
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmunopharmacotherapeutic advancements in addressing methamphetamine abuse
Jinny Claire
Lee
 and
Kim D.
Janda
*
and
Kim D.
Janda
*
Department of Chemistry, Department of Immunology and Microbial Science, The Skaggs Institute for Chemical Biology, The WIRM Institute for Research & Medicine, The Scripps Research Institute, 10550 North Torrey Pines Rd, La Jolla, CA 92037, USA. E-mail: kdjanda@scripps.edu
First published on 8th October 2020
Abstract
Methamphetamine (METH) is an illicit psychostimulant that is known to account for substance abuse disorders globally, second only to opioids, yet has no approved pharmacotherapies. Traditional therapies employ small molecule agonists or antagonists for substance use disorders or overdose reversal by targeting drug-specific receptors in the brain. However, the comprehensive mechanism of METH on multiple sites within the central nervous system (CNS) implies its receptors lack the high affinity and specificity required for an “ideal” drug target. The alternative to pharmacotherapies is to sequester abused drugs in the periphery, effectively eliminating the effects from CNS receptor occupation through pharmacokinetic antagonism. This review presents updates on immunopharmacotherapeutic advancements in addressing methamphetamine abuse by focusing on the cultivation of research optimization strategies regarding hapten chemistry, carrier proteins, and adjuvants implemented in active immunization. Furthermore, we discuss necessary developments for each component of active immunopharmacotherapies and the future of active vaccines in treating METH use disorder.
 Jinny Claire Lee | Jinny Claire Lee obtained her BSE degree in Bio-Convergence from Yonsei University in 2018, where she focused on Alzheimer disease research by implementing chemical biology tools to synthesize peptides for incorporation into a screening system, in order to discover diagnostic and therapeutic small molecules. She is currently pursuing her PhD in Chemical Biology under the supervision of Dr. Kim D. Janda at The Scripps Research Institute. Her doctoral research interest is focused on the development of immunopharmacotherapies for drugs of abuse by working at the interface of medicinal chemistry, molecular biology, and immunology. |
 Kim D. Janda | Prof. Kim D. Janda is the Ely R. Callaway, Jr Chaired Professor in the Departments of Chemistry, Immunology and Microbial Science at The Scripps Research Institute and is the Director of the Worm Institute of Research and Medicine. He is well known for his bridging of the fields of chemistry and immunology, where he has shown how antibodies could be transformed from simple binding entities to catalysts. He pioneered treating drug addiction through immunopharmacotherapy, the creation of synthetic diversity methods, including phage display, and small molecule DNA encoding methodologies. He has published over 550 peer-reviewed papers. |
Introduction
Illicit compounds abused in substance use disorders are lipophilic molecules capable of promptly penetrating the blood–brain barrier in order to activate the mesocorticolimbic system, thus extending dopamine activity at the nucleus accumbens and reinforcing drug dependence.1–3 Methamphetamine (METH) is a potent psychostimulant that is able to affect a multitude of neurotransmitters,4,5 such as dopamine, norepinephrine, serotonin, γ-aminobutyric acid, and histamine, through the inhibition of vesicular monoamine transporters and dysregulation of transmitters at nerve terminals.6–9 In addition to the drug's extensive effects on the central nervous system (CNS), it is also capable of eliciting peripheral effects due to its sympathomimetic nature stimulating stress response processes,5,10 effectively increasing body temperature and heart rate. Acute METH exposure can also disrupt the blood–brain barrier, contributing to structural and functional neural anomalies.9,11,12 Due to METH's comprehensive and complicated pharmacology involving numerous neurotransmitters, traditional therapeutic approaches for drug abuse through receptor modulation have failed to emerge.In the absence of effective pharmacotherapies, the only modern remedies available for METH addiction are psychosocial treatments consisting of cognitive behavioral therapy, residential rehabilitation, contingency management, exercise, matrix model, or repetitive transcranial magnetic stimulation.6,13,14 While contingency management and cognitive behavioral therapy have shown the most promise,10,14,15 these interventions cannot protect the patient from the compulsion to use METH outside of counseling and some note the difficulty of implementing this approach as a standalone treatment due to high relapse rates.16–19 Thus, there is a continuing, concerted effort to establish a therapeutic approach targeting the physiological repercussions of substance abuse, effectively augmenting the psychological benefits obtained from psychosocial remedies. Although there are no approved or efficacious pharmacotherapies for METH dependence,8,14 some pharmaceuticals have been proposed to attenuate the severe psychological effects of METH use.7,20
Clinical trials examining dopaminergic agents, serotonergic agents, γ-aminobutyric acid agents, glutamatergic agents, cholinesterase inhibitors, benzoquinolizine derivatives, opioid agents, and nicotinic agents have been observed with varying results in regards to their effects on METH users.7,13,21 Dopamine agonists, modafinil22–26 and bupropion,27–34 have demonstrated beneficial responses in METH-dependent patients, while naltrexone,35–39 an opioid antagonist, seemed to reduce amphetamine's reinforcement effects. Despite the promising start, naltrexone revealed conflicting results with some studies showing no differences in METH use between treatment and placebo groups.40,41 Modafinil demonstrated potential benefits in alleviating the physiological side effects instigated by METH abuse, such as high blood pressure, but the compound lacked clear evidence in its ability to reduce METH dependence.23 As for bupropion, efficacy comparisons between light and heavy METH users showed the small molecule could aid in reducing drug usage, but only for members within the former group.30
While some pharmaceutical agents demonstrate promising results, their clinical studies consist of small, biased sample sizes and study protocol completion is often low. These factors contribute to apprehension towards recommending pharmacotherapies as effective measures against METH use disorder. In addition to obstacles in research, pharmacological treatments for substance abuse commonly create issues related to high cost, limited availability, compliance difficulties, medication abuse, and relapse to addiction after discontinued treatment.42–44 Since small molecules target the same neural pathways as the abused drug, these therapeutic tools could potentially cause addiction as well.6,45,46 With no promising pharmaceutics and copious potential impediments, the alternative is to obstruct the abused drug peripherally, prior to it acting centrally.
Immunopharmacotherapy accomplishes this alternative approach by using high affinity antibodies to sequester the abused drug while it is in the circulatory system.47 Antibody therapy possesses the potential to promote enduring drug abstinence while producing less of the aforementioned limitations exhibited by pharmacotherapy, since creation of the antibody–drug complex obviates drug interaction with the CNS. While small molecules are routinely used to supplement psychosocial therapies for substance use disorders, their various limitations and potential side effects cause concern. Immunopharmacotherapy can augment psychosocial interventions when administered throughout a comprehensive addiction recovery plan and maintain multiple advantages over the best pharmacotherapies.
One such advantage is that immunopharmacotherapy vaccines could aid in increasing patient compliance since treatment would consist of a few injections over a long period of time, rather than daily or weekly administration of pharmaceuticals.5 Also, antidrug antibodies cannot traverse the blood–brain barrier due to their large size, effectively limiting off-target effects that are prevalent in traditional pharmacotherapies.42,48 Principally, immunopharmacotherapy can assist in relapse prevention, should patients self-administer METH during recovery, by circulating antibodies targeting and sequestering free drug molecules, which would diminish METH's adverse effects and reduce reinforcing drug responses.5,17 The high specificity, minimal side effects, absence of potential abuse and addiction, and enduring protection against the drug of abuse contributes to the major therapeutic benefit immunopharmacotherapy maintains over traditional pharmacotherapies.
During immunopharmacotherapy, anti-METH antibodies can be generated by either active immunization through vaccine stimulation or passive immunization, where engineered monoclonal antibodies are obtained from hybridoma generation or phage display.48,49 In order to determine which mode of immunization is suitable for the patient, the individual's risk of recidivism and overall health is important to consider. For high risk individuals or patients with compromised immune systems, passive immunization is beneficial due to the fact that it provides immediate protection against the drug of abuse and can be used in acute situations, such as a drug overdose.5,6 The antibody can be administered to the patient's needs by implementing more frequent and concentrated doses during difficult times throughout recovery. This method of therapy is also advantageous because antibody formulation within the administered vaccine can be controlled by quality and homogeneity, which is crucial for METH users who are not able to produce adequate levels of antibody titers due to immune suppression from long-term drug usage.50 Passive immunization can diminish METH self-administration,51 reduce locomotor side effects,52–54 and disrupt drug discriminative stimuli.55 Although passive administration is advantageous for these numerous reasons, it is limited as a therapeutic tool since it is expensive to produce and effects are more transient, complicating patient compliance during recovery.
On the other hand, active immunization is less costly to manufacture and can work as a prophylactic treatment since its duration of effect is longer lasting than monoclonal antibodies.5 For patients capable of abstaining from METH until significant antibody titers can be generated, active administration would be a viable, cost-effective, and beneficial option. Active immunization can generate an immune response against the drug of abuse through repeat administration of a drug-like hapten conjugated to a carrier protein, where booster injections prompt increasingly rapid responses due to the long-lasting immunological memory formed from the hapten–protein conjugates. Immediate inhibition of psychological and physiological drug effects provided by passive immunization is valuable, but active immunization is promising due to its potential to inhibit drug reinforcement and prevent relapse by individuals motivated to quit.
While active immunization is ideal, preclinical investigations implementing anti-METH vaccines convey contradictory results. These issues emerge from the fact that active vaccines contain several components consisting of the carrier protein, hapten, hapten–protein linkage, adjuvant, and administration route. Each element can contribute to drastic differences in research results and require careful consideration during active vaccine formulation. Thus, it is imperative to review the various advancements regarding each of these components. This review will focus on immunopharmacotherapeutic advancements against methamphetamine abuse by examining the consequences of hapten chemistry, carrier proteins, and adjuvants in active vaccine formulations. Here we consolidate the enduring efforts to establish and implement the use of an effective and clinically approved therapy for an expanding epidemic.
Conjugate vaccine formulation, function, and implementation
The low molecular weight, limited chemical epitopes for antibody interaction, and structural similarity to endogenous monoamines makes METH a challenging compound for immunopharmacotherapy.19,56 With a molecular weight of 149 Da, METH lacks immunostimulatory abilities because immune responses are only capable of eliciting antibodies against molecules that are greater than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da.45 Therefore, small molecule haptens can be conjugated to a larger immunogenic carrier to provide additional T cell epitopes for antibody production against the abused drug.20,42,45 The hapten–protein conjugate can be formed by covalently attaching the hapten, a chemical derivative of the abused drug,57 with an immunologically inert linker arm. Linker design and placement on the hapten along with the choice of carrier protein can affect hapten molecular orientation and density on the protein surface, which are critical factors that require careful management for optimal and reproducible research results. Subsequently, the conjugate is purified to remove free haptens and mixed with appropriate adjuvants, which help boost the innate immune response in order to generate higher immunoglobulin G (IgG)-mediated antibody titers.5,20 IgG antibodies consist of two potential antigen binding sites;8 therefore, antibody–drug complexes can simultaneously interact with one or two drug molecules. The overall success of active immunization vaccines is determined by antibody titer, concentration, specificity, and affinity.
000 Da.45 Therefore, small molecule haptens can be conjugated to a larger immunogenic carrier to provide additional T cell epitopes for antibody production against the abused drug.20,42,45 The hapten–protein conjugate can be formed by covalently attaching the hapten, a chemical derivative of the abused drug,57 with an immunologically inert linker arm. Linker design and placement on the hapten along with the choice of carrier protein can affect hapten molecular orientation and density on the protein surface, which are critical factors that require careful management for optimal and reproducible research results. Subsequently, the conjugate is purified to remove free haptens and mixed with appropriate adjuvants, which help boost the innate immune response in order to generate higher immunoglobulin G (IgG)-mediated antibody titers.5,20 IgG antibodies consist of two potential antigen binding sites;8 therefore, antibody–drug complexes can simultaneously interact with one or two drug molecules. The overall success of active immunization vaccines is determined by antibody titer, concentration, specificity, and affinity.
Once efficient and effective antibody generation is achieved, individuals exposed to the specific drug of abuse will theoretically be able to avoid the compound's reinforcing psychoactive properties. This phenomenon is fostered by pharmacokinetic antagonism43,58 in which these therapeutic antibodies are capable of reducing the speed of CNS penetration by illicit drugs—primarily by sequestering the compounds within the circulatory system,42,43,45 effectively altering the drug concentration gradient and producing a cascade effect that reduces the amount of drugs at various sites of action across multiple organ systems, most significantly the brain (Fig. 1).6,42 Some studies support pharmacokinetic antagonism as active immunization's primary mechanism of action by demonstrating how immunized murine models initially increased self-administration of drugs in order to compensate for the lack of psychoactive effects elicited by the rapid isolation of METH in the periphery, which is a behavioral response observed in the use of some small molecule antagonists.59–61
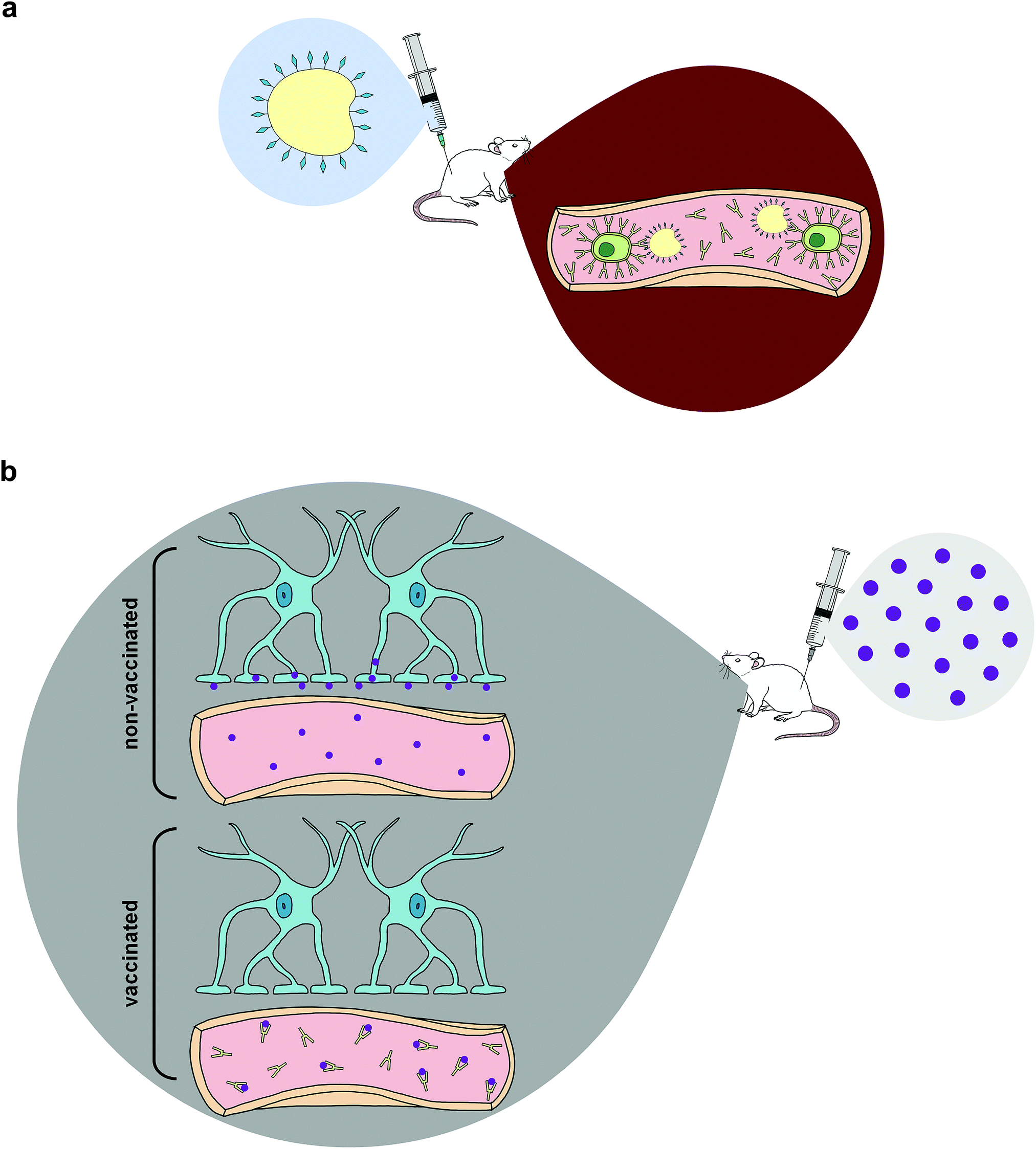 | ||
| Fig. 1 Immunopharmacotherapy mechanism (a) The animal is vaccinated with the hapten–protein conjugate. This immunogen can interact with B-cells in the blood to produce antibodies against the drug of abuse. (b) When the drug of abuse is injected (purple) into the vaccinated animal, the antibodies present in the periphery are able to bind and sequester the free drug before it can pass through the blood–brain barrier and cause psychostimulatory effects. In non-vaccinated animals, there are no such antibodies present, thus drug molecules can freely pass through the blood–brain barrier and interact with receptors in the brain. | ||
Despite potential increases in self-administration, several studies have found reduced METH concentrations in the brain after administering drugs to vaccinated animals and a consequential elevation in drug concentrations in sera,56,62–64 since ingested METH would be confined to the periphery within vaccinated groups. Although a limitation of active immunization arises from the initial waiting period required to generate protective antibody titers, studies show METH use during the immunization period does not negatively impact the antibody affinity or titer elicited in response to the vaccine.65,66 This is crucial since individuals attempting to undergo drug cessation treatment will potentially have a lapse and use drugs of abuse while developing appropriate immune responses to active immunopharmacotherapies.
In addition to observing the physiological consequences by which active vaccines function, researchers can study the behavioral ramifications of drug use during immunopharmacotherapy. Due to the fact that METH increases locomotor activity, this assessment is easily examined using a commercially available apparatus. Effective vaccination is determined by the ability to inhibit hyperlocomotion after moderate drug challenge, as well as prevent METH-induced stereotypic responses.56 Stereotypy is repetitive movement, such as head bobbing, sniffling, or aggressive face-cleaning, that often presents in response to high concentrations of drug administration.56,67–70 Other means to evaluate the addictive properties of METH can be achieved by implementing conditioned place preference or operant self-administration, where both trials can assess acquisition and reinforcement behaviors.5,71 In conditioned place preference, drug administration and vehicle are paired with different contexts and animals that spend more time in the drug-paired context are alleged to experience drug reward conditions. Additional functional analyses include body temperature measurements, since METH use can contribute to hyperthermia. These functional studies allow for researchers to cultivate a more comprehensive understanding of how specific active immunopharmacotherapeutic formulations can impact behavioral models of addiction or relapse.
Ultimately, an effective immunopharmacotherapy should exhibit no adverse side effects in murine models, while also producing copious amounts of high affinity antibodies that specifically interact with the native drug of interest, rather than inactive drug metabolites. In order to achieve these conditions, vaccine formulation requires an optimized hapten, immunogenically inert linker, suitable carrier protein, and robust adjuvant. Hence, exhaustive synthetic organic chemistry is required to obtain such ideal antigens for potential active immunopharmacotherapies.
Consequences of methamphetamine hapten chemistry
Considering the structure of METH and potential positions for linker insertion, anti-METH haptens can be classified into three categories based on molecule modification sites. Immunogenic protein conjugation can be achieved via either the phenyl,64–66,72–74 methyl,56 or N-methyl62,63,75–81 substituents of the drug. Although most studies aim to conserve the remaining overall structure of METH when modifying a single substituent, some design haptens that mimic METH and are structurally divergent from the original compound.75 While the native structure is not preserved, such haptens have still produced high affinity anti-METH antibodies (Table 1, vaccine 15).| Mod | Vaccine | Hapten structure | Carrier protein | Adjuvant | Admin | Vaccinations/overall time | Functional tests | Results | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Abbreviations: mod, modification; admin, administration; ref, reference; SC, subcutaneous; IP, intraperitoneal; IM, intramuscular; KLH, keyhole limpet hemocyanin; TT, tetanus toxoid; DT, diphtheria toxoid; OVA, ovalbumin; BSA, bovine serum albumin; CpG ODN 1826, CpG oligodeoxynucleotide; MPLA, monophosphoryl Lipid A. | |||||||||
| Phenyl | 1 |
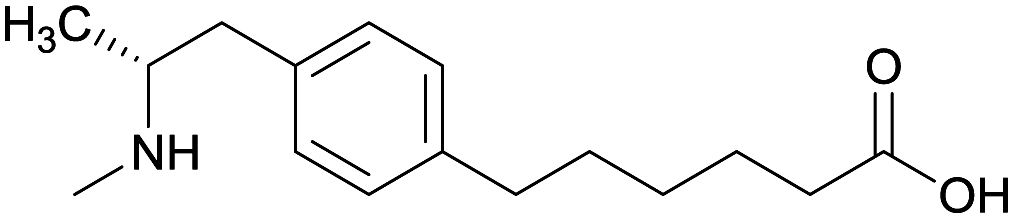
|
KLH | Alum | SC | 3 injections/6 weeks | Locomotor activity | No differences between METH-challenged and non-challenged groups in generating antibody titer and affinity. Failed to attenuate hyperlocomotion | 65 |
| 2 |
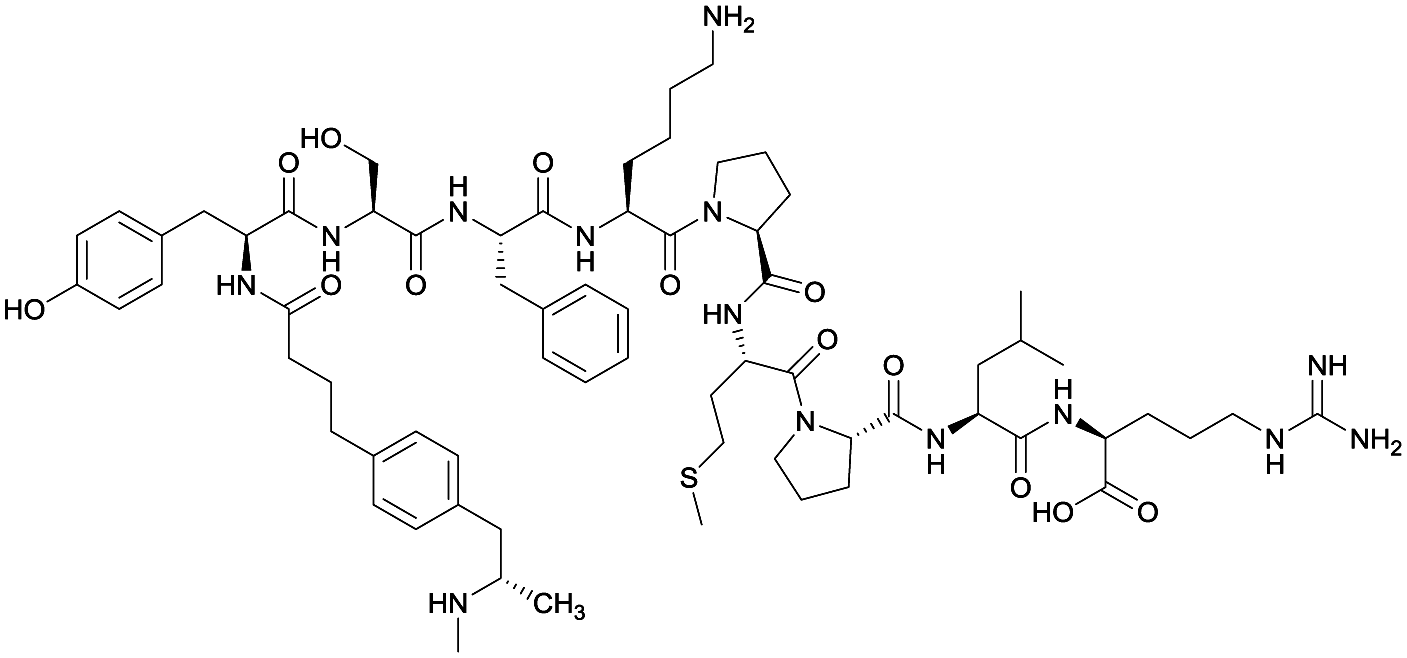
|
None | EP54 (peptide) | SC and IP | 5 injections/5 weeks | Self-administration | Antibody titers were highest in vaccine 3, then vaccine 5, followed by similar levels generated for vaccine 2 and 4. The antibodies generated for vaccine 3 showed high specificity. Self-administration studies showed compensatory effects in vaccinated animals, leading to increased drug intake | 72 | |
| 3 |
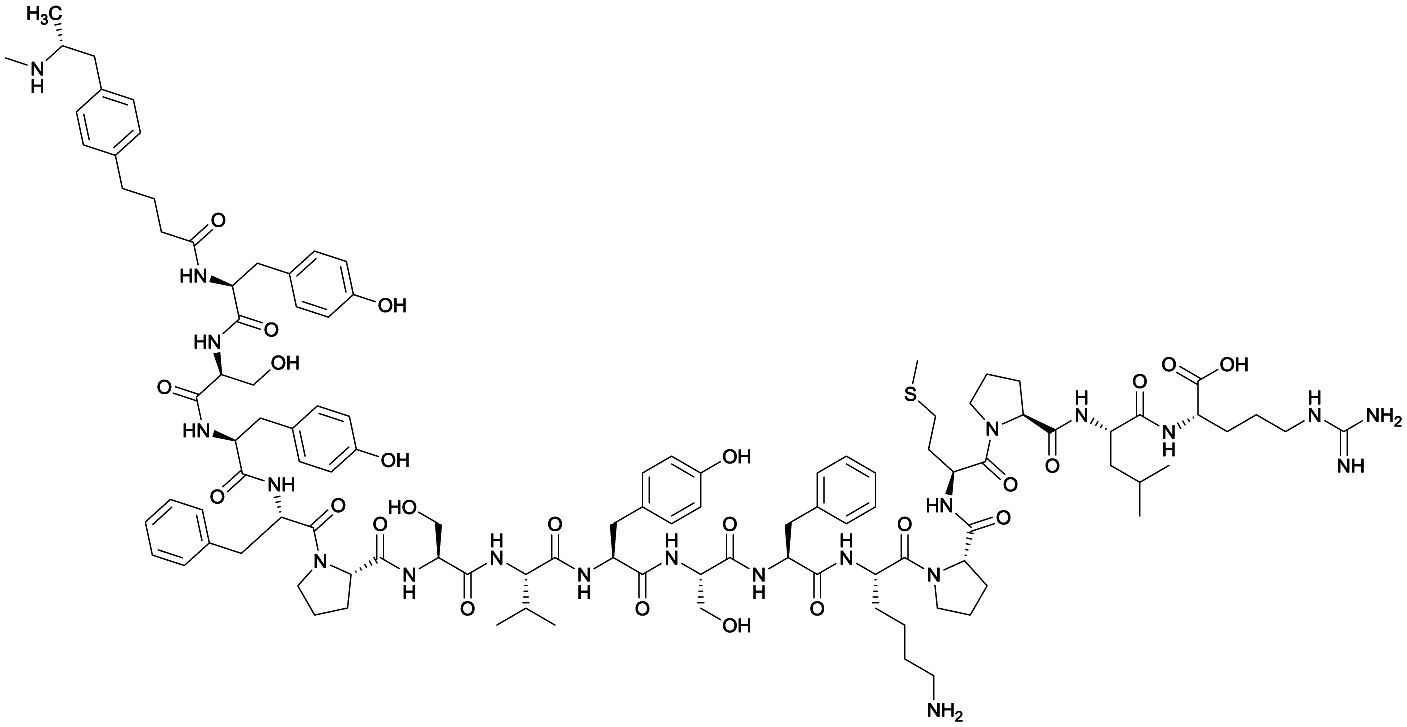
|
TT | |||||||
| 4 |
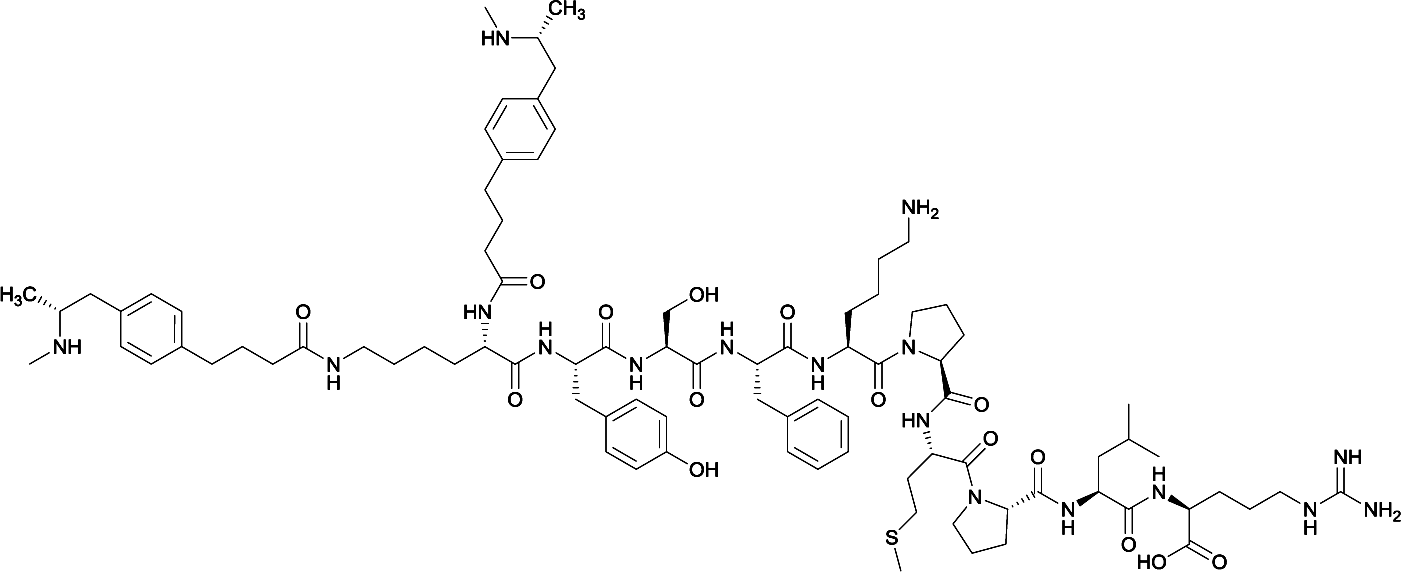
|
None | |||||||
| 5 |
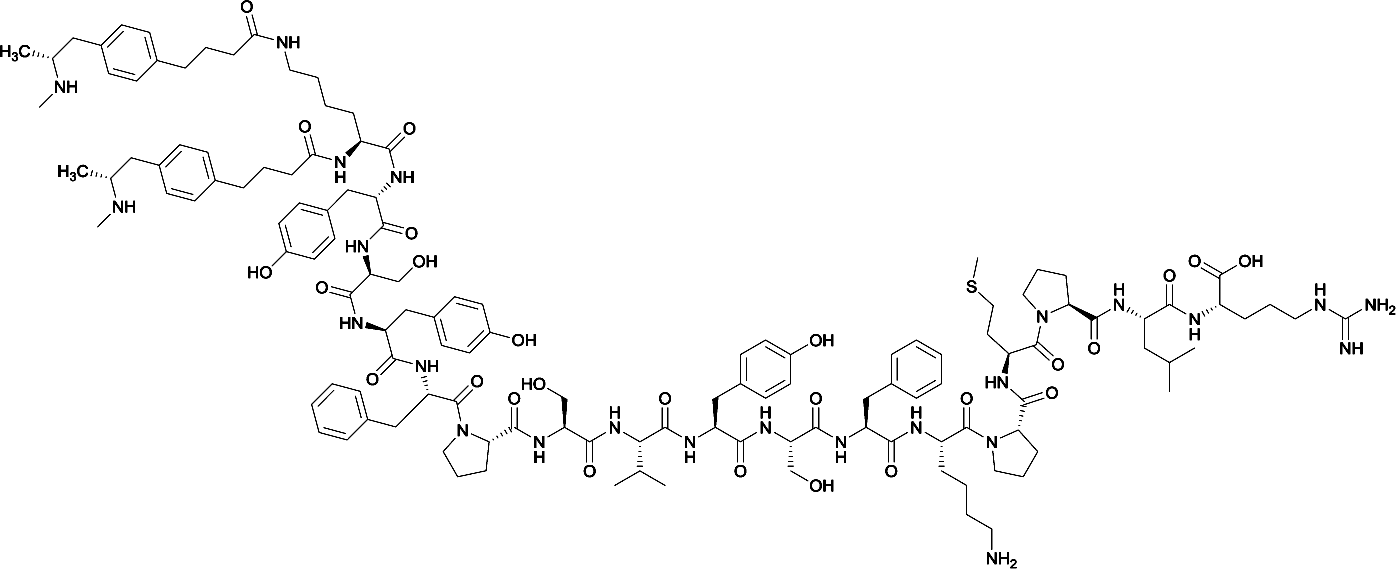
|
TT | |||||||
| 6 |
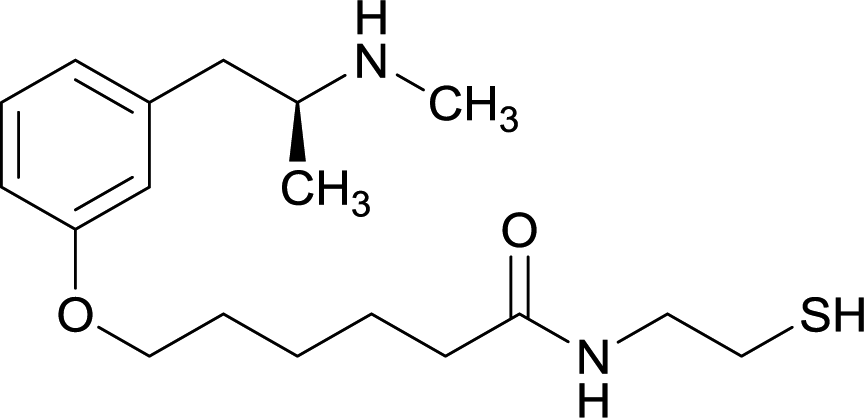
|
OVA and BSA | Freund's complete adjuvant | SC | 3 injections/6 weeks | None | Vaccine 6 produced a significantly higher immune response by providing high epitope densities on maleimide-activated proteins. | 73 | |
| 7 | Same structure as vaccine 6 | KLH | Alhydrogel | SC | 4 injections/12 weeks | Food maintained behaviour | The vaccine reduced effects induced by high doses of METH through high affinity antibodies | 66 | |
| 8 | Same structure as vaccine 6 | KLH | Alhydrogel vs. Sigma Adjuvant System vs. GLA-SE | IM | 3 injections/9 weeks | None | GLA-SE produced double the antibody concentration with enhanced affinities against METH. Significantly, these results were sustained without boosts | 74 | |
| 9 | Same structure as vaccine 6 | KLH | Sigma Adjuvant System | SC | 4 injections/15 weeks | Blood–brain biodistribution | Vaccine 10 showed significant increases in METH sera concentrations in vaccinated animals and decreases in brain concentrations, compared to passive immunization | 64 | |
| 10 |
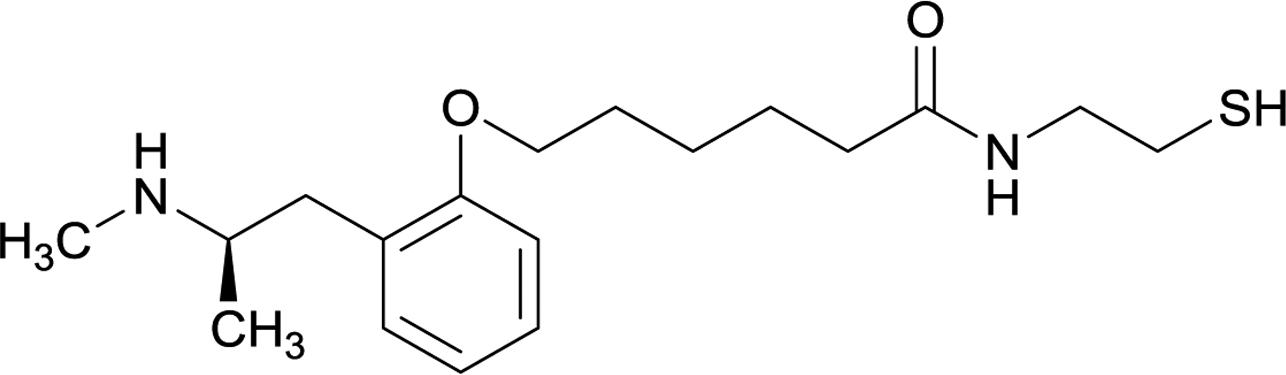
|
||||||||
| Methyl | 11 |
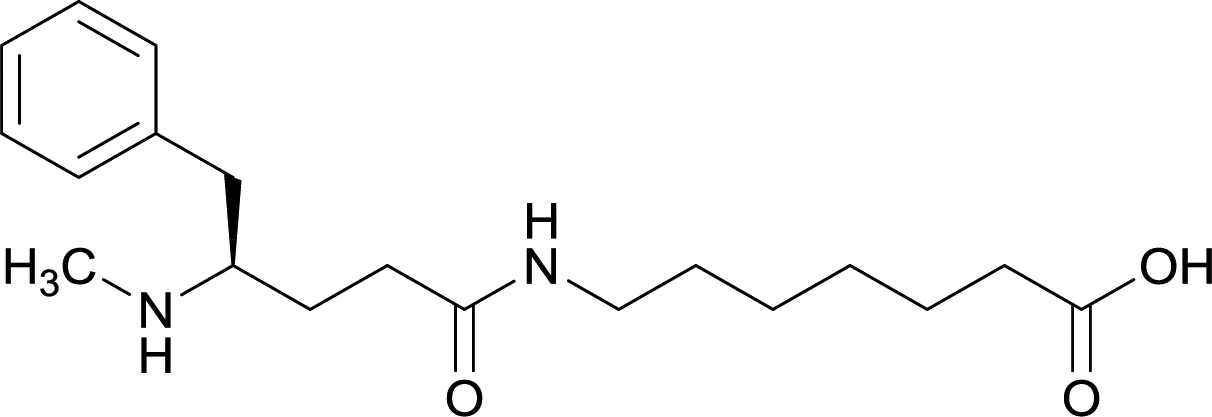
|
TT | Alum and CpG ODN 1826 | IP | 3 injections/4 weeks | Locomotor activity, blood–brain biodistribution, and lethality challenge | All stereoisomers generated antibodies that could affect blood–brain drug partitioning. However, vaccine 12 produced antibodies with 10-fold higher affinities, compared to single isomers and racemate. Only vaccine 12 effectively attenuated METH-induced hyperlocomotion. Lethality studies for vaccine 11, 13, and 12 exhibited survival rates of 16.7%, 33.3% and 83.3%, respectively | 56 |
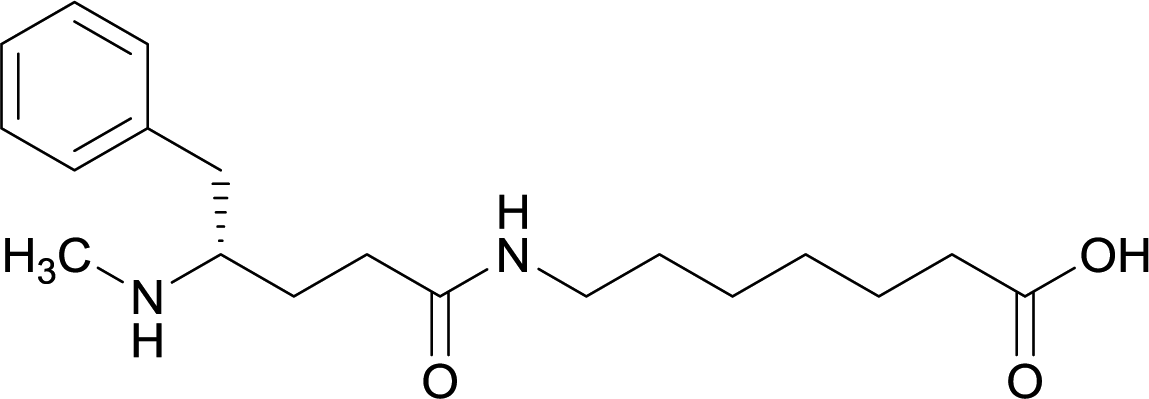
| 12 |
|
||||||||
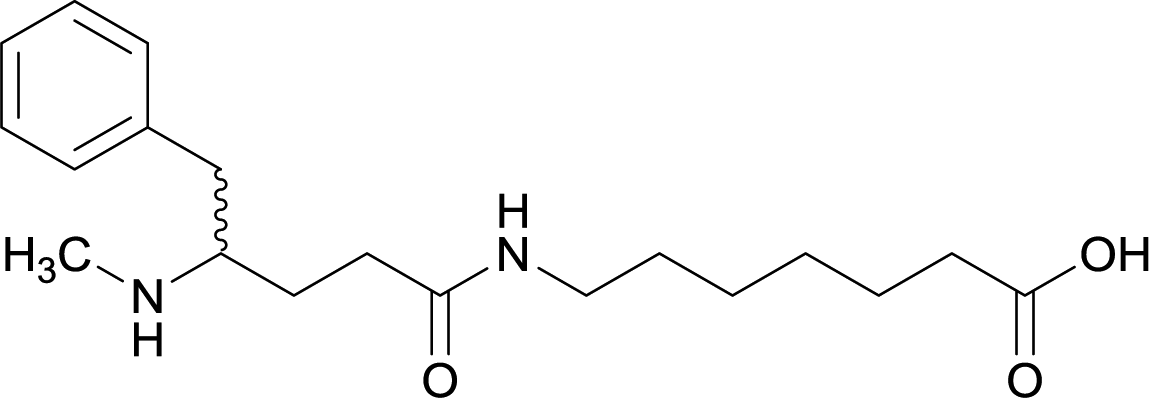
| 13 |
|
||||||||
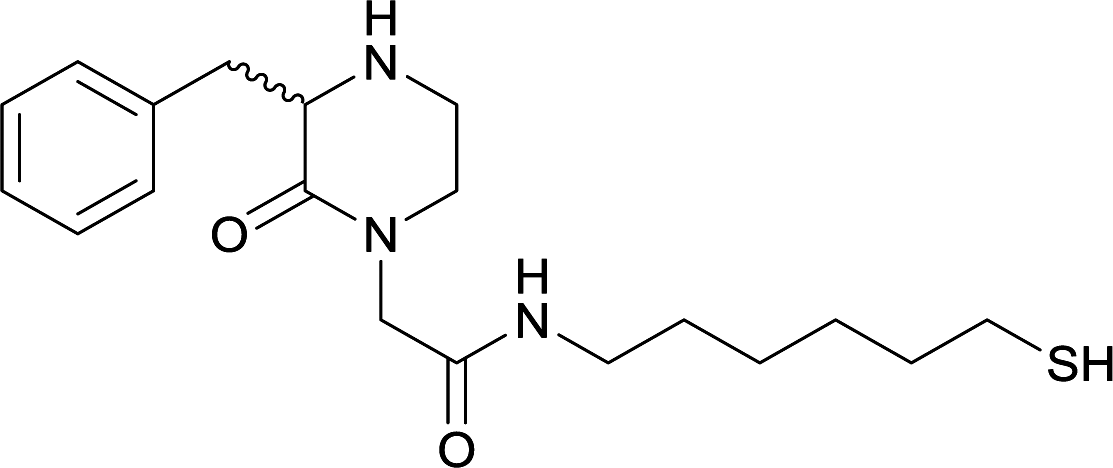
| N-methyl | 14 |
|
KLH or BSA | Sigma Adjuvant System | IP | 3 injections/5 weeks | None | Vaccines 15, 18, and 19 were able to successfully induce increased levels of high affinity antibodies in murine models, with vaccine 15 and 19 producing antibodies with specificities toward METH and amphetamine. Vaccine 18 and 19 produced the highest titers | 75 |
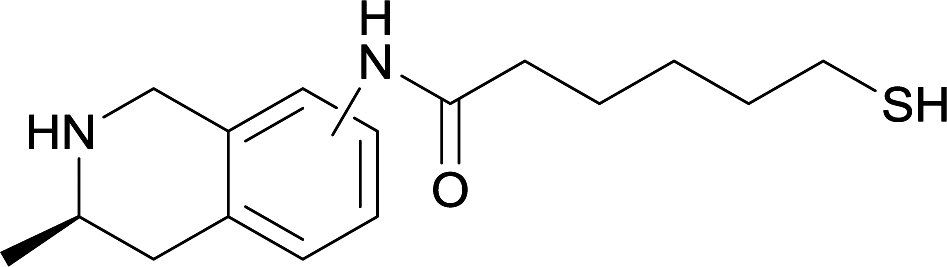
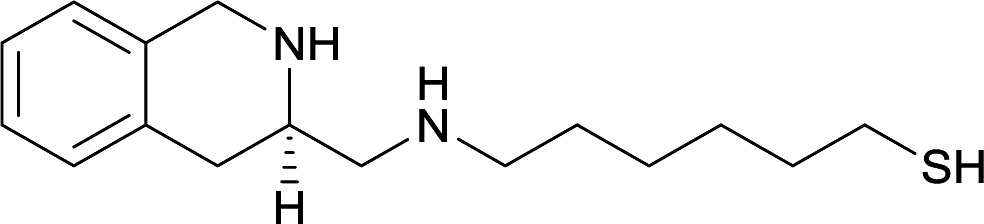
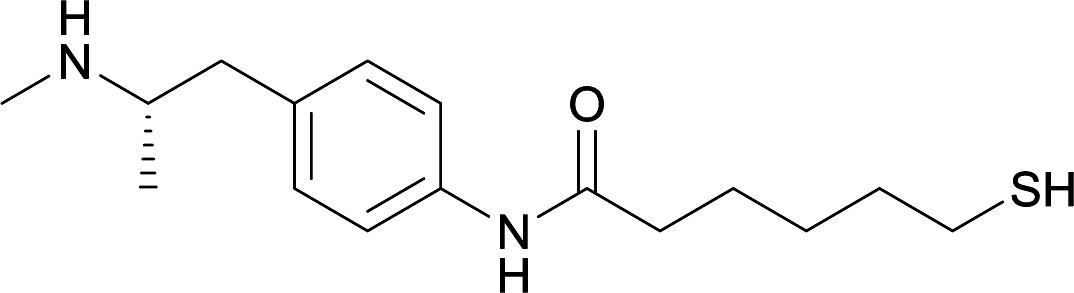
| 15 |
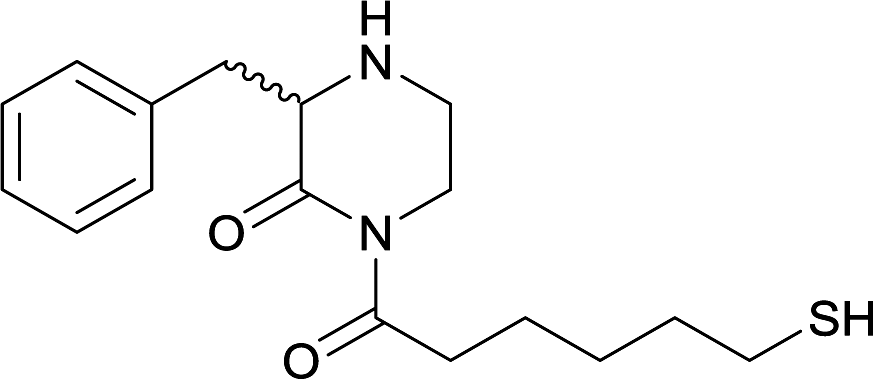
|
||||||||
| 16 |
|
||||||||
| 17 |
|
||||||||
| 18 |
|
||||||||
| 19 |
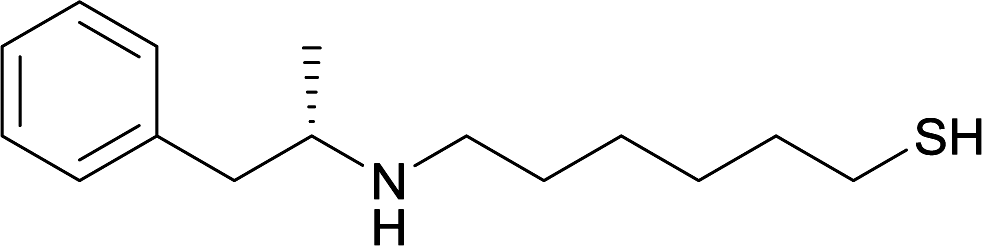
|
||||||||
| 20 | Same structure as vaccine 19 | KLH | Sigma Adjuvant System | SC and IP | 4 injections/9 weeks | Thermoregulation, wheel activity, locomotor activity, and blood–brain biodistribution | The vaccine attenuated METH-induced thermoregulatory and locomotor responses. Pharmacokinetics showed the vaccine produced higher drug concentrations in sera and lower METH presence in the brain after exposure to drugs | 62 | |
| 21 | Same structure as vaccine 19 | KLH | Sigma adjuvant system | SC and IP | 3 injections/5 weeks or 5 injections/13 weeks | Self-administration and acute challenge | The vaccine attenuated self-administration at 0.1 mg per kg. Plasma METH concentrations were increased in vaccinated groups after drug challenges | 76 | |
| 22 |
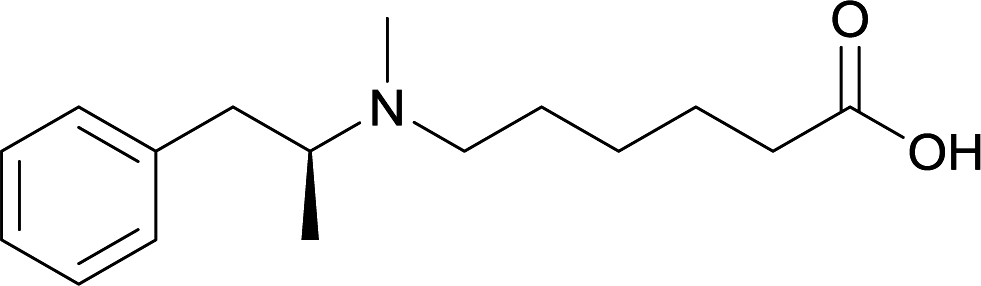
|
DT and BSA | Lipid Tucaresol | SC | 3 injections/5 weeks | None | Vaccines with lipid tucaresol induced antibodies with higher specificity | 77 | |
| 23 | Same structure as vaccine 22 | DT | Sigma adjuvant system | SC | 3 injections/5 weeks | Locomotor activity | Vaccines 24 and 25 produced antibodies with comparable affinities, but vaccine 25, containing peptidic linkers, enhanced antibody concentration. Vaccine 29 elicited higher affinity antibodies, but produced drastically lower titers. Thus, the vaccine was optimized by changing the carrier protein and adjuvant cocktail, which generated antibodies with exceptionally high titers and abilities to attenuate METH-induced locomotor effects | 78 | |
| 24 |
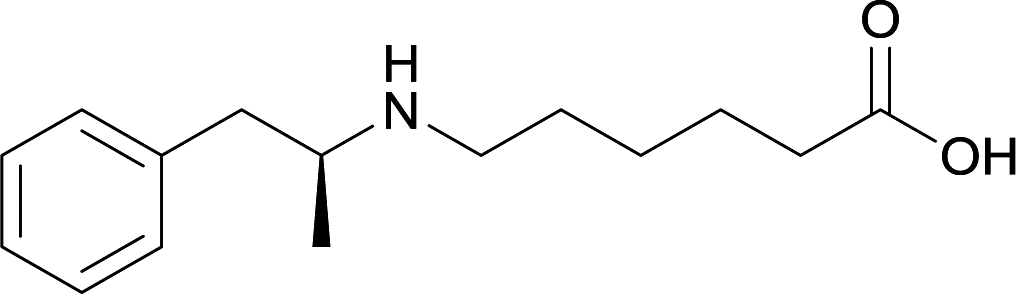
|
||||||||
| 25 |
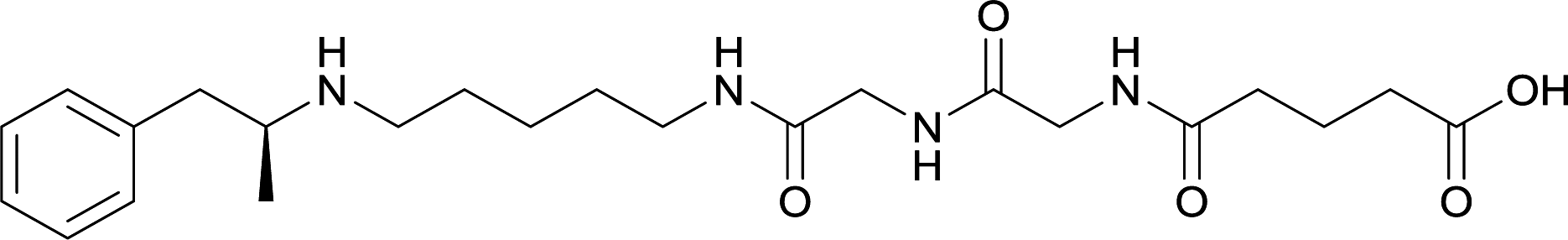
|
||||||||
| 26 |
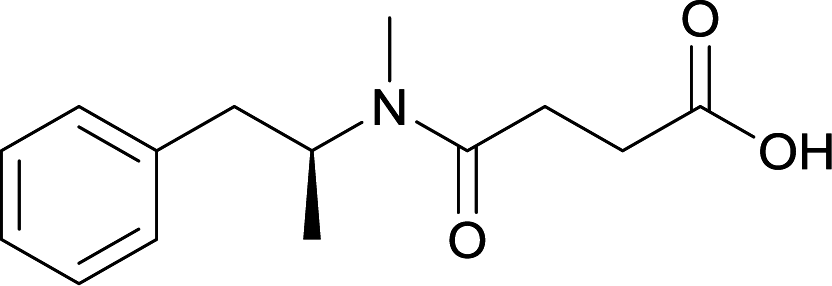
|
||||||||
| 27 |
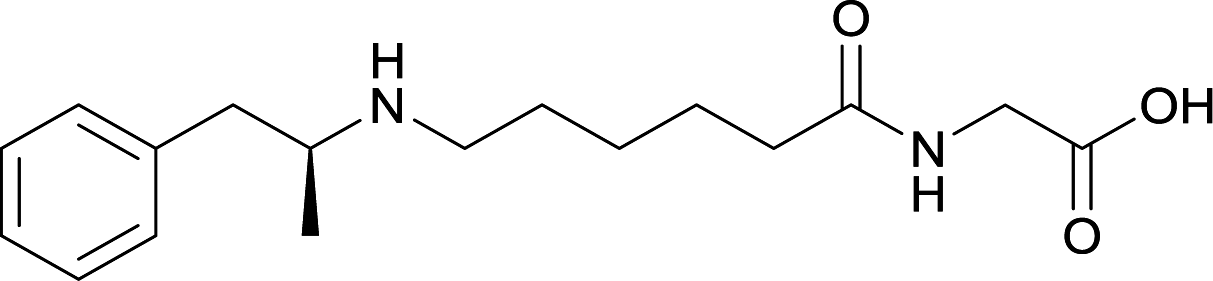
|
||||||||
| 28 |
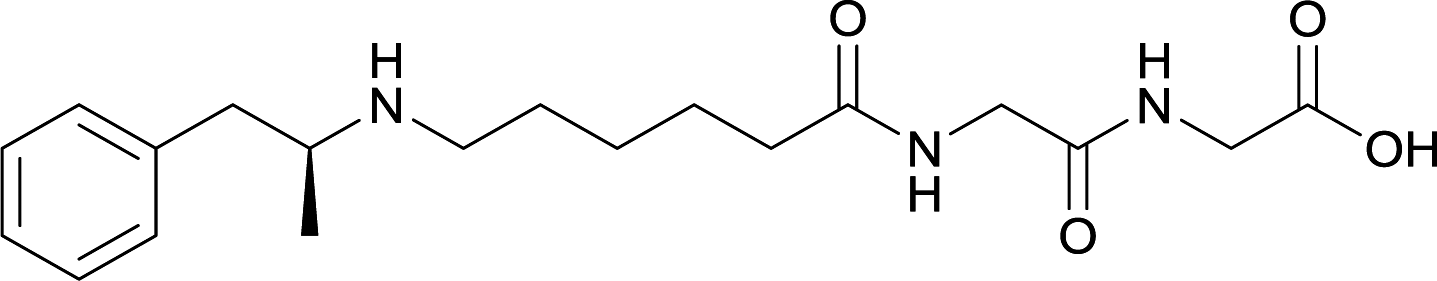
|
||||||||
| 29 |
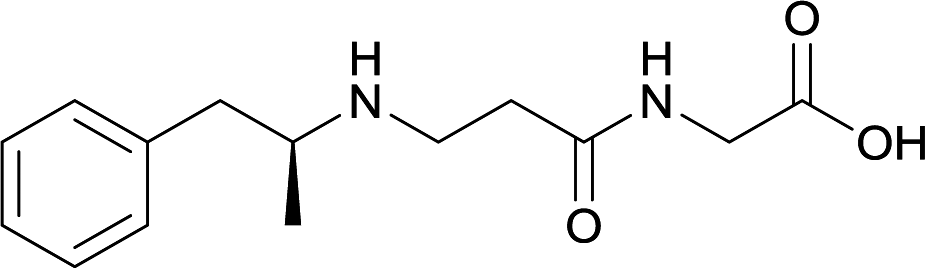
|
||||||||
| 30 | Same structure as vaccine 29 | TT | Alum and CpG ODN 1826 | 3 injections/4 weeks | |||||
| 31 |
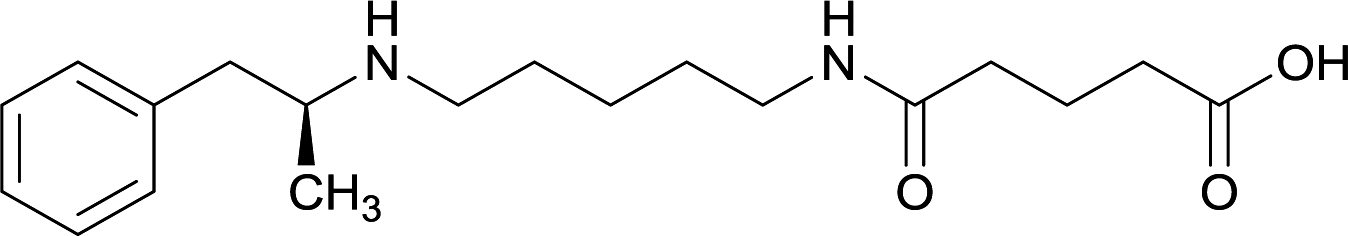
|
KLH or TT | Alum and CpG ODN 1826 | IP | 3 injections/4 weeks | Blood–brain biodistribution | Vaccines 32, 33, 34, and 35 induced antibodies with increased drug affinity, but lower concentrations than control vaccine 31. Since vaccine 35 generated the highest titers, vaccine 36 and 37 were probed for antibody effects. Both elicited antibodies with similar affinities to vaccine 35, but generated increased titers. Vaccine 36 was the most efficient in attenuating drug CNS interaction | 79 | |
| 32 |
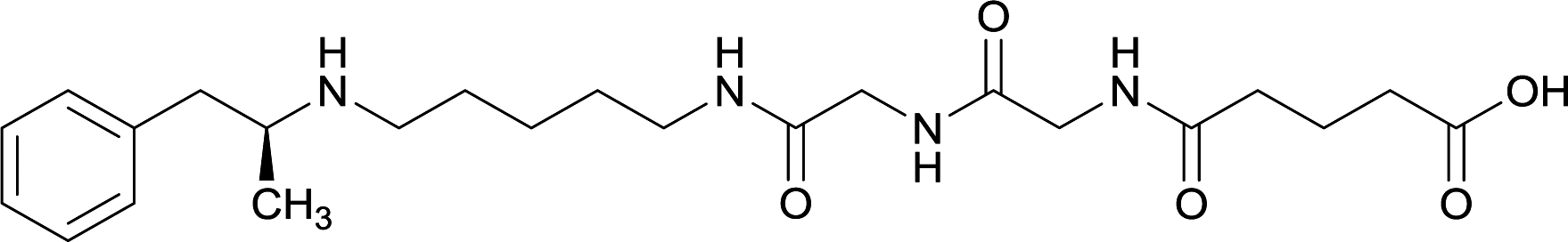
|
||||||||
| 33 |
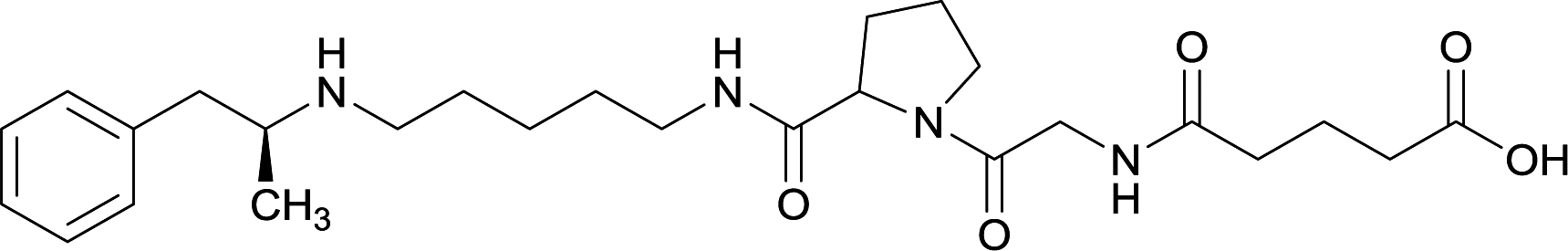
|
||||||||
| 34 |
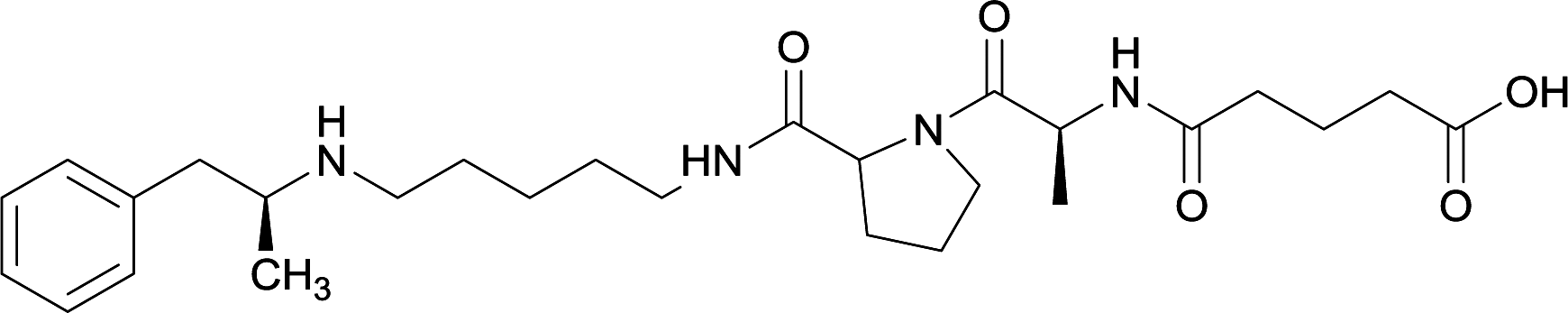
|
||||||||
| 35 |
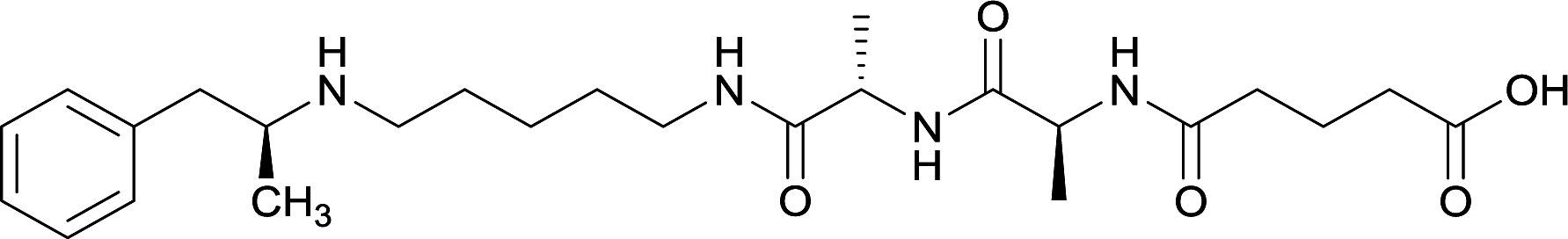
|
||||||||
| 36 |
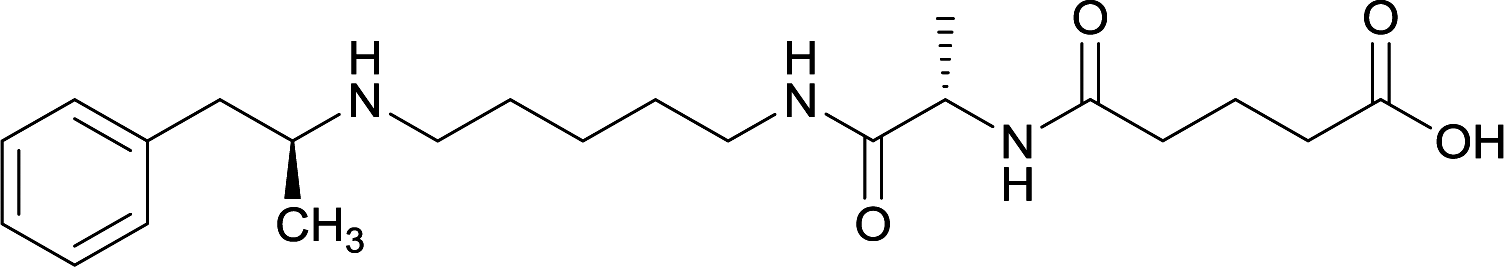
|
||||||||
| 37 |

|
||||||||
| 38 |
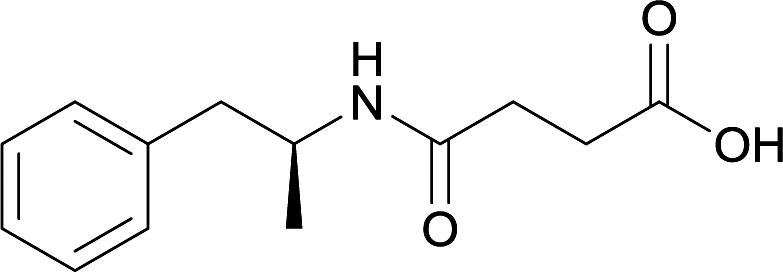
|
KLH | MPLA | SC | 3 injections/20 weeks | Locomotor activity and conditioned place preference | The vaccine generated antibodies at high titers, reduced drug-conditioned approach behaviours, and attenuated METH-induced locomotion at varying doses | 80 | |
| 39 | same structure as vaccine 38 | TT | Alum | IM | 2 injections/3 weeks | conditioned place preference | The vaccine produced higher and sustained antibody concentrations that decreased drug entry into the brain and inhibited METH acquisition and reinstatement | 63 | |
| 40 | Same structure as vaccine 38 | TT | E6020 and Alum | SC | 3 injections/6 weeks | Locomotor activity and blood–brain biodistribution | Vaccines with E6020 produced x3 increase in titers, nanomolar affinities, attenuated METH-induced hyperlocomotion, and decreased brain METH levels while increasing drug sera concentration | 81 | |
Immunopharmacotherapeutic efforts against METH abuse initially focused on the modification of the hapten at the aromatic ring of the parent molecule; however, behavioral studies demonstrated dismal results.65,72 Thus, there was a paradigm shift in 2011, where linker attachments were made at the N-methyl substituent with enhanced results successfully demonstrating active vaccines inhibiting METH-induced locomotor and thermoregulatory disturbances in murine models.62,75,80 A multitude of METH haptens have been reported since its first synthesis in 2001, but the immunopharmacotherapeutic advancement of the field struggles due to the vast variability of several important methodological factors. In this section, research pertaining to hapten synthesis will be reviewed at length in order to analyze interstudy hapten design.
Linker location – phenyl
Preclinical studies for immunopharmacotherapies against METH began with hapten synthesis based on modifications at the aromatic ring. Byrnes-Blake et al. developed vaccine 1 by altering the para position of the phenyl structure, where vaccinated murine models generated and maintained anti-METH antibody titers despite repeat drug exposure during immunization protocols.65 Although the vaccine failed to show efficacy in attenuating METH-induced hyperlocomotion, the study was significant in demonstrating that active anti-METH vaccines could generate sufficient antibody responses despite early drug exposure prior to attaining protective antibody titers. Based on this research, four METH hapten vaccine formulations were conceived by covalently attaching one or two hapten molecules to peptide constructs containing the molecular adjuvant, EP54.72 Of the monovalent (vaccines 2 and 3) and divalent (vaccines 4 and 5) inoculations, vaccine 3 generated antibodies with the highest titers and specificity. Self-administration studies showed an increase in drug ingestion, which indicated compensatory behavior due to pharmacokinetic antagonistic effects generated by the vaccine.Other immunopharmacotherapeutic methods have developed from modifying the meta position of the aromatic ring. Carroll et al. developed vaccine 6 in order to discern a novel method to prepare METH-conjugate vaccines using maleimide activated proteins, which proved to be significantly better than previously reported haptens in producing a substantially enhanced immune response in murine models by the modification providing higher epitope densities.73 Then, the same hapten was covalently coupled to an immunocyanin carrier protein and adjuvanted with Alhydrogel.66 The optimized carrier protein and adjuvant promoted the generation of high affinity anti-METH antibodies that permitted vaccinated animals to maintain normal habits of food consumption after high-dose drug administration.
In a 2015 study, the hapten developed by Carroll et al. was compared to another METH hapten that utilized the same modification, but at the ortho position of the phenyl group.64 This study combined active and passive immunopharmacotherapies and found that compared to passively immunized groups, vaccine 10 and combination antibody therapy significantly increased METH sera concentrations with decreased levels in the brain after drug administration. Although these studies showed promising physiological results, researchers began to probe other potential substructures of the drug for more efficacious linker attachments due to the mediocre behavioral effects.
Linker location – methyl
Early modifications of METH haptens were primarily directed at the phenyl substituent due to the potential complications that could arise from incorporating linker moieties near the stereogenic carbon. Some believed integrating a linker in proximity to the chiral center would result in antibodies that were not selective between target drug enantiomers, (+)-METH and (−)-METH.64 The (+)-METH isomer is the infamous potent psychostimulant that is readily abused, while (−)-METH lacks amphetamine metabolization and CNS interaction.82,83 Considering optimum biological drug potency is obtained when the N-methyl substituent around the chiral carbon is in the (S)-configuration, hapten design is formulated to mimic the (+)-isomers. Thus, some studies incorporate synthetic strategies in order to repress racemization of the stereogenic carbon during hapten synthesis.56,64,79Although the importance in preserving hapten stereochemistry was assumed, Olson et al. was the first to observe and compare the effect of enantiomeric and racemic haptens—affirming the importance of employing (S)-stereochemistry at the N-methyl substituent during hapten design.56 The linker was incorporated at the methyl substituent and (S), (R), and (R/S) haptens all successfully generated antibodies that could affect blood–brain drug partitioning. However, only active vaccine formulations for the (S)-isomer exhibited antibody affinities that were ten-fold higher than the other formulations, while effectively attenuating METH-induced hyperlocomotion and lethality. With such promising physiological and behavioral results obtained from this novel modification, hapten development at this site is bound to expand as thoroughly as the past decade of research dedicated to varying hapten design at the N-methyl substituent.
Linker location – N-methyl
Modifications in this region were initially attempted by Moreno et al. in 2011, where hapten structures drastically different from METH were synthesized by designing low-energy, conformationally constrained haptens in silico.75 The magnitude of antibody titers remained highest in vaccines with unconstrained hapten variants (vaccines 18 and 19), with vaccine 19 demonstrating antibody specificities toward METH and amphetamine. Vaccine 19 not only attenuated physiological and psychomotor drug effects, but also displayed higher drug concentrations in sera with lower METH presence in the brain after drug exposure.62 Behavior studies investigating the efficacy of the immunopharmacotherapy to inhibit self-administration showed the active vaccine could mitigate METH self-administration, with fewer than 17% of the vaccinated animals achieving acquisition compared to 75% of controls.76 These results were the first to demonstrate the efficacy of an active anti-METH vaccine in decreasing the reinforcement effects elicited by METH abuse.The culmination of results from numerous studies seem to convey that successful METH hapten design relies on the preservation of the secondary amine, which is protonated under physiological pH conditions. This can be distinctly observed through the research conducted by Collins et al., where methylation of the secondary amine (vaccine 24) to a tertiary amine (vaccine 23) led to a drastic decrease in antibody affinity without eliciting differences in antibody titer generation.78 Moreover, conversion of the tertiary amine to a tertiary amide (vaccine 26) managed to reduce antibody drug affinity in murine models, while also significantly reducing antibody generation.
While modification of the native secondary amine to a tertiary amide reduced antibody generation and affinity, Shen et al. developed a hapten with a secondary amide that successfully generated antibodies at high titers.80 The active vaccine was able to reduce drug-conditioned approach behaviors, while attenuating METH-induced hypolocomotion at low doses and hyperlocomotion at high doses. This hapten was further optimized by changing the carrier protein and adjuvant formulation.63 These improvements allowed for the active vaccine to produce enhanced and sustained levels of anti-METH antibodies, inhibit METH acquisition and reinstatement, and decrease drug entry into the brain. The crucial nature of carefully considering the site of linker attachment can be observed through the aforementioned cumulative results obtained from the concerted effort to address METH abuse with innovative hapten synthesis for active immunopharmacotherapy.
Linker length
In addition to linker location, which impacts hapten steric configuration and metabolism, linker length and immunological inertness are other critical factors that can be manipulated. Generally, longer spacer arms between the drug moieties and protein-coupling sites have generated broader recognition of METH-like structures.6 Therefore, researchers have inserted various amino acids to alkyl linkers in order to achieve optimal lengths for vaccine efficacy.78,79 The respective effects of embedded amino acids were comprehensively observed in 2017 by Gooyit et al., where di-glycine (vaccine 32), proline-glycine (vaccine 33), proline-alanine (vaccine 34), di-alanine (vaccine 35), mono-alanine (vaccine 36), and tri-alanine (vaccine 37) were attached to alkyl linkers. Dipeptide linker incorporation (vaccines 32, 33, 34, and 35) induced antibodies with increased drug affinity, but concentrations were lower to those of control vaccine 31. However, of the dipeptide haptens, vaccine 35 generated the highest titers. Thus, mono-alanine and tri-alanine variants were probed for antibody affects, revealing both immunopharmacotherapies elicited antibodies with similar affinities to their dipeptide counterpart, but with higher titers analogous to vaccine 31. The mono-alanine hapten exhibited the most efficacy in obviating CNS drug interaction, compared to the other alanine-incorporated haptens and control.The study conducted by Collins et al., vide supra, designed their initial hapten (vaccine 25) by using a diglycine linker. Vaccines 24 and 25 elicited comparable affinities to METH; however, the peptidic linker in vaccine 25 aided in enhancing antibody concentration. Based on these preliminary results, the hapten was optimized by altering the linker to a shorter alkyl spacer between the METH moiety and glycine unit. A glycine or diglycine substituent was incorporated into the hapten from vaccine 24 in order to formulate vaccines 27 and 28, respectively. The hapten for vaccine 29 was developed by attaching a glycine linker to an alkylated amphetamine molecule, where it elicited antibodies with significantly higher affinities than the other formulations. Thus, the culmination of these two studies indicate that the inclusion of a single, structurally simple amino acid into linker design can benefit the functional response produced by METH haptens.
Conjugation chemistry
Depending on the functional group of the linker, hapten–protein conjugation can be achieved through two approaches: carbodiimide conjugation or maleimide conjugation. Small molecule haptens and larger carrier proteins can consist of primary amines, carboxylic groups, or sulfhydryls that can be manipulated for conjugation. Since most carrier proteins contain primary amines, through exposed lysine residues, immunogen formation using carbodiimide crosslinker 1-ethyl-3-(-3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) is the most facile and effective method for hapten–protein conjugation (Fig. 2a). This crosslinker reacts with free carboxyl groups on the hapten linker to form an active intermediate, which reacts with a primary amine on the carrier protein to form an amide bond. | ||
| Fig. 2 Hapten–protein conjugation chemistry (a) haptens (H) with a free carboxyl group form an active intermediate upon reaction with ethyl-3-(-3-dimethylaminopropyl)carbodiimide hydrochloride (EDC). Introduction of the carrier protein (P) to this active complex allows for the primary amine on the protein to form an amide bond with the hapten. (b) Carrier proteins (P) have many surface amines, which can react with sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC) to functionalize maleimide groups onto the surface of the protein. Maleimide-activated proteins can react with haptens (H) containing a terminal sulfhydryl group in order to form a stable thioether bond. | ||
While this method proved effectual, Carroll et al. applied a novel chemical pathway to METH hapten conjugation.73 They found hapten coupling via Michael addition of a thiol to a maleimide-activated protein produced reactions that were high yielding and efficient. Maleimide conjugation is achieved in a two phase strategy, where at first the carrier protein is activated separately in excess sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC). Numerous SMCC molecules attach to the carrier protein as the N-hydroxysuccinimide-ester groups are displaced. After removal of excess crosslinkers and byproducts through desalting, the purified carrier will possess reactive maleimide groups across its surface. Thus, haptens with a terminal sulfhydryl group can be added to the maleimide-activated carrier protein, which will readily form stable thioether bonds between the two molecules (Fig. 2b). Although this method yields efficient hapten–protein conjugation, the stability of the vaccine is questionable since degradation due to retro-Michael reactions may occur during periods of extended storage.84
Contemplation of carrier proteins
The importance of linker location, length, and conjugation arise from these factors improving immunopharmacotherapeutic effects by presenting haptens in a more effective manner on the surface of carrier proteins. Although little is known regarding the specificity of the immune response to the carrier protein,85 enhancing hapten density is established as a crucial criterion because minimal hapten presence on the carrier protein will potentiate a poor immunostimulatory response.19,73 The most common carrier proteins used in immunopharmacotherapy are bovine serum albumin (BSA), ovalbumin (OVA), keyhole limpet hemocyanin (KLH), diphtheria toxoid (DT), and tetanus toxoid (TT). Depending on the carrier protein used, the potential reactive residues available for hapten conjugation can vary drastically (Table 2). While hapten conjugation with BSA is more often used to analyze antibody titers, OVA–hapten conjugates are implemented as well. BSA is an ideal carrier protein in comparison because it contains 26 to 35 functional lysine ε-amine groups for potential conjugations and remains highly soluble even after extensive modification with haptens. On the other hand, OVA contains 20 reactive residues and is sensitive to denaturation and subsequent precipitation.86 The most prevalently used carrier protein is hemocyanin KLH, which is an immense, multi-subunit protein that exhibits increased immunogenicity when dissociated into more soluble individual subunits.87,88 This carrier protein in its multi-subunit formulation boasts over 2,000 reactive amines, while commercially-available subunits consist of 160–320 lysine residues.86| Carrier proteins | MW (kDa) | Reactive residues |
|---|---|---|
| Abbreviations: MW, molecular weight; OVA, ovalbumin; BSA, bovine serum albumin; KLH, keyhole limpet hemocyanin; TT, tetanus toxoid; CRM197, cross-reacting material variant of diphtheria toxoid. | ||
| OVA | 43–45 | 20 |
| BSA | 67 | 26–35 |
| BSA (maleimide) | 67 | 5–15 |
| KLH | 450–13000 |
>2000 |
| KLH (maleimide) | 350–390 | 160–320 |
| TT | 152 | 27–32 |
| CRM197 | 58 | 21–23 |
Toxoid proteins are variants of biologically inactivated native toxins and these carrier proteins have proven to be useful in various human pertussis vaccines89,90 and murine immunopharmacotherapies.56,63,78,81 DT is obtained from a protein secreted by Corynebacterium diphtheriae and is detoxified with formaldehyde; however, a genetically modified cross-reacting material (CRM197) derived from DT has gained more notoriety. CRM197 lacks toxicity and enzymatic activity due to a single point mutation substituting glycine with glutamic acid at amino acid 52.91 Although CRM197 is antigenically indistinguishable from DT, it is more advantageous due to its lack in toxicity and enhanced number of lysyl reactive residues, presenting 21 to 23 amine groups. On the other hand, tetanus toxin can be chemically inactivated to TT, where the number of amines accessible for hapten attachment ranges from 27 to 32.92
The impact of carrier protein selection on antibody generation for active immunopharmacotherapies can be observed through numerous studies. Research conducted by Gooyit et al., vide supra, converted the carrier protein in vaccines 31, 35, and 36 from KLH to TT. These new active vaccines were dosed at half the amount of the original KLH formulations, yet results showed the new TT conjugated immunotherapies elicited antibodies with exceptionally high titers with some changes in affinities. Vaccine 29, developed by Collins et al., elicited antibodies with significantly higher affinities than other hapten designs, but lower titers than vaccines 24 and 25. The promising results obtained from vaccine 29 led to its optimization by changing the carrier protein from DT to TT and formulating the adjuvant system with CpG oligodeoxynucleotide (CpG ODN) and alum, which elicited exceptionally high titers and demonstrated efficacy in attenuating METH-induced locomotor activities. In the study conducted by Duryee et al., vaccines 3 and 5 contained a tetanus toxin T-cell epitope (YSYFPSV) covalently attached to molecular adjuvant EP54. This epitope could engage CD4+ helper T-cells to aid in B cell antibody production during hapten interaction by dendritic cells activated by the molecular adjuvant. Due to this synergistic influence, vaccine 3 presented the most promising immunopharmacotherapeutic effects. Therefore, optimization of immunopharmacotherapy can be achieved through careful consideration of various carrier proteins.
Advancements in adjuvanticity
The necessity for potent adjuvants in the quest for inducing high antibody titers is pronounced by the observation that many individuals immunized with candidate vaccines fail to produce sufficient antibody responses.93–96 Adjuvants help potentiate immune responses to antigens and can modulate the system toward desired effects, which is critical since haptens cannot induce memory recall responses and high, prolonged antibody presence is essential for an effective vaccine. The only adjuvant that has been used in human clinical trials for immunopharmacotherapies has been aluminum hydroxide gel; however, their adjuvanticity has proven to be too weak to allow for the development of an efficacious vaccine.43,96 Preclinical studies often employ potent and novel adjuvants that have yet to be employed within humans, which provide promising results with diminished optimism for successful translation into human clinical trials. Therefore, there is exigency for an adjuvant or adjuvant cocktail that is safe, effective, accessible, and cost-efficient.There are only six adjuvants that are implemented in licensed vaccines: aluminum salts, oil emulsions, liposomes, monophosphoryl lipid A (MPLA), CpG ODN, and virosomes.97 Although aluminum salts have been a reliable adjuvant for other varieties of vaccines, this adjuvant has demonstrated weak or inconsistent results for immunopharmacotherapeutic purposes.74,78,96 Thus, it is imperative to examine the efficacies of the other five adjuvants and their pertinence to immunopharmacotherapy. In numerous studies, Sigma Adjuvant System (SAS), an oil-in-water emulsion adjuvant, has been used in formulating immunopharmacotherapies with different optimized haptens and carrier proteins.62,64,75,76,78 Due to these varying vaccine components, the adjuvanticity of SAS could not be properly determined. Until a 2016 study compared the efficacy of SAS against CpG ODN with alum in active vaccine formulations containing an optimized hapten and carrier protein.78 This investigation found formulation with SAS failed to provide adequate protection in vaccinated mice when challenged with METH for locomotion studies, while CpG ODN with alum effectively reduced METH-induced hyperlocomotion. Another novel oil-in-water emulsion, GLA-SE, has been previously employed in human studies of influenza vaccines, where it displayed a safe and strong immune response in participants.98,99 Applications to anti-METH immunopharmacotherapies have shown this adjuvant could produce double the concentration of antibodies with enhanced affinities against the drug of abuse, compared to SAS and Alhydrogel.74
Some adjuvants are capable of enhancing the immune system through toll-like receptor 4 (TLR-4) agonism, which initiates a signal cascade that produces pro-inflammatory cytokines, effector CD4+ T-cells, and memory CD8+ T-cells.100,101 One such adjuvant, MPLA, is the only non-alum adjuvant approved for human vaccine formulations within America and Europe.102,103 Another TLR-4 agonist is E6020, a synthetic phospholipid dimer that is derived from Gram negative bacteria and exhibits potent immunostimulatory effects, while maintaining safety in several animal models.104–106 This novel adjuvant produces significantly enhanced IgG concentrations in murine models when co-administered with aluminum hydroxide. Thus, in a 2019 study, hapten chemistry and carrier protein optimization of the vaccine developed by Shen et al. was further improved by incorporating the adjuvant cocktail E6020 and alum.81 This novel formulation produced antibodies with threefold higher titers, nanomolar affinities, METH-induced hyperlocomotion attenuation, and drug partitioning capabilities. Another adjuvant known to primarily stimulate immune responses through cell-mediated pathways is CpG ODN. Several studies have shown CpG ODN as an effective adjuvant for immunopharmacotherapy in murine models.56,78,79
In addition to the frequently employed six adjuvant subtypes, anti-METH immunopharmacotherapies have implemented some novel adjuvant systems such as EP54 and lipid tucaresol. Molecular adjuvant EP54 is able to stimulate the immune system by targeting covalently attached antigens, thus activating the antigen-processing and presentation activities of dendritic cells, which is an antigen presenting cell that contains C5a receptors.72,107,108 Another novel adjuvant, lipid tucaresol, demonstrated abilities to induce antibodies with higher specificities than active vaccines formulated with MPLA.77 Tucaresol mediates cellular and humoral immune responses by enhancing T helper (Th) cell priming, compared to alum.109 Since this immunomodulator is associated with disease-promoting Th2 and disease-suppressing Th1,110 researchers have produced a lipid analogue of tucaresol for liposomal formulation because the dual-targeted response seemed ideal for application to immunopharmacotherapies.
Conclusions
Substance abuse is a global epidemic, where METH use is incessantly increasing with no established effective therapeutics. Researchers have attempted to discover novel pharmacotherapies to address the psychostimulatory effects instigated by METH abuse. However, the comprehensive and convoluted mechanism of METH on neural networks have complicated efforts to manipulate a drug-specific target. With no small molecule interventions available for METH addiction, immunopharmacotherapies have compensated for the dearth in therapeutic developments. Numerous hapten designs, carrier protein couplings, and adjuvant formulations have culminated in the credence that hapten–protein combinations substantially impact antibody affinities, while adjuvants primarily affect antibody concentrations.Anti-METH immunopharmacotherapies have the potential to become beneficial clinical tools, yet certain factors require optimization. As indicated in various studies, haptens can be improved in regard to both the linker placement and design, since antibodies elicited by the immunopharmacotherapy should bind preferentially to the drug moiety rather than the linker molecule. This potent psychostimulant has a longer-half life than cocaine and is partially metabolized to amphetamine, another powerful stimulant.8,20 Thus, potential developments for immunopharmacotherapies demonstrate the ability to elicit antibodies with enhanced cross-reactivity for amphetamine. Furthermore, conjugation chemistry should be carefully considered because hapten density, which is rarely reported in most studies, has the potential to impact vaccine efficacy in generating antibodies. Various carrier proteins, ranging from BSA to KLH, have been utilized in immunopharmacotherapies without explicitly determining their respective efficacies. More comprehensive comparative studies are necessary, since some advanced carriers have adjuvant properties that can enhance immunogenicity. A potential carrier protein of interest is N meningitidis outer membrane protein complex (OMPC), which has shown adjuvanticity due to its lipopolysaccharide composition.111 In terms of adjuvants, optimization is ideally achieved by implementing ones that are approved for use in humans or by formulating a combination of adjuvants to attain a compounded effect in generating enhanced antibody concentrations. It is ideal to find a formulation that is nontoxic, yet potent in stimulating an enhanced immune response.
Another crucial factor requiring reformation is the application of single-dose concentrations for behavioral drug challenges. Implementing one concentration when challenging an animal can pose problems in analyzing behavioral results since vaccinated animals demonstrate attenuated behavioral responses to target drugs, such as sensitization and tolerance.5 These two potential reactions can complicate or interfere with effectively analyzing behavioral results, thus it is critical to challenge the efficacy of a vaccine with multiple doses of the drug at varying concentrations. Finally, immunopharmacotherapy can be further improved by refining the minutiae of vaccine administration, such as modes of delivery. Current studies have only considered the traditional route of injecting vaccine formulations subcutaneously or intramuscularly. It would be pertinent to consider other modes of delivery, such as intranasal or aerosol administration, as this route would be easier and more accessible for application as an active vaccine method. Although various advancements in anti-METH immunopharmacotherapies were discussed in this review, none have yet to be established or implemented in human clinical trials. METH use disorder is a mounting epidemic and psychosocial interventions alone are ineffective in reducing drug relapse and abuse.
The extensive abuse of METH leads to increased risk for cerebrovascular and cardiovascular pathologies, as well as the protracted potential for overdose.6,9,112,113 Immunopharmacotherapy can prevent the abuse and relapse potential of METH by peripherally isolating the drug prior to it interacting with the CNS and instigating psychostimulatory effects. Due to their lack of influence on the CNS and low interaction with other small molecules, immunopharmacotherapies are ideal for integration with behavioral approaches. The success of rehabilitation for drugs of abuse depend on the motivation of the abuser and their environment, thus this combinatorial therapeutic approach would be ideal for patients who are motivated to adhere to a comprehensive medical regimen in order to improve their overall health and welfare, but struggle to discontinue METH use due to its potent stimulatory and reinforcing effects. Immunopharmacotherapy would provide such patients with the opportunities to actively choose to seek counseling, avoid drug use, disrupt negative behavioral patterns, and strive for improvements in health and sustainable living. This therapeutic tool could also be used to monitor patients during treatment programs, since METH would remain bound to circulating antibodies if drug use occurred. Such methods of surveillance would permit professionals to identify patients with increased potential for recidivism at earlier time points before more drastic consequences transpire.
Advancements in immunopharmacotherapy allow for the potential to parallel the proliferation of novel drugs with a rapid-response system that could effectively treat modern addiction disorders—ending the expanding epidemic. This therapeutic tool could prevent a patient's temporary lapse in judgement from developing into a relapse to drug use. If an individual could be inoculated from the physiological and psychological factors of drug abuse, then the reinforcing bonds of addiction may be more easily broken. This provides further exigency to establish clinically applicable immunopharmacotherapies for METH, in order to shift treatment outcomes from overdose reversal to successful social integration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Skaggs Institute for Chemical Biology. All images are created by the authors of this manuscript.References
- R. A. Wise, Nat. Rev. Neurosci., 2004, 5, 483–494 CrossRef CAS.
- T. Nakagawa and S. Kaneko, J. Pharmacol. Sci., 2008, 106, 2–8 CrossRef CAS.
- N. D. Volkow, G.-J. Wang, J. S. Fowler, D. Tomasi and F. Telang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15037–15042 CrossRef CAS.
- D. Sulzer, M. S. Sonders, N. W. Poulsen and A. Galli, Prog. Neurobiol., 2005, 75, 406–433 CrossRef CAS.
- T. R. Kosten and T. A. Kosten, Biologics to Treat Substance Use Disorders, Springer, 2016, pp. 65–74 Search PubMed.
- W. B. Gentry, D. Rüedi-Bettschen and S. M. Owens, Hum. Vaccin., 2009, 5, 206–213 CrossRef CAS.
- L. Karila, A. Weinstein, H.-J. Aubin, A. Benyamina, M. Reynaud and S. L. Batki, Br. J. Clin. Pharmacol., 2010, 69, 578–592 CrossRef CAS.
- F. M. Orson, B. M. Kinsey, R. A. K. Singh, Y. Wu, T. Gardner and T. R. Kosten, Ann. N. Y. Acad. Sci., 2008, 1141, 257–269 CrossRef CAS.
- T. J. Abbruscato and P. C. Trippier, ACS Chem. Neurosci., 2018, 9, 2373–2378 CrossRef CAS.
- T. R. Kosten, T. F. Newton, R. De La Garza II and C. N. Haile, Cocaine and methamphetamine dependence: advances in treatment, American Psychiatric Pub, 2012 Search PubMed.
- J. F. Bowyer, B. Robinson, S. Ali and L. C. Schmued, Synapse, 2008, 62, 193–204 CrossRef CAS.
- E. A. Kiyatkin, P. L. Brown and H. S. Sharma, Eur. J. Neurosci., 2007, 26, 1242–1253 CrossRef.
- S. Ciketic, M. R. Hayatbakhsh, C. M. Doran, J. M. Najman and R. McKetin, J. Subst. Use, 2012, 17, 363–383 CrossRef.
- P. V. AshaRani, A. Hombali, E. Seow, W. J. Ong, J. H. Tan and M. Subramaniam, Drug Alcohol Depend., 2020, 212, 108060 CrossRef CAS.
- N. K. Lee and R. A. Rawson, Drug Alcohol Rev., 2008, 27, 309–317 CrossRef.
- D. M. Donovan and E. A. Wells, Addiction, 2007, 102, 121–129 CrossRef.
- S. M. Owens, W. T. Atchley, M. D. Hambuchen, E. C. Peterson and W. B. Gentry, CNS Neurol. Disord. Drug Targets, 2011, 10, 892–898 CrossRef CAS.
- R. McKetin, J. M. Najman, A. L. Baker, D. I. Lubman, S. Dawe, R. Ali, N. K. Lee, R. P. Mattick and A. Mamun, Addiction, 2012, 107, 1998–2008 CrossRef.
- T. Xiaoshan, Y. Junjie, W. Wenqing, Z. Yunong, L. Jiaping, L. Shanshan, N. Kutty Selva and C. Kui, Drug Discovery Today, 2020, 25, 610–619 CrossRef.
- B. M. Kinsey, D. C. Jackson and F. M. Orson, Immunol. Cell Biol., 2009, 87, 309–314 CrossRef CAS.
- K. J. Siefried, L. S. Acheson, N. Lintzeris and N. Ezard, CNS Drugs, 2020, 34, 337–365 CrossRef CAS.
- M. C. McElhiney, J. G. Rabkin, R. Rabkin and E. V. Nunes, Am. J. Drug Alcohol Abuse, 2009, 35, 34–37 CrossRef.
- J. Shearer, S. Darke, C. Rodgers, T. Slade, I. Van Beek, J. Lewis, D. Brady, R. McKetin, R. P. Mattick and A. Wodak, Addiction, 2009, 104, 224–233 CrossRef.
- A. L. Anderson, S. H. Li, K. Biswas, F. McSherry, T. Holmes, E. Iturriaga, R. Kahn, N. Chiang, T. Beresford, J. Campbell, W. Haning, J. Mawhinney, M. McCann, R. Rawson, C. Stock, D. Weis, E. Yu and A. M. Elkashef, Drug Alcohol Depend., 2012, 120, 135–141 CrossRef CAS.
- K. G. Heinzerling, A.-N. Swanson, S. Kim, L. Cederblom, A. Moe, W. Ling and S. Shoptaw, Drug Alcohol Depend., 2010, 109, 20–29 CrossRef CAS.
- N. Lee, A. Pennay, R. Hester, R. McKetin, S. Nielsen and J. Ferris, Drug Alcohol Rev., 2013, 32, 88–95 CrossRef.
- T. F. Newton, J. D. Roache, R. De La Garza, T. Fong, C. L. Wallace, S.-H. Li, A. Elkashef, N. Chiang and R. Kahn, Psychopharmacology, 2005, 182, 426–435 CrossRef CAS.
- T. F. Newton, J. D. Roache, R. De La Garza, 2nd, T. Fong, C. L. Wallace, S. H. Li, A. Elkashef, N. Chiang and R. Kahn, Neuropsychopharmacology, 2006, 31, 1537–1544 CrossRef CAS.
- A. M. Elkashef, R. A. Rawson, A. L. Anderson, S.-H. Li, T. Holmes, E. V. Smith, N. Chiang, R. Kahn, F. Vocci, W. Ling, V. J. Pearce, M. McCann, J. Campbell, C. Gorodetzky, W. Haning, B. Carlton, J. Mawhinney and D. Weis, Neuropsychopharmacology, 2008, 33, 1162–1170 CrossRef CAS.
- S. Shoptaw, K. G. Heinzerling, E. Rotheram-Fuller, T. Steward, J. Wang, A.-N. Swanson, R. De La Garza, T. Newton and W. Ling, Drug Alcohol Depend., 2008, 96, 222–232 CrossRef CAS.
- K. G. Heinzerling, J. Gadzhyan, H. van Oudheusden, F. Rodriguez, J. McCracken and S. Shoptaw, J. Adolesc. Health, 2013, 52, 502–505 CrossRef.
- K. G. Heinzerling, A.-N. Swanson, T. M. Hall, Y. Yi, Y. Wu and S. J. Shoptaw, Addiction, 2014, 109, 1878–1886 CrossRef.
- T. M. Winhusen, G. S. Brigham, F. Kropp, R. Lindblad, J. G. Gardin, 2nd, P. Penn, C. Hodgkins, T. M. Kelly, A. Douaihy, M. McCann, L. D. Love, E. DeGravelles, K. Bachrach, S. C. Sonne, B. Hiott, L. Haynes, G. Sharma, D. F. Lewis, P. VanVeldhuisen, J. Theobald and U. Ghitza, J. Clin. Psychiatry, 2014, 75, 336–343 CrossRef CAS.
- A. L. Anderson, S.-H. Li, D. Markova, T. H. Holmes, N. Chiang, R. Kahn, J. Campbell, D. L. Dickerson, G. P. Galloway, W. Haning, J. D. Roache, C. Stock and A. M. Elkashef, Drug Alcohol Depend., 2015, 150, 170–174 CrossRef CAS.
- N. Jayaram-Lindström, P. Wennberg, O. Beck and J. Franck, Nord. J. Psychiatry, 2005, 59, 167–171 CrossRef.
- N. Jayaram-Lindström, M. Konstenius, S. Eksborg, O. Beck, A. Hammarberg and J. Franck, Neuropsychopharmacology, 2008, 33, 1856–1863 CrossRef.
- N. Jayaram-Lindström, A. Hammarberg, O. Beck and J. Franck, Am. J. Psychiatry, 2008, 165, 1442–1448 CrossRef.
- M. Kohno, L. E. Dennis, H. McCready, D. L. Schwartz, W. F. Hoffman and P. T. Korthuis, Drug Alcohol Depend., 2018, 192, 186–192 CrossRef CAS.
- J. Tiihonen, E. Krupitsky, E. Verbitskaya, E. Blokhina, O. Mamontova, J. Föhr, P. Tuomola, K. Kuoppasalmi, V. Kiviniemi and E. Zwartau, Am. J. Psychiatry, 2012, 169, 531–536 CrossRef.
- V. Runarsdottir, I. Hansdottir, T. Tyrfingsson, M. Einarsson, K. Dugosh, C. Royer-Malvestuto, H. Pettinati, J. Khalsa and G. E. Woody, J. Addict. Med., 2017, 11, 197–204 CrossRef CAS.
- P. O. Coffin, G.-M. Santos, J. Hern, E. Vittinghoff, D. Santos, T. Matheson, G. Colfax and S. L. Batki, Addiction, 2018, 113, 268–278 CrossRef.
- O. Ohia-Nwoko, T. A. Kosten and C. N. Haile, in International Review of Neurobiology, ed. R. L. Bell and S. Rahman, Academic Press, 2016, vol. 126, pp. 263–291 Search PubMed.
- C. R. Alving, G. R. Matyas, O. Torres, R. Jalah and Z. Beck, Vaccine, 2014, 32, 5382–5389 CrossRef CAS.
- T. Kosten, C. Domingo, F. Orson and B. Kinsey, Br. J. Clin. Pharmacol., 2014, 77, 368–374 CrossRef CAS.
- T. Kosten and S. M. Owens, Pharmacol. Ther., 2005, 108, 76–85 CrossRef CAS.
- W. M. Compton, C. M. Jones and G. T. Baldwin, N. Engl. J. Med., 2016, 374, 154–163 CrossRef CAS.
- A. Y. Moreno and K. D. Janda, Pharmacol., Biochem. Behav., 2009, 92, 199–205 CrossRef CAS.
- Y. H. Chen and C. H. Chen, Curr. Gene Ther., 2013, 13, 39–50 CrossRef CAS.
- J. Bazan, I. Całkosiński and A. Gamian, Hum. Vaccin. Immunother., 2012, 8, 1817–1828 CrossRef CAS.
- Q. Yu, D. F. Larson and R. R. Watson, Life Sci., 2003, 73, 129–140 CrossRef CAS.
- D. E. McMillan, W. C. Hardwick, M. Li, M. G. Gunnell, F. I. Carroll, P. Abraham and S. M. Owens, J. Pharmacol. Exp. Ther., 2004, 309, 1248–1255 CrossRef CAS.
- K. A. Byrnes-Blake, E. M. Laurenzana, F. I. Carroll, P. Abraham, W. B. Gentry, R. D. Landes and S. M. Owens, Eur. J. Pharmacol., 2003, 461, 119–128 CrossRef CAS.
- K. A. Byrnes-Blake, E. M. Laurenzana, R. D. Landes, W. B. Gentry and S. M. Owens, Eur. J. Pharmacol., 2005, 521, 86–94 CrossRef CAS.
- W. B. Gentry, E. M. Laurenzana, D. K. Williams, J. R. West, R. J. Berg, T. Terlea and S. M. Owens, Int. Immunopharmacol., 2006, 6, 968–977 CrossRef CAS.
- D. E. McMillan, W. C. Hardwick, M. Li and S. M. Owens, Behav. Pharmacol., 2002, 13, 465–473 CrossRef CAS.
- M. E. Olson, T. Sugane, B. Zhou and K. D. Janda, J. Am. Chem. Soc., 2019, 141, 14089–14092 CrossRef CAS.
- K. Landsteiner, The Specificity of Serological Reactions, Courier Corporation, 2013 Search PubMed.
- T. R. Kosten and C. B. Domingo, Expert Opin. Biol. Ther., 2013, 13, 1093–1097 CrossRef CAS.
- G. F. Koob, F. Weiss, V. Deroche and R. Weissenborn, Psychopharmacology, 1996, 126, 311–322 CrossRef.
- A. C. Barrett, J. R. Miller, J. M. Dohrmann and S. B. Caine, Neuropharmacology, 2004, 47, 256–273 CrossRef CAS.
- R. Y. Depoortere, D. H. Li, J. D. Lane and M. W. Emmett-Oglesby, Pharmacol., Biochem. Behav., 1993, 45, 539–548 CrossRef CAS.
- M. L. Miller, A. Y. Moreno, S. M. Aarde, K. M. Creehan, S. A. Vandewater, B. D. Vaillancourt, M. J. Wright, Jr., K. D. Janda and M. A. Taffe, Biol. Psychiatry, 2013, 73, 721–728 CrossRef CAS.
- C. N. Haile, T. A. Kosten, X. Y. Shen, P. W. O'Malley, K. J. Winoske, B. M. Kinsey, Y. Wu, Z. Huang, E. D. Lykissa, N. Naidu, J. A. Cox, R. Arora, T. R. Kosten and F. M. Orson, Am. J. Addict., 2015, 24, 748–755 CrossRef.
- M. D. Hambuchen, F. I. Carroll, D. Rüedi-Bettschen, H. P. Hendrickson, L. J. Hennings, B. E. Blough, L. E. Brieaddy, R. R. Pidaparthi and S. M. Owens, J. Med. Chem., 2015, 58, 4665–4677 CrossRef CAS.
- K. A. Byrnes-Blake, F. I. Carroll, P. Abraham and S. M. Owens, Int. Immunopharmacol., 2001, 1, 329–338 CrossRef CAS.
- D. Rüedi-Bettschen, S. L. Wood, M. G. Gunnell, C. M. West, R. R. Pidaparthi, F. I. Carroll, B. E. Blough and S. M. Owens, Vaccine, 2013, 31, 4596–4602 CrossRef.
- K. Antoniou, E. Kafetzopoulos, Z. Papadopoulou-Daifoti, T. Hyphantis and M. Marselos, Neurosci. Biobehav. Rev., 1998, 23, 189–196 CrossRef CAS.
- J. F. Brien, J. C. Kitney, J. E. Peachey and B. J. Rogers, Res. Commun. Chem. Pathol. Pharmacol., 1978, 22, 313–328 CAS.
- E. H. Ellinwood and R. L. Balster, Eur. J. Pharmacol., 1974, 28, 35–41 CrossRef CAS.
- R. Kuczenski and D. Segal, J. Neurosci., 1989, 9, 2051–2065 CrossRef CAS.
- Y. Shaham, U. Shalev, L. Lu, H. de Wit and J. Stewart, Psychopharmacology, 2003, 168, 3–20 CrossRef CAS.
- M. J. Duryee, R. A. Bevins, C. M. Reichel, J. E. Murray, Y. Dong, G. M. Thiele and S. D. Sanderson, Vaccine, 2009, 27, 2981–2988 CrossRef CAS.
- F. I. Carroll, B. E. Blough, R. R. Pidaparthi, P. Abraham, P. K. Gong, L. Deng, X. Huang, M. Gunnell, J. O. Lay, Jr., E. C. Peterson and S. M. Owens, J. Med. Chem., 2011, 54, 5221–5228 CrossRef CAS.
- M. W. Stevens, M. G. Gunnell, R. Tawney and S. M. Owens, Int. Immunopharmacol., 2016, 35, 137–141 CrossRef CAS.
- A. Y. Moreno, A. V. Mayorov and K. D. Janda, J. Am. Chem. Soc., 2011, 133, 6587–6595 CrossRef CAS.
- M. L. Miller, S. M. Aarde, A. Y. Moreno, K. M. Creehan, K. D. Janda and M. A. Taffe, Drug Alcohol Depend., 2015, 153, 29–36 CrossRef CAS.
- K. C. Collins, J. E. Schlosburg, J. W. Lockner, P. T. Bremer, B. A. Ellis and K. D. Janda, Chem. Commun., 2014, 50, 4079–4081 RSC.
- K. C. Collins, J. E. Schlosburg, P. T. Bremer and K. D. Janda, J. Med. Chem., 2016, 59, 3878–3885 CrossRef CAS.
- M. Gooyit, P. O. Miranda, C. J. Wenthur, A. Ducime and K. D. Janda, ACS Chem. Neurosci., 2017, 8, 468–472 CrossRef CAS.
- X. Y. Shen, T. A. Kosten, A. Y. Lopez, B. M. Kinsey, T. R. Kosten and F. M. Orson, Drug Alcohol Depend., 2013, 129, 41–48 CrossRef CAS.
- R. Arora, C. N. Haile, T. A. Kosten, Y. Wu, M. Ramakrishnan, L. D. Hawkins, F. M. Orson and T. R. Kosten, Am. J. Addict., 2019, 28, 119–126 CrossRef.
- R. West, A. Pesce, C. Mikel, J. Velasco, E. Gonzales, Z. Dizon, P. Almazan and S. Latyshev, J. Pain., 2012, 13, S89 CrossRef.
- J. Caldwell and S. J. Mule, Amphetamines and related stimulants: chemical, biological, clinical, and sociological aspects, CRC Press, 2019 Search PubMed.
- P. A. Szijj, C. Bahou and V. Chudasama, Drug Discovery Today Technol., 2018, 30, 27–34 CrossRef.
- T. Gefen, J. Vaya, S. Khatib, I. Rapoport, M. Lupo, E. Barnea, A. Admon, E. D. Heller, E. Aizenshtein and J. Pitcovski, Immunology, 2015, 144, 116–126 CrossRef CAS.
- G. T. Hermanson, in Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press, Boston, 3rd edn, 2013, pp. 839–865 Search PubMed.
- A. H. Bartel and D. H. Campbell, Arch. Biochem. Biophys., 1959, 82, 232–234 CrossRef CAS.
- T. T. Herskovits, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1988, 91, 597–611 CrossRef CAS.
- S. A. Halperin, S. McNeil, J. Langley, M. Blatter, M. Dionne, J. Embree, R. Johnson, T. Latiolais, W. Meekison, F. Noya, S. Senders, P. Zickler and D. R. Johnson, Vaccine, 2011, 29, 8459–8465 CrossRef CAS.
- P. L. McCormack, Drugs, 2012, 72, 1765–1791 CrossRef CAS.
- G. Giannini, R. Rappuoli and G. Ratti, Nucleic Acids Res., 1984, 12, 4063–4069 CrossRef CAS.
- R. Jalah, O. B. Torres, A. V. Mayorov, F. Li, J. F. G. Antoline, A. E. Jacobson, K. C. Rice, J. R. Deschamps, Z. Beck, C. R. Alving and G. R. Matyas, Bioconjugate Chem., 2015, 26, 1041–1053 CrossRef CAS.
- D. Hatsukami, D. Jorenby, D. Gonzales, N. Rigotti, E. Glover, C. Oncken, D. Tashkin, V. Reus, R. Akhavain, R. Fahim, P. Kessler, M. Niknian, M. Kalnik and S. Rennard, Clin. Pharmacol. Ther., 2011, 89, 392–399 CrossRef CAS.
- J. Cornuz, S. Zwahlen, W. F. Jungi, J. Osterwalder, K. Klingler, G. van Melle, Y. Bangala, I. Guessous, P. Müller, J. Willers, P. Maurer, M. F. Bachmann and T. Cerny, PLoS One, 2008, 3, e2547 CrossRef.
- B. A. Martell, F. M. Orson, J. Poling, E. Mitchell, R. D. Rossen, T. Gardner and T. R. Kosten, Arch. Gen. Psychiatry, 2009, 66, 1116–1123 CrossRef CAS.
- M. Haney, E. W. Gunderson, H. Jiang, E. D. Collins and R. W. Foltin, Biol. Psychiatry, 2010, 67, 59–65 CrossRef CAS.
- M. L. Mbow, E. De Gregorio, N. M. Valiante and R. Rappuoli, Curr. Opin. Immunol., 2010, 22, 411–416 CrossRef CAS.
- J. J. Treanor, B. Essink, S. Hull, S. Reed, R. Izikson, P. Patriarca, K. L. Goldenthal, R. Kohberger and L. M. Dunkle, Vaccine, 2013, 31, 5760–5765 CrossRef CAS.
- R. N. Coler, S. L. Baldwin, N. Shaverdian, S. Bertholet, S. J. Reed, V. S. Raman, X. Lu, J. DeVos, K. Hancock, J. M. Katz, T. S. Vedvick, M. S. Duthie, C. H. Clegg, N. Van Hoeven and S. G. Reed, PLoS One, 2010, 5, e13677 CrossRef.
- N. J. Steers, C. R. Alving and M. Rao, Vaccine, 2008, 26, 2849–2859 CrossRef CAS.
- N. J. Steers, K. K. Peachman, S. McClain, C. R. Alving and M. Rao, Vaccine, 2009, 27, 6939–6949 CrossRef CAS.
- S. Thoelen, N. De Clercq and N. Tornieporth, Vaccine, 2001, 19, 2400–2403 CrossRef CAS.
- D. M. Harper, E. L. Franco, C. Wheeler, D. G. Ferris, D. Jenkins, A. Schuind, T. Zahaf, B. Innis, P. Naud, N. S. De Carvalho, C. M. Roteli-Martins, J. Teixeira, M. M. Blatter, A. P. Korn, W. Quint and G. Dubin, The Lancet, 2004, 364, 1757–1765 CrossRef CAS.
- G. L. Morefield, L. D. Hawkins, S. T. Ishizaka, T. L. Kissner and R. G. Ulrich, Clin. Vaccine Immunol., 2007, 14, 1499–1504 CrossRef CAS.
- S. T. Ishizaka and L. D. Hawkins, Expert Rev. Vaccines, 2007, 6, 773–784 CrossRef CAS.
- M. M. Giuliani, J. Adu-Bobie, M. Comanducci, B. Aricò, S. Savino, L. Santini, B. Brunelli, S. Bambini, A. Biolchi, B. Capecchi, E. Cartocci, L. Ciucchi, F. Di Marcello, F. Ferlicca, B. Galli, E. Luzzi, V. Masignani, D. Serruto, D. Veggi, M. Contorni, M. Morandi, A. Bartalesi, V. Cinotti, D. Mannucci, F. Titta, E. Ovidi, J. A. Welsch, D. Granoff, R. Rappuoli and M. Pizza, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10834–10839 CrossRef CAS.
- G. V. Hegde, E. Meyers-Clark, S. S. Joshi and S. D. Sanderson, Int. Immunopharmacol., 2008, 8, 819–827 CrossRef CAS.
- R. M. Tempero, M. A. Hollingsworth, M. D. Burdick, A. M. Finch, S. M. Taylor, S. M. Vogen, E. L. Morgan and S. D. Sanderson, J. Immunol., 1997, 158, 1377–1382 CAS.
- J. Rhodes, H. Chen, S. R. Hall, J. E. Beesley, D. C. Jenkins, P. Collins and B. Zheng, Nature, 1995, 377, 71–75 CrossRef CAS.
- A. C. Smith, V. Yardley, J. Rhodes and S. L. Croft, Antimicrob. Agents Chemother., 2000, 44, 1494–1498 CrossRef CAS.
- M. Pérez-Melgosa, H. D. Ochs, P. S. Linsley, J. D. Laman, M. V. Meurs, R. A. Flavell, R. K. Ernst, S. I. Miller and C. B. Wilson, Eur. J. Immunol., 2001, 31, 2373–2381 CrossRef.
- S. Moeller, H. B. Huttner, T. Struffert and H. H. Müller, Alcohol. Drug Addict., 2016, 29, 39–41 CrossRef.
- S. Won, R. A. Hong, R. V. Shohet, T. B. Seto and N. I. Parikh, Clin. Cardiol., 2013, 36, 737–742 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |