amyloidosis.325
A limiting problem in brain gene therapy is brain delivery. This limitation is due to the high complexity of the brain and the impermeability of the blood–brain barrier. Formulations have been developed that are able to cross the BBB efficiently and accumulate in the brain parenchyma.317,318 In addition, the transient opening of the BBB by the use of focused ultrasounds may further enhance the accumulation of formulations in the brain.314 These tools should now be investigated for the delivery of gene therapies in the brain.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. acknowledges the European Research Council Starting Grant (ERC-StG-2019-848325).References
- S. Jenks, JNCI, J. Natl. Cancer Inst., 2000, 92, 98–100 CrossRef CAS.
- J. L. Fox, Nat. Biotechnol., 2000, 18, 1136–1136 CrossRef CAS.
- S. W. J. McPhee, C. G. Janson, C. Li, R. J. Samulski, A. S. Camp, J. Francis, D. Shera, L. Lioutermann, M. Feely, A. Freese and P. Leone, J. Gene Med., 2006, 8, 577–588 CrossRef CAS.
- W. J. Marks, J. L. Ostrem, L. Verhagen, P. A. Starr, P. S. Larson, R. A. E. Bakay, R. Taylor, D. A. Cahn-Weiner, A. J. Stoessl, C. W. Olanow and R. T. Bartus, Lancet Neurol., 2008, 7, 400–408 CrossRef.
- M. A. Kotterman, T. W. Chalberg and D. V. Schaffer, Annu. Rev. Biomed. Eng., 2015, 17, 63–89 CrossRef CAS.
- A. Jagannath and M. Wood, Briefings Funct. Genomics, 2007, 6, 40–49 CrossRef CAS.
- R. K. M. Leung and P. A. Whittaker, Pharmacol. Ther., 2005, 107, 222–239 CrossRef CAS.
- J. K. W. Lam, M. Y. T. Chow, Y. Zhang and S. W. S. Leung, Mol. Ther.–Nucleic Acids, 2015, 4, e252 CrossRef CAS.
- A. G. Bader, D. Brown and M. Winkler, Cancer Res., 2010, 70, 7027 CrossRef CAS.
- D. B. T. Cox, R. J. Platt and F. Zhang, Nat. Med., 2015, 21, 121–131 CrossRef CAS.
- F. D. Urnov, E. J. Rebar, M. C. Holmes, H. S. Zhang and P. D. Gregory, Nat. Rev. Genet., 2010, 11, 636–646 CrossRef CAS.
- A. J. Bogdanove and D. F. Voytas, Science, 2011, 333, 1843–1846 CrossRef CAS.
- A. M. Scharenberg, P. Duchateau and J. Smith, Curr. Gene Ther., 2013, 13, 291–303 CrossRef CAS.
- P. D. Hsu, E. S. Lander and F. Zhang, Cell, 2014, 157, 1262–1278 CrossRef CAS.
- V. J. McCurdy, H. E. Rockwell, J. R. Arthur, A. M. Bradbury, A. K. Johnson, A. N. Randle, B. L. Brunson, M. Hwang, H. L. Gray-Edwards, N. E. Morrison, J. A. Johnson, H. J. Baker, N. R. Cox, T. N. Seyfried, M. Sena-Esteves and D. R. Martin, Gene Ther., 2015, 22, 181–189 CrossRef CAS.
- S. Ramaswamy, J. L. McBride, I. Han, E. M. Berry-Kravis, L. Zhou, C. D. Herzog, M. Gasmi, R. T. Bartus and J. H. Kordower, Neurobiol. Dis., 2009, 34, 40–50 CrossRef CAS.
- S. R. Murphy, C. C. Y. Chang, G. Dogbevia, E. Y. Bryleva, Z. Bowen, M. T. Hasan and T. Y. Chang, Mol. Ther., 2013, 21, 1497–1506 CrossRef CAS.
- D. Saha, R. L. Martuza and S. D. Rabkin, Immunotherapy, 2018, 10, 779–786 CrossRef CAS.
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS.
- S. Ingusci, G. Verlengia, M. Soukupova, S. Zucchini and M. Simonato, Front. Pharmacol., 2019, 10, 724 CrossRef CAS.
- B. E. Deverman, B. M. Ravina, K. S. Bankiewicz, S. M. Paul and D. W. Sah, Nat. Rev. Drug Discovery, 2018, 17, 641–659 CrossRef CAS.
- F. Schlachetzki, Y. Zhang, R. J. Boado and W. M. Pardridge, Neurology, 2004, 62, 1275–1281 CrossRef CAS.
- M. Simonato, J. Bennett, N. M. Boulis, M. G. Castro, D. J. Fink, W. F. Goins, S. J. Gray, P. R. Lowenstein, L. H. Vandenberghe and T. J. Wilson, Nat. Rev. Neurol., 2013, 9, 277–291 CrossRef CAS.
- W. M. Pardridge, Neuron, 2002, 36, 555–558 CrossRef CAS.
- M.-L. Rogers and R. A. Rush, J. Controlled Release, 2012, 157, 183–189 CrossRef CAS.
- T. B. Lentz, S. J. Gray and R. J. Samulski, Neurobiol. Dis., 2012, 48, 179–188 CrossRef CAS.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS.
- M. A. Kay, J. C. Glorioso and L. Naldini, Nat. Med., 2001, 7, 33–40 CrossRef CAS.
- B. L. Davidson and X. O. Breakefield, Nat. Rev. Neurosci., 2003, 4, 353–364 CrossRef CAS.
- S. R. Choudhury, E. Hudry, C. A. Maguire, M. Sena-Esteves, X. O. Breakefield and P. Grandi, Neuropharmacology, 2017, 120, 63–80 CrossRef CAS.
- W. T. J. M. C. Hermens and J. Verhaagen, Prog. Neurobiol., 1998, 55, 399–432 CrossRef CAS.
- M. Giacca and S. Zacchigna, J. Controlled Release, 2012, 161, 377–388 CrossRef CAS.
- M. Hocquemiller, L. Giersch, M. Audrain, S. Parker and N. Cartier, Hum. Gene Ther., 2016, 27, 478–496 CrossRef CAS.
- H. Fu and D. M. McCarty, Curr. Opin. Virol., 2016, 21, 87–92 CrossRef CAS.
- L. Costantini, J. Bakowska, X. Breakefield and O. Isacson, Gene Ther., 2000, 7, 93–109 CrossRef CAS.
- K. Lundstrom, Diseases, 2018, 6, 42 CrossRef.
- X. Danthinne and M. Imperiale, Gene Ther., 2000, 7, 1707–1714 CrossRef CAS.
- R. Alba, A. Bosch and M. Chillon, Gene Ther., 2005, 12, S18–S27 CrossRef CAS.
- M. Candolfi, J. F. Curtin, W.-D. Xiong, K. M. Kroeger, C. Liu, A. Rentsendorj, H. Agadjanian, L. Medina-Kauwe, D. Palmer and P. Ng, Mol. Ther., 2006, 14, 371–381 CrossRef CAS.
- M. Candolfi, G. E. Pluhar, K. Kroeger, M. Puntel, J. Curtin, C. Barcia, A. G. Muhammad, W. Xiong, C. Liu and S. Mondkar, Neuro-Oncology, 2007, 9, 245–258 CrossRef CAS.
- A. K. M. G. Muhammad, M. Puntel, M. Candolfi, A. Salem, K. Yagiz, C. Farrokhi, K. M. Kroeger, W. Xiong, J. F. Curtin, C. Liu, K. Lawrence, N. S. Bondale, J. Lerner, G. J. Baker, D. Foulad, R. N. Pechnick, D. Palmer, P. Ng, P. R. Lowenstein and M. G. Castro, Clin. Pharmacol. Ther., 2010, 88, 204–213 CrossRef CAS.
- Human Gene Therapy Methods, 2012, vol. 23, pp. 271–284, ISSN: 1946–6536, https://www.liebertpub.com/toc/hgtb/23/4 Search PubMed.
- M. J. Bellini, C. B. Hereñú, R. G. Goya and L. M. Garcia-Segura, J. Neuroinflammation, 2011, 8, 21 CrossRef CAS.
- X.-R. Zheng, S.-S. Zhang, Y.-J. Yang, F. Yin, X. Wang, L. Zhong and X.-H. Yu, Brain Res. Bull., 2010, 81, 372–377 CrossRef CAS.
- A. Alonso, E. Reinz, B. Leuchs, J. Kleinschmidt, M. Fatar, B. Geers, I. Lentacker, M. G. Hennerici, S. C. de Smedt and S. Meairs, Mol. Ther.–Nucleic Acids, 2013, 2, e73 CrossRef.
- M. J. Benskey, N. C. Kuhn, J. J. Galligan, J. Garcia, S. E. Boye, W. W. Hauswirth, C. Mueller, S. L. Boye and F. P. Manfredsson, Mol. Ther., 2015, 23, 488–500 CrossRef CAS.
- D. Wang, L. Zhong, M. Li, J. Li, K. Tran, L. Ren, R. He, J. Xie, R. P. Moser and C. Fraser, Mol. Ther.–Methods Clin. Dev., 2018, 11, 65–72 CrossRef CAS.
- N. Nayerossadat, T. Maedeh and P. A. Ali, Adv. Biomed. Res., 2012, 1, 1–11 CrossRef.
- M. A. Kotterman and D. V. Schaffer, Nat. Rev. Genet., 2014, 15, 445–451 CrossRef CAS.
- L. Shen, J. Miao, F. Yuan, Y. Zhao, Y. Tang, Y. Wang and G. Yang, Gene Ther., 2013, 20, 93–101 CrossRef CAS.
- F. Shen, R. Kuo, M. Milon-Camus, Z. Han, L. Jiang, W. L. Young and H. Su, Stroke, 2013, 44, 252–254 CrossRef CAS.
- D. Doycheva, N. Xu, J. Tang and J. Zhang, J. Neuroinflammation, 2019, 16, 174 CrossRef.
- M. S. Rafii, M. H. Tuszynski, R. G. Thomas, D. Barba, J. B. Brewer, R. A. Rissman, J. Siffert, P. S. Aisen and f. t. A.-N. S. Team, JAMA Neurol., 2018, 75, 834–841 CrossRef.
- Y.-C. Chen, N.-X. Ma, Z.-F. Pei, Z. Wu, F. H. Do-Monte, S. Keefe, E. Yellin, M. S. Chen, J.-C. Yin, G. Lee, A. Minier-Toribio, Y. Hu, Y.-T. Bai, K. Lee, G. J. Quirk and G. Chen, Mol. Ther., 2020, 28, 217–234 CrossRef CAS.
- M. S. Rafii, T. L. Baumann, R. A. Bakay, J. M. Ostrove, J. Siffert, A. S. Fleisher, C. D. Herzog, D. Barba, M. Pay and D. P. Salmon, Alzheimer's Dementia, 2014, 10, 571–581 CrossRef.
- T. R. Doeppner, M. Doehring, E. Bretschneider, A. Zechariah, B. Kaltwasser, B. Müller, J. C. Koch, M. Bähr, D. M. Hermann and U. Michel, Acta Neuropathol., 2013, 126, 251–265 CrossRef CAS.
- D. Bhere, K. Tamura, H. Wakimoto, S. H. Choi, B. Purow, J. Debatisse and K. Shah, Neuro-Oncology, 2017, 20, 215–224 CrossRef.
- R. Martier, M. Sogorb-Gonzalez, J. Stricker-Shaver, J. Hübener-Schmid, S. Keskin, J. Klima, L. J. Toonen, S. Juhas, J. Juhasova, Z. Ellederova, J. Motlik, E. Haas, S. van Deventer, P. Konstantinova, H. P. Nguyen and M. M. Evers, Mol. Ther.–Methods Clin. Dev., 2019, 15, 343–358 CrossRef CAS.
- J. Miniarikova, V. Zimmer, R. Martier, C. C. Brouwers, C. Pythoud, K. Richetin, M. Rey, J. Lubelski, M. M. Evers and S. J. van Deventer, Gene Ther., 2017, 24, 630–639 CrossRef CAS.
- D. Bhere, N. Arghiani, E. R. Lechtich, Y. Yao, S. Alsaab, F. Bei, M. M. Matin and K. Shah, Sci. Rep., 2020, 10, 1779 CrossRef CAS.
- I. Zafir-Lavie, S. Sherbo, H. Goltsman, F. Badinter, E. Yeini, P. Ofek, R. Miari, O. Tal, A. Liran and T. Shatil, J. Controlled Release, 2018, 291, 80–89 CrossRef CAS.
- M. Shimada, S. Abe, T. Takahashi, K. Shiozaki, M. Okuda, H. Mizukami, D. M. Klinman, K. Ozawa and K. Okuda, PLoS One, 2013, 8, e57606 CrossRef CAS.
- F. Moda, C. Vimercati, I. Campagnani, M. Ruggerone, G. Giaccone, M. Morbin, L. Zentilin, M. Giacca, I. Zucca and G. Legname, Prion, 2012, 6, 383–390 CrossRef CAS.
- S. Bäck, J. Necarsulmer, L. R. Whitaker, L. M. Coke, P. Koivula, E. J. Heathward, L. V. Fortuno, Y. Zhang, C. G. Yeh, H. A. Baldwin, M. D. Spencer, C. A. Mejias-Aponte, J. Pickel, A. F. Hoffman, C. E. Spivak, C. R. Lupica, S. M. Underhill, S. G. Amara, A. Domanskyi, J. E. Anttila, M. Airavaara, B. T. Hope, F. K. Hamra, C. T. Richie and B. K. Harvey, Neuron, 2019, 102, 105–119 CrossRef.
- J.-Z. Wang, P. Wu, Z.-M. Shi, Y.-L. Xu and Z.-J. Liu, Brain Dev., 2017, 39, 547–556 CrossRef.
- J. Nishiyama, T. Mikuni and R. Yasuda, Neuron, 2017, 96, 755–768 CrossRef CAS.
- B. György, C. Lööv, M. P. Zaborowski, S. Takeda, B. P. Kleinstiver, C. Commins, K. Kastanenka, D. Mu, A. Volak, V. Giedraitis, L. Lannfelt, C. A. Maguire, J. K. Joung, B. T. Hyman, X. O. Breakefield and M. Ingelsson, Mol. Ther.–Nucleic Acids, 2018, 11, 429–440 CrossRef.
- C. M. Fuentes and D. V. Schaffer, Curr. Opin. Biomed. Eng., 2018, 7, 33–41 CrossRef.
- J. Hartmann, F. B. Thalheimer, F. Höpfner, T. Kerzel, K. Khodosevich, D. García-González, H. Monyer, I. Diester, H. Büning, J. E. Carette, P. Fries and C. J. Buchholz, Mol. Ther.–Methods Clin. Dev., 2019, 14, 252–260 CrossRef CAS.
- P.-H. Hsu, K.-C. Wei, C.-Y. Huang, C.-J. Wen, T.-C. Yen, C.-L. Liu, Y.-T. Lin, J.-C. Chen, C.-R. Shen and H.-L. Liu, PLoS One, 2013, 8, e57682 CrossRef CAS.
- J. M. Mudannayake, A. Mouravlev, D. M. Fong and D. Young, Mol. Ther.–Methods Clin. Dev., 2016, 3, 16075 CrossRef.
- S. Powell, N. Khan, C. Parker, R. Samulski, G. Matsushima, S. Gray and T. McCown, Gene Ther., 2016, 23, 807–814 CrossRef CAS.
- G. von Jonquieres, D. Fröhlich, C. B. Klugmann, X. Wen, A. E. Harasta, R. Ramkumar, Z. H. Spencer, G. D. Housley and M. Klugmann, Front. Mol. Neurosci., 2016, 9, 13 Search PubMed.
- B. Caffery, J. S. Lee and A. A. Alexander-Bryant, Nanomaterials, 2019, 9, 105 CrossRef.
- N. Nayerossadat, T. Maedeh and P. A. Ali, Adv. Biomed. Res., 2012, 1, 27–27 CrossRef.
- J. Connolly, Gene Ther., 2002, 9, 1730–1734 CrossRef CAS.
- Z. Lai and R. O. Brady, J. Neurosci. Res., 2002, 67, 363–371 CrossRef CAS.
- J. Jakobsson, C. Ericson, M. Jansson, E. Björk and C. Lundberg, J. Neurosci. Res., 2003, 73, 876–885 CrossRef CAS.
- J. Jakobsson and C. Lundberg, Mol. Ther., 2006, 13, 484–493 CrossRef CAS.
- D. Blessing and N. Déglon, Curr. Opin. Virol., 2016, 21, 61–66 CrossRef CAS.
- J. Jakobsson, T. T. Nielsen, K. Staflin, B. Georgievska and C. Lundberg, Gene Ther., 2006, 13, 966–973 CrossRef CAS.
- A. Colin, M. Faideau, N. Dufour, G. Auregan, R. Hassig, T. Andrieu, E. Brouillet, P. Hantraye, G. Bonvento and N. Déglon, Glia, 2009, 57, 667–679 CrossRef.
- M. Humbel, M. Ramosaj, V. Zimmer, S. Regio, L. Aeby, S. Moser, A. Boizot, M. Sipion, M. Rey and N. Déglon, Gene Ther., 2020, 1–14 Search PubMed.
- R. Schlimgen, J. Howard, D. Wooley, M. Thompson, L. R. Baden, O. O. Yang, D. C. Christiani, G. Mostoslavsky, D. V. Diamond, E. G. Duane, K. Byers, T. Winters, J. A. Gelfand, G. Fujimoto, T. W. Hudson and J. M. Vyas, J. Occup. Environ. Med., 2016, 58, 1159–1166 CrossRef.
- H. Miyoshi, U. Blömer, M. Takahashi, F. H. Gage and I. M. Verma, J. Virol., 1998, 72, 8150–8157 CrossRef CAS.
- M. C. Milone and U. O'Doherty, Leukemia, 2018, 32, 1529–1541 CrossRef CAS.
- M. Åkerblom, R. Sachdeva, L. Quintino, E. E. Wettergren, K. Z. Chapman, G. Manfre, O. Lindvall, C. Lundberg and J. Jakobsson, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- L. Zeng, X. He, Y. Wang, Y. Tang, C. Zheng, H. Cai, J. Liu, Y. Fu and G. Yang, Gene Ther., 2014, 21, 37–43 CrossRef CAS.
- M. K. Sayeg, B. H. Weinberg, S. S. Cha, M. Goodloe, W. W. Wong and X. Han, ACS Synth. Biol., 2015, 4, 788–795 CrossRef CAS.
- L. Chen, K. Zhang, Z. Shi, A. Zhang, Z. Jia, G. Wang, P. Pu, C. Kang and L. Han, Oncol. Rep., 2014, 31, 1573–1580 CrossRef CAS.
- M. Ghasemi-Kasman, H. Baharvand and M. Javan, Biomed. Pharmacother., 2017, 96, 1222–1229 CrossRef CAS.
- C.-D. Liu, Q. Wang, D.-K. Zong, S.-C. Pei, Y. Yan, M.-L. Yan, L.-L. Sun, Y.-Y. Hao, M. Mao, W.-J. Xing, H. Ren and J. Ai, Neurobiol. Aging, 2016, 45, 76–87 CrossRef CAS.
- Y. Zhang, B. Han, Y. He, D. Li, X. Ma, Q. Liu and J. Hao, Neurochem. Int., 2017, 107, 182–190 CrossRef CAS.
- J. Zhang, Y. Wang, P. Zhu, X. Wang, M. Lv and H. Feng, J. Neurol. Sci., 2012, 320, 6–11 CrossRef CAS.
- P. Kumar, H. Wu, J. L. McBride, K.-E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39–43 CrossRef CAS.
- O. Singer, R. A. Marr, E. Rockenstein, L. Crews, N. G. Coufal, F. H. Gage, I. M. Verma and E. Masliah, Nat. Neurosci., 2005, 8, 1343–1349 CrossRef CAS.
- J.-L. Dreyer, Mol. Biotechnol., 2011, 47, 169–187 CrossRef CAS.
- P. Zhao, C. Wang, Z. Fu, Y. You, Y. Cheng, X. Lu, A. Lu, N. Liu, P. Pu and C. Kang, Int. J. Oncol., 2007, 31, 361–368 CAS.
- M. K. Sapru, J. W. Yates, S. Hogan, L. Jiang, J. Halter and M. C. Bohn, Exp. Neurol., 2006, 198, 382–390 CrossRef CAS.
- C. Van den Haute, K. Eggermont, B. Nuttin, Z. Debyser and V. Baekelandt, Hum. Gene Ther., 2003, 14, 1799–1807 CrossRef CAS.
- T. H. Hutson, E. Foster, J. M. Dawes, R. Hindges, R. J. Yáñez-Muñoz and L. D. Moon, J. Gene Med., 2012, 14, 299–315 CrossRef CAS.
- H.-F. Wu, J.-S. Cen, Q. Zhong, L. Chen, J. Wang, D. Y. B. Deng and Y. Wan, Biomaterials, 2013, 34, 1686–1700 CrossRef CAS.
- Q. Hu, C. Chen, N. H. Khatibi, L. Li, L. Yang, K. Wang, J. Han, W. Duan, J. H. Zhang and C. Zhou, Brain Res., 2011, 1367, 347–359 CrossRef CAS.
- B. Kantor, R. M. Bailey, K. Wimberly, S. N. Kalburgi and S. J. Gray, in Advances in genetics, Elsevier, 2014, vol. 87, pp. 125–197 Search PubMed.
- S. De Silva and W. J. Bowers, Viruses, 2009, 1, 594–629 CrossRef CAS.
- K. A. Cassady and J. N. Parker, Open Virol. J., 2010, 4, 103 CAS.
- P. Marconi, D. Krisky, T. Oligino, P. L. Poliani, R. Ramakrishnan, W. F. Goins, D. J. Fink and J. C. Glorioso, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11319 CrossRef CAS.
- S. Artusi, Y. Miyagawa, W. F. Goins, J. B. Cohen and J. C. Glorioso, Diseases, 2018, 6, 74 CrossRef CAS.
- B. K. Berges, J. H. Wolfe and N. W. Fraser, Mol. Ther., 2007, 15, 20–29 CrossRef CAS.
- G. Zhang, G. Jin, X. Nie, R. Mi, G. Zhu, W. Jia and F. Liu, PLoS One, 2014, 9, e95872 CrossRef.
- W. Zhang, G. Fulci, H. Wakimoto, T. A. Cheema, J. S. Buhrman, D. S. Jeyaretna, A. O. S. Rachamimov, S. D. Rabkin and R. L. Martuza, Neoplasia, 2013, 15, 591–599 CrossRef CAS.
- D. M. Patel, P. M. Foreman, L. B. Nabors, K. O. Riley, G. Y. Gillespie and J. M. Markert, Hum. Gene Ther.: Clin. Dev., 2016, 27, 69–78 CAS.
- J. C. Corona, A. Gimenez-Cassina, F. Lim and J. Díaz-Nido, J. Neurosci. Res., 2010, 88, 1943–1950 CAS.
- H. Uchida, M. Marzulli, K. Nakano, W. F. Goins, J. Chan, C.-S. Hong, L. Mazzacurati, J. Y. Yoo, A. Haseley, H. Nakashima, H. Baek, H. Kwon, I. Kumagai, M. Kuroki, B. Kaur, E. A. Chiocca, P. Grandi, J. B. Cohen and J. C. Glorioso, Mol. Ther., 2013, 21, 561–569 CrossRef CAS.
- C. Passaro, Q. Alayo, I. DeLaura, J. McNulty, K. Grauwet, H. Ito, V. Bhaskaran, M. Mineo, S. E. Lawler and K. Shah, Clin. Cancer Res., 2019, 25, 290–299 CrossRef CAS.
- H. Decker, S. Jürgensen, M. F. Adrover, J. Brito-Moreira, T. R. Bomfim, W. L. Klein, A. L. Epstein, F. G. De Felice, D. Jerusalinsky and S. T. Ferreira, J. Neurochem., 2010, 115, 1520–1529 CrossRef CAS.
- S. T. Lim, M. Airavaara and B. K. Harvey, Pharmacol. Res., 2010, 61, 14–26 CrossRef CAS.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS.
- Y. Zhou, Z. Peng, E. S. Seven and R. M. Leblanc, J. Controlled Release, 2018, 270, 290–303 CrossRef CAS.
- R. D. Jayant, D. Sosa, A. Kaushik, V. Atluri, A. Vashist, A. Tomitaka and M. Nair, Expert Opin. Drug Delivery, 2016, 13, 1433–1445 CrossRef CAS.
- M. Ramamoorth and A. Narvekar, J. Clin. Diagn. Res., 2015, 9, GE01–GE06 Search PubMed.
- S. Wohlfart, S. Gelperina and J. Kreuter, J. Controlled Release, 2012, 161, 264–273 CrossRef CAS.
- T.-T. Zhang, W. Li, G. Meng, P. Wang and W. Liao, Biomater. Sci., 2016, 4, 219–229 RSC.
- X. Wei, X. Chen, M. Ying and W. Lu, Acta Pharm. Sin. B, 2014, 4, 193–201 CrossRef.
- H. Gao, Acta Pharm. Sin. B, 2016, 6, 268–286 CrossRef.
- K. F. Timbie, B. P. Mead and R. J. Price, J. Controlled Release, 2015, 219, 61–75 CrossRef CAS.
- J. M. Bergen, I.-K. Park, P. J. Horner and S. H. Pun, Pharm. Res., 2008, 25, 983–998 CrossRef CAS.
- A. E. Aly and B. L. Waszczak, Expert Opin. Drug Delivery, 2015, 12, 1923–1941 CrossRef CAS.
- J. B. Wolinsky, Y. L. Colson and M. W. Grinstaff, J. Controlled Release, 2012, 159, 14–26 CrossRef CAS.
- D. S. Hersh, A. S. Wadajkar, N. B. Roberts, J. G. Perez, N. P. Connolly, V. Frenkel, J. A. Winkles, G. F. Woodworth and A. J. Kim, Curr. Pharm. Des., 2016, 22, 1177–1193 CrossRef CAS.
- D. B. Vieira and L. F. Gamarra, Int. J. Nanomed., 2016, 11, 5381 CrossRef CAS.
- M. Agrawal, Ajazuddin, D. K. Tripathi, S. Saraf, S. Saraf, S. G. Antimisiaris, S. Mourtas, M. Hammarlund-Udenaes and A. Alexander, J. Controlled Release, 2017, 260, 61–77 CrossRef CAS.
- E. Quagliarini, R. Di Santo, S. Palchetti, G. Ferri, F. Cardarelli, D. Pozzi and G. Caracciolo, Pharmaceutics, 2020, 12, 1–13 CrossRef.
- L. Wasungu and D. Hoekstra, J. Controlled Release, 2006, 116, 255–264 CrossRef CAS.
- D. B. Vieira and L. F. Gamarra, Int. J. Nanomed., 2016, 11, 5381–5414 CrossRef CAS.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS.
- G. Erel-Akbaba, L. A. Carvalho, T. Tian, M. Zinter, H. Akbaba, P. J. Obeid, E. A. Chiocca, R. Weissleder, A. G. Kantarci and B. A. Tannous, ACS Nano, 2019, 13, 4028–4040 CrossRef CAS.
- J. Jin, K. H. Bae, H. Yang, S. J. Lee, H. Kim, Y. Kim, K. M. Joo, S. W. Seo, T. G. Park and D.-H. Nam, Bioconjugate Chem., 2011, 22, 2568–2572 CrossRef CAS.
- N. K. Yaghi, J. Wei, Y. Hashimoto, L.-Y. Kong, K. Gabrusiewicz, E. K. Nduom, X. Ling, N. Huang, S. Zhou and B. C. P. Kerrigan, Neuro-Oncology, 2017, 19, 372–382 CAS.
- Z. R. Cohen, S. Ramishetti, N. Peshes-Yaloz, M. Goldsmith, A. Wohl, Z. Zibly and D. Peer, ACS Nano, 2015, 9, 1581–1591 CrossRef CAS.
- D. Yu, O. F. Khan, M. L. Suvà, B. Dong, W. K. Panek, T. Xiao, M. Wu, Y. Han, A. U. Ahmed and I. V. Balyasnikova, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6147–E6156 CrossRef CAS.
- P. M. Costa, A. L. Cardoso, C. Custódia, P. Cunha, L. P. de Almeida and M. C. P. de Lima, J. Controlled Release, 2015, 207, 31–39 CrossRef CAS.
- T. Tsujiuchi, A. Natsume, K. Motomura, G. Kondo, M. Ranjit, R. Hachisu, I. Sugimura, S. Tomita, I. Takehara, M. Woolley, N. U. Barua, S. S. Gill, A. S. Bienemann, Y. Yamashita, S. Toyokuni and T. Wakabayashi, Am. J. Transl. Res., 2014, 6, 169–178 Search PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS.
- T. Merdan, J. Kopeček and T. Kissel, Adv. Drug Delivery Rev., 2002, 54, 715–758 CrossRef CAS.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS.
- Z. Liu, Z. Zhang, C. Zhou and Y. Jiao, Prog. Polym. Sci., 2010, 35, 1144–1162 CrossRef CAS.
- I.-K. Park, K. Singha, R. B. Arote, Y.-J. Choi, W. J. Kim and C.-S. Cho, Macromol. Rapid Commun., 2010, 31, 1122–1133 CrossRef CAS.
- S. An, Y. Kuang, T. Shen, J. Li, H. Ma, Y. Guo, X. He and C. Jiang, Biomaterials, 2013, 34, 8949–8959 CrossRef CAS.
- J. Shen, Z. Zhao, W. Shang, C. Liu, B. Zhang, Z. Xu and H. Cai, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 894–903 CrossRef CAS.
- Y. Liu, Y. Guo, S. An, Y. Kuang, X. He, H. Ma, J. Li, J. Lv, N. Zhang and C. Jiang, PLoS One, 2013, 8, e62905 CrossRef CAS.
- P. Wang, X. Zheng, Q. Guo, P. Yang, X. Pang, K. Qian, W. Lu, Q. Zhang and X. Jiang, J. Controlled Release, 2018, 279, 220–233 CrossRef CAS.
- T. Yin, P. Wang, J. Li, R. Zheng, B. Zheng, D. Cheng, R. Li, J. Lai and X. Shuai, Biomaterials, 2013, 34, 4532–4543 CrossRef CAS.
- C. Zhang, Z. Gu, L. Shen, X. Liu and H. Lin, Curr. Pharm. Biotechnol., 2017, 18, 1124–1131 CAS.
- M. Van Woensel, T. Mathivet, N. Wauthoz, R. Rosière, A. D. Garg, P. Agostinis, V. Mathieu, R. Kiss, F. Lefranc and L. Boon, Sci. Rep., 2017, 7, 1–14 CrossRef CAS.
- B. M. Godinho, J. R. Ogier, R. Darcy, C. M. O'Driscoll and J. F. Cryan, Mol. Pharm., 2013, 10, 640–649 CrossRef CAS.
- S. Son, J. Jang, H. Youn, S. Lee, D. Lee, Y.-S. Lee, J. M. Jeong, W. J. Kim and D. S. Lee, Biomaterials, 2011, 32, 4968–4975 CrossRef.
- C. Helmschrodt, S. Höbel, S. Schöniger, A. Bauer, J. Bonicelli, M. Gringmuth, S. A. Fietz, A. Aigner, A. Richter and F. Richter, Mol. Ther.–Nucleic Acids, 2017, 9, 57–68 CrossRef CAS.
- C.-Q. Mao, J.-Z. Du, T.-M. Sun, Y.-D. Yao, P.-Z. Zhang, E.-W. Song and J. Wang, Biomaterials, 2011, 32, 3124–3133 CrossRef CAS.
- K. L. Kozielski, S. Y. Tzeng, B. A. Hurtado De Mendoza and J. J. Green, ACS Nano, 2014, 8, 3232–3241 CrossRef CAS.
- V. Sava, O. Fihurka, A. Khvorova and J. Sanchez-Ramos, Nanomedicine, 2020, 24, 102119 CrossRef CAS.
- S. An, D. He, E. Wagner and C. Jiang, Small, 2015, 11, 5142–5150 CrossRef CAS.
- I.-D. Kim, C.-M. Lim, J.-B. Kim, H. Y. Nam, K. Nam, S.-W. Kim, J.-S. Park and J.-K. Lee, J. Controlled Release, 2010, 142, 422–430 CrossRef CAS.
- A. P. Mann, P. Scodeller, S. Hussain, J. Joo, E. Kwon, G. B. Braun, T. Mölder, Z.-G. She, V. R. Kotamraju and B. Ranscht, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- H. Lopez-Bertoni, K. L. Kozielski, Y. Rui, B. Lal, H. Vaughan, D. R. Wilson, N. Mihelson, C. G. Eberhart, J. Laterra and J. J. Green, Nano Lett., 2018, 18, 4086–4094 CrossRef CAS.
- S. Watanabe, K. Hayashi, K. Toh, H. J. Kim, X. Liu, H. Chaya, S. Fukushima, K. Katsushima, Y. Kondo and S. Uchida, Nat. Commun., 2019, 10, 1–13 CrossRef CAS.
- Z. Shatsberg, X. Zhang, P. Ofek, S. Malhotra, A. Krivitsky, A. Scomparin, G. Tiram, M. Calderón, R. Haag and R. Satchi-Fainaro, J. Controlled Release, 2016, 239, 159–168 CrossRef CAS.
- M. Van Woensel, N. Wauthoz, R. Rosière, V. Mathieu, R. Kiss, F. Lefranc, B. Steelant, E. Dilissen, S. W. Van Gool and T. Mathivet, J. Controlled Release, 2016, 227, 71–81 CrossRef CAS.
- Y. Yang, T. Du, J. Zhang, T. Kang, L. Luo, J. Tao, Z. Gou, S. Chen, Y. Du and J. He, Adv. Sci., 2017, 4, 1600491 CrossRef.
- Y.-E. Seo, H.-W. Suh, R. Bahal, A. Josowitz, J. Zhang, E. Song, J. Cui, S. Noorbakhsh, C. Jackson and T. Bu, Biomaterials, 2019, 201, 87–98 CrossRef CAS.
- X. J. Loh, T.-C. Lee, Q. Dou and G. R. Deen, Biomater. Sci., 2016, 4, 70–86 RSC.
- F. Yin, B. Gu, Y. Lin, N. Panwar, S. C. Tjin, J. Qu, S. P. Lau and K.-T. Yong, Coord. Chem. Rev., 2017, 347, 77–97 CrossRef CAS.
- M. S. Shim and Y. J. Kwon, Adv. Drug Delivery Rev., 2012, 64, 1046–1059 CrossRef CAS.
- E. Keles, Y. Song, D. Du, W.-J. Dong and Y. Lin, Biomater. Sci., 2016, 4, 1291–1309 RSC.
- I.-D. Kim, E. Sawicki, H.-K. Lee, E.-H. Lee, H. J. Park, P.-L. Han, K. Kim, H. Choi and J.-K. Lee, Nanomedicine, 2016, 12, 1219–1229 CrossRef CAS.
- U. K. Sukumar, R. J. C. Bose, M. Malhotra, H. A. Babikir, R. Afjei, E. Robinson, Y. Zeng, E. Chang, F. Habte, R. Sinclair, S. S. Gambhir, T. F. Massoud and R. Paulmurugan, Biomaterials, 2019, 218, 119342 CrossRef CAS.
- B. Yoo, M. A. Ifediba, S. Ghosh, Z. Medarova and A. Moore, Mol. Imaging Biol., 2014, 16, 680–689 CrossRef.
- W. Y. Tong, M. Alnakhli, R. Bhardwaj, S. Apostolou, S. Sinha, C. Fraser, T. Kuchel, B. Kuss and N. H. Voelcker, J. Nanobiotechnol., 2018, 16, 38 CrossRef.
- J. Jung, A. Solanki, K. A. Memoli, K.-i. Kamei, H. Kim, M. A. Drahl, L. J. Williams, H.-R. Tseng and K. Lee, Angew. Chem., Int. Ed., 2010, 49, 103–107 CrossRef CAS.
- C. Saraiva, J. Paiva, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 291–305 CrossRef CAS.
- F. M. Kievit, K. Wang, T. Ozawa, A. W. Tarudji, J. R. Silber, E. C. Holland, R. G. Ellenbogen and M. Zhang, Nanomedicine, 2017, 13, 2131–2139 CrossRef CAS.
- D. Z. Liu, Y. Cheng, R. Q. Cai, W. W. Wang Bd, H. Cui, M. Liu, B. L. Zhang, Q. B. Mei and S. Y. Zhou, Nanomedicine, 2018, 14, 991–1003 CrossRef CAS.
- L. L. Israel, A. Galstyan, E. Holler and J. Y. Ljubimova, J. Controlled Release, 2020, 320, 45–62 CrossRef CAS.
- C. Qiao, J. Yang, Q. Shen, R. Liu, Y. Li, Y. Shi, J. Chen, Y. Shen, Z. Xiao, J. Weng and X. Zhang, Adv. Mater., 2018, 30, e1705054 CrossRef.
- H. W. Yang, C. Y. Huang, C. W. Lin, H. L. Liu, C. W. Huang, S. S. Liao, P. Y. Chen, Y. J. Lu, K. C. Wei and C. C. Ma, Biomaterials, 2014, 35, 6534–6542 CrossRef CAS.
- M. Zhou, N. Jiang, J. Fan, S. Fu, H. Luo, P. Su, M. Zhang, H. Shi, J. Zeng, Y. Huang, Y. Li, H. Shen, A. Zhang and R. Li, J. Controlled Release, 2019, 310, 24–35 CrossRef CAS.
- E. Dirren, J. Aebischer, C. Rochat, C. Towne, B. L. Schneider and P. Aebischer, Ann. Clin. Transl. Neurol., 2015, 2, 167–184 CrossRef CAS.
- M. K. Keaveney, H.-a. Tseng, T. L. Ta, H. J. Gritton, H.-Y. Man and X. Han, Cell Rep., 2018, 24, 294–303 CrossRef CAS.
- Y. Shi, Y. Jiang, J. Cao, W. Yang, J. Zhang, F. Meng and Z. Zhong, J. Controlled Release, 2018, 292, 163–171 CrossRef CAS.
- M. S. Sands and B. L. Davidson, Mol. Ther., 2006, 13, 839–849 CrossRef CAS.
- T. Ohashi, J. Hum. Genet., 2019, 64, 139–143 CrossRef.
- E. L. Aronovich and P. B. Hackett, Mol. Genet. Metab., 2015, 114, 83–93 CrossRef CAS.
- G. Massaro, M. P. Hughes, S. M. Whaler, K.-L. Wallom, D. A. Priestman, F. M. Platt, S. N. Waddington and A. A. Rahim, Hum. Mol. Genet., 2020, 29, 1933–1949 CrossRef.
- C. McIntyre, A. L. K. Derrick-Roberts, S. Byers and D. S. Anson, J. Gene Med., 2014, 16, 374–387 CrossRef CAS.
- M. Tardieu, M. Zérah, M. L. Gougeon, J. Ausseil, S. de Bournonville, B. Husson, D. Zafeiriou, G. Parenti, P. Bourget, B. Poirier, V. Furlan, C. Artaud, T. Baugnon, T. Roujeau, R. G. Crystal, C. Meyer, K. Deiva and J. M. Heard, Lancet Neurol., 2017, 16, 712–720 CrossRef CAS.
- R. J. Chandler, I. M. Williams, A. L. Gibson, C. D. Davidson, A. A. Incao, B. T. Hubbard, F. D. Porter, W. J. Pavan and C. P. Venditti, Hum. Mol. Genet., 2017, 26, 52–64 CAS.
- M. P. Hughes, D. A. Smith, L. Morris, C. Fletcher, A. Colaco, M. Huebecker, J. Tordo, N. Palomar, G. Massaro, E. Henckaerts, S. N. Waddington, F. M. Platt and A. A. Rahim, Hum. Mol. Genet., 2018, 27, 3079–3098 CrossRef CAS.
- W. Poewe, K. Seppi, C. M. Tanner, G. M. Halliday, P. Brundin, J. Volkmann, A. E. Schrag and A. E. Lang, Nat. Rev. Dis. Primers, 2017, 3, 17013 CrossRef.
- W. L. Hwu, S. I. Muramatsu, S. H. Tseng, K. Y. Tzen, N. C. Lee, Y. H. Chien, R. O. Snyder, B. J. Byrne, C. H. Tai and R. M. Wu, Sci. Transl. Med., 2012, 4, 134ra61 Search PubMed.
- K. Kojima, T. Nakajima, N. Taga, A. Miyauchi, M. Kato, A. Matsumoto, T. Ikeda, K. Nakamura, T. Kubota, H. Mizukami, S. Ono, Y. Onuki, T. Sato, H. Osaka, S. I. Muramatsu and T. Yamagata, Brain, 2019, 142, 322–333 CrossRef.
- K. S. Bankiewicz, J. L. Eberling, M. Kohutnicka, W. Jagust, P. Pivirotto, J. Bringas, J. Cunningham, T. F. Budinger and J. Harvey-White, Exp. Neurol., 2000, 164, 2–14 CrossRef CAS.
- A. Nagarajan, R. Bodhicharla, J. Winter, C. Anbalagan, K. Morgan, M. Searle, A. Nazir, A. Adenle, A. Fineberg, D. Brady, K. Vere, J. Richens, P. O'Shea, D. Bell and D. de-Pomerai, CNS Neurol. Disord.: Drug Targets, 2015, 14, 1054–1068 CrossRef CAS.
- M. Takahashi, M. Suzuki, M. Fukuoka, N. Fujikake, S. Watanabe, M. Murata, K. Wada, Y. Nagai and H. Hohjoh, Mol. Ther.–Nucleic Acids, 2015, 4, e241 CrossRef.
- J. H. Kordower, Isacson, Brooks, Olanow, Hunot, Stocchi, Jenner, Rascol and Marek, Ann. Neurol., 2003, 53 DOI:10.1002/ana.10485.
- J. S. Zheng, L. L. Tang, S. S. Zheng, R. Y. Zhan, Y. Q. Zhou, J. Goudreau, D. Kaufman and A. F. Chen, Mol. Brain Res., 2005, 134, 155–161 CrossRef CAS.
- P. Barroso-Chinea, I. Cruz-Muros, D. Afonso-Oramas, J. Castro-Hernandez, J. Salas-Hernandez, A. Chtarto, D. Luis-Ravelo, M. Humbert-Claude, L. Tenenbaum and T. Gonzalez-Hernandez, Neurobiol. Dis., 2016, 88, 44–54 CrossRef CAS.
- C. Warren Olanow, R. T. Bartus, T. L. Baumann, S. Factor, N. Boulis, M. Stacy, D. A. Turner, W. Marks, P. Larson, P. A. Starr, J. Jankovic, R. Simpson, R. Watts, B. Guthrie, K. Poston, J. M. Henderson, M. Stern, G. Baltuch, C. G. Goetz, C. Herzog, J. H. Kordower, R. Alterman, A. M. Lozano and A. E. Lang, Ann. Neurol., 2015, 78, 248–257 CrossRef CAS.
- W. J. Marks Jr., R. T. Bartus, J. Siffert, C. S. Davis, A. Lozano, N. Boulis, J. Vitek, M. Stacy, D. Turner, L. Verhagen, R. Bakay, R. Watts, B. Guthrie, J. Jankovic, R. Simpson, M. Tagliati, R. Alterman, M. Stern, G. Baltuch, P. A. Starr, P. S. Larson, J. L. Ostrem, J. Nutt, K. Kieburtz, J. H. Kordower and C. W. Olanow, Lancet Neurol., 2010, 9, 1164–1172 CrossRef.
- B. Hampoelz, A. Schwarz, P. Ronchi, H. Bragulat-Teixidor, C. Tischer, I. Gaspar, A. Ephrussi, Y. Schwab and M. Beck, Cell, 2019, 179, 671–686 CrossRef CAS.
- M. T. Heneka, M. J. Carson, J. E. Khoury, G. E. Landreth, F. Brosseron, D. L. Feinstein, A. H. Jacobs, T. Wyss-Coray, J. Vitorica, R. M. Ransohoff, K. Herrup, S. A. Frautschy, B. Finsen, G. C. Brown, A. Verkhratsky, K. Yamanaka, J. Koistinaho, E. Latz, A. Halle, G. C. Petzold, T. Town, D. Morgan, M. L. Shinohara, V. H. Perry, C. Holmes, N. G. Bazan, D. J. Brooks, S. Hunot, B. Joseph, N. Deigendesch, O. Garaschuk, E. Boddeke, C. A. Dinarello, J. C. Breitner, G. M. Cole, D. T. Golenbock and M. P. Kummer, Lancet Neurol., 2015, 14, 388–405 CrossRef CAS.
- W. V. Graham, A. Bonito-Oliva and T. P. Sakmar, Annu. Rev. Med., 2017, 68, 413–430 CrossRef CAS.
- H. Zheng and E. H. Koo, Mol. Neurodegener., 2011, 6, 27 CrossRef CAS.
- D. E. Hurtado, L. Molina-Porcel, J. C. Carroll, C. Macdonald, A. K. Aboagye, J. Q. Trojanowski and V. M. Y. Lee, J. Neurosci., 2012, 32, 7392–7402 CrossRef CAS.
- M. H. Tuszynski, L. Thal, M. Pay, D. P. Salmon, H. Sang, U. R. Bakay, P. Patel, A. Blesch, H. L. Vahlsing, G. Ho, G. Tong, S. G. Potkin, J. Fallon, L. Hansen, E. J. Mufson, J. H. Kordower, C. Gall and J. Conner, Nat. Med., 2005, 11, 551–555 CrossRef CAS.
- M. H. Tuszynski, J. H. Yang, D. Barba, U. Hoi-Sang, R. A. E. Bakay, M. M. Pay, E. Masliah, J. M. Conner, P. Kobalka, S. Roy and A. H. Nagahara, JAMA Neurol., 2015, 72, 1139–1147 CrossRef.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS.
- S. L. Ginn, A. K. Amaya, I. E. Alexander, M. Edelstein and M. R. Abedi, J. Gene Med., 2018, 20, e3015–e3015 CrossRef.
- H. Ito, T. Kanzawa, T. Miyoshi, S. Hirohata, S. Kyo, A. Iwamaru, H. Aoki, Y. Kondo and S. Kondo, Hum. Gene Ther., 2005, 16, 685–698 CrossRef CAS.
- T. Ozawa, J. L. Hu, L. J. Hu, E. L. Kong, A. W. Bollen, K. R. Lamborn and D. F. Deen, Cancer Gene Ther., 2005, 12, 449–455 CrossRef CAS.
- H. Tsurushima, X. Yuan, L. E. Dillehay and K. W. Leong, Cancer Gene Ther., 2007, 14, 706–716 CrossRef CAS.
- N. Ji, D. Weng, C. Liu, Z. Gu, S. Chen, Y. Guo, Z. Fan, X. Wang, J. Chen, Y. Zhao, J. Zhou, J. Wang, D. Ma and N. Li, Oncotarget, 2016, 7, 4369–4378 CrossRef.
- C. N. Chao, Y. H. Yang, M. S. Wu, M. C. Chou, C. Y. Fang, M. C. Lin, C. K. Tai, C. H. Shen, P. L. Chen, D. Chang and M. Wang, Sci. Rep., 2018, 8, 1–11 CrossRef.
- T. A. Cheema, R. Kanai, G. W. Kim, H. Wakimoto, B. Passer, S. D. Rabkin and R. L. Martuza, Clin. Cancer Res., 2011, 17, 7383–7393 CrossRef CAS.
- J. Martinez-Quintanilla, D. He, H. Wakimoto, R. Alemany and K. Shah, Mol. Ther., 2015, 23, 108–118 CrossRef CAS.
- A. J. Brenner, K. B. Peters, J. Vredenburgh, F. Bokstein, D. T. Blumenthal, S. Yust-Katz, I. Peretz, B. Oberman, L. S. Freedman, B. M. Ellingson, T. F. Cloughesy, N. Sher, Y. C. Cohen, N. Lowenton-Spier, T. Rachmilewitz Minei, N. Yakov, I. Mendel, E. Breitbart and P. Y. Wen, Neuro-Oncology, 2019, 22, 694–704 CrossRef.
- Z. Zhang, Z. Zhu, K. Watabe, X. Zhang, C. Bai, M. Xu, F. Wu and Y. Y. Mo, Cell Death Differ., 2013, 20, 1558–1568 CrossRef CAS.
- R. El Fatimy, S. Subramanian, E. J. Uhlmann and A. M. Krichevsky, Mol. Ther., 2017, 25, 368–378 CrossRef CAS.
- S. Ito, A. Natsume, S. Shimato, M. Ohno, T. Kato, P. Chansakul, T. Wakabayashi and S. U. Kim, Cancer Gene Ther., 2010, 17, 299–306 CrossRef CAS.
- D. GuhaSarkar, J. Neiswender, Q. Su, G. Gao and M. Sena-Esteves, Mol. Oncol., 2017, 11, 180–193 CrossRef CAS.
- D. E. Hurtado, L. Molina-Porcel, J. C. Carroll, C. MacDonald, A. K. Aboagye, J. Q. Trojanowski and V. M.-Y. Lee, J. Neurosci., 2012, 32, 7392–7402 CrossRef CAS.
- Y.-X. Yong, Y.-M. Li, J. Lian, C.-M. Luo, D.-X. Zhong and K. Han, J. Cell. Biochem., 2019, 120, 368–379 CrossRef CAS.
- M. M. Evers, J. Miniarikova, S. Juhas, A. Vallès, B. Bohuslavova, J. Juhasova, H. K. Skalnikova, P. Vodicka, I. Valekova, C. Brouwers, B. Blits, J. Lubelski, H. Kovarova, Z. Ellederova, S. J. van Deventer, H. Petry, J. Motlik and P. Konstantinova, Mol. Ther., 2018, 26, 2163–2177 CrossRef CAS.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. A. Wood, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS.
- W. Y. Tong, M. Alnakhli, R. Bhardwaj, S. Apostolou, S. Sinha, C. Fraser, T. Kuchel, B. Kuss and N. H. Voelcker, J. Nanobiotechnol., 2018, 16, 38 CrossRef.
- M. Campbell, F. Hanrahan, O. L. Gobbo, M. E. Kelly, A.-S. Kiang, M. M. Humphries, A. T. H. Nguyen, E. Ozaki, J. Keaney, C. W. Blau, C. M. Kerskens, S. D. Cahalan, J. J. Callanan, E. Wallace, G. A. Grant, C. P. Doherty and P. Humphries, Nat. Commun., 2012, 3, 849 CrossRef.
- Y.-E. Seo, H.-W. Suh, R. Bahal, A. Josowitz, J. Zhang, E. Song, J. Cui, S. Noorbakhsh, C. Jackson, T. Bu, A. Piotrowski-Daspit, R. Bindra and W. M. Saltzman, Biomaterials, 2019, 201, 87–98 CrossRef CAS.
- M. Rodriguez, J. Lapierre, C. R. Ojha, A. Kaushik, E. Batrakova, F. Kashanchi, S. M. Dever, M. Nair and N. El-Hage, Sci. Rep., 2017, 7, 1862 CrossRef.
- J. F. Alterman, L. M. Hall, A. H. Coles, M. R. Hassler, M.-C. Didiot, K. Chase, J. Abraham, E. Sottosanti, E. Johnson, E. Sapp, M. F. Osborn, M. Difiglia, N. Aronin and A. Khvorova, Mol. Ther.–Nucleic Acids, 2015, 4, e266 CrossRef CAS.
- H. Zhang, Y. Lin, Y. Qu and Q. Chen, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 2372–2379 Search PubMed.
- C. L. Esposito, S. Nuzzo, S. Catuogno, S. Romano, F. de Nigris and V. de Franciscis, Mol. Ther.–Nucleic Acids, 2018, 10, 398–411 CrossRef CAS.
- F. D. Urnov, J. C. Miller, Y.-L. Lee, C. M. Beausejour, J. M. Rock, S. Augustus, A. C. Jamieson, M. H. Porteus, P. D. Gregory and M. C. Holmes, Nature, 2005, 435, 646–651 CrossRef CAS.
- J. Boch, H. Scholze, S. Schornack, A. Landgraf, S. Hahn, S. Kay, T. Lahaye, A. Nickstadt and U. Bonas, Science, 2009, 326, 1509–1512 CrossRef CAS.
- J. C. Miller, S. Tan, G. Qiao, K. A. Barlow, J. Wang, D. F. Xia, X. Meng, D. E. Paschon, E. Leung, S. J. Hinkley, G. P. Dulay, K. L. Hua, I. Ankoudinova, G. J. Cost, F. D. Urnov, H. S. Zhang, M. C. Holmes, L. Zhang, P. D. Gregory and E. J. Rebar, Nat. Biotechnol., 2011, 29, 143–148 CrossRef CAS.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS.
- P. Mali, L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville and G. M. Church, Science, 2013, 339, 823–826 CrossRef CAS.
- T. Gaj, S. J. Sirk, S.-L. Shui and J. Liu, Cold Spring Harbor Perspect. Biol., 2016, 8, a023754 CrossRef.
- M. Heidenreich and F. Zhang, Nat. Rev. Neurosci., 2016, 17, 36–44 CrossRef CAS.
- K. Rodgers and M. McVey, J. Cell Physiol., 2016, 231, 15–24 CrossRef CAS.
- M. Garriga-Canut, C. Agustín-Pavón, F. Herrmann, A. Sánchez, M. Dierssen, C. Fillat and M. Isalan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E3136–E3145 CrossRef CAS.
- B. Zeitler, S. Froelich, K. Marlen, D. A. Shivak, Q. Yu, D. Li, J. R. Pearl, J. C. Miller, L. Zhang, D. E. Paschon, S. J. Hinkley, I. Ankoudinova, S. Lam, D. Guschin, L. Kopan, J. M. Cherone, H. B. Nguyen, G. Qiao, Y. Ataei, M. C. Mendel, R. Amora, R. Surosky, J. Laganiere, B. J. Vu, A. Narayanan, Y. Sedaghat, K. Tillack, C. Thiede, A. Gartner, S. Kwak, J. Bard, L. Mrzljak, L. Park, T. Heikkinen, K. K. Lehtimaki, M. M. Svedberg, J. Haggkvist, L. Tari, M. Toth, A. Varrone, C. Halldin, A. E. Kudwa, S. Ramboz, M. Day, J. Kondapalli, D. J. Surmeier, F. D. Urnov, P. D. Gregory, E. J. Rebar, I. Munoz-Sanjuan and H. S. Zhang, Nat. Med., 2019, 25, 1131–1142 CrossRef CAS.
- C. Agustin-Pavon, M. Mielcarek, M. Garriga-Canut and M. Isalan, Mol. Neurodegener., 2016, 11, 64 CrossRef.
- M. Zahur, J. Tolö, M. Bähr and S. Kügler, Front. Mol. Neurosci., 2017, 10, 142–142 CrossRef.
- M. J. Moscou and A. J. Bogdanove, Science, 2009, 326, 1501 CrossRef CAS.
- L. Cong, R. Zhou, Y.-C. Kuo, M. Cunniff and F. Zhang, Nat. Commun., 2012, 3, 968–968 CrossRef.
- M. L. Maeder, S. J. Linder, D. Reyon, J. F. Angstman, Y. Fu, J. D. Sander and J. K. Joung, Nat. Methods, 2013, 10, 243–245 CrossRef CAS.
- V. M. Bedell, Y. Wang, J. M. Campbell, T. L. Poshusta, C. G. Starker, R. G. Krug, 2nd, W. Tan, S. G. Penheiter, A. C. Ma, A. Y. Leung, S. C. Fahrenkrug, D. F. Carlson, D. F. Voytas, K. J. Clark, J. J. Essner and S. C. Ekker, Nature, 2012, 491, 114–118 CrossRef CAS.
- Z. Qiu, M. Liu, Z. Chen, Y. Shao, H. Pan, G. Wei, C. Yu, L. Zhang, X. Li, P. Wang, H. Y. Fan, B. Du, B. Liu, M. Liu and D. Li, Nucleic Acids Res., 2013, 41, e120 CrossRef CAS.
- F. A. Ran, L. Cong, W. X. Yan, D. A. Scott, J. S. Gootenberg, A. J. Kriz, B. Zetsche, O. Shalem, X. Wu, K. S. Makarova, E. V. Koonin, P. A. Sharp and F. Zhang, Nature, 2015, 520, 186–191 CrossRef CAS.
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef.
- L. A. Gilbert, M. H. Larson, L. Morsut, Z. Liu, G. A. Brar, S. E. Torres, N. Stern-Ginossar, O. Brandman, E. H. Whitehead, J. A. Doudna, W. A. Lim, J. S. Weissman and L. S. Qi, Cell, 2013, 154, 442–451 CrossRef CAS.
- L. A. Gilbert, M. A. Horlbeck, B. Adamson, J. E. Villalta, Y. Chen, E. H. Whitehead, C. Guimaraes, B. Panning, H. L. Ploegh, M. C. Bassik, L. S. Qi, M. Kampmann and J. S. Weissman, Cell, 2014, 159, 647–661 CrossRef CAS.
- Y. Zheng, W. Shen, J. Zhang, B. Yang, Y.-N. Liu, H. Qi, X. Yu, S.-Y. Lu, Y. Chen, Y.-Z. Xu, Y. Li, F. H. Gage, S. Mi and J. Yao, Nat. Neurosci., 2018, 21, 447–454 CrossRef CAS.
- H. Zhou, J. Liu, C. Zhou, N. Gao, Z. Rao, H. Li, X. Hu, C. Li, X. Yao, X. Shen, Y. Sun, Y. Wei, F. Liu, W. Ying, J. Zhang, C. Tang, X. Zhang, H. Xu, L. Shi, L. Cheng, P. Huang and H. Yang, Nat. Neurosci., 2018, 21, 440–446 CrossRef CAS.
- S. M. Heman-Ackah, A. R. Bassett and M. J. A. Wood, Sci. Rep., 2016, 6, 28420–28420 CrossRef CAS.
- F. K. Ekman, D. S. Ojala, M. M. Adil, P. A. Lopez, D. V. Schaffer and T. Gaj, Mol. Ther.–Nucleic Acids, 2019, 17, 829–839 CrossRef CAS.
- J. W. Shin, K.-H. Kim, M. J. Chao, R. S. Atwal, T. Gillis, M. E. MacDonald, J. F. Gusella and J.-M. Lee, Hum. Mol. Genet., 2016, 25, 4566–4576 CAS.
- A. M. Monteys, S. A. Ebanks, M. S. Keiser and B. L. Davidson, Mol. Ther., 2017, 25, 12–23 CrossRef CAS.
- S. Yang, R. Chang, H. Yang, T. Zhao, Y. Hong, H. E. Kong, X. Sun, Z. Qin, P. Jin, S. Li and X.-J. Li, J. Clin. Invest., 2017, 127, 2719–2724 CrossRef.
- B. György, C. Lööv, M. P. Zaborowski, S. Takeda, B. P. Kleinstiver, C. Commins, K. Kastanenka, D. Mu, A. Volak, V. Giedraitis, L. Lannfelt, C. A. Maguire, J. K. Joung, B. T. Hyman, X. O. Breakefield and M. Ingelsson, Mol. Ther.–Nucleic Acids, 2018, 11, 429–440 CrossRef.
- H. Park, J. Oh, G. Shim, B. Cho, Y. Chang, S. Kim, S. Baek, H. Kim, J. Shin, H. Choi, J. Yoo, J. Kim, W. Jun, M. Lee, C. J. Lengner, Y. K. Oh and J. Kim, Nat. Neurosci., 2019, 22, 524–528 CrossRef CAS.
- B. Lee, K. Lee, S. Panda, R. Gonzales-Rojas, A. Chong, V. Bugay, H. M. Park, R. Brenner, N. Murthy and H. Y. Lee, Nat. Biomed. Eng., 2018, 2, 497–507 CrossRef CAS.
- S. C. Vermilyea, A. Babinski, N. Tran, S. To, S. Guthrie, J. H. Kluss, J. K. Schmidt, G. J. Wiepz, M. G. Meyer, M. E. Murphy, M. R. Cookson, M. E. Emborg and T. G. Golos, Sci. Rep., 2020, 10, 3447 CrossRef CAS.
- J. Arias-Fuenzalida, J. Jarazo, X. Qing, J. Walter, G. Gomez-Giro, S. L. Nickels, H. Zaehres, H. R. Scholer and J. C. Schwamborn, Stem Cell Rep., 2017, 9, 1423–1431 CrossRef CAS.
- G. Murlidharan, K. Sakamoto, L. Rao, T. Corriher, D. Wang, G. Gao, P. Sullivan and A. Asokan, Mol. Ther.–Nucleic Acids, 2016, 5, e338–e338 CrossRef.
- N. Kalebic, E. Taverna, S. Tavano, F. K. Wong, D. Suchold, S. Winkler, W. B. Huttner and M. Sarov, EMBO Rep., 2016, 17, 338–348 CrossRef CAS.
- S. Back, J. Necarsulmer, L. R. Whitaker, L. M. Coke, P. Koivula, E. J. Heathward, L. V. Fortuno, Y. Zhang, C. G. Yeh, H. A. Baldwin, M. D. Spencer, C. A. Mejias-Aponte, J. Pickel, A. F. Hoffman, C. E. Spivak, C. R. Lupica, S. M. Underhill, S. G. Amara, A. Domanskyi, J. E. Anttila, M. Airavaara, B. T. Hope, F. K. Hamra, C. T. Richie and B. K. Harvey, Neuron, 2019, 102, 105–119 CrossRef CAS.
- H. Sun, S. Fu, S. Cui, X. Yin, X. Sun, X. Qi, K. Cui, J. Wang, L. Ma, F.-Y. Liu, F.-F. Liao, X.-H. Wang, M. Yi and Y. Wan, Sci. Adv., 2020, 6, eaay6687 CrossRef CAS.
- W. Yang, Y. Liu, Z. Tu, C. Xiao, S. Yan, X. Ma, X. Guo, X. Chen, P. Yin, Z. Yang, S. Yang, T. Jiang, S. Li, C. Qin and X. J. Li, Cell Res., 2019, 29, 334–336 CrossRef CAS.
- T. Mikuni, J. Nishiyama, Y. Sun, N. Kamasawa and R. Yasuda, Cell, 2016, 165, 1803–1817 CrossRef CAS.
- K. Suzuki, Y. Tsunekawa, R. Hernandez-Benitez, J. Wu, J. Zhu, E. J. Kim, F. Hatanaka, M. Yamamoto, T. Araoka, Z. Li, M. Kurita, T. Hishida, M. Li, E. Aizawa, S. Guo, S. Chen, A. Goebl, R. D. Soligalla, J. Qu, T. Jiang, X. Fu, M. Jafari, C. R. Esteban, W. T. Berggren, J. Lajara, E. Nuñez-Delicado, P. Guillen, J. M. Campistol, F. Matsuzaki, G.-H. Liu, P. Magistretti, K. Zhang, E. M. Callaway, K. Zhang and J. C. I. Belmonte, Nature, 2016, 540, 144–149 CrossRef CAS.
- L. Swiech, M. Heidenreich, A. Banerjee, N. Habib, Y. Li, J. Trombetta, M. Sur and F. Zhang, Nat. Biotechnol., 2015, 33, 102–106 CrossRef CAS.
- B. T. Staahl, M. Benekareddy, C. Coulon-Bainier, A. A. Banfal, S. N. Floor, J. K. Sabo, C. Urnes, G. A. Munares, A. Ghosh and J. A. Doudna, Nat. Biotechnol., 2017, 35, 431–434 CrossRef CAS.
- J. Nishiyama, T. Mikuni and R. Yasuda, Neuron, 2017, 96, 755–768 CrossRef CAS.
- X. Xu, T. Wan, H. Xin, D. Li, H. Pan, J. Wu and Y. Ping, J. Gene Med., 2019, 21, e3107 CrossRef.
- D. P. Dever, S. G. Scharenberg, J. Camarena, E. J. Kildebeck, J. T. Clark, R. M. Martin, R. O. Bak, Y. Tang, M. Dohse, J. A. Birgmeier, K. A. Jagadeesh, G. Bejerano, A. Tsukamoto, N. Gomez-Ospina, N. Uchida and M. H. Porteus, iScience, 2019, 15, 524–535 CrossRef CAS.
- H. Yang, H. Wang, C. S. Shivalila, A. W. Cheng, L. Shi and R. Jaenisch, Cell, 2013, 154, 1370–1379 CrossRef CAS.
- T. Horii, Y. Arai, M. Yamazaki, S. Morita, M. Kimura, M. Itoh, Y. Abe and I. Hatada, Sci. Rep., 2014, 4, 4513–4513 CrossRef.
- M. Zuckermann, V. Hovestadt, C. B. Knobbe-Thomsen, M. Zapatka, P. A. Northcott, K. Schramm, J. Belic, D. T. W. Jones, B. Tschida, B. Moriarity, D. Largaespada, M. F. Roussel, A. Korshunov, G. Reifenberger, S. M. Pfister, P. Lichter, D. Kawauchi and J. Gronych, Nat. Commun., 2015, 6, 7391 CrossRef CAS.
- M. Kumar, B. Keller, N. Makalou and R. E. Sutton, Hum. Gene Ther., 2001, 12, 1893–1905 CrossRef CAS.
- S. Incontro, C. S. Asensio, R. H. Edwards and R. A. Nicoll, Neuron, 2014, 83, 1051–1057 CrossRef CAS.
- R. J. Platt, S. Chen, Y. Zhou, M. J. Yim, L. Swiech, H. R. Kempton, J. E. Dahlman, O. Parnas, T. M. Eisenhaure, M. Jovanovic, D. B. Graham, S. Jhunjhunwala, M. Heidenreich, R. J. Xavier, R. Langer, D. G. Anderson, N. Hacohen, A. Regev, G. Feng, P. A. Sharp and F. Zhang, Cell, 2014, 159, 440–455 CrossRef CAS.
- N. Kumar, W. Stanford, C. de Solis, Aradhana, N. D. Abraham, T.-M. J. Dao, S. Thaseen, A. Sairavi, C. U. Gonzalez and J. E. Ploski, Front. Mol. Neurosci., 2018, 11, 413–413 CrossRef CAS.
- G. Chen, A. A. Abdeen, Y. Wang, P. K. Shahi, S. Robertson, R. Xie, M. Suzuki, B. R. Pattnaik, K. Saha and S. Gong, Nat. Nanotechnol., 2019, 14, 974–980 CrossRef CAS.
- K. Y. Chan, M. J. Jang, B. B. Yoo, A. Greenbaum, N. Ravi, W. L. Wu, L. Sanchez-Guardado, C. Lois, S. K. Mazmanian, B. E. Deverman and V. Gradinaru, Nat. Neurosci., 2017, 20, 1172–1179 CrossRef CAS.
- K. S. Hanlon, B. P. Kleinstiver, S. P. Garcia, M. P. Zaborowski, A. Volak, S. E. Spirig, A. Muller, A. A. Sousa, S. Q. Tsai, N. E. Bengtsson, C. Loov, M. Ingelsson, J. S. Chamberlain, D. P. Corey, M. J. Aryee, J. K. Joung, X. O. Breakefield, C. A. Maguire and B. Gyorgy, Nat. Commun., 2019, 10, 4439 CrossRef.
- H. C. Verdera, K. Kuranda and F. Mingozzi, Mol. Ther., 2020, 28, 723–746 CrossRef CAS.
- N. Merienne, G. Vachey, L. de Longprez, C. Meunier, V. Zimmer, G. Perriard, M. Canales, A. Mathias, L. Herrgott, T. Beltraminelli, A. Maulet, T. Dequesne, C. Pythoud, M. Rey, L. Pellerin, E. Brouillet, A. L. Perrier, R. du Pasquier and N. Déglon, Cell Rep., 2017, 20, 2980–2991 CrossRef CAS.
- S. Yang, S. Li and X.-J. Li, Cell Rep., 2018, 25, 2653–2659 CrossRef CAS.
- G. Murlidharan, K. Sakamoto, L. Rao, T. Corriher, D. Wang, G. Gao, P. Sullivan and A. Asokan, Mol. Ther.–Nucleic Acids, 2016, 5, e338–e338 CrossRef.
- H. Yin, C.-Q. Song, J. R. Dorkin, L. J. Zhu, Y. Li, Q. Wu, A. Park, J. Yang, S. Suresh, A. Bizhanova, A. Gupta, M. F. Bolukbasi, S. Walsh, R. L. Bogorad, G. Gao, Z. Weng, Y. Dong, V. Koteliansky, S. A. Wolfe, R. Langer, W. Xue and D. G. Anderson, Nat. Biotechnol., 2016, 34, 328–333 CrossRef CAS.
- W. Sun, W. Ji, J. M. Hall, Q. Hu, C. Wang, C. L. Beisel and Z. Gu, Angew. Chem., Int. Ed., 2015, 54, 12029–12033 CrossRef CAS.
- S. Ramakrishna, A.-B. Kwaku Dad, J. Beloor, R. Gopalappa, S.-K. Lee and H. Kim, Genome Res., 2014, 24, 1020–1027 CrossRef CAS.
- K. Lee, M. Conboy, H. M. Park, F. Jiang, H. J. Kim, M. A. Dewitt, V. A. Mackley, K. Chang, A. Rao, C. Skinner, T. Shobha, M. Mehdipour, H. Liu, W.-c. Huang, F. Lan, N. L. Bray, S. Li, J. E. Corn, K. Kataoka, J. A. Doudna, I. Conboy and N. Murthy, Nat. Biomed. Eng., 2017, 1, 889–901 CrossRef CAS.
- P. Gee, M. S. Y. Lung, Y. Okuzaki, N. Sasakawa, T. Iguchi, Y. Makita, H. Hozumi, Y. Miura, L. F. Yang, M. Iwasaki, X. H. Wang, M. A. Waller, N. Shirai, Y. O. Abe, Y. Fujita, K. Watanabe, A. Kagita, K. A. Iwabuchi, M. Yasuda, H. Xu, T. Noda, J. Komano, H. Sakurai, N. Inukai and A. Hotta, Nat. Commun., 2020, 11, 1334 CrossRef CAS.
- R. Shahbazi, G. Sghia-Hughes, J. L. Reid, S. Kubek, K. G. Haworth, O. Humbert, H.-P. Kiem and J. E. Adair, Nat. Mater., 2019, 18, 1124–1132 CrossRef CAS.
- A. Kaushik, A. Yndart, V. Atluri, S. Tiwari, A. Tomitaka, P. Gupta, R. D. Jayant, D. Alvarez-Carbonell, K. Khalili and M. Nair, Sci. Rep., 2019, 9, 3928 CrossRef.
- A. M. Monteys, S. A. Ebanks, M. S. Keiser and B. L. Davidson, Mol. Ther., 2017, 25, 12–23 CrossRef CAS.
- F. K. Ekman, D. S. Ojala, M. M. Adil, P. A. Lopez, D. V. Schaffer and T. Gaj, Mol. Ther.–Nucleic Acids, 2019, 17, 829–839 CrossRef CAS.
- L. Ye, J. J. Park, M. B. Dong, Q. Yang, R. D. Chow, L. Peng, Y. Du, J. Guo, X. Dai, G. Wang, Y. Errami and S. Chen, Nat. Biotechnol., 2019, 37, 1302–1313 CrossRef CAS.
- C. Straub, A. J. Granger, J. L. Saulnier and B. L. Sabatini, PLoS One, 2014, 9, e105584–e105584 CrossRef.
- Y. Shinmyo, S. Tanaka, S. Tsunoda, K. Hosomichi, A. Tajima and H. Kawasaki, Sci. Rep., 2016, 6, 20611 CrossRef CAS.
- C. Praça, A. Rai, T. Santos, A. C. Cristovão, S. L. Pinho, R. Cecchelli, M.-P. Dehouck, L. Bernardino and L. S. Ferreira, J. Controlled Release, 2018, 284, 57–72 CrossRef.
- N. Lipsman, Y. Meng, A. J. Bethune, Y. Huang, B. Lam, M. Masellis, N. Herrmann, C. Heyn, I. Aubert, A. Boutet, G. S. Smith, K. Hynynen and S. E. Black, Nat. Commun., 2018, 9, 2336 CrossRef.
- T.-E. Park, N. Mustafaoglu, A. Herland, R. Hasselkus, R. Mannix, E. A. FitzGerald, R. Prantil-Baun, A. Watters, O. Henry, M. Benz, H. Sanchez, H. J. McCrea, L. C. Goumnerova, H. W. Song, S. P. Palecek, E. Shusta and D. E. Ingber, Nat. Commun., 2019, 10, 2621 CrossRef.
- C. Praça, S. C. Rosa, E. Sevin, R. Cecchelli, M.-P. Dehouck and L. S. Ferreira, Stem Cell Rep., 2019, 13, 599–611 CrossRef.
- J. Feng, S. Lepetre-Mouelhi, A. Gautier, S. Mura, C. Cailleau, F. Coudore, M. Hamon and P. Couvreur, Sci. Adv., 2019, 5, eaau5148 CrossRef CAS.
- A. Joseph, C. Contini, D. Cecchin, S. Nyberg, L. Ruiz-Perez, J. Gaitzsch, G. Fullstone, X. Tian, J. Azizi, J. Preston, G. Volpe and G. Battaglia, Sci. Adv., 2017, 3, e1700362 CrossRef.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT04167540, acessed 22 July 2020.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03634007, acessed 22 July 2020.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT04133454, acessed 22 July 2020.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03952637, acessed 22 July 2020.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03725670, acessed 22 July 2020.
- E. A. Stadtmauer, J. A. Fraietta, M. M. Davis, A. D. Cohen, K. L. Weber, E. Lancaster, P. A. Mangan, I. Kulikovskaya, M. Gupta, F. Chen, L. Tian, V. E. Gonzalez, J. Xu, I.-y. Jung, J. J. Melenhorst, G. Plesa, J. Shea, T. Matlawski, A. Cervini, A. L. Gaymon, S. Desjardins, A. Lamontagne, J. Salas-Mckee, A. Fesnak, D. L. Siegel, B. L. Levine, J. K. Jadlowsky, R. M. Young, A. Chew, W.-T. Hwang, E. O. Hexner, B. M. Carreno, C. L. Nobles, F. D. Bushman, K. R. Parker, Y. Qi, A. T. Satpathy, H. Y. Chang, Y. Zhao, S. F. Lacey and C. H. June, Science, 2020, 367, eaba7365 CrossRef CAS.
- Alnylam, https://www.alnylam.com/, acessed 23 July 2020.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |