
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in nanotechnology-based drug carriers for celastrol delivery
Ling
Guo
ab,
Yongping
Zhang
ab and
Khuloud T.
Al-Jamal
 *b
*b
aGuizhou Engineering Technology Research Center for Processing and Preparation of Traditional Chinese Medicine and Ethnic Medicine, College of Pharmaceutical Sciences, Guizhou University of Traditional Chinese Medicine, Dongqing South Road, Huaxi University City, Guiyang, Guizhou 550025, P.R. China
bInstitute of Pharmaceutical Science, Faculty of Life Sciences & Medicine, King's College London, Franklin-Wilkins Building, 150 Stamford Street, London SE1 9NH, UK. E-mail: khuloud.al-jamal@kcl.ac.uk
First published on 16th August 2021
Abstract
Celastrol (CLT) is an active ingredient that was initially discovered and extracted from the root of Tripterygium wilfordii. The potential pharmacological activities of CLT in cancer, obesity, and inflammatory, auto-immune, and neurodegenerative diseases have been demonstrated in recent years. However, CLT's clinical application is extremely restricted by its low solubility/permeability, poor bioavailability, and potential off-target toxicity. The advent of nanotechnology provides a solution to improve the oral bioavailability, therapeutic effects or tissue-targeting ability of CLT. This review focuses on the most recent advances, improvements, inventions, and updated literature of various nanocarrier systems for CLT.
1. Introduction
Traditional Chinese herbal plants have long been utilized to help researchers find new active ingredients. Celastrol (CLT), also known as tripterine, is one of the most abundant bioactive compounds obtained from the root bark of Tripterygium wilfordii. During the last few decades, CLT has been described as a component of traditional medicine that can be applied to treat a number of disorders, including cancer, obesity, and inflammatory, auto-immune, and neurodegenerative diseases by modulating various molecular targets.1 Evaluation of the atomic orbital energy reveals that carbons C2 on ring A and C6 on ring B of CLT are highly susceptible toward a nucleophilic attack2 (Fig. 1). CLT can react with the nucleophilic thiol groups of cysteine residues and form covalent Michael adducts. The possible key mechanism is that CLT can exert a multitude of putative therapeutic effects by affecting the activities of a broad range of proteins. To date, although all the studies or trials on CLT were carried out in the pre-clinical stage, the impressive biological activities and promising experimental results would undoubtedly support the growing interest in CLT as one of the top five natural active substances of the 21st century for future drug development.3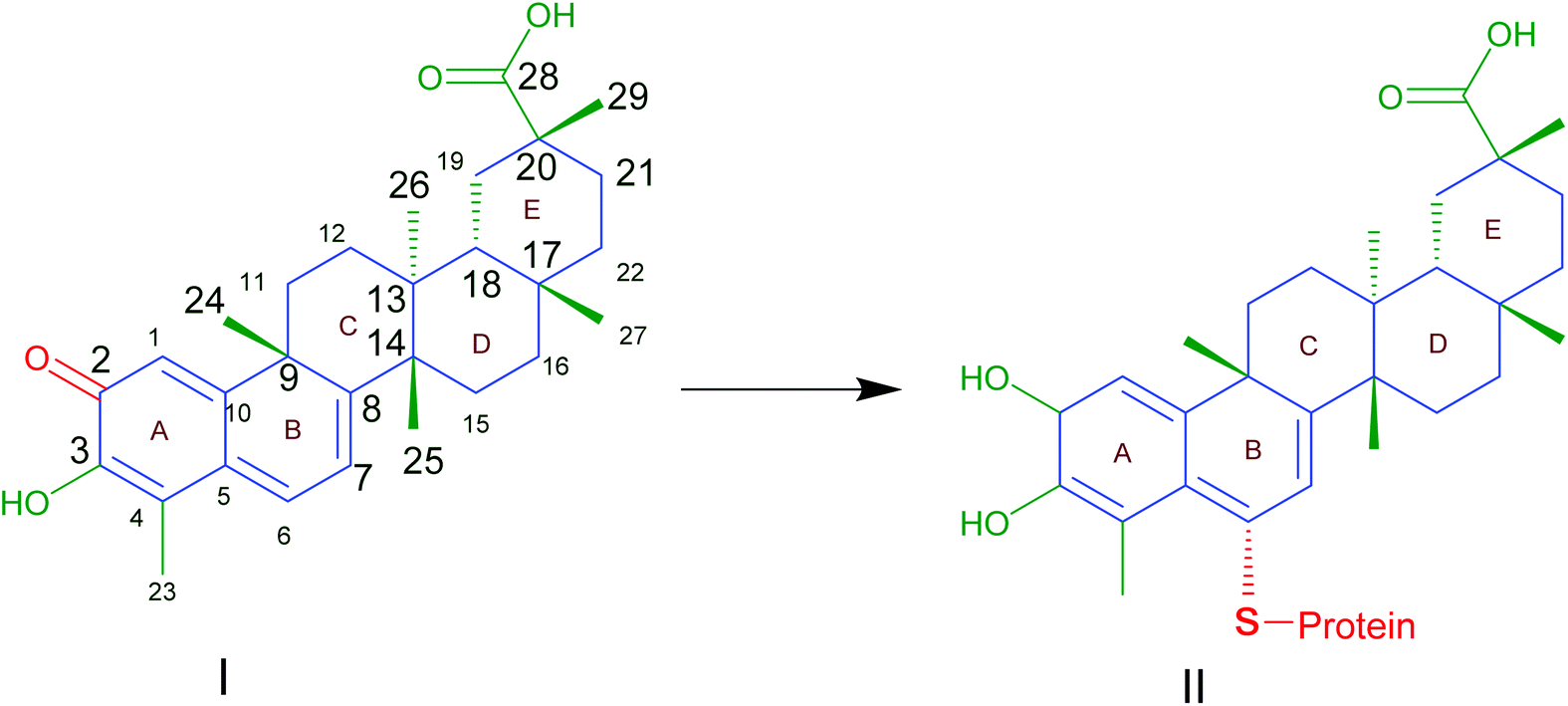 | ||
| Fig. 1 Celastrol (I) contains electrophilic sites at position C2 of ring A and C6 of ring B responsible for the binding affinity to thiol groups of cysteine residues forming covalent Michael adducts (II) which could affect the function of proteins. Reproduced from ref. 1 with permission from Elsevier, copyright 2019. | ||
CLT has been shown in vitro and in vivo to suppress a broad variety of cancers, such as liver cancer,4 lung cancer,5 pancreatic cancer,6 breast cancer,7 prostate cancer,8 colorectal cancer,9 ovarian cancer,10 and skin cancer.11 The induction of cell apoptosis, the suppression of cell proliferation and cell cycle arrest are the key mechanisms for CLT in cancer treatment.1 CLT has the potential to facilitate the apoptosis of tumor cells by the regulation of nuclear factor kappa B (NF-κB) signaling12 or by the development of reactive oxygen species (ROS)13 and downstream activation of c-Jun N-terminal kinases.14 By inhibiting the AKT/mTOR/P70S6K signaling pathway, and lowering the regulation of vascular endothelial growth factor receptors (VEGFRs), CLT can inhibit cell signaling associated with angiogenesis.15 In addition, it was also demonstrated that CLT can prevent tumor invasion and cancer metastasis by downregulating the signal transducer and activator of transcription 6 (STAT6) and CIP2 A/c-MYC signaling pathways,16 or by inhibiting M2-like polarization of tumor-associated macrophages (TAMs)17 and matrix metalloproteinase-9 (MMP-9).18 Overall, it is concluded that CLT exhibits its anticancer activity through multiple pathways.
CLT has been investigated for its preventive benefits and anti-inflammatory effects in several inflammatory and auto-immune disorders,19 including allergic asthma,20 rheumatoid arthritis,21 systemic lupus erythematosus,22 inflammatory bowel disease,23 osteoarthritis,24 and skin inflammation.25 CLT mainly prevents the production of pro-inflammatory cytokines, NF-κB, and adhesion molecules,14,26 which are all involved in inflammation. CLT can also minimize inflammation by promoting the autophagy of mitochondria through the nuclear receptor Nur77.27 Besides, the efficacy of CLT in animal models of Alzheimer's diseases (AD)28 and Parkinson's diseases (PD)29 has been reported. CLT's encouraging effect on heat shock proteins (HSP), which has been related to the refolding and reactivation of proteins, as well as the prevention of aggregate formation, can lead to the alleviation of AD symptoms.30,31 Also, CLT can prevent cell death in PD through decreasing the production of ROS, cytochrome c and apoptosis promotion factors, and reducing the α-synuclein protein which is considered to be responsible for the pathogenesis of PD by the activation of autophagy.32
In recent years, CLT has also been exploited to treat obesity and type 2 diabetes mellitus.33 However, despite the excellent anti-tumor, anti-inflammatory, neuroprotective, anti-obesity and anti-diabetic activities of CLT, its further application in the clinic remains to be restricted by several main drawbacks including low water solubility (11 μg mL−1 at pH 7.4),34 poor permeability (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 5.63),35 reduced oral bioavailability (absolute bioavailability of 17.06%),36 and off-target side effects (cardiotoxicity, hepatotoxicity, and neurotoxicity).37 Numerous conjugates and derivatives of CLT have been introduced and tested to improve effectiveness and mitigate toxicity.38 In addition, several types of nanotechnology-based delivery approaches (Fig. 2), such as liposomes,39 phytosomes,40 bilosomes,41 niosomes,42 exosomes,43 polymeric nanoparticles,44 lipid nanoparticles,45 protein nanoparticles,37 mesoporous silica nanoparticles (MSNs),46 gold nanoparticles (AuNPs),47 quantum dots (QDs),48 dendrimers,49 micelles,50 micro/nanoemulsions,51 nanocrystals52 and inclusion complexes,53 have been found to be suitable for encapsulating or loading of CLT to provide better solubility and bioavailability,54,55 better permeability,56,57 stronger therapeutic efficacy and lower toxicities.37,58 A year-by-year diagram illustrating the research efforts made over the last ten years in this direction is shown in Fig. 3. This review will narrow our attention on the fabrication and development of various CLT-based nanocarriers. CLT nanoformulations might provide new opportunities to improve the bioavailability and biological activity of CLT.
P = 5.63),35 reduced oral bioavailability (absolute bioavailability of 17.06%),36 and off-target side effects (cardiotoxicity, hepatotoxicity, and neurotoxicity).37 Numerous conjugates and derivatives of CLT have been introduced and tested to improve effectiveness and mitigate toxicity.38 In addition, several types of nanotechnology-based delivery approaches (Fig. 2), such as liposomes,39 phytosomes,40 bilosomes,41 niosomes,42 exosomes,43 polymeric nanoparticles,44 lipid nanoparticles,45 protein nanoparticles,37 mesoporous silica nanoparticles (MSNs),46 gold nanoparticles (AuNPs),47 quantum dots (QDs),48 dendrimers,49 micelles,50 micro/nanoemulsions,51 nanocrystals52 and inclusion complexes,53 have been found to be suitable for encapsulating or loading of CLT to provide better solubility and bioavailability,54,55 better permeability,56,57 stronger therapeutic efficacy and lower toxicities.37,58 A year-by-year diagram illustrating the research efforts made over the last ten years in this direction is shown in Fig. 3. This review will narrow our attention on the fabrication and development of various CLT-based nanocarriers. CLT nanoformulations might provide new opportunities to improve the bioavailability and biological activity of CLT.
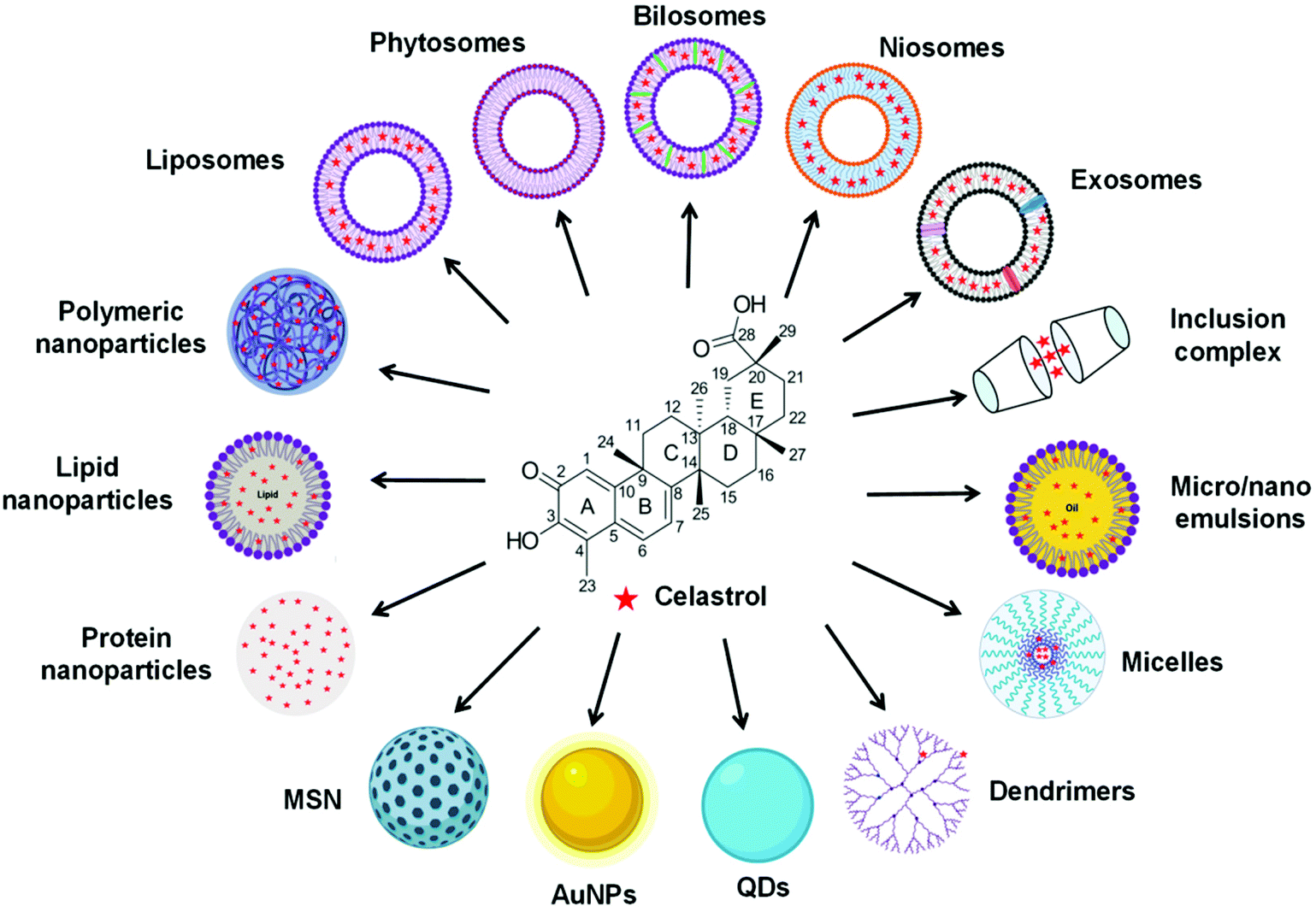 | ||
| Fig. 2 Various types of celastrol-loaded nanoformulations. | ||
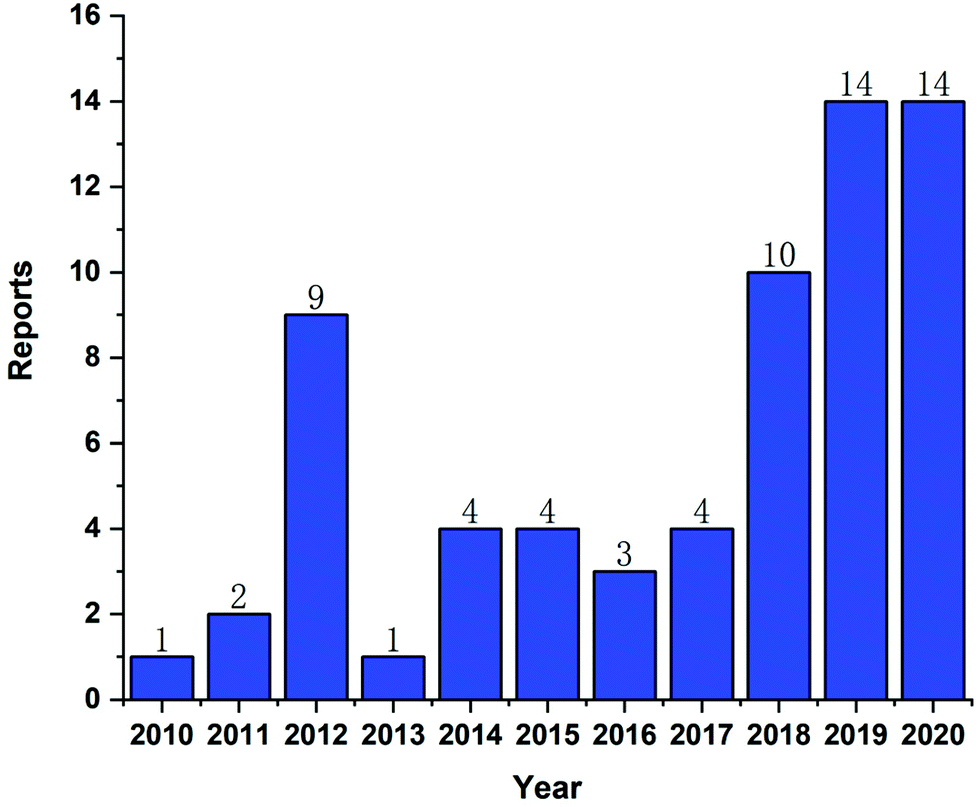 | ||
| Fig. 3 Various types of celastrol nanoformulations developed over the past ten years. | ||
2. Nanocarrier based approaches for CLT delivery
2.1 Vesicular carrier systems
Vesicles are colloidal particles in which an aqueous compartment is surrounded by a concentric bilayer that is composed of amphiphilic molecules.59 Vesicular systems have been applied as promising carriers to improve the bioavailability of encapsulated drugs and provide controlled and sustained therapeutic action. Several types of vesicular systems including liposomes, phytosomes, bilosomes, niosomes and exosomes have been explored for CLT delivery (Table 1).| Classification | Formulation ± co-treatment | Composition ± targeting moiety | Preparation technique | Physico-chemical characteristics | Used cell line/animal model | Route of administration/treatment dosage | Major outcome | Ref. |
|---|---|---|---|---|---|---|---|---|
| PS: particle size; ZP: zeta potential; EE: entrapment efficiency; DL: drug loading; n.a.: not applicable; i.v.: intravenous; i.p.: intraperitoneal; SPC: soybean phosphatidylcholine; DOPC: 1,2-dioleoyl-sn-glycero-3-phospholcholine; DSPC: 1,2-distearoyl-sn-glycero-3-phosphocholine; DSPEmPEG2000: 1,2-distearoyl-phosphatidylethanolamine-methylpolyethyleneglycolconjugate-2000; Gala-PEG2000-DSPE: galactose-modified 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy(polyethylene glycol)-2000; CREKA: Cys-Arg-Glu-Lys-Ala; mPEG2000-DSPE: methoxyl terminated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy (polyethylene glycol)-2000; Mal-PEG2000-DSPE: maleimide-PEG2000-DSPE: maleimide-terminated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy(polyethylene glycol)-2000; UUO: unilateral ureteral obstruction; LFA-1 I domain: lymphocyte function associated antigen-1 integrin I domain; DPPC: 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DPPE: 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine; PEG1000-PE: 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-methoxy(polyethylene glycol)-1000; DOGS-NTA(Ni): 1,2-dioleoyl-sn-glycero-3-(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl; FA: folic acid; STS: sodium tanshinone IIA sulfonate; CS: chitosan; HPMC: hydroxypropyl methylcellulose; HA: hyaluronic acid; DOTAP: 1,2-stearoyl-3-trimethylammonium-propane; SDC: sodium deoxycholate. | ||||||||
| Liposomes | Liposomes | Soybean phospholipids, cholesterol, Tween 80 | Ethanol injection method | PS: ∼90 nm; ZP: ∼−88 mV; EE: ∼98% | In situ rat intestinal perfusion model; mouse lung carcinoma xenograft model | In situ; oral (CLT dose: 2 mg kg−1 for 16 consecutive days) | Improved solubility and permeability; enhanced anti-cancer efficacy | 35 |
| Liposomes | SPC, sodium deoxycholate | Thin-film hydration method | PS: ∼128 nm; ZP: n.a.; EE: ∼72% | Glioma cell lines (U251, SHG44, and C6); human glioma xenograft model | In vitro; i.p. (CLT dose: 1 and 4 mg kg−1 day−1, 5 days per week for 4 weeks) | Increased bioavailability; enhanced anti-cancer efficacy; reduced toxicity | 61 | |
| PEGylated liposomes | DOPC, cholesterol, DSPEmPEG2000/DSPC, cholesterol, DSPEmPEG2000 | Thin-film hydration method | PS: ∼75 nm/∼104 nm; ZP: ∼−7 mV/∼−10 mV; EE: ∼70%/∼87% | Prostate cancer cell line (VCaP) | In vitro | Decreased viability of prostate cancer cells, while eliminating the need for toxic solubilising agents | 62 | |
| Galactose-modified PEGylated liposomes | SPC, cholesterol, Gala-PEG2000-DSPE | Thin-film hydration method | PS: ∼139 nm; ZP: n.a.; EE: ∼91% | Human hepatocellular carcinoma cell line (HepG2); AKT/c-Met-induced hepatocellular carcinoma mouse models | In vitro; i.v. (CLT dose: 2 mg kg−1, once every other day for 2 weeks) | Improved solubility; increased liver-targeted delivery; enhanced anti-cancer efficacy; reduced the side effects | 39 | |
| LFA-1 I domain conjugated PEGylated liposomes | DPPC, DPPE, cholesterol, PEG1000-PE, DOGS-NTA(Ni), LFA-1 I domain | Thin-film hydration method | PS: ∼115 nm; ZP: n.a.; DL: ∼0.8% | Human dermal microvascular endothelial cells (HMEC-1); human monocytic leukemia cells (THP-1); mouse brain microvascular endothelial cells (bEnd.3) | In vitro | Increased inflamed site-targeted delivery; enhanced anti-inflammatory efficacy | 63 | |
| CREKA-coupled PEGylated liposomes | Lipoid S100, cholesterol, mPEG2000-DSPE, Mal-PEG2000-DSPE, CREKA | Thin-film hydration method | PS: ∼112 nm; ZP: ∼20.33 mV; EE: ∼94%; DL: ∼5% | Normal rat renal fibroblasts (NRK-49F); mouse UUO model | In vitro; i.v. (CLT dose: 1 mg kg−1, every other day, for a total of 5 times) | Increased renal fibroblast-targeted delivery; enhanced anti-fibrotic efficacy; lessened toxicity | 64 | |
| FA-conjugated PEGylated liposomes (co-deliver with irinotecan) | DPPC, cholesterol, DSPE-PEG-FA | Thin-film hydration method | PS: ∼174 nm; ZP: ∼−9 mV; DL: ∼29% | The folate receptor-positive breast cancer cell lines (MCF-7, MDA-MB-231), and folate receptor-negative lung cancer cell line (A549); human breast cancer xenograft model | In vitro; i.v. (CLT dose: n.a) | Increased tumor-targeted delivery; enhanced anti-cancer efficacy; reduced side effects | 65 | |
| Multicomponent-based liposomes (co-delivered with STS) | Coix oil, RH40, PEG400, SPC, Cholesterol | Thin-film hydration method | PS: ∼110 nm; ZP: ∼−27 mV; EE: ∼75% | Human breast cancer cell line (MCF-7) and normal human liver cell line (L-02); human breast cancer xenograft model | In vitro; i.p. (CLT dose: 2 mg kg−1) | Enhanced anti-breast cancer activity; diminished systemic toxicity | 66 | |
| Phytosomes | Phytosomes | SPC | Solvent evaporation method | PS: ∼178 nm; ZP: ∼−39 mV; DL or EE: n.a. | New Zealand white male rabbits | Oral | Enhanced bioavailability; improved pharmacokinetics | 54 |
| Phytosomes (co-delivered with selenium) | SPC, sodium oleate, Na2SeO3, reduced GSH | Melting-hydration/in situ reduction method | PS: ∼126 nm; ZP: n.a.; EE: ∼99% | Caco-2 cells; adjuvant-induced arthritis rat model | In vitro; oral (CLT dose: 10 mg kg−1 for 2 weeks) | Improved permeability; enhanced antiarthritic effects | 40 | |
| Laminated sponge loaded with protamine decorated phytosomes | SPC, protamine, CS, HPMC, PEG 400, mannitol, ethyl cellulose | Solvent evaporation method/simple titration/lyophilization | PS: ∼250 nm; ZP: ∼+22 mV; DL or EE: n.a. | Chicken pouch mucosa; New Zealand white male rabbits | Ex vivo; topical | Superior mucopermeation profile; higher bioavailability | 68 | |
| Bilosomes | HA-functionalized bilosomes | SPC, DOTAP, SDC, HA | Thin film hydration method | PS: ∼119 nm; ZP: ∼−34 mV; EE: ∼100%; DL: ∼8% | Murine macrophage cell line (Raw264.7); collagen antibody induced arthritis mouse model | In vitro; i.v. (CLT dose: n.a.) | Enhanced bioavailability and antiarthritic efficacy | 41 |
| Niosomes | Niosomes | Span ® 20, Span ® 60, cholesterol | Thin film hydration method | PS: ∼147 nm; ZP: ∼−49 mV; EE: ∼90% | Psoriatic skin; imiquimod-induced psoriatic mouse models | In vitro; topical (CLT dose: 4 mg kg−1, once a day for 5 days) | Improved topical permeation and anti-psoriasis activity | 42 |
| Exosomes | Exosomes | Exosomes isolated from bovine raw milk | Simple mixing method | PS: ∼106 nm; ZP: n.a.; DL: ∼20% | Non-small cell-lung carcinoma (H1299 and A549); lung cancer xenograft model | In vitro; oral (CLT dose: 8 mg kg−1) | Enhanced antiproliferative and anti-cancer activities | 75 |
CLT was encapsulated into liposomes to address the issue of limited bioavailability.35 In this study, the permeability efficiency of free CLT and its liposomal formulation was compared in a four-site perfusion rat intestinal model, and the results showed that CLT-loaded liposomes displayed greater absorption potential in all intestine segments. Moreover, oral liposomes encapsulating CLT gave a higher tumor inhibition rate in a lung carcinoma xenograft model than free drug. In another study,61 it was found that liposome-based CLT formulation also presented an enhanced bioavailability with higher Cmax and AUC, longer t1/2, and greater biodistribution than free CLT. Treatment with liposomal CLT could suppress the growth of glioma cell lines (U251, SHG44, and C6), resulting in better in vivo antitumor activity and less serious side effects than the same dose of free CLT. Wolfram et al.62 fabricated PEGylated DOPC and DSPC liposomal formulations and compared the stability and encapsulation efficiency between formulations. The results displayed that liposomes prepared using DSPC exhibited improved shelf-life stability and higher encapsulation efficiency than those prepared with DOPC. However, contrary to other studies, the authors revealed that the DMSO-solubilized raw CLT and liposomal CLT caused a similar reduction in prostate cancer cell viability and demonstrated similar cellular uptake patterns. It should be stated that DMSO is not recommended for in vivo delivery due to its non-selective toxicity.
Chen et al.39 designed a novel CLT-loaded liposome coated with galactose molecules which binds to the asialoglycoprotein receptor enriched in hepatic parenchymal plasma membranes to enhance CLT delivery to human hepatocellular carcinoma for therapeutic purposes. In vitro studies showed that CLT loaded galactosylated liposome (C-GPL) improved the cellular uptake of CLT via receptor–ligand interaction, thereby increasing its cytotoxicity against tumor cells. The in vivo antitumor efficacy of C-GPL was further tested in an AKT/c-Met-triggered hepatocellular carcinoma mouse model. It was observed that C-GPL treatment substantially minimized the harm to liver and effectively prevented the growth of hepatocellular carcinoma by inhibiting AKT activation, inducing cell apoptosis, and retarding cell proliferation. Moreover, C-GPL did not cause significant weight loss and damage to normal organs.
Kang et al.63 developed liposomes encapsulating CLT and coated with a lymphocyte function associated antigen inserted domain (LFA-1 I domain) to target ICAM-1 that is overexpressed in both inflamed immune and vascular cells. They found the optimization in the affinity and avidity of the LFA-1 I domain on the surface of liposomes could increase the specificity to inflamed endothelial cells and monocytes with a high upregulation of ICAM-1. Moreover, these ICAM-1-selected liposomal CLT could shield endothelial cells from lipopolysaccharides challenges, inhibit the infiltration of monocytes and leukocytes in inflammatory sites, and prevent the amplification of inflammatory signals. It has been reported that myofibroblasts in fibrotic kidney overexpress fibronectin, and that the linear pentapeptide Cys-Arg-Glu-Lys-Ala (CREKA) can bind specifically to fibronectin. Based on the knowledge, Li et al.64 recently designed CREKA-coupled liposomes for the specific delivery of CLT to the renal interstitium, thereby attenuating unilateral ureteral obstruction mediated renal fibrosis and reducing the systemic toxicity of CLT.
Liposomes are able to encapsulate both hydrophilic and lipophilic drugs for the combination chemotherapy of cancers. The synergistic effect of drugs could improve their activity towards tumor cells while maintaining low cytotoxicity towards normal cells. CLT and irinotecan were co-encapsulated in folic acid-conjugated liposomes (Lipo/Cs/Ir-FA) by Soe et al.65 for the specific treatment of breast cancer. The in vitro cell assay displayed that Lipo/Cs/Ir-FA had significantly improved apoptosis in folate receptor-positive breast cancer cells (MCF-7 and MDA-MB-231), but not in folate receptor-negative lung cancer cells (A549). After treatment with Lipo/Cs/Ir-FA, breast cancer xenograft mice showed the greatest tumor growth suppression as compared to control mice and mice treated with free drugs or non-folate targeting formulations. Moreover, mice given Lipo/Cs/Ir-FA did not display body weight loss or organ toxicity.
Qu et al.66 fabricated multicomponent liposomes (T/CM-L) co-encapsulating sodium tanshinone IIA sulfonate (STS) and a small-sized microemulsion of CLT (CM) to enhance anti-breast cancer therapy. T/CM-L showed synergistic anti-breast cancer activity through the initial release of STS to normalize the dysfunctional tumor microenvironment, followed by the release of CM (and its payloads) to kill tumor cells. T/CM-L displayed greater cytotoxicity against MCF-7 cells and better suppression of xenograft tumor development than CLT monotherapy or the standard CLT/STS combination therapy. Most importantly, as compared to CLT alone, T/CM-L demonstrated lower systemic toxicity.
Phytosomal CLT formed by the complexation of CLT with phosphatidylcholine has been reported to result in enhanced bioavailability and an improved pharmacokinetic profile.54 In the study, the oral administration of phytosomal CLT to rabbits caused 4-fold and 5-fold increase in AUC0–8 and Cmax, respectively, as compared to crude CLT. In another study, Zhu et al.40 fabricated selenium-deposited phytosomes (Se@Tri-PTs) loaded with CLT to incorporate two anti-inflammatory components into one single nanosystem to treat arthritis through a synergistic process. In contrast to selenium-free phytosomes, Se@Tri-PTs displayed highly efficient uptake by Caco-2 cells and preferable intestinal epithelial permeability and significantly enhanced antiarthritic effects.
Freag et al.68 prepared protamine-modified phytosomal CLT nanocarriers (PRT-TRI-PHY) and then loaded them into laminated chitosan/hydroxypropyl methylcellulose composite sponges to control drug release and permeation through buccal mucosa. The ex vivo permeation study using chicken pouch mucosa showed that sponges loaded with PRT-TRI-PHY displayed a superior permeation profile with 2.3-fold higher flux value than the uncoated TRI-PHY counterparts. In a rabbit in vivo pharmacokinetic study, PRT-TRI-PHY had a ∼244% higher bioavailability than TRI-PHY. This study demonstrated that a buccal mucoadhesive system containing phytosomes can be used to deliver CLT penetrating the buccal mucosa to enhance its bioavailability.
Yang et al.41 developed hyaluronic acid (HA)-decorated bilosomes for delivering CLT to inflamed joints selectively to enhance the bioavailability and anti-arthritic efficacy. CLT-loaded bilosomes (CLT-BLs) were first fabricated using a thin film hydration process, and then coated with HA to form HA@CLT-BLs. The in vivo pharmacokinetic studies demonstrated that HA@CLT-BLs significantly improved the systemic and intra-arthritic bioavailability of CLT. As expected, HA@CLT-BLs showed remarkably superior antiarthritic efficacy to uncoated CLT-BLs.
A recent study on CLT-loaded niosomes was reported to enhance topical permeation and anti-psoriasis activity.42 The prepared CLT-loaded niosomes had approximately a particle size of 147 nm and a yield of up to 90%. The in vitro permeation studies indicated that CLT-loaded niosomes had enhanced water-solubility and skin permeation. When evaluating the therapeutic effect of CLT-loaded niosomes in psoriasis mouse models, HE-stained skin sections showed that CLT-loaded niosome treatment significantly alleviated the infiltration of inflammatory cells, hyperkeratosis and parakeratosis, thus displaying a much lower scaling and erythema score than the free CLT group.
2.2 Nanoparticulate carrier systems
Nanoparticulate carrier systems are colloidal dispersions with a size ranging from 10 to 1000 nm.76 Nanoparticulate carrier systems could be based on organic or inorganic materials or hybrid structures and have been used as an effective strategy to secure the entity of drugs in the blood circulation, modify in vivo biodistribution of drugs and deliver drugs to the site of action in a controlled and sustained manner.76 In recent years, various nanoparticulate carrier systems for CLT have been produced, such as polymeric nanoparticles, lipid-based nanoparticles, albumin nanoparticles, silk fibroin nanoparticles, MSN, AuNPs, QDs, dendrimers and nanocrystals. A summary of nanoparticulate carrier systems for CLT is given in Table 2.| Classification | Formulation ± co-treatment | Composition ± targeting moiety | Preparation technique | Physico-chemical characteristics | Used cell line/animal model | Route of administration/treatment dosage | Major outcome | Ref. |
|---|---|---|---|---|---|---|---|---|
| PS: particle size; ZP: zeta potential; EE: entrapment efficiency; DL: drug loading; n.a.: not applicable; i.v.: intravenous; i.p.: intraperitoneal; PCL: polycaprolactone; RES: reticuloendothelial system; PEG: polyethylene glycol; LFA-1 I domain: lymphocyte function associated antigen (LFA)-1 integrin I domain; UAN: urethane acrylate nonionomer; Ni-NTA: nickel-nitrilotriacetic acid; APS: aminoethylanisamide-polymer-disulfide bond; MIT: mitoxantrone; PEG-PLGA: poly(ethylene glycol) methyl ether-block-poly(lactic-co-glycolic acid); NLCs: nanostructured lipid carriers; SLT: soybean lecithin; TPGS: D-Į-tocopherol polyethylene glycol succinate; Gb: glyceryl behenate; IPM: isopropyl myristate; MCTs: medium-chain triglycerides; CPPs: cell-penetrating peptides; HSA: human serum albumin; MsPGN: mesangial proliferative glomerulonephritis; AIA: adjuvant-induced arthritis; PAB: palmitic acid-modified bovine serum albumin; HA: hyaluronic acid; MT: 1-methyltryptophan; MSN: mesoporous silica nanoparticles; CTAB: cetyltrimethylammonium bromide; NH4F: ammonium fluoride; TEOS: tetraethylorthosilicate; PLD-b-PEG: poly(L-aspartic acid)-b-poly(ethylene glycol); DPPC: 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DSPE-PEG2000: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]; CTAC; cetyltrimethylammonium chloride; APTMS: amino propyl trimethoxy silane; PEI: poly(ethylene imine); TMOS: tetramethyl orthosilicate; CTACl: cetyltrimethyl ammonium chloride; AuNPs: gold nanoparticles; PVP: PVP-co-2-dimethylaminoethyl methacrylate; Cys-modified CdTe QDs: cysteamine-modified cadmium–tellurium quantum dots; G4 PAMAM: generation 4 poly(amidoamine); G4 PAMAM-NH2: amino terminus G4 PAMAM dendrimers; G4 PAMAM-OH: hydroxyl terminus G4 PAMAM dendrimers; EpCAM: epithelial cell adhesion molecule; G5 PAMAM: generation 5 poly(amidoamine); DOX: doxorubicin. | ||||||||
| Polymeric nanoparticles | PCL nanoparticles | PCL, Pluronic F-127 | Nanoprecipitation method | PS: ∼176 nm; ZP: ∼−16 mV; EE: ∼65%; DL: ∼3% | Human prostate carcinoma cell lines (LNCaP, DU-145, and PC3) | In vitro | Higher cytotoxicity | 44 |
| PCL nanoparticles | PCL, Tween 80 | Low-energy emulsification/evaporation method | PS: ∼75 nm; ZP: n.a.; EE: ∼65% | Prostatic cancer cell line (LNCaP); LNCaP xenograft tumor-bearing mouse model | In vitro; i.v. (CLT dose: 1 mg kg−1 every other day) | Improved pharmacokinetics and enhanced antitumor effect via RES saturation | 79 | |
| PEG nanoparticles | PEG | Synthesis method | PS: ∼119 nm; ZP: ∼−15 mV; DL or EE: n.a. | Human non-small cell lung cancer cell line (A549); human breast cell line (MDA-MB-231); human hepatocarcinoma cell line (SMMC-7221); A549 xenograft tumor-bearing mouse model | In vitro; i.v. (CLT dose: 4.8 mg kg−1) | Improved solubility; better bioactivity and safety | 80 | |
| LFA-1 I domain modified UAN nanoparticles | UAN, Ni-NTA, LFA-1 I domain | Synthesis method | PS: ∼44 nm; ZP: n.a.; DL or EE: n.a. | Human cervical cancer cell line (HeLa) | In vitro | Enhanced tumor cells targeting; higher cytotoxicity | 81 | |
| APS nanoparticles (co-deliver with MIT) | APS copolymer | Solvent evaporation method | PS: ∼112 nm; ZP: ∼−16 mV (pH 7.4); ZP: ∼+30 mV (pH 6.5); EE: 78%; DL: 7% | Desmoplastic melanoma cell line (BPD6); normal fibroblasts (3T3); TGF-β activated fibroblast (3T3-T); murine macrophage cell line (Raw264.7); BPD6 tumor-bearing mouse model | In vitro; i.v. (CLT dose: 2 mg kg−1) | Enhanced cancer-specific chemo-immunotherapy efficacy; fewer side effects | 82 | |
| Neutrophil membrane-coated PEG-PLGA nanoparticles | PEG-PLGA copolymer, deoxycholic acid sodium, solutol HS15 | Emulsion/solvent evaporation method, followed by the direct extrusion method | PS: ∼167 nm; ZP: ∼−15 mV; EE: ∼85% | Murine macrophage cell line (Raw264.7); murine pancreatic cancer cell line (Panc02); mouse fibroblasts (L929); human umbilical vein endothelial cells (HUVEC); orthotopic and ectopic pancreatic cancer model | In vitro; i.v. (CLT dose: 1 mg kg−1, every 7 days for consecutive 4 doses) | Improved tumor targetability; improved cytotoxicity; enhanced anti-cancer efficacy; fewer side effects | 58 | |
| Neutrophil membrane-coated PEG-PLGA nanoparticles | PEG-PLGA copolymer, deoxycholic acid sodium, solutol HS15 | Emulsion/solvent evaporation method, followed by direct extrusion method | PS: ∼167 nm; ZP: ∼−15 mV; EE: ∼85% | Murine melanoma cell (B16F10); B16F10 xenograft tumor-bearing mouse model | In vitro; i.v. (CLT dose: 1 mg kg−1, every other day for consecutive 4 times) | Improved tumor targetability; improved cytotoxicity; enhanced anti-cancer efficacy; fewer side effects | 84 | |
| Neutrophil membrane-coated PEG-PLGA nanoparticles | PEG-PLGA copolymer, deoxycholic acid sodium, solutol HS15 | Emulsion/solvent evaporation method, followed by direct extrusion method | PS: ∼167 nm; ZP: ∼−15 mV; EE: ∼85% | Acute pancreatitis rat model | i.v. (CLT dose: 1 mg kg−1) | Specific drug delivery to pancreas; enhanced therapeutic efficacy; reduced systemic toxicity | 85 | |
| Lipid-nanoparticles | NLCs | Precirol ATO-5, labrasol, SLT, TPGS, Pluronic F68 | Solvent evaporation method | PS: ∼132 nm; ZP: ∼−27 mV; EE: ∼89% | Human immortal keratinocyte cell line (HaCaT) | In vitro | Delayed drug release; improved drug deposition in the skin | 87 |
| Cationic NLCs; anionic NLCs; neutral NLCs | Stearylamine, IPM, SLT, TPGS; Precirol ATO-5, IPM, SLT, TPGS; Gb, MCTs, SLT, TPGS | Solvent evaporation method | PS: ∼90 nm; ∼88 nm; 85 nm; ZP: ∼+26 mV; ∼−24 mV; ∼−2.7 mV; EE: ∼64%; ∼68%; ∼73% | Human immortal keratinocyte cell line (HaCaT); melanoma cell line (B16BL6); B16F10 xenograft tumor-bearing mouse model | In vitro; topical (CLT dose: 6 mg kg−1 day−1 for 16 consecutive days) | Cationic NLCs enhanced the percutaneous penetration and antimelanoma efficacy of CLT | 88 | |
| NLCs | Stearic acid, isopropyl myristate, SLT, TPGS | Solvent evaporation method | PS: ∼110 nm; ZP: ∼−30 mV; EE: ∼79% | In situ rat intestinal perfusion model; Caco-2 cells | In situ; in vitro | Improved absorption; better biocompatibility | 45 | |
| CPP coated NLCs | CPPs, Precirol ATO-5, Labrafil M1944CS, SLT, TPGS, Pluronic F68 | Solvent evaporation method | PS: ∼127 nm; ZP: ∼+29 mV; EE: ∼73% | In situ rat intestinal perfusion model; Caco-2 cells; beagles | In situ; in vitro; oral | Improved absorption; enhanced bioavailability | 56 | |
| CPP coated NLCs | CPPs, Precirol ATO-5, Labrafil M1944CS, SLT, TPGS, Pluronic F68 | Solvent evaporation method | PS: ∼127 nm; ZP: ∼+29 mV; EE: ∼73% | Human prostatic carcinoma cell line (PC-3); mouse prostate carcinoma cell line (RM-1); prostate tumor-bearing mouse model | In vitro; oral (CLT dose: 2 and 4 mg kg−1 every day for 15 days) | Improved cytotoxicity; enhanced anti-cancer efficacy; fewer side effects | 89 | |
| Lipid nanospheres | Lecithin, sodium oleate, soybean oil | Solvent diffusion method | PS: ∼150 nm; ZP: n.a.; EE: ∼100% | In situ rat intestinal perfusion model; Sprague-Dawley rats | In situ; oral | Improved gut wall permeability and post-enterocyte lymphatic transport; enhanced bioavailability | 55 | |
| Broccoli lipid nanoparticles | Broccoli derived lipids, Pluronic F68 | Solvent diffusion method | PS: ∼75 nm; ZP: n.a.; EE: ∼100% | In situ rat intestinal perfusion model; MDCK-II cells; Sprague-Dawley rats | In situ; in vitro; oral | Improved permeability; enhanced bioavailability | 57 | |
| Protein nanoparticles | HSA-nanoparticles | HSA | Microfluidic co-flow method | PS: ∼123 nm; ZP: n.a.; EE: ∼75% | Murine macrophage cell line (Raw264.7) | In vitro | Enhanced solubility; reduced cellular toxicity | 91 |
| HSA-nanoparticles | HSA, soybean oil | Ultrasonication emulsification method | PS: ∼96 nm; ZP: ∼−23 mV; EE: ∼95%; DL: ∼2% | Rat glomerular mesangial cell line (HBZY-1); reversible and irreversible rat model of MsPGN | In vitro; i.v. (CLT dose: 1 mg kg−1 once every other day until nephrectomy) | Increased mesangial cell-targetability; enhanced anti-glomerulonephritis efficacy; reduced systemic toxicities | 37 | |
| HSA-Kolliphor HS 15 nanoparticles | HSA, soybean oil, solutol HS 15 | Ultrasonication emulsification method | PS: ∼79 nm; ZP: ∼−1 mV; EE: ∼95%; DL: ∼2% | Murine macrophage cell line (Raw264.7); human fibroblast-like synoviocytes (HFLS); AIA arthritis rat model | In vitro; i.v. (CLT dose: 1 mg kg−1 given on days 14, 16, 18 and 20 after induction of arthritis | Enhanced accumulation in inflamed joints and therapeutic effects against rheumatoid arthritis; reduced systemic toxicities | 92 | |
| PAB nanoparticles | PAB, soybean oil | Ultrasonication emulsification method | PS: ∼99 nm; ZP: ∼−30 mV; EE: ∼94%; DL: ∼3% | Murine macrophage cell line (Raw264.7); human fibroblast-like synoviocytes (HFLS); mouse melanoma cells (B16F10); human renal tubular epithelial cells (HK-2); human umbilical vein endothelial cells (HUVEC); AIA arthritis rat model | In vitro; i.v. (CLT dose: 0.6 mg kg−1 given on days 14, 16, 18 and 20 after induction of arthritis) | Enhanced accumulation in inflamed joints and therapeutic effects against rheumatoid arthritis; improved pharmacokinetic profile and biosafety | 93 | |
| HA coated cationic albumin nanoparticles (co-deliver with MT) | Cationic bovine serum albumin, soybean oil, HA | Ultrasonication emulsification method followed by high pressure homogenization | PS: ∼313 nm; ZP: ∼−34 mV; DL or EE: n.a. | Murine pancreatic cancer cell line (Panc02); Panc02 xenograft and orthotopic pancreatic cancer mice models | In vitro; i.v. (CLT dose: 0.5 mg kg−1 once every four days for consecutive 4 times) | Improved tumor targetability; enhanced chemoimmunotherapy against pancreatic cancer; reduced systemic toxicities | 94 | |
| Silk fibroin nanoparticles | Silk fibroin solution | Desolvation method | PS: ∼291 nm; ZP: ∼−25 mV; EE: ∼81% | Sprague-Dawley rats | i.v. | Improved pharmacokinetic profile | 96 | |
| Silk fibroin nanoparticles | Silk fibroin solution | Modified desolvation method | PS: ∼170 nm; ZP: ∼−26 mV; EE: ∼87% | Pancreatic cancer cell lines (MIA PaCa-2, PANC-1) | In vitro | Better anticancer efficacy of combination nanoparticle therapy | 97 | |
| Inorganic nanoparticles | PEGylated polyaminoacid-capped MSN | CTAB, NH4F, TEOS, PLD-b-PEG | Simple mixing in a two-step process | PS: ∼150 nm; ZP: n.a.; EE: ∼100%; DL: ∼21% | Murine squamous cell carcinoma cell line (SCC7); subline of SK-N-SH human neuroblastoma cell line (SH-SY5Y); human breast carcinoma cell line (BT-474); SCC-7 xenograft tumor-bearing mouse model | In vitro; i.v. (CLT dose: 1 mg kg−1 every 3 days for 4 times) | Enhanced anti-cancer efficacy; fewer side effects | 46 |
| Inorganic nanoparticles | PEGylated lipid bilayer-supported MSN (co-deliver with axitinib) | CTAB, NH4F, TEOS, DPPC, DSPE-PEG2000, cholesterol | Simple mixing and thin-film hydration method | PS: ∼120 nm; ZP: n.a.; EE: ∼100%; DL: >21% | Murine squamous cell carcinoma cell line (SCC7); subline of SK-N-SH human neuroblastoma cell line (SH-SY5Y); human breast carcinoma cell line (BT-474); SCC-7 xenograft tumor-bearing mouse model | In vitro; i.v. (CLT dose: 1 mg kg−1 every 3 days for 4 times) | Enhanced synergistic anti-cancer efficacy | 101 |
| Glucose-functionalized MSN | CTAC, APTMS TEOS, PEI, glucose | PEI mediated conjugation | PS: ∼615 nm; ZP: ∼+50 mV; DL: ∼2% | Human cervical cancer cell line (HeLa); human non-small cell lung cancer cell line (A549); mouse embryonic fibroblasts (MEFs) | In vitro | Enhanced target-specific anti-cancer efficacy; minimal off-target side effects | 102 | |
| FA conjugated MSN | TMOS, APTMS, CTACl, PEI, FA | PEI mediated conjugation | PS: ∼300 nm; ZP: ∼+40 mV; DL: ∼4% | Human cervical cancer cell lines (HeLa); human non-small cell lung cancer cell line (A549) | In vitro | Targeted induction of the heat shock response in HeLa cells expressing folate receptors | 103 | |
| FA-modified AuNPs | AuNPs, PVP, FA | PVP mediated conjugation | PS: ∼22 nm; ZP: n.a.; EE: ∼94%; DL: ∼50% | 2D and 3D breast cancer models | In vitro | Enhanced anticancer activity | 47 | |
| Cys-CdTe QDs nanocomposites | Cys-CdTe QDs | Simple mixing followed by centrifugation | PS: ∼6 nm; ZP: ∼+10 mV; EE: ∼85%; DL: ∼34% | Human leukemia cancer cell line (K562) and its multidrug-resistant cell line (K562/A02) | In vitro | Cellular labeling and pH-sensitive responsive-drug release; increased cytotoxicity | 48 | |
| Dendrimers | Dendrimers | G4 PAMAM-NH2 dendrimers, G4 PAMAM-OH dendrimers | Simple mixing | PS: n.a.; ZP: n.a.; DL or EE: n.a. | Murine microglia cell line (N9) | In vitro | G4 PAMAM-OH dendrimers had greater anti-inflammatory activity and minimal cytotoxicity | 108 |
| EpCAM aptamer conjugated dendrimers | EpCAM aptamer, PEG, G5 PAMAM dendrimers | Covalent conjugation method | PS: ∼300 nm; ZP: ∼−7 mV; DL or EE: n.a. | Human normal embryonic kidney cell line (AD293); human colorectal cancer cell line (SW620); zebrafish embryos model; SW620 xenograft tumor-bearing mouse model | In vitro; i.v. (CLT dose: 0.4 mg kg−1 every 2 day for a total of 6 injections) | Enhanced targeted anticancer activity; reduced local and systemic toxicity | 49 | |
| Nanocrystals | Nanocrystals (co-deliver with DOX) | CLT and DOX | Precipitation method | PS: ∼126 nm; ZP: ∼+24 mV; DL or EE: n.a. | Human breast adenocarcinoma cell line (MCF-7); multidrug-resistant MCF-7 cells (MCF-7/ADR); 3D multicellular tumor spheroids | In vitro | Synergistic combination chemotherapy | 52 |
Sanna et al.44 prepared CLT-loaded poly(ε-caprolactone) nanoparticles by a nanoprecipitation method showing significantly increased antiproliferative effects on prostate carcinoma cell lines with respect to free CLT. Plain CLT-loaded nanoparticles show non-specific distribution after injection and are easily captured by the reticuloendothelial system (RES), which inherently results in poor in vivo delivery efficiency. To achieve better pharmacokinetic and antitumor efficacy of CLT, Yin et al.79 designed a RES saturation strategy involving the injection of blank nanoparticles first, followed by CLT-loaded nanoparticles. It was found that preinjection of blank nanoparticles could prolong the systemic circulation of CLT-loaded nanoparticles, and thus could significantly improve the pharmacokinetics and biodistribution of CLT. In LNCaP tumor xenograft mice, CLT-loaded nanoparticles treated with RES saturation outperformed free CLT and CLT-loaded nanoparticles alone in terms of antitumor efficacy. With the aim of improving the water-solubility and drug-ability of CLT, Shan et al.80 synthesized a sequence of PEG-CLT derivatives (PEGCs) by conjugating various types of PEGs to the carboxyl of CLT through the Mitsunobu reaction. Interestingly, among these PEGCs, DC1000 obtained by conjugating PEG with a molecular weight of 1000 and CLT could be easily dispersed in water to form self-assembled nanoparticles. It showed the greatest cytotoxicity against A549, MDA-MB-231 and SMMC-7221 tumor cells, and possessed a significantly reduced inhibition rate without causing death and edema in A549 cell xenograft-bearing nude mice. These findings revealed that DC1000 was much safer and more active than free CLT. In another study, Park et al.81 incorporated CLT into amphiphilic urethane acrylate nonionomer polymeric nanoparticles functionalized with the lymphocyte function-associated antigen-1 I domain (LFA-1 I domain) and found that these CLT-loaded nanoparticles could be taken up selectively by ICAM-1-expressing HeLa cells, displaying greater cytotoxicity compared with free CLT.
Combinational therapy, which combines chemotherapy and immunotherapy, is becoming increasingly common in cancer therapy. Liu et al.82 developed polymeric nanoparticles based on the amino ethylanisamide (AEAA)-polymer-disulfide bond (APS) for the co-delivery of mitoxantrone (an inducer of immunogenic cell death) and CLT (an anticancer and antifibrotic agent) for the chemoimmunotherapy of desmoplastic melanoma. AEAA is a Sigma receptor ligand with a high affinity for melanoma and tumor-associated fibroblasts. In vivo studies demonstrated that APS nanoparticles targeting the Sigma receptor resulted in considerably more drug distribution in the tumor than free drug distribution. Furthermore, the codelivery system greatly promoted immunogenic cell death-mediated immunotherapeutic effects and normalized the fibrotic and immunosuppressive tumor microenvironment in diseased mice.
Nanoparticles camouflaged by the cell plasma membrane provide a novel strategy to selectively deliver therapeutic payloads for treating cancer and inflammatory disorders.83 Cao et al.58 prepared a neutrophil camouflaged nanosystem (NNPs) by coating membranes of neutrophils on CLT loaded PEG-PLGA nanoparticles for the targeted treatment of pancreatic carcinoma. CLT-loaded NNPs could distribute specifically to the tumor site and significantly inhibit tumor growth of pancreatic ductal adenocarcinoma-bearing mice. Compared with nanoparticles without cell membrane coating and free CLT, CLT-NNPs showed the most prolonged survival and minimized liver metastasis. Similarly, Zhou et al.84 demonstrated the targeted therapy of CLT-loaded NNPs against malignant melanoma. CLT-loaded NNPs displayed significantly enhanced cytotoxic activity and apoptosis-inducing effect in the B16F10 melanoma cell line and dramatically increased antitumor efficacy in B16F10 tumor-bearing mice xenografts. During inflammation, neutrophils are able to mobilize rapidly to the site of inflammation in response to migration signals.83 Based on the natural merits of the neutrophil membrane, the same research group utilized CLT-loaded NNPs for targeted acute pancreatitis (AP) treatment.85 CLT-loaded NNPs could accumulate preferentially in inflamed pancreatic tissue. As compared to the other three control groups, CLT-loaded NNPs greatly decreased the levels of pancreatic myeloperoxidase and serum amylase in AP rats. In addition, AP rats treated with CLT-loaded NNP showed the largest reduction in ascites weight and wet/dry ratio. More importantly, the systemic toxicity of CLT in AP rats was significantly reduced by utilizing NNPs as the delivery carrier.
Nanostructured lipid carriers (NLCs) were developed as a topical delivery vehicle to enhance the skin penetration of CLT.87 It was found that encapsulating CLT into NLCs resulted in a prolonged release profile and enhanced drug accumulation into the upper skin layers. To optimize CLT-loaded NLCs for topical antimelanoma therapy, the effect of the surface charge of NLCs on in vitro skin penetration and the in vivo therapeutic efficacy of CLT were carefully evaluated by Chen et al.88 They used different solid and liquid matrices to prepare cationic, anionic, and neutral NLCs. The skin permeation experiment indicated that the cationic NLCs delivered the most CLT skin permeation amount, which was 1.35 and 1.95 times higher than the neutral and anionic NLCs, respectively. In vitro cellular assays displayed that the cationic NLCs had a higher uptake by B16BL6 cells and better cytotoxicity than the neutral and anionic NLCs. Similarly, percutaneous administration of the cationic NLCs provided greater in vivo antimelanoma efficacy in a B16 melanoma tumor model. These findings suggest that cationic NLCs are promising delivery systems of CLT for topical anti-melanoma treatment.
Zhou et al.45 reported that encapsulating CLT into NLCs could not only decrease the cytotoxicity of CLT against Caco-2 cells, but also improve drug absorption across intestinal epithelia. Another research by the same group revealed that the oral bioavailability of CLT has been further improved by coating cell-penetrating peptides on the surface of NLCs.56 The results indicated that cell-penetrating peptide-coated CLT-loaded NLCs (CT-NLC) outperformed the uncoated NLCs for the oral delivery of CLT. Yuan et al.89 found that CT-NLC effectively inhibited prostate tumor cell proliferation in vitro. Furthermore, CT-NLC showed greatly increased antitumor efficacy while causing less toxicity in the mouse model of prostate cancer.
Apart from the above reported NLCs, CLT-loaded lipid nanospheres constituted of soybean oil, lecithin and sodium oleate also notably improved the oral bioavailability and intestinal permeability of CLT.55 In another study, the lipids extracted from broccoli were used to fabricate CLT-loaded lipid nanoparticles (CLT-BLNs) for enhancing the oral bioavailability of CLT.57 As compared to CLT-loaded common lipid nanoparticles (CLT-CLNs), oral administration of CLT-BLNs exhibited superior intestinal permeability and higher bioavailability. The study provided new insight that food-originated materials could be used to construct nanocarriers for better oral delivery of drugs with poor bioavailability.
Hakala et al.91 utilized a microfluidic co-flow method to generate CLT-loaded HSA nanoparticles with a homogeneous size distribution by adjusting the flow rates. The obtained albumin nanoparticles enhanced the water solubility of CLT and decreased its cellular toxicity. Our previous study found CLT had potent effects against mesangioproliferative glomerulonephritis, but its off-target distributions resulted in severe cardiotoxicity, hepatotoxicity and neurotoxicity.37 To improve its efficacy and safety, we attempted to deliver CLT selectively to mesangial cells by encapsulating CLT into HSA nanoparticles (CLT-AN).37 Our results showed that CLT-AN, with an optimal size of 95 nm, could cross the fenestrations of the endothelium and accumulate in mesangial cells, thus possessing better capacity than CLT to ameliorate the acute and progressive glomerular injuries in anti-Thy1.1 nephritic rats. More importantly, CLT-AN presented lower drug accumulation in non-target organs and tissues than free CLT, reducing CLT-related systemic toxicities. Gong et al.92 constructed HSA-Kolliphor HS 15 nanoparticles (HSA-HS15 NPs) for selectively delivering CLT to inflamed sites to treat rheumatoid arthritis. CLT-HSA-HS15 NPs greatly improved the pharmacokinetic profile of CLT, with significantly higher plasma concentration, prolonged blood circulation time and increased AUC0−t as compared to free CLT and even CLT-HSA NPs. Moreover, compared with liposomes and HSA NPs, HSA-HS15 NPs exhibited longer and greater retention in inflamed joints due to the inflammatory targetability of albumin, the added HS15 and the ELVIS effect of nanoparticles. However, the interesting results were that adding HS15 could significantly enhance safety but had no substantial contribution to improving efficacy. The authors suspected that HS15 shielded the affinity between HSA and inflammatory cells in inflamed sites, which might limit the efficacy of CLT-HSA-HS15 NPs. In their other work,93 they found for the first time that palmitic acid-functionalized bovine serum albumin nanoparticles (PAB NPs) could target activated macrophages more effectively via scavenger receptor-A than bovine serum albumin nanoparticles (BSA NPs) targeting the same cells via CD44. In an adjuvant-induced arthritis rat model, CLT-PAB NPs greatly alleviated rheumatoid arthritis symptoms at a lower CLT dose with better safety as compared to CLT-BSA NPs. Hu et al.94 reported an innovative work about hyaluronic acid coated cationic albumin nanoparticles (HNPs) that were designed for the co-delivery of CLT and 1-methyltryptophan for pancreatic cancer chemoimmunotherapy. HNP had a unique hollow structure with an average size of ∼300 nm, which could be reduced to a smaller particle size in the presence of hyaluronidase. Based on the size decreasing effect and the CD44 mediated interaction, HNP showed significantly enhanced accumulation in both the xenograft pancreatic tumor and the orthotopic pancreatic tumor site, and thereby had remarkably enhanced tumor inhibition in both tumor models through downregulation of the immunosuppressive tumor microenvironment.
Silk fibroin (SF), derived primarily from silkworms, was accepted as a biomaterial by the US FDA in 1993.95 SF based nanoparticles have attracted much attention to be used as a carrier due to its outstanding mechanical properties, biocompatibility, biodegradability, and structural re-adjustment flexibility. Onyeabor et al.96 encapsulated CLT into SF nanoparticles by the desolvation method and achieved dramatically improved pharmacokinetic properties. In another study, Ding et al.97 produced triptolide and CLT loaded silk fibroin nanoparticles (TPL-SFNPs and CLT-SFNPs) and evaluated their cytotoxicity and synergistic effect in MIA PaCa-2 and PANC-1 human pancreatic cells. TPL-SFNPs and CLT-SFNPs hindered colony formation and induced apoptosis more effectively than free triptolide and CLT. Moreover, combination treatment with TPL-SFNPs and CLT-SFNPs showed considerably greater anticancer efficacy compared with TPL-SFNPs or CLT-SFNPs alone.
MSN has gotten a lot of recognition for its application in drug delivery and biomedicine due to its excellent mesoporous structure, flexible pore size, and easy of surface modification.100 Choi et al.46 reported PEGylated polyaminoacid-capped CLT-loaded MSN (CMSN-PEG) for mitochondria-targeted drug delivery. CMSN-PEG exhibited a pH-responsive, slower, and continuous release pattern. It could induce significant apoptosis in BT-474, SCC7 and SH-SY5Y cells via its sustained release characteristics. As compared to free CLT, CMSN-PEG therapy markedly increased the expression of the apoptosis proteins corresponding to mitochondrial apoptosis and displayed excellent in vivo antitumor activity, as evidenced by the remarkable increment in the apoptotic markers, and diminished proliferation markers. Another study conducted by the same research group successfully prepared anticancer drug combination nanoparticles (ACML) with CLT encapsulated in the MSN and axitinib in PEGylated lipidic bilayers for multi-targeted cancer therapy.101 The results showed that ACML had synergistic and superior antitumor efficacy and involved more efficiently improved effects on angiogenesis and mitochondrial function than either drug administered alone in both tumor cell lines in vitro and tumor-xenograft mouse model in vivo. Niemelä et al.102 modified glucose moieties, as affinity ligands, onto the surface of CLT loaded MSN (MSN-Glu) for maximizing the cellular uptake by cancer cells. Cellular uptake assays confirmed that MSN-Glu could efficiently enhance cellular uptake by the targeted HeLa and A549 cancer cells but not healthy MEF cells. More importantly, when CLT was loaded into MSN-Glu, its apoptosis induction effects were significantly enhanced. Similarly, they utilized folic acid functionalized CLT-loaded MSN for specific targeting.103 The authors reported that MSN could deliver CLT specifically to folate receptor positive HeLa cells, with only minor off-target effects in folate receptor negative A549 cells.
AuNPs are one of the most commonly used inorganic nanoparticles. The tuneable sizes, easy synthesis, and good optical properties make them a promising tool to develop drug delivery and imaging systems for therapeutic and diagnostic applications.104 Law et al.47 had recently engineered folic acid decorated CLT loaded AuNPs (FCA) to enhance the anticancer activity of CLT. It was found that FCA exhibited higher cellular uptake efficiency and caused more severe apoptosis than the CLT AuNPs and CLT alone in both 2D and 3D breast cancer models. QDs are widely utilized for labeling, imaging, targeted drug delivery, and photodynamic therapy.105 Li et al.48 developed fluorescent nanocomposites based on CLT and cysteamine modified cadmium-tellurium QDs (Cys-CdTe QDs) for cancer cell labeling and targeted treatment. Cys-CdTe QDs with the positively charged surfaces could interact with electronegative CLT molecules to self-assemble and form the drug nanocomposite CLT-Cys-CdTe. The in vitro release study showed that CLT-Cys-CdTe nanocomposites had a sensitive pH triggered release profile with relatively slow and sustained release at pH 7.4 but fast release at pH 6.0. The CdTe QDs were found to be easily internalized into cancer cells for real-time labeling and tracing. CLT-Cys-CdTe nanocomposites significantly enhanced drug accumulation in K562 cells and K562/A02 cells, thereby increasing CLT's cytotoxicity. CLT-Cys-CdTe nanocomposites also could greatly overcome the multidrug resistance of K562/A02 cells by arresting the cell cycle at the G2/M-phase and promoting cell apoptosis.
In a study by Boridy et al.108 CLT was conjugated to the amino termini of PAMAM dendrimers (CLT/G4-NH2) or hydroxyl termini of PAMAM dendrimers (CLT/G4-OH) to aid its solubilization, reduce cytotoxicity, and increase anti-inflammatory potency in lipopolysaccharide-stimulated microglial cells. The authors showed that G4-NH2 increased the solubility CLT more effectively than G4-OH, and CLT/G4-NH2 displayed stronger cytotoxicity against microglia than CLT alone while exhibiting minimal anti-inflammatory activity. In contrast, CLT/G4-OH could inhibit the release of proinflammatory cytokines without reducing microglial cell viability. These findings support that the G4-OH dendrimer is a better nanocarrier for delivering CLT to microglia that are overly activated in the pathological brain.
Ge et al.49 reported a smart bioconjugated nanosystem composed of epithelial cell adhesion molecule (EpCAM) aptamer, PEG, and G5 PAMAM dendrimers to selectively deliver CLT into EpCAM-upregulation tumors to enhance the antitumor effectiveness and reduce the toxicity. Taking advantage of the enhanced permeability and retention (EPR) effect and the active targeting effect of dendrimers, the multifunctional dendrimer-CLT bioconjugates produced superior apoptosis-inducing effects than free CLT in EpCAM-positive SW620 colorectal cancer cells both in vitro and in nude mice. Moreover, the biosafety of bioconjugate testing in xenograft mice and zebrafish models revealed significantly reduced local and systemic toxicity.
CLT has been reported with the ability of overcoming multidrug resistance (MDR) by suppress the activation of P-glycoprotein (P-gp)110 and promoting tumor cell apoptosis. To enhance the solubility and bioavailability of CLT and overcome the MRD of doxorubicin (DOX), CLT was selected by Xiao et al.52 to conjugate with DOX to synthesize a carrier-free nanocrystal for potential synergistic combination chemotherapy. At an optimal molar ratio of 1:4 (DOX to CLT), the CLT/DOX nanocrystal with a spherical morphology demonstrated fabulous stability and tumor acidic microenvironment sensitive drug release. It was found that the CLT/DOX nanocrystal could maximize the water-solubility of CLT and dramatically increased cellular drug accumulation by suppressing P-gp expression. As compared to free DOX, free CLT, and the CLT/DOX mixture, the CLT/DOX nanocrystal caused considerably more apoptosis and autophagy, resulting in a more efficiently synergistic therapeutic effect.
2.3 Micellar systems
Polymeric micelles are macromolecular assemblies with a hydrophobic inner core and a hydrophilic outer shell that form spontaneously in aqueous solution from block copolymers or graft copolymers at a concentration above the critical micelle concentration.111 Polymeric micelles have been extensively explored as a novel form of drug delivery carriers, especially for the effective delivery of hydrophobic drugs that can be covalently bound or physically entrapped in the center of the micelles.112 The summary of CLT-loaded polymeric micelles is given in Table 3.| Block copolymer | Preparation technique | Physico-chemical characteristics | Used cell line/animal model | Route of administration/treatment dosage | Major outcome | Ref. |
|---|---|---|---|---|---|---|
| PS: particle size; ZP: zeta potential; EE: entrapment efficiency; DL: drug loading; n.a.: not applicable; i.v.: intravenous; i.p.: intraperitoneal; CNV: corneal neovascularization; mPEG-PLL: methoxyl poly (ethylene glycol)-b-poly(L-lysine); PEG-b-PPS: poly(ethylene glycol)-b-poly(propylene sulfide); CIA: collagen-induced arthritis; mPEG-PCL: methoxy poly(ethylene glycol)-block-poly(ε-caprolactone); CLT-PEG-G Rh2: celastrol-polyethylene glycol-ginsenoside Rh2; TET-CSOSA: tetraiodothyroacetic acid-chitosan oligosaccharide-stearic acid; CTPP-CSOSA: (4-carboxybutyl) triphenylphosphonium bromide-chitosan oligosaccharide-stearic acid. | ||||||
| Carboxyl-terminated Pluronic P123 | Thin-film hydration method | PS: ∼117 nm; ZP: ∼−2 mV; EE: ∼100%; DL: ∼23% | n.a. | n.a. | Improved solubility; extended release | 34 |
| Thin-film hydration method | PS: ∼50 nm; ZP: n.a.; EE: ∼100%; DL: ∼5% | OVA-induced mouse model of allergic inflammation | Subcutaneous (CLT dose: 10 μg) | Enhanced anti-allergic effects | 50 | |
| Thin-film hydration method | PS: ∼93 nm; ZP: n.a.; DL: ∼21% | Human basophil cell line (KU812); immature human mast cell line (HMC-1); allergic asthma and passive cutaneous anaphylaxis mouse models | In vitro; i.p. (CLT dose: 0.8 mg kg−1 for 5 consecutive days) and i.v. (CLT dose: 1.5 mg kg−1) | Increased mast cell and basophil-targeted drug delivery; enhanced anti-allergic effects | 113 | |
| PEG-PCL | Ultrasonic agitation method | PS: ∼48 nm; ZP: n.a.; DL: ∼7% | Human umbilical vein endothelial cell (HUVEC, EA.hy 926); suture-induced CNV rat model | In vitro; subconjunctival (CLT dose: 5.44 mg mL−1, 100 μL per injection) | Improved solubility; extended release; enhanced antiangiogenic effect | 114 |
| Ultrasonic agitation method | PS: ∼48 nm; ZP: n.a.; DL: ∼7% | Human umbilical vein endothelial cell (HUVEC, EA.hy 926); macrophages (NR8383); activated macrophage-induced CNV rat model | In vitro; implanted in rat corneal pocket | Improved solubility; extended release; enhanced antiangiogenic effect | 115 | |
| Ultrasonic agitation method | PS: ∼48 nm; ZP: n.a.; DL: ∼7% | Human retinoblastoma cell line (SO-Rb 50); human retinoblastoma xenograft model | In vitro; i.p. (CLT dose: 27.2 mg per kg per 2 days) | Improved solubility; extended release; enhanced anti-cancer efficacy | 116 | |
| Ultrasonic agitation method | PS: ∼48 nm; ZP: n.a.; DL: ∼7% | Human umbilical vein endothelial cell (HUVEC, EA.hy 926); human retinoblastoma cell line (SO-Rb 50); human retinoblastoma xenograft model | In vitro; i.p. (CLT dose: 27.2 mg per kg per 2 days) | Enhanced antiangiogenic and anti-cancer effects | 117 | |
| Nanoprecipitation method | PS: 50–70 nm; ZP: 0 mV; EE: ∼1%; DL: ∼28% | Diet-induced obese C57BL/6N male mice | Oral (CLT dose: 5 mg kg−1 day−1 or 7.5 mg kg−1 day−1 for consecutive 21 days) | Effective anti-obesity activity; reduced side toxicities | 118 | |
| mPEG-PLL | In situ chemical conjugation-induced self-assembly method | PS: ∼65 nm; ZP: n.a.; EE: ∼54%; DL: ∼10% | Murine melanoma cell (B16F10); B16F10 xenograft tumor-bearing mouse model | In vitro; i.p. (CLT dose: 2 and 4 mg kg−1 every 2 days) | Improved solubility; extended release; enhanced anti-cancer efficacy; reduced side toxicities | 119 |
| PEG-PPS | Thin-film hydration method | PS: 15–18 nm; ZP: n.a.; EE: 30–100% | Murine macrophage cell line (RAW 264.7); high-fat diet ldlr−/− mice | In vitro; i.v. (CLT dose: 200 ng mL−1, 100 μL per injection) | Enhanced anti-inflammatory and anti-atherosclerotic effects | 120 |
| PEG-PPS | Solvent evaporation method | PS: ∼135 nm; ZP: n.a.; DL: ∼5% | Murine macrophage cell line (Raw 264.7); CIA arthritis rat model | In vitro; i.v. (CLT dose: 10 mg kg−1 given on days 30 and 40 after induction of arthritis) | Enhanced accumulation in inflamed joints and therapeutic effects against rheumatoid arthritis; reduced toxicity | 121 |
| CLT-PEG-G Rh2 derivative | Ethanol injection dialysis method | PS: ∼122 nm; ZP: ∼−23 mV; EE: ∼85% | Human non-small cell lung cancer cell line (A549); Sprague-Dawley rats | In vitro; i.v. | Enhanced anti-cancer efficacy; improved pharmaceutical profile | 122 |
| Poly[thioctic acid-graftedPEG/(benzyl amine)] | Solvent displacement dialysis method | PS: ∼69 nm; DL: ∼9% | Human retinoblastoma cell line (Y79) | In vitro | Enhanced apoptosis induction effect | 123 |
| TET-CSOSA conjugates | Solvent displacement dialysis method | PS: ∼83 nm; ZP: ∼23 mV; EE: ∼73%; DL: ∼14% | Mouse breast cell line (4T1); human breast cell line (MDA-MB-231); human umbilical vein endothelial cell (HUVEC, EA.hy 926); 4T1 cellular spheroids; primary 4T1 tumor-bearing mouse model | In vitro; i.v. (CLT dose: 2 mg kg−1 every 2 days for 5 times) | Increased tumor-targeted delivery; enhanced anti-cancer and anti-metastasis efficacy | 124 |
| CTPP-CSOSA conjugates | Solvent displacement dialysis method | PS: ∼64 nm; ZP: ∼23 mV; EE: ∼92%; DL: ∼12% | Human breast adenocarcinoma cell line (MCF-7); human non-small cell lung cancer cell line (A549); human normal liver cell line (L-02); MCF-7 xenograft tumor-bearing mouse model | In vitro; i.v. (CLT dose: 2 mg kg−1, every other day for the first 9 days) | Increased mitochondria-targeted drug delivery; enhanced anti-cancer efficacy; reduced side toxicities | 125 |
| Zein-chondroitin sulphate conjugate (co-deliver with sulfasalazine) | Solvent evaporation method | PS: ∼154 nm; ZP: ∼35 mV; EE: ∼87%; DL: ∼13% | Human breast adenocarcinoma cell line (MCF-7); primary breast cancer mouse model | In vitro; i.v. (CLT dose: 2 mg kg−1, 2 times weekly for 3 weeks) | Increased tumor-targeted delivery; enhanced anti-cancer efficacy | 126 |
CLT-loaded polymeric micelles made of CLT and the methoxyl poly (ethylene glycol)-b-poly(L-lysine) polymer were developed by a simple “in situ chemical conjugation-induced self-assembly” technique.119 The CLT-loaded polymeric micelles could slowly release CLT and about 60% of the CLT was released after 48 h. The CLT-loaded polymeric micelles were efficiently taken up by B16F10 tumor cells, resulting in fabulous cytotoxicity. The in vivo antitumor evaluation indicated that the CLT-loaded polymeric micelles had remarkably stronger antitumor activity and lower side effects in B16F10 tumor-bearing mice compared to free CLT. Allen et al.120 reported that CLT-loaded poly(ethylene glycol)-b-poly(propylene sulfide) (PEG-b-PPS) micelles had high NF-κB inhibition with a half maximal effective concentration (EC50) nearly 50000 times lower than that of free CLT. Furthermore, CLT-loaded micelles could significantly reduce TNF-α secretion by RAW 264.7 cells after LPS stimulation, and inhibited the infiltration of neutrophils and monocytes within atherosclerotic plaques of ldlr−/− mice. In another work, An et al.121 utilized PEG-PPS micelles for CLT delivery (C-PEPS) to treat rheumatoid arthritis. It was found that C-PEPS was discovered to be capable of inhibiting LPS-activated inflammatory responses in RAW264.7 cells by blocking the Notch1 and NF-κB pathways. Moreover, C-PEPS could accumulatively distribute in the inflamed joints and alleviated the major rheumatoid arthritis-associated symptoms without inducing obvious toxicity in major organs.
Li et al. synthesized CLT and a ginsenoside Rh2-conjugated PEG derivative (CLT-PEG-G Rh2) which was able to form CLT-loaded polymeric micelles (CG-M) when reaching its critical micellar concentration (1 × 10−5 M).122 Under physiological, conditions, CG-M was stable, but it can readily disassemble and release CLT and G Rh2 in acidic and enzymatic environments. In pharmacokinetics analysis, the parameters t1/2 and AUC for the CG-M were 1.03- and 2.44-times higher than those for non-micelle control, respectively, indicating that the CG-M had a longer circulation period and thereby showed a potentially enhanced EPR effect. In cellular studies, CG-M displayed significantly higher cellular uptake, greater apoptosis induction and stronger antiproliferative activity against A549 cells.
A reduction-sensitive polymeric micellar system was developed by Guo et al.123 based on a copolymer poly[thioctic acid-grafted poly(ethylene glycol)/(benzyl amine)] (PTEB), for retinoblastoma cell-targeted CLT delivery. The cell assays indicated that CLT-PTEB micelles with excellent reduction-responsive behavior could effectively accelerate the uptake of CLT by the human retinoblastoma cells and cause enhanced cell apoptosis.
Tan et al.125 reported CLT-loaded (4-carboxybutyl) triphenylphosphonium bromide conjugated stearic acid-g-chitosan oligosaccharide polymeric micelles (CTPP-CSOSA/CLT) for mitochondrial targeting and pH-sensitive drug release to treat cancer. Owing to grafting CTPP, CTPP-CSOSA/CLT displayed more effective mitochondria targeting as well as faster drug release in mitochondria. CTPP-CSOSA/CLT greatly enhanced ROS levels, allowing ΔΨm to decrease and more cytochrome c to be released into the cytoplasm, and then induced the substantial apoptosis of tumor cells. The in vivo distribution study showed that the distribution of CTPP-CSOSA micelles in tumor tissue was also improved by modifying the lipophilic cation CTPP. CTPP-CSOSA/CLT micelles were further found to inhibit tumor growth most effectively with no noticeable abnormalities in the CTPP-CSOSA/CLT micelle treated mice.
Elhasany et al.126 designed a multi-functional micellar nanomedicine (SPIONs/CS-loaded SFZ/zein-ChS PMs) by encapsulating CLT and sulfasalazine (SFZ), and oleic acid-capped superparamagnetic iron oxide nanoparticles (SPIONs) into the hydrophobic core of zein-chondroitin sulphate (zein-ChS) micelles. The combination of magnetic targeting and the active targeting effect of ChS increased the uptake of the micelles by MCF-7 cancer cells, resulting in a higher cytotoxicity against MCF-7 breast cancer cells. Moreover, in the in vivo experiments, SPIONs/CS-loaded SFZ/zein-ChS PMs showed superior anti-tumor activity compared to non-magnetically micelles-, free drug-treated and positive control groups.
2.4 Emulsified drug delivery systems
Micro/nanoemulsions are transparent thermodynamically stable dispersions made of water, oil, surfactant and co-surfactant having droplet sizes smaller than 100 nm.127 Micro/nanoemulsions systems are simple to manufacture and can incorporate hydrophobic drugs into the oil phase, thereby enhancing their solubility and bioavailability. The summary of CLT-loaded micro/nanoemulsions is given in Table 4.| Formulation ± co-treatment | Composition ± targeting moiety | Preparation technique | Physico-chemical characteristics | Used cell line/animal model | Route of administration/treatment dosage | Major outcome | Ref. |
|---|---|---|---|---|---|---|---|
| ZP: zeta potential; EE: entrapment efficiency; DL: drug loading; n.a.: not applicable; i.v.: intravenous; i.p.: intraperitoneal; SMEDDS: self-microemulsifying drug delivery system; OP-10: octyl polyethylene glycol phenyl ether; PEG 400: polyethylene glycol 400; DSPE-PEG-Tf: transferrin modified 1,2-distearoyl-sn-glycero-3-phosphoethanolamine N-[amino (polyethylene glycol)]. | |||||||
| SMEDDS dispersible tablets | Ethyl oleate, OP-10, transcutol P, microcrystalline cellulose KG 802 | Wet granulation compression method | PS: ∼25 nm; ZP: n.a.; DL: ∼10% | Sprague-Dawley rats | Oral | Improved solubility; enhanced bioavailability | 128 |
| Transferrin-modified microemulsions (co-delivered with coix seed) | 40Cremophor® RH 40, PEG 400, transferrin | Emulsification method | PS: ∼40 nm; ZP: ∼−14 mV; EE: ∼95%; LC: ∼41% | Cervical cancer cell lines (HeLa cells); HeLa 3D tumor spheroids | In vitro | Enhanced anti-cancer efficacy | 51 |
| Transferrin-modified microemulsions (co-delivered with coix seed) | 40Cremophor® RH 40, PEG 400, transferrin | Emulsification method | PS: ∼28 nm; ZP: n.a.; DL or EE: n.a. | Cervical cancer cell lines (HeLa cells); HeLa xenograft tumor-bearing mouse model | In vitro; i.p. (CLT dose: 1.8 mg kg−1, every other day) | Enhanced anti-cancer efficacy; reduced toxicity | 129 |
| Transferrin-modified microemulsions (co-delivered with β-elemene) | Labrafil® M 1944CS, Kolliphor® HS15, PEG 400, DSPE-PEG-transferrin | Emulsification method | PS: ∼69 nm; ZP: ∼−19 mV; EE: ∼95% | Lung cancer cell lines (A549 and Lewis cells); A549-xenograft nude mouse tumor model | In vitro; i.v. (CLT dose: 2 mg kg−1) | Enhanced anti-cancer efficacy; reduced toxicity | 130 |
| Transferrin and cell-penetrating peptide SA-R6H4 modified microemulsions (co-delivered with brucea oil and glycyrrhizin) | Glycyrrhizin, Brucea oil, Linoleic acid, RH40, PEG400, DSPE-PEG-NH2, SA-R6H4, transferrin | Emulsification method | PS: ∼69 nm; ZP: ∼−19 mV; EE: ∼95% | Human ovarian carcinoma cell line (SKOV3); SKOV-3 xenograft nude mouse tumor model | In vitro; i.v. (CLT dose: 5 mg kg−1 given on days 8, 11, 14, 19, 22 and 25 after tumor inoculation | Enhanced anti-cancer efficacy; reduced toxicity | 131 |
Qi et al.128 formulated a liquid self-microemulsifying drug delivery system (SMEDDS) containing CLT, which was then built up into the solid dispersible tablets using microcrystalline cellulose KG 802 as the adsorbent by a wet granulation compression method. The solid tablets could disperse in distilled water within ∼3 min After oral administration to rats, the relative bioavailability of CLT SMEDDS and SMEDDS dispersible tablets compared to the aqueous suspensions of CLT was 569 ± 7.07% and 558 ± 6.77%, respectively.
A transferrin-functionalized microemulsion encapsulating coix seed oil and CLT (Tf-CT-MEs) was fabricated by Chen et al. for the improved treatment of cervical cancer.51In vitro cellular assays demonstrated that Tf-CT-MEs increased the cellular uptake, thus allowing for enhanced cytotoxicity towards HeLa cells. Due to the advantages of small particle size (40.02 ± 0.21 nm) and transferrin modification, Tf-CT-MEs displayed improved tumor penetration and greater anti-cervical cancer activity in HeLa 3D tumor spheroids. Guo et al.129 further evaluated the in vivo therapeutic effectiveness of Tf-CT-MEs in HeLa tumor-bearing xenograft mouse models. In the study, Tf-CT-MEs demonstrated superior anticancer potency while displaying lower toxicity in vital organs. Similarly, intravenously injectable, transferrin-modified microemulsions carrying both β-elemene and CLT (Tf-EC-MEs) have been reported for the combination therapy of lung cancer.130 The codelivery system exhibited synergistic antiproliferative effects on cultured cells in vitro, as well as improved efficacy in A549-bearing xenograft mouse tumor models via active tumor targeting. Moreover, Tf-EC-MEs did not result in the apparent toxicity associated with free CLT therapy. Zhao et al.131 incorporated CLT, brucea oil, and glycyrrhizin into a single microemulsion system, which was then decorated dually with transferrin and SA-R6H4 cell-penetration peptide (Tf/SA-R6H4-TBG-MEs) to enhance ovarian cancer therapy. It was demonstrated that Tf/SA-R6H4-TBG-MEs had the most potent inhibition of in vitro cell proliferation and in vivo tumor growth, as well as the longest survival period among all the treatment groups. Moreover, Tf/SA-R6H4-TBG-MEs efficiently reduced liver and kidney toxicity.
2.5 Other nano-formulations
Inclusion complexes are made up of a mixture of drug molecules that are located in the cavity of host molecules. To improve the solubility of CLT, the inclusion complexes of CLT with monopolyamine-modified β-cyclodextrin (β-CD) (H1) and the polyamine-bridged bis(β-CD) (H2) were produced by Yang et al.53 Complexation with H1 and H2 dramatically increased CLT's water-solubility, as evidenced by a 52.1- and 60.4-fold improvement over CLT, respectively. Moreover, as compared to the positive control of cisplatin, the H1/CLT inclusion complex produced superior cytotoxicity in five tumor cells, and CLT related side effects were slightly diminished when it was complexed with H1. The study offers the possibility of enhancing the physicochemical properties of CLT, which would be useful in its final clinical trials for cancer treatment.3. Discussions and future perspectives
CLT holds great promise among natural agents due to its pleiotropic biological activities. However, its unfavorable physicochemical and pharmacokinetic properties such as poor solubility, weak bioavailability, and systemic toxicity compromise its therapeutic advantages hindering its clinical application. To overcome these drawbacks, it seems that encapsulation into specific nanocarriers can be of great interest and will fully exploit its potential applications. This comprehensive literature survey on CLT loaded nanocarriers explicitly indicates that this approach could enhance the physicochemical properties, bioavailability, biological activities and/or reduce the toxicity of CLT.The improved bioavailability effects of various types of CLT nanoformulations given orally, intravenously and intraperitoneally are shown in Fig. 4 and Table 5. Overall, various CLT-loaded nanocarriers administered through different routes maintain CLT plasma concentrations for a more extended period of time and improve other pharmacokinetic parameters, thus providing better bioavailability. CLT is water insoluble34 and has a LogP of 5.63,35 indicating that it is a Biopharmaceutics Classification System (BCS) Class IV-type drug with low solubility/permeability, suggesting CLT may be poorly absorbed through the intestinal mucosa. As shown in Fig. 4, compared to other nanocarriers administered orally, lipid-based nanoparticles have been the most representatively used nanocarriers to improve CLT delivery to overcome the issues of poor absorption and limited oral bioavailability. The high affinity of the lipid material used here with the cell membrane, which effectively increases the transmembrane influx and caveolae/clathrin-mediated endocytosis, is thought to be responsible for the improved absorption of CLT in lipid-based nanoparticles.55,56 Furthermore, the favored lymphatic transport of lipid nanocarriers that allows them to bypass the liver metabolism is another advantage for better absorption.55,57,128
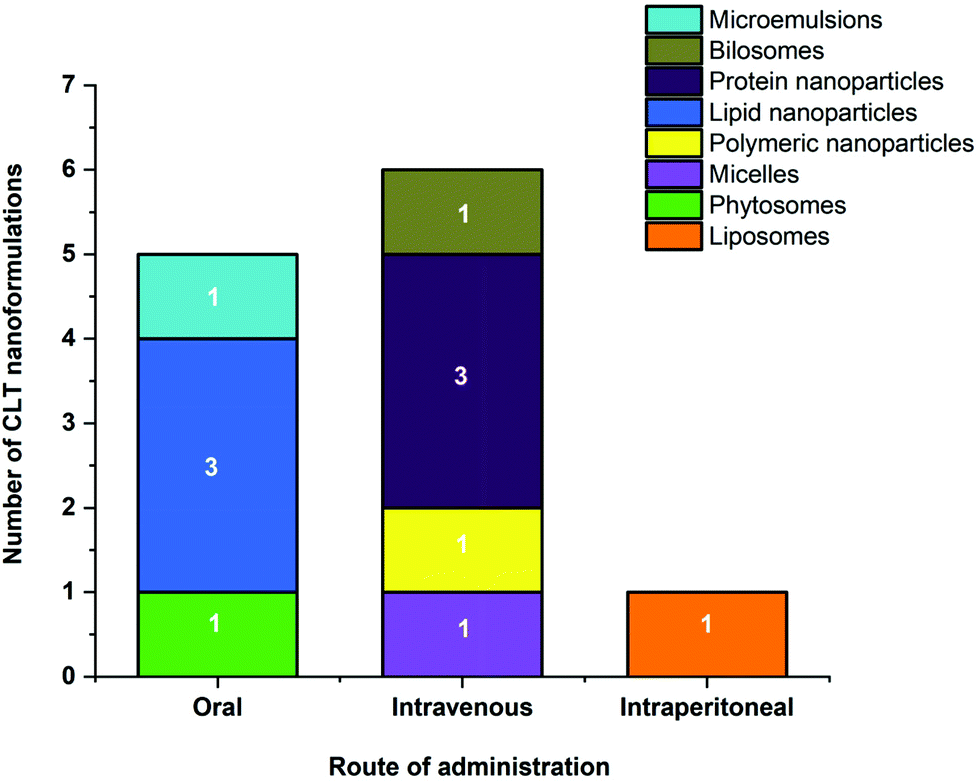 | ||
| Fig. 4 Number of different nanoformulations with their route of administration for the improved bioavailability of CLT. | ||
| Classification | Formulation | In vivo model | Route of administration | CLT dose | Major outcome | Ref. |
|---|---|---|---|---|---|---|
| AUC0−∞: area under the concentration time-curve from time zero to infinity; AUC0−t: area under the concentration time-curve from time zero to the last measurable drug concentration; T1/2: half-life; Cmax: the maximal drug concentration; Tmax: the time of the Cmax observed; Cl: clearance rate; MRT: mean residence time; i.v.: intravenous; i.p.: intraperitoneal; HA: hyaluronic acid; PCL: polycaprolactone; CPPs: cell-penetrating peptides; NLCs: nanostructured lipid carriers; HSA: human serum albumin; PAB: palmitic acid-modified bovine serum albumin; PEG: poly(ethylene glycol); SMEDDS: self-microemulsifying drug delivery system; *FREE CLT: comparatively to free CLT; *SUSP: comparatively to free CLT suspension; *SOL: comparatively to free CLT solution: *SOL(PEG): Comparatively to free CLT solubilized using PEG 300; *SOL(COSOLVENT): comparatively to free CLT solubilized in a cosolvent comprising propanediol/ethanol/water = 5/3/2 (v/v); *SOL(DMSO): comparatively to free CLT solubilized in a DMSO: saline mixture; *SOL(ETHANOL): comparatively to free CLT solubilized in a 75% ethanol: water mixture. | ||||||
| Liposomes | Liposomes | Healthy Kunming mice | i.p. | 4 mg kg−1 | Improved bioavailability (Cmax, Tmax, T1/2, MRT and AUC0−∞ were, respectively, 1.59-fold, 3.75-fold, 3.08-fold, 2.91-fold, and 3.73-fold increase*SOL(DMSO)) | 61 |
| Phytosomes | Phytosomes | Healthy New Zealand white male rabbits | Oral | 40 mg kg−1 | Improved bioavailability (T1/2, MRT, AUC0–8 h and Cmax values were, respectively, 10.52-fold, 5.83-fold, 9.20-fold, and 5.00-fold increase*SUSP) | 54 |
| Bilosomes | HA-functionalized bilosomes | Established arthritic Sprague-Dawley rats | i.v. | 10 mg kg−1 | Improved bioavailability (Cmax and AUC0−∞ were, respectively, 1.12-fold and 3.13-fold increase*SOL(ETHANOL); Cl (3.13-fold decrease*SOL(ETHANOL) | 41 |
| Polymeric nanoparticles | PCL nanoparticles | Healthy Sprague-Dawley rats | i.v. | 2.5 mg kg−1 | Improved bioavailability (AUC0−t, T1/2 and MRT were, respectively, 10.86-fold, 15.25-fold, and 15.24-fold increase*SOL(COSOLVENT)); Cl (11.04-fold decrease*SOL(COSOLVENT)) | 79 |
| Lipid based nanoparticles | CPPs coated NLCs | Healthy Beagles | Oral | 1.5 mg kg−1 | Improved bioavailability (T1/2, MRT, AUC0−∞ and Cmax values were, respectively, 1.93-fold, 2.47-fold, 4.11-fold, and 2.16-fold increase*SUSP) | 56 |
| Lipid nanospheres | Healthy Sprague-Dawley rats | Oral | 50 mg kg−1 | The absolute bioavailability of the CLT loaded in lipid nanospheres and CLT dispersed in suspensions were 30.01% and 13.35%, respectively; the relative bioavailability of CLT loaded in lipid nanospheres to CLT suspensions was 224.88% | 55 | |
| Broccoli lipid nanoparticles | Healthy Sprague-Dawley rats | Oral | 40 mg kg−1 | The relative bioavailability of the CLT loaded in broccoli lipid nanoparticles was 494.13% compared with CLT suspensions; Cmax (4.43-fold increase*SUSP) | 57 | |
| Protein nanoparticles | HSA-Kolliphor HS 15 nanoparticles | Healthy Sprague-Dawley rats | i.v. | 2 mg kg−1 | Improved bioavailability (Cmax, T1/2 and AUC0–24 h were, respectively, 3.09-fold, 3.61-fold and 2.56-fold increase*SOL) | 92 |
| PAB nanoparticles | Healthy Sprague-Dawley rats | i.v. | 2 mg kg−1 | Improved bioavailability (Cmax, T1/2 and AUC0–24 h were, respectively, 1.68-fold, 4.72-fold and 4.66-fold increase*SOL) | 93 | |
| Silk fibroin nanoparticles | Healthy Sprague-Dawley rats | i.v. | 1 mg kg−1 | Improved bioavailability (MRT, AUC0−∞ and Cmax values were, respectively, 1.96-fold, 2.61-fold, and 4.36-fold increase*SOL(PEG)); Cl (2.51-fold decrease*SOL(PEG)) | 96 | |
| Micelles | CLT-PEG-G Rh2 micelles | Healthy Sprague-Dawley rat | i.v. | 3 mg kg−1 | Improved bioavailability (T1/2, MRT, AUC0−∞ and Cmax values were, respectively, 2.03-fold, 2.13-fold, 3.44-fold, and 1.32-fold increase*SOL) | 122 |
| Microemulsions | SMEDDS dispersible tablets | Healthy Sprague-Dawley rats | Oral | 4 mg kg−1 | Improved bioavailability (Cmax and AUC0−∞ were, respectively, 5.24-fold and 5.58-fold increase*SUSP) | 128 |
The present review also addressed findings on the biological activities of CLT after encapsulation into nanocarriers (Fig. 5). Nanocarriers encapsulating CLT have increased its anti-inflammatory properties in acute pancreatitis,85 glomerulonephritis,37 renal fibrosis,64 arthritis,40,92 psoriasis,42 and allergic asthma.50,113 Furthermore, encapsulated CLT also reveals remarkable advancements in anti-atherosclerotic,120 antiangiogenic,114,115 and anti-obesity properties.118 Likewise, nanocarriers have been most commonly used to enhance the anticancer activity of CLT against lung cancer, breast cancer, prostate cancer, pancreatic cancer, colorectal cancer, cervical cancer, ovarian cancer, glioma hepatocellular carcinoma, melanoma, squamous cell carcinoma, leukemia and retinoblastoma. Among all these cancer diseases, lung cancer and breast cancer seem to be the most studied, followed by prostate cancer and melanoma (Fig. 5). Different CLT nanoformulations with their route of administration for in vivo improved antitumor efficacy of CLT are shown in Fig. 6. As a promising nanocarrier, polymer micelles have been most widely applied to enhance the anti-tumor therapeutic effect of CLT. For the tumor treatment of CLT nanoformulations, intravenous administration remains the most commonly used route by which maximum bioavailability could be obtained.
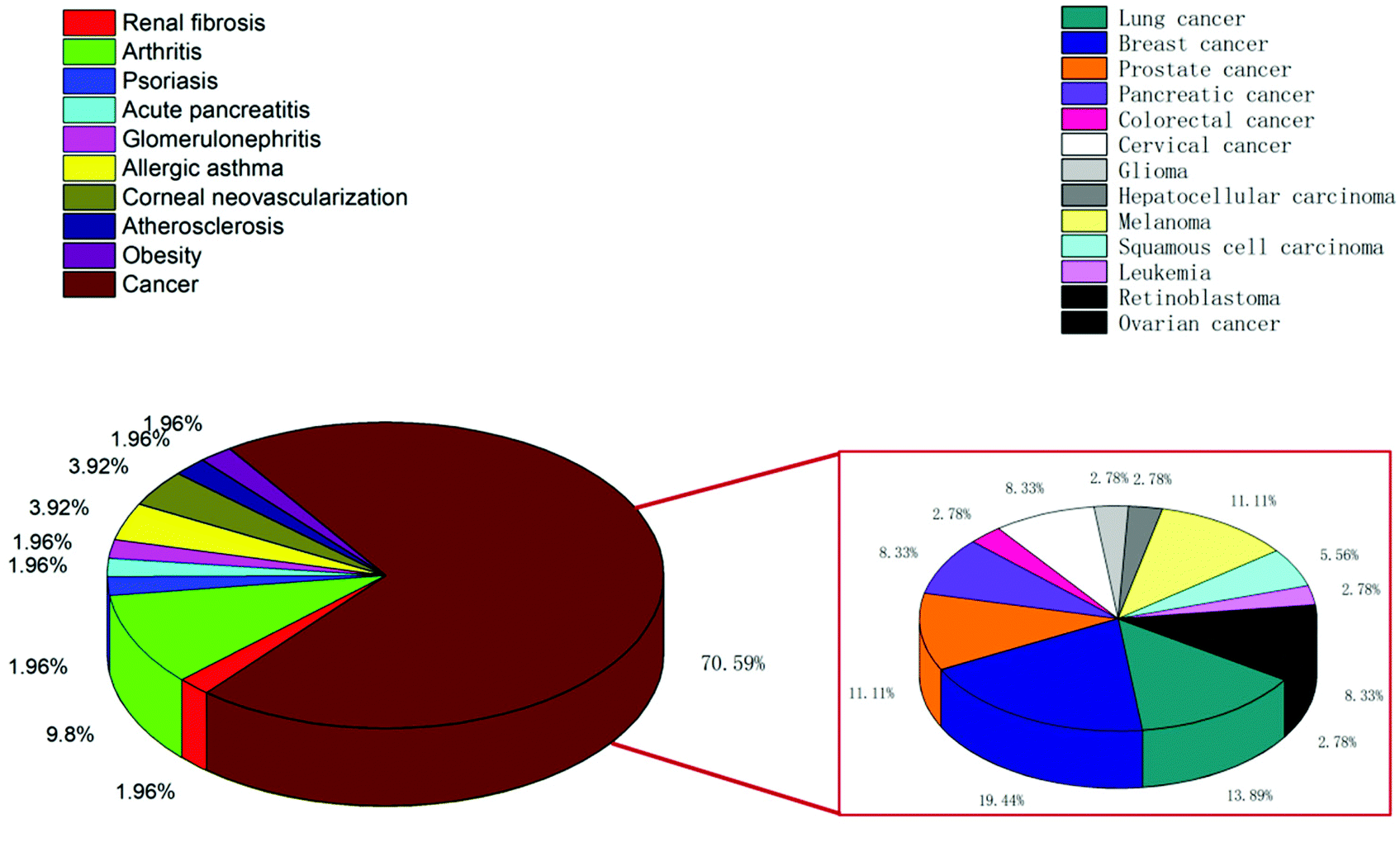 | ||
| Fig. 5 Various diseases for which therapeutic activity of celastrol nanoformulations have been reported. | ||
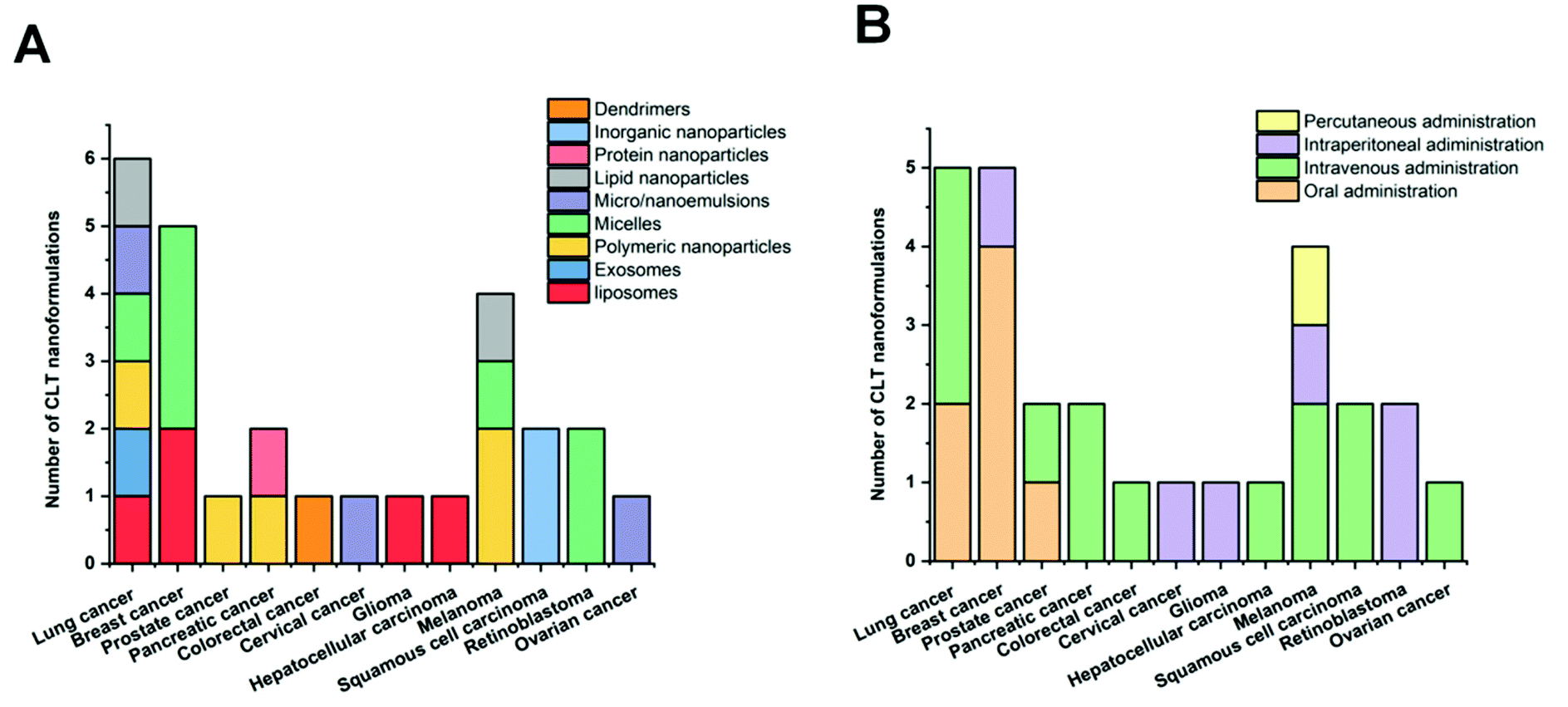 | ||
| Fig. 6 Different CLT nanoformulations (A) with their route of administration (B) for improved in vivo antitumor efficacy of CLT. | ||
The nanocarriers described above also allowed for the simultaneous delivery of other bioactive ingredients besides CLT, such as other phytochemicals and conventional chemotherapeutic agents. In addition, the surface of the nanocarriers could be decorated with ligands to receptors and other specific targets excessively upregulated in targeted cells, enabling the active targeted delivery of CLT and thereby showing enhanced efficacy and decreased off-target toxicity. Stimuli-responsive and triggered release systems based on the differences in the pH of blood and intracellular mitochondria have been designed to realize rapid CLT release at mitochondrial target sites to increase the tumor inhibition effectiveness. Several recent reports also highlight novel cell membrane biomimetic nanoparticles for the delivery of CLT to achieve enhanced inflammation and cancer therapy. Given that CLT also possesses remarkable neuroprotective effects, and still no reports on CLT nanoformulations on this aspect to date, we believe the combination of CLT with nanotechnology will be explored further as potential therapeutic approaches for neurodegenerative disorders in the future.
Overall, the current comprehensible evidence on drug delivery systems collected from the above reports undoubtedly open up a new avenue for poorly bioavailable drugs like CLT. Many questions, however, remain unanswered. For example, a wide range of CLT nanoformulations with different features are available, but the detailed characterization including particle size, zeta potential, entrapment efficiency, drug-loading efficiency or storage stability of these nanoformulations remains largely unexplored. Thorough characterization of the CLT nanoformulations should be carefully carried out in the future. Some reported studies also have seemed to have some shortcomings, such as the lack of in vivo experiments. Given that in vitro findings do not always give rise to comparable outcomes in vivo, further research is needed to ensure that the nanoformulations perform as well in vivo. In addition, there is also a lack of details on the pharmacokinetics, biodistribution, toxicity and biocompatibility of certain CLT nanoformulations. As a result, for advance research on CLT nanoformulations, the gap in this field must be filled.
4 Conclusions
In summary, advances in nanodelivery of CLT over the past 10 years shed light on the promise CLT carries when it is appropriately delivered. They equally highlighted the need for a thorough characterization of the physicochemical and biopharmaceutical properties of such nanoformulations. Their safety and efficacy profiles should be extensively studied in an in vivo experimental setup before their clinical application for the treatment of a particular disease can be realized.Author contributions
Conceptualization, Ling Guo and Khuloud T. Al-Jamal; writing – original draft preparation, Ling Guo; writing – review and editing, Ling Guo, Khuloud T. Al-Jamal; supervision, Yongping Zhang, Khuloud T. Al-Jamal; funding acquisition, Ling Guo, Yongping Zhang, Khuloud T. Al-Jamal. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Pharmaceutical Experimental Teaching Center Platform Construction Project (Guizhou Higher Education Development [2017] No. 158), the Guizhou Province High-level Innovative Talent Cultivation “Hundred Levels” Project ([2015]4030), and the National Natural Science Foundation of China (81960650).References
- S. W. Ng, Y. Chan, D. K. Chellappan, T. Madheswaran, F. Zeeshan and Y. L. Chan, et al., Molecular modulators of celastrol as the keystones for its diverse pharmacological activities, Biomed. Pharmacother., 2019, 109, 1785–1792 CrossRef CAS PubMed.
- S. Sreeramulu, S. L. Gande, M. Göbel and H. Schwalbe, Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone–cochaperone, by triterpene celastrol, Angew. Chem., Int. Ed., 2009, 48(32), 5853–5855 CrossRef CAS PubMed.
- T. W. Corson and C. M. Crews, Molecular understanding and modern application of traditional medicines: triumphs and trials, Cell, 2007, 130(5), 769–774 CrossRef CAS PubMed.
- P. Rajendran, F. Li, M. K. Shanmugam, R. Kannaiyan, J. N. Goh and K. F. Wong, et al., Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo, Cancer Prev. Res., 2012, 5(4), 631–643 CrossRef CAS PubMed.
- H. Mou, Y. Zheng, P. Zhao, H. Bao, W. Fang and N. Xu, Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria-and Fas/FasL-mediated pathways, Toxicol. in Vitro, 2011, 25(5), 1027–1032 CrossRef CAS PubMed.
- X. Zhao, S. Gao, H. Ren, H. Huang, W. Ji and J. Hao, Inhibition of autophagy strengthens celastrol-induced apoptosis in human pancreatic cancer in vitro and in vivo models, Curr. Mol. Med., 2014, 14(4), 555–563 CrossRef CAS PubMed.
- S. Shrivastava, M. K. Jeengar, V. S. Reddy, G. B. Reddy and V. Naidu, Anticancer effect of celastrol on human triple negative breast cancer: possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3 K/Akt pathways, Exp. Mol. Pathol., 2015, 98(3), 313–327 CrossRef CAS PubMed.
- J. Guo, X. Huang, H. Wang and H. Yang, Celastrol induces autophagy by targeting AR/miR-101 in prostate cancer cells, PLoS One, 2015, 10(10), 1–18 Search PubMed.
- L. Lin, Y. Sun, D. Wang, S. Zheng, J. Zhang and C. Zheng, Celastrol ameliorates ulcerative colitis-related colorectal cancer in mice via suppressing inflammatory responses and epithelial-mesenchymal transition, Front. Pharmacol., 2016, 6, 320 Search PubMed.
- L.-N. Xu, N. Zhao, J.-Y. Chen, P.-P. Ye, X.-W. Nan and H.-H. Zhou, et al., Celastrol inhibits the growth of ovarian cancer cells in vitro and in vivo, Front. Oncol., 2019, 9, 2 CrossRef PubMed.
- J.-H. Lee, Y.-S. Won, K.-H. Park, M.-K. Lee, H. Tachibana and K. Yamada, et al., Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3 K/AKT signaling, Apoptosis, 2012, 17(12), 1275–1286 CrossRef CAS PubMed.
- G. Sethi, K. S. Ahn, M. K. Pandey and B. B. Aggarwal, Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-κB–regulated gene products and TAK1-mediated NF-κB activation, Blood, 2006, 109(7), 2727–2735 CrossRef PubMed.
- P.-P. Li, W. He, P.-F. Yuan, S.-S. Song, J.-T. Lu and W. Wei, Celastrol induces mitochondria-mediated apoptosis in hepatocellular carcinoma Bel-7402 cells, Am. J. Chin. Med., 2015, 43(01), 137–148 CrossRef CAS PubMed.
- H. W. Jung, Y. S. Chung, Y. S. Kim and Y.-K. Park, Celastrol inhibits production of nitric oxide and proinflammatory cytokines through MAPK signal transduction and NF-κB in LPS-stimulated BV-2 microglial cells, Exp. Mol. Med., 2007, 39(6), 715–721 CrossRef CAS PubMed.
- P. Yadav, V. Jaswal, A. Sharma, D. Kashyap, H. S. Tuli and V. K. Garg, et al., Celastrol as a pentacyclic triterpenoid with chemopreventive properties, Pharm. Pat. Anal., 2018, 7(4), 155–167 CrossRef CAS PubMed.
- J. Wu, M. Ding, N. Mao, Y. Wu, C. Wang and J. Yuan, et al., Celastrol inhibits chondrosarcoma proliferation, migration and invasion through suppression CIP2A/c-MYC signaling pathway, J. Pharmacol. Sci., 2017, 134(1), 22–28 CrossRef CAS PubMed.
- Y. Yang, S. Cheng, G. Liang, L. Honggang and H. Wu, Celastrol inhibits cancer metastasis by suppressing M2-like polarization of macrophages, Biochem. Biophys. Res. Commun., 2018, 503(2), 414–419 CrossRef CAS PubMed.
- Y. Kim, H. Kang, S. W. Jang and J. Ko, Celastrol inhibits breast cancer cell invasion via suppression of NF-κB-mediated matrix metalloproteinase-9 expression, Cell. Physiol. Biochem., 2011, 28(2), 175–184 CrossRef CAS PubMed.
- R. Kannaiyan, M. K. Shanmugam and G. Sethi, Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer, Cancer Lett., 2011, 303(1), 9–20 CrossRef CAS PubMed.
- D. Y. Kim, J. W. Park, D. Jeoung and J. Y. Ro, Celastrol suppresses allergen-induced airway inflammation in a mouse allergic asthma model, Eur. J. Pharmacol., 2009, 612(1–3), 98–105 CrossRef CAS PubMed.
- H. Li, Y. Jia, Y. Pan, D. Pan, D. Li and L. Zhang, Effect of tripterine on collagen-induced arthritis in rats, Acta Pharmacol. Sin., 1997, 18(3), 270–273 CAS.
- H. Li, Y.-Y. Zhang, X.-Y. Huang, Y.-N. Sun, Y.-F. Jia and D. Li, Beneficial effect of tripterine on systemic lupus erythematosus induced by active chromatin in BALB/c mice, Eur. J. Pharmacol., 2005, 512(2–3), 231–237 CrossRef CAS PubMed.
- Z. Jia, C. Xu, J. Shen, T. Xia, J. Yang and Y. He, The natural compound celastrol inhibits necroptosis and alleviates ulcerative colitis in mice, Int. Immunopharmacol., 2015, 29(2), 552–559 CrossRef CAS PubMed.
- W. Wang, C. Ha, T. Lin, D. Wang, Y. Wang and M. Gong, Celastrol attenuates pain and cartilage damage via SDF–1/CXCR 4 signalling pathway in osteoarthritis rats, J. Pharm. Pharmacol., 2018, 70(1), 81–88 CrossRef CAS PubMed.
- D. Kim, E. Shin, Y. Kim, B. Lee, J. G. Jun and J. Park, et al., Suppression of inflammatory responses by celastrol, a quinone methide triterpenoid isolated from Celastrus regelii, Eur. J. Clin. Invest., 2009, 39(9), 819–827 CrossRef CAS PubMed.
- V. Joshi, S. H. Venkatesha, C. Ramakrishnan, A. N. N. Urs, V. Hiremath and K. D. Moudgil, et al., Celastrol modulates inflammation through inhibition of the catalytic activity of mediators of arachidonic acid pathway: secretory phospholipase A2 group IIA, 5-lipoxygenase and cyclooxygenase-2, Pharmacol. Res., 2016, 113, 265–275 CrossRef CAS PubMed.
- M. Hu, Q. Luo, G. Alitongbieke, S. Chong, C. Xu and L. Xie, et al., Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy, Mol. Cell, 2017, 66(1), 141–153 CrossRef CAS PubMed.
- D. Paris, N. J. Ganey, V. Laporte, N. S. Patel and M. J. Mullan, Reduction of β-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer's disease, J. Neuroinflammation, 2010, 7(1), 17 CrossRef PubMed.
- C. Cleren, N. Y. Calingasan, J. Chen and M. F. Beal, Celastrol protects against MPTP- and 3-nitropropionic acid–induced neurotoxicity, J. Neurochem., 2005, 94(4), 995–1004 CrossRef CAS PubMed.
- A. M. Chow, D. W. Tang, A. Hanif and I. R. Brown, Induction of heat shock proteins in cerebral cortical cultures by celastrol, Cell Stress Chaperones, 2013, 18(2), 155–160 CrossRef CAS PubMed.
- A. M. Chow, D. W. Tang, A. Hanif and I. R. Brown, Localization of heat shock proteins in cerebral cortical cultures following induction by celastrol, Cell Stress Chaperones, 2014, 19(6), 845–851 CrossRef CAS PubMed.
- Y.-N. Deng, J. Shi, J. Liu and Q.-M. Qu, Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy, Neurochem. Int., 2013, 63(1), 1–9 CrossRef CAS PubMed.
- J. Liu, J. Lee, M. A. S. Hernandez, R. Mazitschek and U. Ozcan, Treatment of obesity with celastrol, Cell, 2015, 161(5), 999–1011 CrossRef CAS PubMed.
- X. Peng, J. Wang, H. Song, D. Cui, L. Li and J. Li, et al., Optimized preparation of celastrol-loaded polymeric nanomicelles using rotatable central composite design and response surface methodology, J. Biomed. Nanotechnol., 2012, 8(3), 491–499 CrossRef CAS PubMed.
- J. Song, F. Shi, Z. Zhang, F. Zhu, J. Xue and X. Tan, et al., Formulation and evaluation of celastrol-loaded liposomes, Molecules, 2011, 16(9), 7880–7892 CrossRef CAS PubMed.
- J. Zhang, C.-Y. Li, M.-J. Xu, T. Wu, J.-H. Chu and S.-J. Liu, et al., Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats, J. Ethnopharmacol., 2012, 144(1), 195–200 CrossRef CAS PubMed.
- L. Guo, S. Luo, Z. Du, M. Zhou, P. Li and Y. Fu, et al., Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis, Nat. Commun., 2017, 8(1), 1–17 CrossRef PubMed.
- W. Hou, B. Liu and H. Xu, Celastrol: Progresses in structure-modifications, structure-activity relationships, pharmacology and toxicology, Eur. J. Med. Chem., 2020, 112081 CrossRef CAS PubMed.
- X. Chen, X. Hu, J. Hu, Z. Qiu, M. Yuan and G. Zheng, Celastrol-loaded galactosylated liposomes effectively inhibit AKT/c-Met-triggered rapid hepatocarcinogenesis in mice, Mol. Pharmaceutics, 2020, 17(3), 738–747 CrossRef CAS PubMed.
- S. Zhu, C. Luo, W. Feng, Y. Li, M. Zhu and S. Sun, et al., Selenium-deposited tripterine phytosomes ameliorate the antiarthritic efficacy of the phytomedicine via a synergistic sensitization, Int. J. Pharm., 2020, 578, 119104 CrossRef CAS PubMed.
- H. Yang, Z. Liu, Y. Song and C. Hu, Hyaluronic acid-functionalized bilosomes for targeted delivery of tripterine to inflamed area with enhancive therapy on arthritis, Drug Delivery, 2019, 26(1), 820–830 CrossRef CAS PubMed.
- S. Meng, L. Sun, L. Wang, Z. Lin, Z. Liu and L. Xi, et al., Loading of water-insoluble celastrol into niosome hydrogels for improved topical permeation and anti-psoriasis activity, Colloids Surf., B, 2019, 182, 110352 CrossRef CAS PubMed.
- R. Munagala, F. Aqil, J. Jeyabalan and R. C. Gupta, Bovine milk-derived exosomes for drug delivery, Cancer Lett., 2016, 371(1), 48–61 CrossRef CAS PubMed.
- V. Sanna, J. C. Chamcheu, N. Pala, H. Mukhtar, M. Sechi and I. A. Siddiqui, Nanoencapsulation of natural triterpenoid celastrol for prostate cancer treatment, Int. J. Nanomed., 2015, 10, 6835 CrossRef CAS PubMed.
- L. Zhou, Y. Chen, Z. Zhang, J. He, M. Du and Q. Wu, Preparation of tripterine nanostructured lipid carriers and their absorption in rat intestine, Pharmazie, 2012, 67(4), 304–310 CAS.
- J. Y. Choi, B. Gupta, T. Ramasamy, J.-H. Jeong, S. G. Jin and H.-G. Choi, et al., PEGylated polyaminoacid-capped mesoporous silica nanoparticles for mitochondria-targeted delivery of celastrol in solid tumors, Colloids Surf., B, 2018, 165, 56–66 CrossRef CAS PubMed.
- S. Law, A. W. Leung and C. Xu, Folic acid-modified celastrol nanoparticles: synthesis, characterization, anticancer activity in 2D and 3D breast cancer models, Artif. Cells, Nanomed., Biotechnol., 2020, 48(1), 542–559 CrossRef CAS PubMed.
- J. Li, L. Shi, Y. Shao, M. Selke, B. Chen and H. Jiang, et al., The cellular labeling and pH-sensitive responsive-drug release of celastrol in cancer cells based on Cys-CdTe QDs, Sci. China: Chem., 2014, 57(6), 833–841 CrossRef CAS.
- P. Ge, B. Niu, Y. Wu, W. Xu, M. Li and H. Sun, et al., Enhanced cancer therapy of celastrol in vitro and in vivo by smart dendrimers delivery with specificity and biosafety, Chem. Eng. J., 2020, 383, 123228 CrossRef CAS.
- X. Peng, Y. Liang, J. Li, L. Lin, J. Wang and Y. Yin, et al., Preventive effects of ovalbumin-conjugated celastrol-loaded nanomicelles’ in a mouse model of ovalbumin-induced allergic airway inflammation, Eur. J. Pharm. Sci., 2020, 143, 105172 CrossRef CAS PubMed.
- Y. Chen, D. Qu, R. Fu, M. Guo, Y. Qin and J. Guo, et al., A Tf-modified tripterine-loaded coix seed oil microemulsion enhances anti-cervical cancer treatment, Int. J. Nanomed., 2018, 13, 7275 CrossRef CAS PubMed.
- Y. Xiao, J. Liu, M. Guo, H. Zhou, J. Jin and J. Liu, et al., Synergistic combination chemotherapy using carrier-free celastrol and doxorubicin nanocrystals for overcoming drug resistance, Nanoscale, 2018, 10(26), 12639–12649 RSC.
- H. Yang, Z. Pan, W. Jin, L. Zhao, P. Xie and S. Chi, et al., Preparation, characterization and cytotoxic evaluation of inclusion complexes between celastrol with polyamine-modified β-cyclodextrins, J. Inclusion Phenom. Macrocyclic Chem., 2019, 95(1–2), 147–157 CrossRef CAS.
- M. S. Freag, W. M. Saleh and O. Y. Abdallah, Self-assembled phospholipid-based phytosomal nanocarriers as promising platforms for improving oral bioavailability of the anticancer celastrol, Int. J. Pharm., 2018, 535(1–2), 18–26 CrossRef CAS PubMed.
- X. Zhang, T. Zhang, X. Zhou, H. Liu, H. Sun and Z. Ma, et al., Enhancement of oral bioavailability of tripterine through lipid nanospheres: preparation, characterization, and absorption evaluation, J. Pharm. Sci., 2014, 103(6), 1711–1719 CrossRef CAS PubMed.
- Y. Chen, L. Yuan, L. Zhou, Z.-H. Zhang, W. Cao and Q. Wu, Effect of cell-penetrating peptide-coated nanostructured lipid carriers on the oral absorption of tripterine, Int. J. Nanomed., 2012, 7, 4581 CAS.
- W. Li, T. Zhang, Y. Ye, X. Zhang and B. Wu, Enhanced bioavailability of tripterine through lipid nanoparticles using broccoli-derived lipids as a carrier material, Int. J. Pharm., 2015, 495(2), 948–955 CrossRef CAS PubMed.
- X. Cao, Y. Hu, S. Luo, Y. Wang, T. Gong and X. Sun, et al., Neutrophil-mimicking therapeutic nanoparticles for targeted chemotherapy of pancreatic carcinoma, Acta Pharm. Sin. B, 2019, 9(3), 575–589 CrossRef PubMed.
- S. Biju, S. Talegaonkar, P. Mishra and R. Khar, Vesicular systems: an overview, Indian J. Pharm. Sci., 2006, 68(2), 141–153 CrossRef CAS.
- D. Sharma, A. A. E. Ali and L. R. Trivedi, An Updated Review on: Liposomes as drug delivery system, PharmaTutor, 2018, 6(2), 50–62 CrossRef CAS.
- Y. Huang, D. Zhou, T. Hang, Z. Wu, J. Liu and Q. Xu, et al., Preparation, characterization, and assessment of the antiglioma effects of liposomal celastrol, Anti-Cancer Drugs, 2012, 23(5), 515–524 CrossRef CAS PubMed.
- J. Wolfram, K. Suri, Y. Huang, R. Molinaro, C. Borsoi and B. Scott, et al., Evaluation of anticancer activity of celastrol liposomes in prostate cancer cells, J. Microencapsulation, 2014, 31(5), 501–507 CrossRef CAS PubMed.
- S. Kang, T. Park, X. Chen, G. Dickens, B. Lee and K. Lu, et al., Tunable physiologic interactions of adhesion molecules for inflamed cell-selective drug delivery, Biomaterials, 2011, 32(13), 3487–3498 CrossRef CAS PubMed.
- R. Li, Y. Li, J. Zhang, Q. Liu, T. Wu and J. Zhou, et al., Targeted delivery of celastrol to renal interstitial myofibroblasts using fibronectin-binding liposomes attenuates renal fibrosis and reduces systemic toxicity, J. Controlled Release, 2020, 320, 32–44 CrossRef CAS PubMed.
- Z. C. Soe, R. K. Thapa, W. Ou, M. Gautam, H. T. Nguyen and S. G. Jin, et al., Folate receptor-mediated celastrol and irinotecan combination delivery using liposomes for effective chemotherapy, Colloids Surf., B, 2018, 170, 718–728 CrossRef CAS PubMed.
- D. Qu, L. Wang, Y. Qin, M. Guo, J. Guo and M. Huang, et al., Non-triggered sequential-release liposomes enhance anti-breast cancer efficacy of STS and celastrol-based microemulsion, Biomater. Sci., 2018, 6(12), 3284–3299 RSC.
- H. A. Pawar and B. D. Bhangale, Phytosome as a novel biomedicine: a microencapsulated drug delivery system, J. Bioanal. Biomed., 2015, 7(1), 06–12 CAS.
- M. S. Freag, W. M. Saleh and O. Y. Abdallah, Laminated chitosan-based composite sponges for transmucosal delivery of novel protamine-decorated tripterine phytosomes: ex vivo mucopenetration and in vivo pharmacokinetic assessments, Carbohydr. Polym., 2018, 188, 108–120 CrossRef CAS PubMed.
- J. Ahmad, M. Singhal, S. Amin, M. Rizwanullah, S. Akhter and M. Amjad Kamal, et al., Bile salt stabilized vesicles (bilosomes): a novel nano-pharmaceutical design for oral delivery of proteins and peptides, Curr. Pharm. Des., 2017, 23(11), 1575–1588 CrossRef CAS PubMed.
- A. Shukla, V. Mishra and P. Kesharwani, Bilosomes in the context of oral immunization: development, challenges and opportunities, Drug Discovery Today, 2016, 21(6), 888–899 CrossRef CAS PubMed.
- A. M. Al-mahallawi, A. A. Abdelbary and M. H. Aburahma, Investigating the potential of employing bilosomes as a novel vesicular carrier for transdermal delivery of tenoxicam, Int. J. Pharm., 2015, 485(1–2), 329–340 CrossRef CAS PubMed.
- Y. Rahimpour and H. Hamishehkar, Niosomes as carrier in dermal drug delivery, Recent Adv. Novel Drug Carrier Syst., 2012, 1(1), 141–164 Search PubMed.
- M. Osanloo, S. Assadpour, A. Mehravaran, M. Abastabar and J. Akhtari, Niosome-loaded antifungal drugs as an effective nanocarrier system: A mini review, Curr. Med. Mycol., 2018, 4(4), 31 Search PubMed.
- M. J. Haney, N. L. Klyachko, Y. Zhao, R. Gupta, E. G. Plotnikova and Z. He, et al., Exosomes as drug delivery vehicles for Parkinson's disease therapy, J. Controlled Release, 2015, 207, 18–30 CrossRef CAS PubMed.
- F. Aqil, H. Kausar, A. K. Agrawal, J. Jeyabalan, A.-H. Kyakulaga and R. Munagala, et al., Exosomal formulation enhances therapeutic response of celastrol against lung cancer, Exp. Mol. Pathol., 2016, 101(1), 12–21 CrossRef CAS PubMed.
- V. Mohanraj and Y. Chen, Nanoparticles-a review, Trop. J. Pharm. Res., 2006, 5(1), 561–573 Search PubMed.
- N. Jawahar and S. Meyyanathan, Polymeric nanoparticles for drug delivery and targeting: A comprehensive review, Int. J. Health Allied Sci., 2012, 1(4), 217 CrossRef.
- F. Masood, Polymeric nanoparticles for targeted drug delivery system for cancer therapy, Mater. Sci. Eng., C, 2016, 60, 569–578 CrossRef CAS PubMed.
- J. Yin, P. Wang, Y. Yin, Y. Hou and X. Song, Optimization on biodistribution and antitumor activity of tripterine using polymeric nanoparticles through RES saturation, Drug Delivery, 2017, 24(1), 1891–1897 CrossRef CAS PubMed.
- W.-G. Shan, H.-G. Wang, R. Wu, Z.-J. Zhan and L.-F. Ma, Synthesis and anti-tumor activity study of water-soluble PEG-celastrol coupling derivatives as self-assembled nanoparticles, Bioorg. Med. Chem. Lett., 2019, 29(5), 685–687 CrossRef CAS PubMed.
- S. Park, S. Kang, A. J. Veach, Y. Vedvyas, R. Zarnegar and J.-Y. Kim, et al., Self-assembled nanoplatform for targeted delivery of chemotherapy agents via affinity-regulated molecular interactions, Biomaterials, 2010, 31(30), 7766–7775 CrossRef CAS PubMed.
- Q. Liu, F. Chen, L. Hou, L. Shen, X. Zhang and D. Wang, et al., Nanocarrier-mediated chemo-immunotherapy arrested cancer progression and induced tumor dormancy in desmoplastic melanoma, ACS Nano, 2018, 12(8), 7812–7825 CrossRef CAS PubMed.
- H. Wang, Y. Liu, R. He, D. Xu, J. Zang and N. Weeranoppanant, et al., Cell membrane biomimetic nanoparticles for inflammation and cancer targeting in drug delivery, Biomater. Sci., 2020, 8(2), 552–568 RSC.
- X. Zhou, R. Yu, X. Cao, Z.-R. Zhang and L. Deng, Bio-Mimicking Nanoparticles for Targeted Therapy of Malignant Melanoma, J. Biomed. Nanotechnol., 2019, 15(5), 993–1004 CrossRef CAS PubMed.
- X. Zhou, X. Cao, H. Tu, Z.-R. Zhang and L. Deng, Inflammation-targeted delivery of Celastrol via neutrophil membrane-coated nanoparticles in the management of acute pancreatitis, Mol. Pharmaceutics, 2019, 16(3), 1397–1405 CrossRef CAS PubMed.
- A. Puri, K. Loomis, B. Smith, J.-H. Lee, A. Yavlovich and E. Heldman, et al., Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic, Crit. Rev. Ther. Drug Carrier Syst., 2009, 26(6), 523–580 CrossRef CAS PubMed.
- Formulation, characterization and evaluation of the in vitro skin permeation of nanostructured lipid carriers encapsulated tripterine, in 2012 International Conference on Biomedical Engineering and Biotechnology, ed. Y. Chen, L. Zhou, L. Yuan, Z. Zhang and Q. Wu, IEEE, 2012 Search PubMed.
- Y. Chen, L. Zhou, L. Yuan, Z.-H. Zhang, X. Liu and Q. Wu, Formulation, characterization, and evaluation of in vitro skin permeation and in vivo pharmacodynamics of surface-charged tripterine-loaded nanostructured lipid carriers, Int. J. Nanomed., 2012, 7, 3023 CAS.
- L. Yuan, C. Liu, Y. Chen, Z. Zhang, L. Zhou and D. Qu, Antitumor activity of tripterine via cell-penetrating peptide-coated nanostructured lipid carriers in a prostate cancer model, Int. J. Nanomed., 2013, 8, 4339 Search PubMed.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, Albumin-based nanoparticles as potential controlled release drug delivery systems, J. Controlled Release, 2012, 157(2), 168–182 CrossRef CAS PubMed.
- T. Hakala, S. Davies, Z. Toprakcioglu, B. Bernardim, G. J. Bernardes and T. Knowles, A microfluidic co–flow route for human serum albumin–drug–nanoparticle assembly, Chem.– Eur. J., 2020, 26(27), 5965–5969 CrossRef CAS PubMed.
- T. Gong, P. Zhang, C. Deng, Y. Xiao, T. Gong and Z. Zhang, An effective and safe treatment strategy for rheumatoid arthritis based on human serum albumin and Kolliphor® HS 15, Nanomedicine, 2019, 14(16), 2169–2187 CrossRef CAS PubMed.
- T. Gong, T. Tan, P. Zhang, H. Li, C. Deng and Y. Huang, et al., Palmitic acid-modified bovine serum albumin nanoparticles target scavenger receptor-A on activated macrophages to treat rheumatoid arthritis, Biomaterials, 2020, 258, 1–13 CrossRef PubMed.
- Y. Hu, X. Chen, Y. Xu, X. Han, M. Wang and T. Gong, et al., Hierarchical assembly of hyaluronan coated albumin nanoparticles for pancreatic cancer chemoimmunotherapy, Nanoscale, 2019, 11(35), 16476–16487 RSC.
- J. Melke, S. Midha, S. Ghosh, K. Ito and S. Hofmann, Silk fibroin as biomaterial for bone tissue engineering, Acta Biomater., 2016, 31, 1–16 CrossRef CAS PubMed.
- F. Onyeabor, A. Paik, S. Kovvasu, B. Ding, J. Lin and M. A. Wahid, et al., Optimization of preparation and preclinical pharmacokinetics of celastrol-encapsulated silk fibroin nanoparticles in the rat, Molecules, 2019, 24(18), 3271 CrossRef CAS PubMed.
- B. Ding, M. A. Wahid, Z. Wang, C. Xie, A. Thakkar and S. Prabhu, et al., Triptolide and celastrol loaded silk fibroin nanoparticles show synergistic effect against human pancreatic cancer cells, Nanoscale, 2017, 9(32), 11739–11753 RSC.
- H.-C. Huang, S. Barua, G. Sharma, S. K. Dey and K. Rege, Inorganic nanoparticles for cancer imaging and therapy, J. Controlled Release, 2011, 155(3), 344–357 CrossRef CAS PubMed.
- R. A. Sperling and W. J. Parak, Surface modification, functionalization and bioconjugation of colloidal inorganic nanoparticles, Philos. Trans. R. Soc., A, 2010, 368(1915), 1333–1383 CrossRef CAS PubMed.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li and J. Liu, et al., Mesoporous silica nanoparticles in drug delivery and biomedical applications, Nanomedicine, 2015, 11(2), 313–327 CrossRef CAS PubMed.
- J. Y. Choi, T. Ramasamy, S. Y. Kim, J. Kim, S. K. Ku and Y. S. Youn, et al., PEGylated lipid bilayer-supported mesoporous silica nanoparticle composite for synergistic co-delivery of axitinib and celastrol in multi-targeted cancer therapy, Acta Biomater., 2016, 39, 94–105 CrossRef CAS PubMed.
- E. Niemelä, D. Desai, Y. Nkizinkiko, J. E. Eriksson and J. M. Rosenholm, Sugar-decorated mesoporous silica nanoparticles as delivery vehicles for the poorly soluble drug celastrol enables targeted induction of apoptosis in cancer cells, Eur. J. Pharm. Biopharm., 2015, 96, 11–21 CrossRef PubMed.
- E. Niemelä, D. Desai, E. Lundsten, J. M. Rosenholm, P. Kankaanpää and J. E. Eriksson, Quantitative bioimage analytics enables measurement of targeted cellular stress response induced by celastrol-loaded nanoparticles, Cell Stress Chaperones, 2019, 24(4), 735–748 CrossRef PubMed.
- N. Elahi, M. Kamali and M. H. Baghersad, Recent biomedical applications of gold nanoparticles: A review, Talanta, 2018, 184, 537–556 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Quantum dot bioconjugates for imaging, labelling and sensing, Nat. Mater., 2005, 4(6), 435–446 CrossRef CAS PubMed.
- S. Svenson, Dendrimers as versatile platform in drug delivery applications, Eur. J. Pharm. Biopharm., 2009, 71(3), 445–462 CrossRef CAS PubMed.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Dendrimer-based drug and imaging conjugates: design considerations for nanomedical applications, Drug Discovery Today, 2010, 15(5–6), 171–185 CrossRef CAS PubMed.
- S. Boridy, G. M. Soliman and D. Maysinger, Modulation of inflammatory signaling and cytokine release from microglia by celastrol incorporated into dendrimer nanocarriers, Nanomedicine, 2012, 7(8), 1149–1165 CrossRef CAS PubMed.
- P. Anita P, J. Patel, S. Patel Khushbu, B. Deshmukh Aaishwarya and R. Mishra Bharat, A review on drug nanocrystal a carrier free drug delivery, Int. J. Res. Ayurveda Pharm., 2011, 2(2), 448–458 Search PubMed.
- T. Nabekura, T. Hiroi, T. Kawasaki and Y. Uwai, Effects of natural nuclear factor-kappa B inhibitors on anticancer drug efflux transporter human P-glycoprotein, Biomed. Pharmacother., 2015, 70, 140–145 CrossRef CAS PubMed.
- M. Yokoyama, Polymeric micelles as a new drug carrier system and their required considerations for clinical trials, Expert Opin. Drug Delivery, 2010, 7(2), 145–158 CrossRef CAS PubMed.
- Y. Lu and K. Park, Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs, Int. J. Pharm., 2013, 453(1), 198–214 CrossRef CAS PubMed.
- X. Peng, J. Wang, X. Li, L. Lin, G. Xie and Z. Cui, et al., Targeting mast cells and basophils with anti-FcεRIα fab-conjugated celastrol-loaded micelles suppresses allergic inflammation, J. Biomed. Nanotechnol., 2015, 11(12), 2286–2299 CrossRef CAS PubMed.
- Z. Li, L. Yao, J. Li, W. Zhang, X. Wu and Y. Liu, et al., Celastrol nanoparticles inhibit corneal neovascularization induced by suturing in rats, Int. J. Nanomed., 2012, 7, 1163 CAS.
- Z. Li, J. Li, L. Zhu, Y. Zhang, J. Zhang and L. Yao, et al., Celastrol nanomicelles attenuate cytokine secretion in macrophages and inhibit macrophage-induced corneal neovascularization in rats, Int. J. Nanomed., 2016, 11, 6135 CrossRef CAS PubMed.
- Z. Li, X. Wu, J. Li, L. Yao, L. Sun and Y. Shi, et al., Antitumor activity of celastrol nanoparticles in a xenograft retinoblastoma tumor model, Int. J. Nanomed., 2012, 7, 2389 CrossRef CAS PubMed.
- Z. Li, Z. Guo, D. Chu, H. Feng, J. Zhang and L. Zhu, et al., Effectively suppressed angiogenesis-mediated retinoblastoma growth using celastrol nanomicelles, Drug Delivery, 2020, 27(1), 358–366 CrossRef CAS PubMed.
- J. Zhao, D. Luo, Z. Zhang, N. Fan, Y. Wang and H. Nie, et al., Celastrol-loaded PEG-PCL nanomicelles ameliorate inflammation, lipid accumulation, insulin resistance and gastrointestinal injury in diet-induced obese mice, J. Controlled Release, 2019, 310, 188–197 CrossRef CAS PubMed.
- J. Li, Y. Jia, P. Zhang, H. Yang, X. Cong and L. An, et al., Celastrol self-stabilized nanoparticles for effective treatment of melanoma, Int. J. Nanomed., 2020, 15, 1205 CrossRef CAS PubMed.
- S. D. Allen, Y.-G. Liu, T. Kim, S. Bobbala, S. Yi and X. Zhang, et al., Celastrol-loaded PEG-b-PPS nanocarriers as an anti-inflammatory treatment for atherosclerosis, Biomater. Sci., 2019, 7(2), 657–668 RSC.
- L. An, Z. Li, L. Shi, L. Wang and J. Li, Inflammation-targeted celastrol nanodrug attenuates collagen-induced arthritis through NF-κB and Notch1 pathways, Nano Lett., 2020, 20(10), 7728–7736 CrossRef CAS PubMed.
- P. Li, X. Zhou, D. Qu, M. Guo, C. Fan and T. Zhou, et al., Preliminary study on fabrication, characterization and synergistic anti-lung cancer effects of self-assembled micelles of covalently conjugated celastrol–polyethylene glycol–ginsenoside Rh2, Drug Delivery, 2017, 24(1), 834–845 CrossRef CAS PubMed.
- Z. Guo, L. Shi, H. Feng, F. Yang, Z. Li and J. Zhang, et al., Reduction-sensitive nanomicelles: delivery celastrol for retinoblastoma cells effective apoptosis, Chin. Chem. Lett., 2020, 32(3), 1046–1050 CrossRef.
- Y. Zhao, Y. Tan, T. Meng, X. Liu, Y. Zhu and Y. Hong, et al., Simultaneous targeting therapy for lung metastasis and breast tumor by blocking the NF-κB signaling pathway using celastrol-loaded micelles, Drug Delivery, 2018, 25(1), 341–352 CrossRef CAS PubMed.
- Y. Tan, Y. Zhu, Y. Zhao, L. Wen, T. Meng and X. Liu, et al., Mitochondrial alkaline pH-responsive drug release mediated by Celastrol loaded glycolipid-like micelles for cancer therapy, Biomaterials, 2018, 154, 169–181 CrossRef CAS PubMed.
- K. A. Elhasany, S. N. Khattab, A. A. Bekhit, D. M. Ragab and A. O. Elzoghby, Combination of magnetic targeting with synergistic inhibition of NF-κB and glutathione via micellar drug nanomedicine enhances its anti-tumor efficacy, Eur. J. Pharm. Biopharm., 2020, 155, 162–176 CrossRef CAS PubMed.
- F. Shakeel, W. Ramadan, M. S. Faisal, M. Rizwan, M. Faiyazuddin and G. Mustafa, et al., Transdermal and topical delivery of anti-inflammatory agents using nanoemulsion/microemulsion: an updated review, Curr. Nanosci., 2010, 6(2), 184–198 CrossRef CAS.
- X. Qi, J. Qin, N. Ma, X. Chou and Z. Wu, Solid self-microemulsifying dispersible tablets of celastrol: Formulation development, charaterization and bioavailability evaluation, Int. J. Pharm., 2014, 472(1–2), 40–47 CrossRef CAS PubMed.
- M. Guo, D. Qu, Y. Qin, Y. Chen, Y. Liu and M. Huang, et al., Transferrin-functionalized microemulsions coloaded with coix seed oil and tripterine deeply penetrate to improve cervical cancer therapy, Mol. Pharmaceutics, 2019, 16(12), 4826–4835 CrossRef CAS PubMed.
- Q. Zhang, X. Tian and X. Cao, Transferrin-functionalised microemulsion co-delivery of β-elemene and celastrol for enhanced anti-lung cancer treatment and reduced systemic toxicity, Drug Delivery Transl. Res., 2019, 9(3), 667–678 CrossRef CAS PubMed.
- H. Zhao, M. Chen, Z. Zhao, L. Zhu and S. Yuan, A multicomponent-based microemulsion for boosting ovarian cancer therapy through dual modification with transferrin and SA-R 6 H 4, Drug Delivery Transl. Res., 2020, 1–14 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |