
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrief survey on organometalated antibacterial drugs and metal-based materials with antibacterial activity
Przemysław
Biegański
 a,
Łukasz
Szczupak
a,
Manuel
Arruebo
bcd and
Konrad
Kowalski
*a
a,
Łukasz
Szczupak
a,
Manuel
Arruebo
bcd and
Konrad
Kowalski
*a
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Łódź, Tamka 12, 91-403 Łódź, Poland. E-mail: kondor15@wp.pl; konrad.kowalski@chemia.uni.lodz.pl; Tel: +48-42-635-5759
bInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain
cDepartment of Chemical Engineering, University of Zaragoza, Campus Río Ebro - Edificio I + D, C/Poeta Mariano Esquillor S/N, 50018 Zaragoza, Spain
dNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine, CIBER-BBN, 28029 Madrid, Spain
First published on 26th January 2021
Abstract
Rising bacterial antibiotic resistance is a global threat. To deal with it, new antibacterial agents and antiseptic materials need to be developed. One alternative in this quest is the organometallic derivatization of well-established antibacterial drugs and also the fabrication of advanced metal-based materials having antibacterial properties. Metal-based agents and materials often show new modes of antimicrobial action which enable them to overcome drug resistance in pathogenic bacterial strains. This review summarizes recent (2017–2020) progress in the field of organometallic-derived antibacterial drugs and metal-based materials having antibacterial activity. Specifically, it covers organometallic derivatives of antibacterial drugs including β-lactams, ciprofloxacin, isoniazid, trimethoprim, sulfadoxine, sulfamethoxazole, and ethambutol as well as non-antibacterial drugs like metformin, phenformin and aspirin. Recent advances and reported clinical trials in the use of metal-based nanomaterials as antibiofouling coatings on medical devices, as photocatalytic agents in indoor air pollutant control, and also as photodynamic/photothermal antimicrobial agents are also summarized.
 Przemysław Biegański | Przemysław Biegański obtained his MSc degree in chemistry in 2020 from the Łódź University of Technology (Poland). He then started his PhD at the University of Łódź under the supervision of Prof. Konrad Kowalski. His PhD project is focused on the chemistry of organometallic bioconjugates and organometallic nucleoside analogues. |
 Łukasz Szczupak | Łukasz Szczupak obtained his MSc degree in chemistry in 2012 from the University of Łódź (Poland). In 2017, he received his PhD degree in chemical sciences working under the supervision of Prof. Konrad Kowalski. In 2017, he came to Mabion biotechnological company and worked on monoclonal antibody therapy. In 2018, he re-joined Prof. K. Kowalski's group at the University of Łódź. His main current interests are in developing new metal-based antimicrobial and anticancer agents. |
 Manuel Arruebo | Manuel Arruebo is a Full Professor in Chemical Engineering at the University of Zaragoza, Spain. He is the co-author of more than 150 scientific publications, including 4 book chapters. A number of those papers have been published in journals having high-impact factors including Nature Nanotechnology, Nature Catalysis, PNAS, Nano Today, Trends in Biotechnology, Biomaterials, Chemistry of Materials, Small, and Hepatology, among others, having in 2020 an h-index of 38 (WoS) and 41 (Google scholar). He was awarded in 2014 an ERC Consolidator Grant (selected from 3673 applicants throughout Europe), which helped him to consolidate his own nanobiotechnology research group. |
 Konrad Kowalski | Konrad Kowalski received his PhD degree in chemical sciences in 2003 at the University of Łódź (Poland). Afterwards, he worked as a postdoctoral fellow at Imperial College London (UK) with Prof. Nicholas J. Long and at the University of Regensburg (Germany) with Prof. Rainer F. Winter. In 2011, he obtained his DSc (habilitation) degree and became Associate Professor at the University of Łódź. In 2020, he was promoted to Full Professor of Chemistry. His current scientific interests include biology-oriented chemistry of organometallic compounds, xeno-nucleic acids and luminescent probes for bioimaging applications. |
1. Introduction
Bacteria are unicellular organisms usually a few micrometers in length which together with archaea (formerly archaebacteria) belong to the prokaryote domain of life. High-resolution microscopy and isotopic analyses of some Archaean (4–2.5 Ga) rocks show that ancestors of modern bacteria existed on earth between 3.47 and 2.7 Ga,1–3 whereas studies on 1.88 Ga old stromatolites showed Fe2O3-mineralized microfossils of bacterial cells.4 Bacteria inhabit all ecological niches including extreme environments like hydrothermal vents,5 hot springs,6 the deep sub-seafloor,7 sub-polar snowpacks,8 the Antarctic desert9 and even nuclear waste.10 The human body also represents an ecological niche which harbors more than 100 trillion bacteria and other microorganisms of microbiota.11 Especially human gut microbiota can influence our physiology and even the risk of non-infectious disease development in organisms.11 This is exemplified by recent studies which show links between the presence of Ruminococcus flavefaciens and hypertension as well as between Clostridium and platelet counts.12 Other studies have revealed links between colon-cancer development and production of colibactin, which is a genotoxic metabolite produced by Escherichia coli.13Interaction between humans and infective bacterial pathogens is as old as the history of mankind,14 although the modern era of antibacterial agents began in 1928 with the discovery of penicillin by Alexander Fleming.15 Penicillin was introduced in hospitals in the 1940s but its effectiveness was soon questioned by the appearance of penicillin-resistant Staphylococcus aureus strains.16 A similar scenario occurred in the case of streptomycin, discovered by Waksman,17 as the first streptomycin-resistant Mycobacterium tuberculosis was reported just a few years after its discovery.18 Likewise, the first methicillin-resistant S. aureus (MRSA) appeared rapidly after the introduction of this antibiotic in 1959.19,20 While time goes by, the progressive appearance of new drug-resistant strains including the emergence and spread of multidrug resistant (MDR) ESKAPE pathogens has continued.21–26 According to the WHO, today MDR bacteria are one of the key problems for health-care systems.27 Thus, severe infection caused by some drug-resistant Gram-negative strains of Pseudomonas aeruginosa, Acinetobacter baumannii, Escherichia coli, Klebsiella pneumoniae, Mycobacterium tuberculosis or Enterobacter spp. may, despite the best medical care, produce a lethal outcome.25,28–36 Also infections caused by some Gram-positive S. aureus (MRSA), vancomycin-resistant enterococci (VRE) and penicillin-resistant Streptococcus pneumoniae (PRSP) pathogenic strains can cause life-threatening problems during treatment.25,37–40
Where does drug-resistance arise and what are the key mechanisms of resistance in bacteria? To answer the first question one has to look back into the long evolutionary history of bacteria. During billions of years of evolution, bacteria developed a number of genetically driven biochemical processes and biomolecules which enabled them to withstand the presence of different competitor microorganisms invading or sharing their environment. In return, the attacked competitors have protected themselves with their own set of molecular mechanisms, which ultimately results in a never ending arms race. Drug-resistance in bacteria originates from this evolutionary molecular competition. Uncontrolled use of antibacterial drugs in medicine, veterinary practice and agriculture increased the selection pressure on bacteria and enabled drug-resistant strains to develop, survive, and spread.26 Drug resistance is a dynamic phenomenon mediated by mobile genetic vectors like R plasmids (containing transposons), bacteriophages or naked DNA following conjugation, transduction, and transformation processes, respectively.41 Transposons are able to move from plasmid to plasmid and back and forth to the bacterial chromosome. Drug resistance can spread within the same bacterial strain as well as horizontally between different bacteria.
To answer the second question – there are many specific mechanisms of resistance in bacteria.22 All of them, however, can be classified into three different general mechanistic groups (Fig. 1).
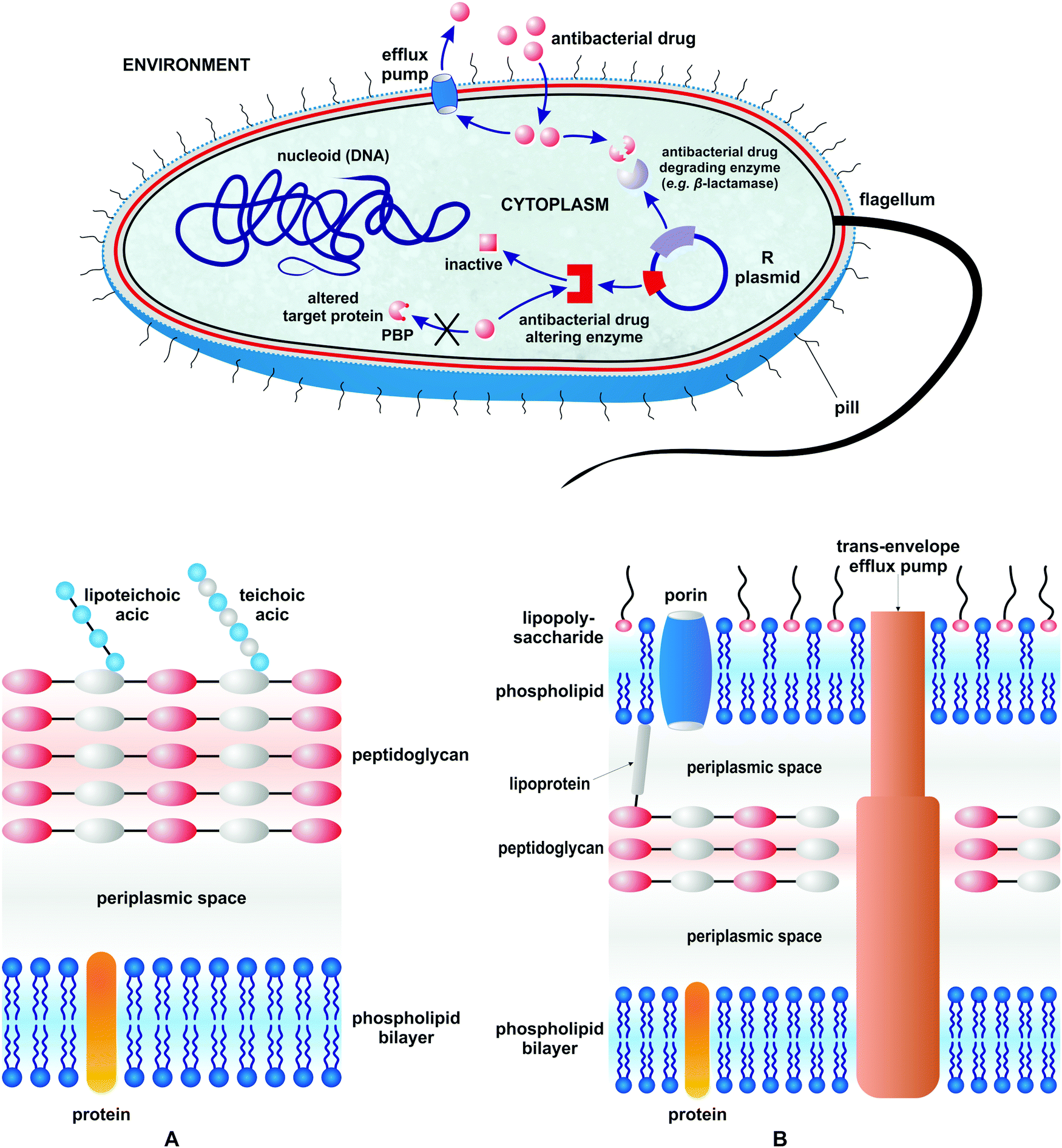 | ||
| Fig. 1 Different mechanisms of drug resistance in bacteria and a schematic representation of the Gram-positive (A) and Gram-negative (B) bacterial cell envelope. | ||
The first mechanism pertains to the bacterial development of enzymes which either degrade or alter antibacterial drugs to make them inactive. The second pertains to the biosynthesis of structurally modified proteins which help them to be protected from inhibition by antibacterial agents (target modification). The last mechanism relates to the modification of the bacterial cell wall to impair antibacterial drug influx and/or development of protein pump systems to enable drug efflux. The most effective way to eliminate drug resistant bacteria from a given ecological niche is to outnumber their population by non-pathogenic (non-resistant) strains in the absence of selecting factors. Obviously this approach cannot be widely applied to cure patients, especially those with acute and fast progressing infections. Thus, antibacterial drugs are a second and clinically most relevant option. They are over 200 marketed antibacterial drugs which belong to over 11 large classes.42,43 Historically, antibacterial agents diverge into those isolated from natural source antibiotics and those of synthetic origin. They either eliminate bacteria (bactericidal effect) or inhibit their growth (bacteriostatic effect). Antibacterial drugs interfere with different targets and biochemical processes in the bacterial cell. Table 1 summarizes the major classes of antibacterial drugs, their mechanisms of action, and the year of their introduction.
| Antibacterial drug class (year of introduction, representative drug(s)) | Mechanism of action |
|---|---|
| Sulfonamides (1935, prontosil) and trimethoprim (1960s) | Inhibitors of folic acid synthesis (inhibitors of dihydropteroate synthase, dihydrofolate reductase) |
| Quinolones (1960s) (ciprofloxacin, ozenoxacin) | Inhibitors of DNA replication (inhibitors of topoisomerase IV, gyrase) |
| Rifamycin (1957, does not belong to the large antibiotic families) | Inhibitor of RNA synthesis |
| β-Lactams (1940s) (penicillins, cephalosporins, carbapenems, monobactams) (selected examples: ampicillin, cefiderocol) | Inhibitors of cell wall synthesis and remodeling (PBP inhibitors) |
| Glycopeptides (1959) (vancomycin, oritavancin), lipopeptides (2003, daptomycin) and other cell wall inhibitors which do not belong to the large antibiotic families: cycloserine (1955), isoniazid (1952) | Inhibitors of cell wall synthesis |
| Phenylpropanoids (1949, chloramphenicol), aminoglycosides (1950, tobramycin), macrolides (1952, erythromycin), polyketides (1949, tetracycline), oxazolidinones (2000, linezolid), streptogramins (1962, quinupristin/dalfopristin), lincosamides (1960s, clindamycin) | Inhibitors of protein synthesis |
| Nitroimidazoles (1960, metronidazole) | Cellular damage of DNA and proteins |
Over the last decades the number of new antibacterial drugs has decreased.43 In 2000–2020 only twenty seven antibacterial drugs have been approved for clinical use.44 In parallel, bacteria have developed resistance to some of the “last resort drugs” like quinupristin/dalfopristin dyad, linezolid and daptomycin.45,46 In the face of vanishing effectiveness of all antibacterial classes, the need for new compounds is immense. In that respect, natural products are still at the forefront of research as exemplified by the isolation of darobactin,47 teixobactin,48 numerous components present in plants49 and other sources of antibacterially active compounds.43,50 Problems with culturing some antibiotic-producing microorganisms51 and limited industrial-scale synthetic accessibility to complex antibacterial natural products are two drawbacks of this approach. Therefore, culture-independent methods capable of identifying and delivering first-in class synthetic antibacterial compounds and derivatization of well-established antibacterial drugs are two attractive alternatives. Other complementary strategies comprise photodynamic (PDT)52–54 and photothermal (PTT) antibacterial therapies in which light is used to produce cytotoxic reactive oxygen species (ROS) and thermal damage, respectively.55 Also, photoactivated carbon monoxide-releasing molecules (PhotoCORMs) are an emerging class of antimicrobials with promising activity against antibiotic resistant bacteria.56–58
One of the alternative approaches to combating bacterial pathogens is based on inorganic59–62 and organometallic compounds.63–78 The latter are defined by the presence of at least one metal–carbon (M–C) bond. The biological activity of organometallic compounds has been a subject of many studies, especially in the field of anticancer therapy.79–83 Organometallic compounds are attractive candidates for medical applications as their mechanisms of action are often multi-modal, and thus not commonly accessible with purely organic pharmacophores. This feature increases the chances for organometallic compounds to overcome drug-resistance in bacteria. Fig. 2 shows the multi-modal mechanisms of action for a hypothesized bioorganometallic compound in a bacterial cell. They comprise: (a) direct protein inhibition, (b) activation by light followed by singlet oxygen generation or CO release, (c) ligand(s) dissociation and macromolecule metalation, and (d) redox activation and ROS/oxidative stress generation.
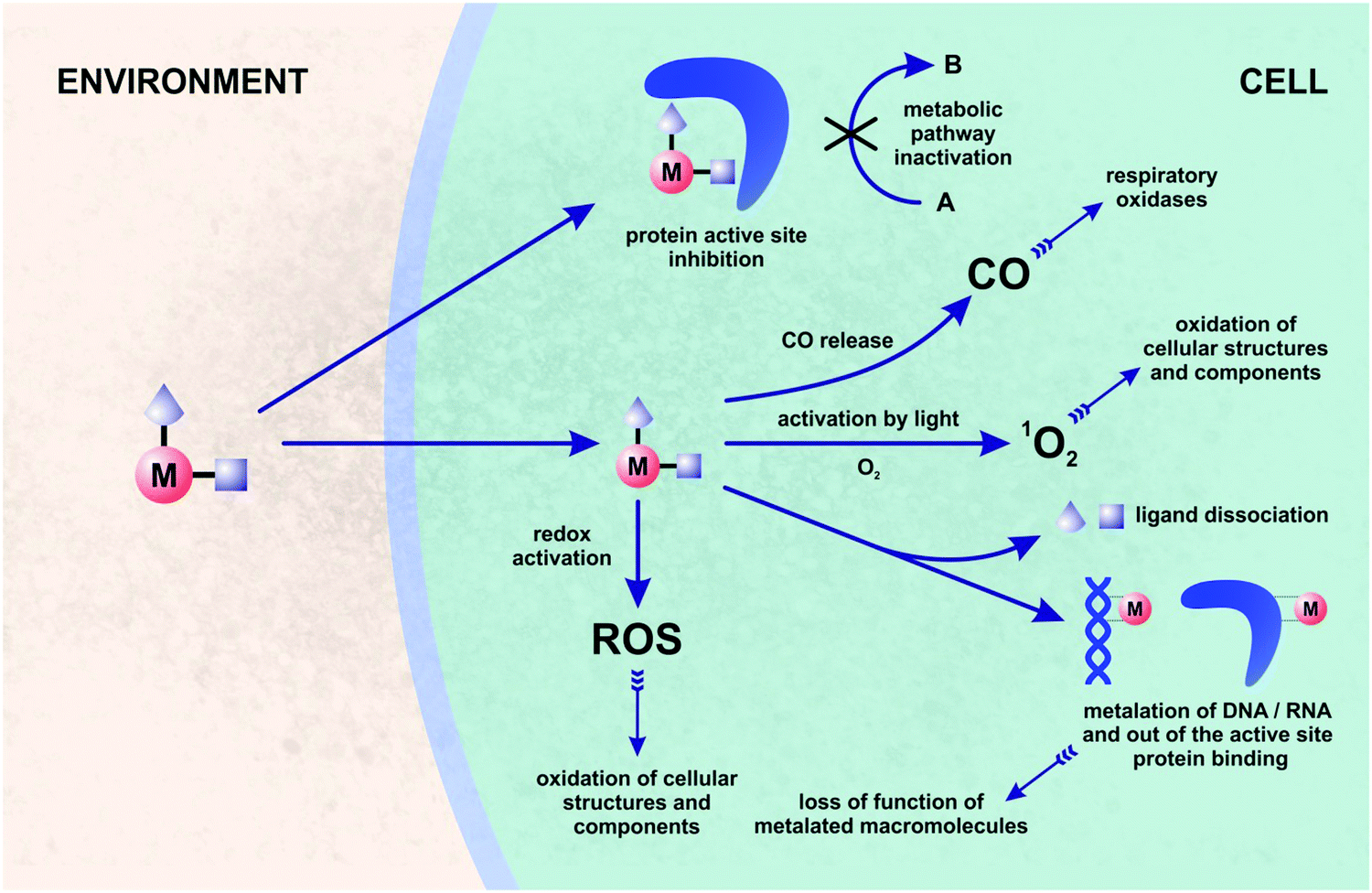 | ||
| Fig. 2 General view on possible mechanisms of action of organometallic compounds in bacteria. | ||
Antibiotics affecting more than one cellular or biomolecular target have been recognized as highly attractive pharmacological options. Fourteen from 27 recently introduced antibacterial drugs have demonstrated synergistic or multi-target mechanisms of action.44 Of note is also the fact that the analysis of the mechanisms of action for well-established antibacterial drugs revealed their ability to generate oxidative stress in bacteria, which increases/complements their target-specific modes of action.84–86
Applications of gold in therapy have long roots in mankind's history.87 However, it was not until Robert Koch's observation of the bacteriostatic activity of K[Au(CN)2] followed by the introduction of salvarsan by Paul Ehrlich that metals entered into the antibacterial field following the scientific method.88,89 Furthermore, the antiproliferative activity of cisplatin was first observed on E. coli strains.90 The next breakthrough was the discovery of ferrocene.91–94 This sandwich, redox active, iron-containing complex rapidly gained importance within organometallic,95 bioorganometallic96–98 and anti-infective organometallic chemistry.66 Beyond ferrocene, a number of other organometallic compounds have been studied as antibacterial agents.63–68 Strategies for the development of antibacterially active organometallic compounds are diverse. A major group relies on the derivatization of well-established antibacterial drugs and natural products,63,97 whereas others focus on the synthesis of entirely new species.99 This short review focuses mainly on organometallic-antibacterial drugs and on metal-based materials with antibacterial activity. It is not exhaustive; we just focus on the most important advancements and, in respect of organometallic compounds, we cover only the recent literature from 2017 to 2020. In respect of antibacterially active materials, a wider time frame was applied, providing a general view on the field. Readers broadly interested in the rapidly burgeoning field of organometallic antibacterial agents are encouraged to read preceding reviews.63–68
2. Organometallic derivatives of antibacterial drugs
2.1. Derivatives of β-lactams
β-lactam antibiotics have been at the forefront of antibacterial therapy since the 1940s. Their target-specific mechanism of action relies on penicillin binding protein (PBP) inhibition. PBPs are enzymes that catalyze the last steps of bacterial cell wall synthesis.100,101 β-lactams inhibit PBPs by irreversible acylation of a serine in a catalytic site.102,103 One of the major resistance mechanisms developed by bacteria against β-lactams involves the production of β-lactam hydrolyzing enzymes – β-lactamases.104 β-lactamases of class A, C and D all share a common Ser residue in an active site, whereas class B are Zn2+-dependent enzymes.105 The first ferrocenyl conjugates of β-lactams were obtained in the 1970s.106 In 2017, Kowalski, Chen and co-workers first reported ferrocenyl (Fc) and ruthenocenyl (Rc) 7-aminodesacetoxycephalosporanic acid (7-ADCA) derivatives 1–6 (Fig. 3).107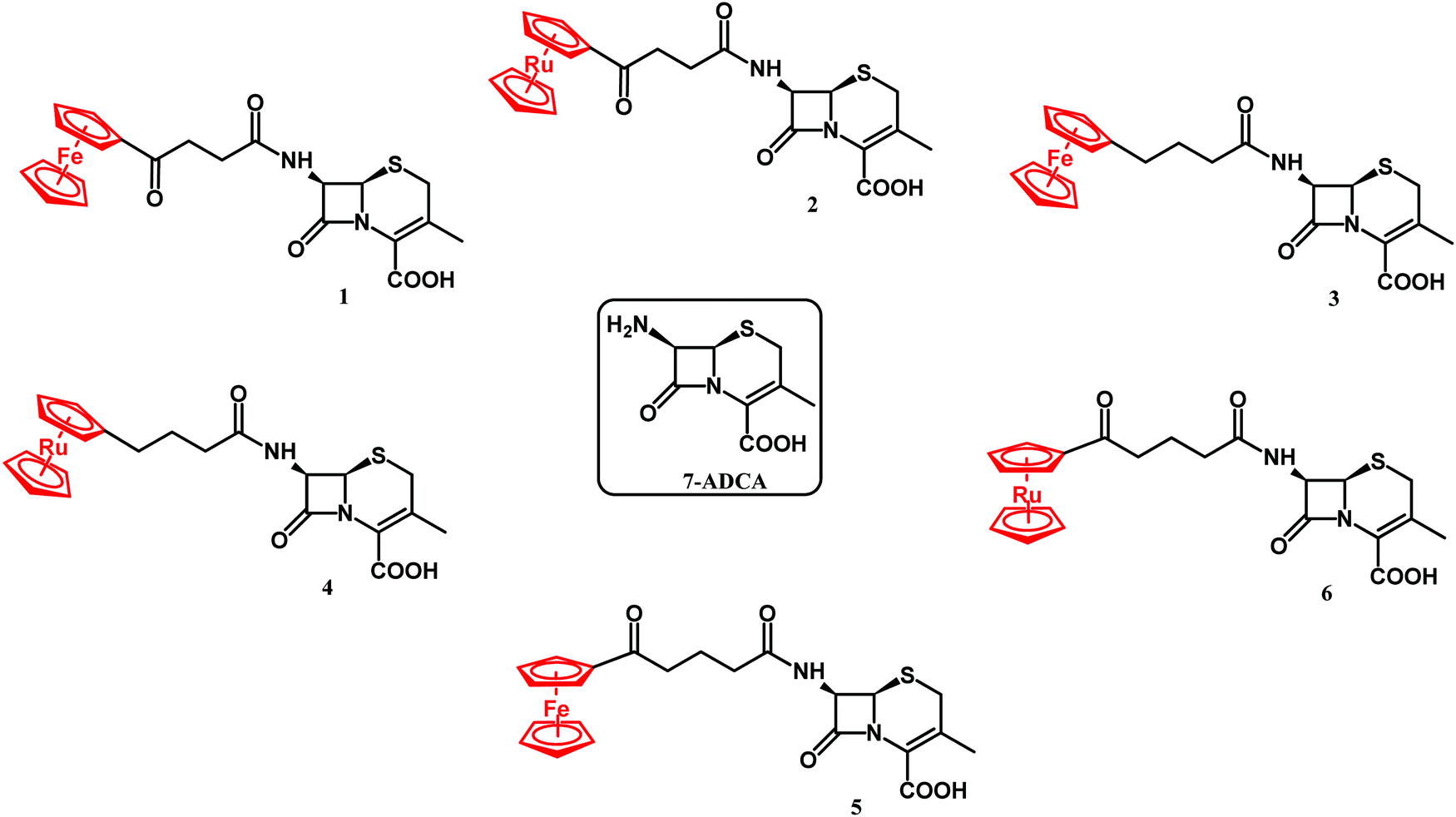 | ||
| Fig. 3 Structures of 7-ADCA and compounds 1–6. | ||
The inhibitory activity of these metallocenyl-β-lactams was examined against DD-carboxypeptidase 64-575 from Saccharopolyspora erythraea 64-575, CTX-M-14 class A β-lactamase and Bacillus cereus 569/H9 class B metallo-β-lactamase. DD-Carboxypeptidase 64-575, similar to PBPs, shows affinity to a number of β-lactam antibiotics and therefore serves as a good model for studies on PBP inhibition. The active site of CTX-M-14 β-lactamase also shares catalytic features with those of PBPs.108,109 In comparison with penicillin G, compounds 1–6 have shown enhanced inhibitory activities, substantiating the role of the metallocenyl entity in protein binding. The highest inhibition found was against DD-carboxypeptidase 64-575 with an inhibitory concentration (IC50) value at the nanomolar level. Remarkably, ruthenocenyl derivatives were better inhibitors than their ferrocenyl congeners. The most active Rc derivative 4 showed an IC50 value of 27 nM against DD-carboxypeptidase 64-575 and 7 μM against CTX-M-14 β-lactamase, whereas the inhibitory properties of compound 2 and 6 were characterized by IC50 values of 52 nM (DD-carboxypeptidase 64-575) and 44 μM (CTX-M-14 β-lactamase), and 81 nM (DD-carboxypeptidase 64-575) and 65 μM (569/H9 metallo-β-lactamase), respectively. Metallocenyl-β-lactams 1–6 showed lower activity than penicillin G and ampicillin in cell-based tests against model Gram-positive strains of methicillin-sensitive Staphylococcus aureus (MSSA), MRSA, vancomycin-intermediate S. aureus (VISA) and S. epidermidis. This feature can be explained by an impaired uptake of bacterial cells. On the other hand, the high inhibitory activities of 1–6 justified further studies aiming to understand the molecular basis of the interaction between the enzyme and the organometallic β-lactams.107 Indeed, X-ray crystallographic analysis of the CTX-M E166A mutant with ruthenocenyl derivative 2 proved the formation of a covalent acyl–enzyme complex between the β-lactam ring carbon of 2 and the Ser70Oγ of the protein. Fig. 4 shows compound 2 bound to the CTX-M E166A enzyme.
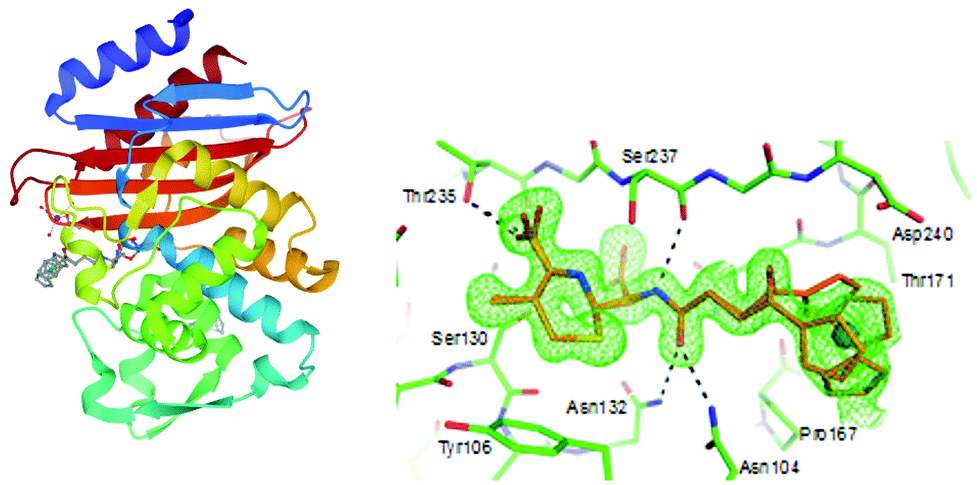 | ||
| Fig. 4 Left: Structure of the acyl–enzyme complex of CTX-M E166A with compound 2 (compound 2 depicted in grey; adopts two alternative conformations). Image from the RCSB PDB (rcsb.org) of PDB ID 5UJO (E. M. Lewandowski, Ł. Szczupak, S. Wong, J. Skiba, A. Guśpiel, J. Solecka, V. Vrček, K. Kowalski and Y. Chen, Organometallics, 2017, 36, 1673–1676). Right: Enlarged view of the acyl–enzyme complex nested in the protein binding site. The protein and compound are shown in green and gold, respectively. The unbiased Fo–Fc density map is shown in green at 2σ. The figure was reproduced from ref. 107 with permission from the American Chemical Society. | ||
XRD analysis revealed also an intact compound 2 captured at the crystal-packing interface.107 Finding a non-hydrolyzed 2 in the crystal was unique as metallocenyl β-lactams eluded small-molecule crystallographic characterization. Recently, Kowalski, Chen and co-workers reported on ferrocenyl and ruthenocenyl 7-aminocephalosporanic acid (7-ACA) derivatives 7 and 8 (Fig. 5).110
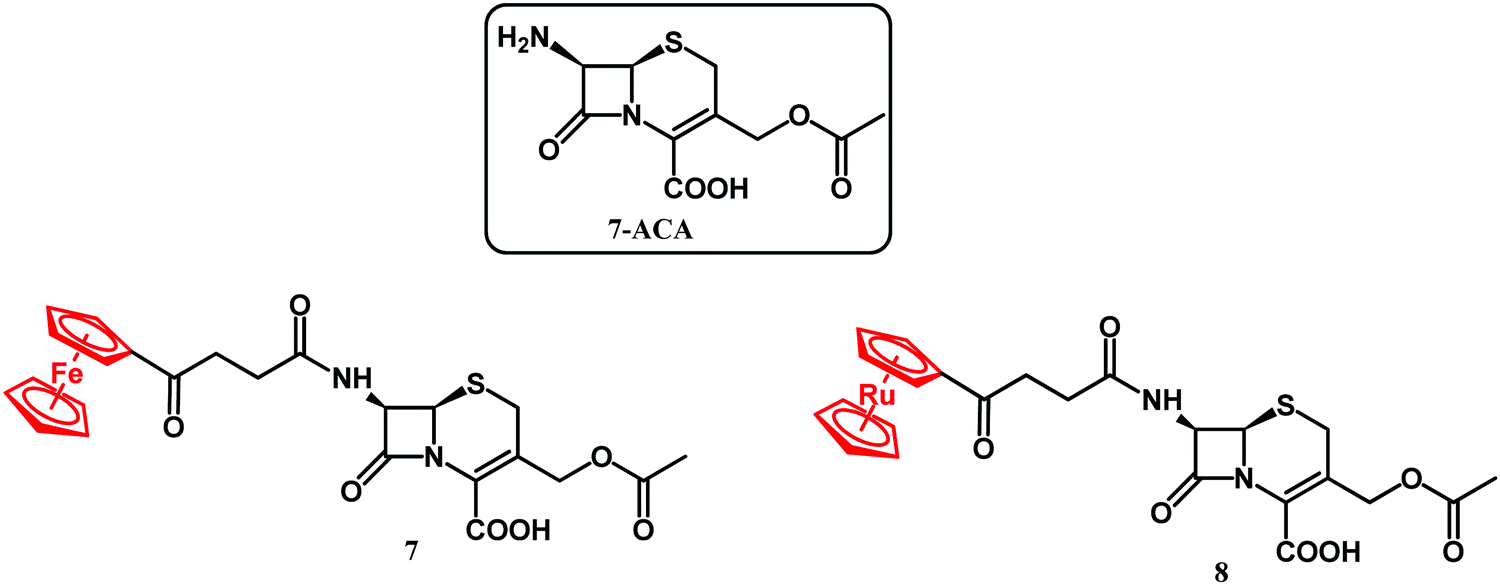 | ||
| Fig. 5 Structures of 7-ACA and compounds 7 and 8. | ||
The antibacterial activity of the two conjugates was assayed against reference Gram-negative bacterial strains (Escherichia coli ATCC 25922, E. coli NCTC 8196, Proteus vulgaris ATCC 49990, and Pseudomonas aeruginosa NCTC 6749), Gram-positive bacterial strains (S. aureus ATCC 6538, S. aureus ATCC 29213, S. aureus ATCC 25923, S. epidermidis ATCC 12228, and Enterococcus faecalis ATCC 29212), two methicillin-resistant S. aureus bacterial strains, and twelve clinical isolates of S. aureus, including two MRSA isolates. No antibacterial activity against Gram-negative strains and significant activity against Gram-positive Staphylococci was observed. Ruthenocenyl compound 8 showed MIC values 2–8 times lower than that of the ferrocenyl derivative 7. Compared with the activity of metallocenyl-7-ADCA compounds 1–6 against the ATCC 29213 strain, the 7-ACA compounds 7 and 8 had reduced the MIC values by 64× and 16× for the Fc and Rc conjugates, respectively. Compounds 7 and 8 showed particularly high antibacterial activity against clinical strains of S. aureus isolated from the naso-pharynx and from ulcers/furuncles. In all these cases, the activity of ruthenocenyl compound 8 (MIC value 0.85 μM) was higher than that of 7 (MIC value 15 μM) and ampicillin. In the case of drug-resistant bone isolates of S. aureus, the ruthenocenyl derivative 8 again showed higher activity than its Fc counterpart and ampicillin. Furthermore, conjugate 8 showed two-fold higher antibacterial activity than ampicillin did against the MecA positive strain of S. aureus EDCC 5443. A lack of toxicity toward mammalian cells is the essential feature of any new antibacterial drug candidate. To address this issue, the cytotoxicity of 7 and 8 was tested in vitro against mouse murine fibroblast L929 and human cervical epithelioid carcinoma HeLa cells. Both tested compounds showed negligible toxicity with IC50 higher than 440 μM. The antibacterial activity studies on 7 and 8 were further augmented by the identification of the complex crystal structure of compound 8 with CTX-M-14 β-lactamase and by scanning electron microscopy (SEM) micrographs.110 In conclusion, the higher antibacterial activity of the ruthenocenyl β-lactams compared to the ferrocenyl counterparts can be ascribed to favorable interactions of the Rc entity with amino acid residues in the active site of PBP enzymes.
In 2020, Mislin and co-workers reported on Au(I)–ampicillin complexes 9–14 (Fig. 6).111 The antibacterial activity of compounds 9–14 was tested against Gram-positive S. aureus, S. epidermidis, E. faecalis and E. faecium as well as against Gram-negative E. coli strains. It turned out that the size of the gold-coordinated phosphine has a key role in the activity, whereas the nature of the X linker has not. Accordingly, the bulkiest triphenylphosphine derivative 12 was less active than the rest of that series as well as ampicillin. On the contrary, less sterically hindered triethylphosphine compounds 10, 13 and 14 all showed improved (or comparable) activity compared to ampicillin against Gram-positive strains. The most potent against Staphylococcus species and several Enterococcus strains compound 10 was tested to evaluate its toxicity in healthy human hepatocytes. It did not affect eukaryotic cell viability at concentrations up to 10 μM. Above that concentration the compound was cytotoxic. Despite this drawback, compound 10 remained as a good starting point for the design of new gold-based β-lactam antibacterial agents.
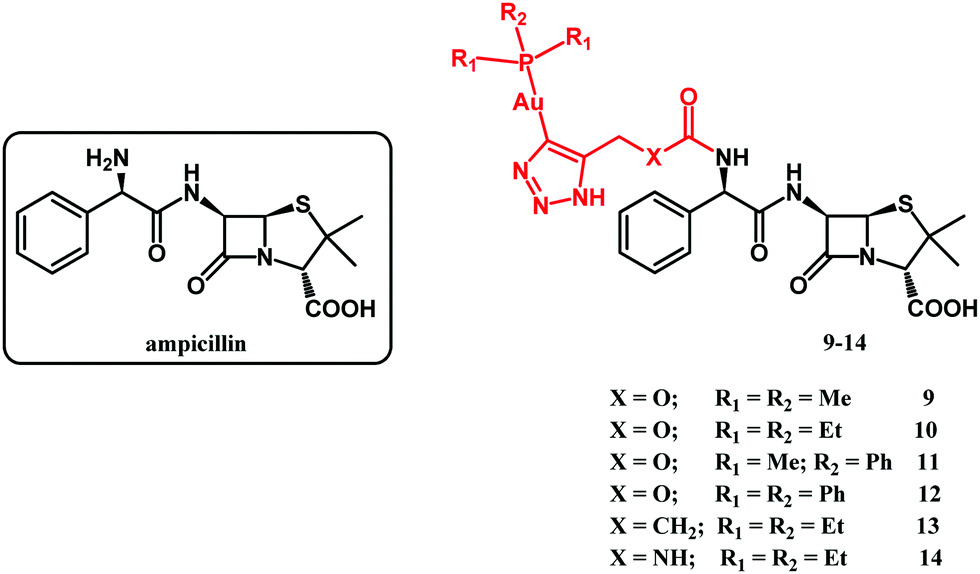 | ||
| Fig. 6 Structures of ampicillin and compounds 9–14. | ||
2.2. Derivatives of ciprofloxacin
Quinolones are by far the most successful synthetic antibacterial drugs on a global scale. The mechanism of antibacterial activity of quinolones involves inhibition of type II bacterial DNA topoisomerases; namely gyrase (the primary target in G-negative bacteria) and topoisomerase IV (the primary target in G-positive bacteria).112,113 Both of these proteins are crucial for bacterial physiology and are broadly distributed. Topoisomerase inhibition by quinolones collides with bacterial replication forks and transcription complexes, which ultimately leads to SOS system induction (SOS after international telegraph (or optical) distress signal “SOS” in the Morse alphabet). When DNA strand breaks surpass the SOS response, the bacterial cell dies. Kowalski, Stączek and co-workers reported on six organometallic ciprofloxacin derivatives 9–14 (Fig. 7).114 | ||
| Fig. 7 Structures of ciprofloxacin and compounds 9–14. | ||
Antibacterial activity studies of 9–14 showed that conjugation of the organometallic moiety to the ciprofloxacin scaffold enhances the bactericidal effect. Accordingly, the N-alkyl derivatives 12–14 were the most active compounds, although derivatives 9–11 also showed significant activity. Compounds 13 and 14 were substantially more active against the Gram-negative E. coli ATCC 25922 strain than ciprofloxacin. Their MIC values were 0.0006 and 0.0001 μM, respectively, while the MIC of ciprofloxacin against the same strain was 0.01 μM. Thus, in the case of the cymantrenyl derivative 14, the MIC value was 100 times lower than that of ciprofloxacin. Furthermore, compound 14 was more active than ciprofloxacin against the S. aureus ATCC 6538 and K. pneumoniae ATCC 13883 strains with MICs of 0.4 and 0.001 μM, respectively. For the latter, the MIC value of 14 was 50 times lower than that of ciprofloxacin. Cymantrenyl derivative 11 overcame drug-resistance in two clinical bone isolates of S. aureus (MRSA) strains. Another feature of organometallic ciprofloxacin derivatives was their ability to eradicate pathogenic bacteria in the stationary growth phase. In this regard, the most active was cymantrene conjugate 14. It decreased the viability of the E. coli ATCC strain from 3.3 × 108 (untreated control) to 2.2 × 106 CFU mL−1. The remarkable antibacterial activity of compounds 9–14 stems from their dual mechanism of action. With the exception of 9, all compounds inhibited the introduction of supercoils by E. coli gyrase and the decatenation process by S. aureus topoisomerase IV. This is the first mechanism of action and it originated from the ciprofloxacin portion of the conjugates. The second mechanism of action pertains to the ability of oxidative stress induction in bacterial cells and stems from the organometallic portion of those conjugates. The synergistic effect between the two mechanisms enables them to overcome drug resistance in bacteria as well as to eliminate them even in the stationary phase of growth. Fig. 8 shows the structural changes in the bacterial morphology upon treatment with the organometallic ciprofloxacin derivatives 12–14.
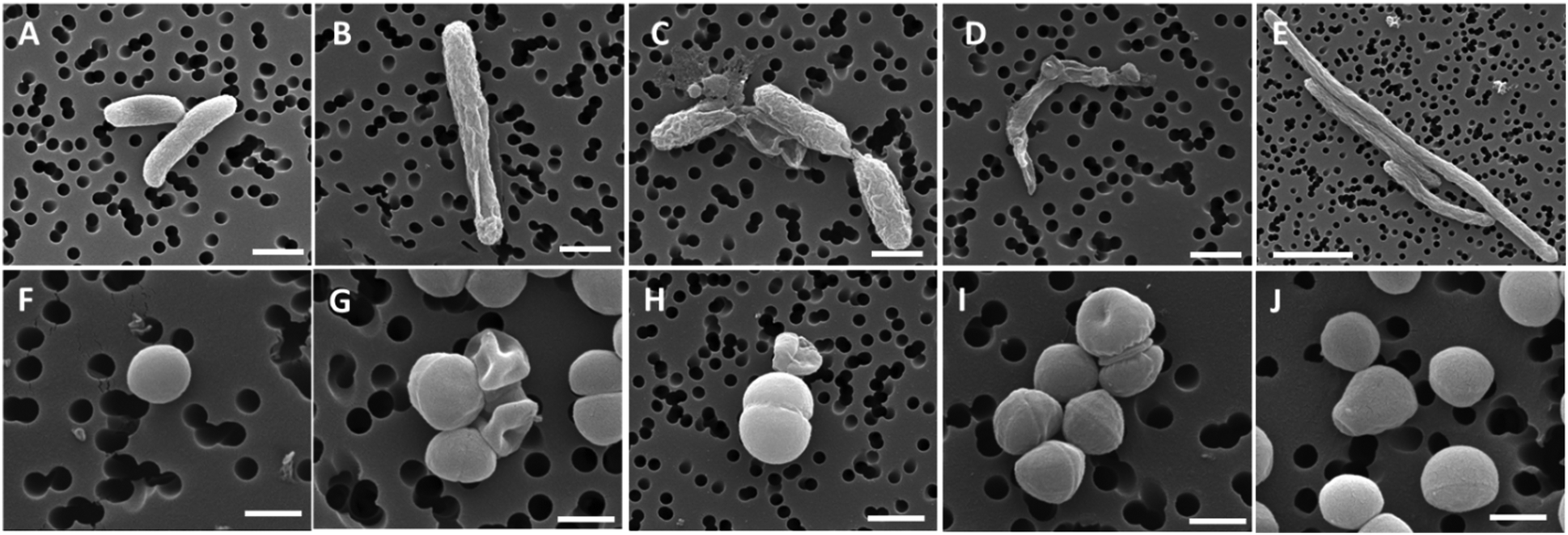 | ||
| Fig. 8 SEM micrographs of the bacterial cell morphology after exposure to compounds 12–14 and ciprofloxacin. Top panels, E. coli cells; bottom panels, S. aureus cells. (A and F) control, non-treated samples; and bacteria treated with compounds 12 (B and G), 13 (C and H), 14 (D and I), and ciprofloxacin (E and J). Scale bars: 500 nm in F, G, I, and J; 1 μm in A–D and H; and 3 μm in E. Reproduced from ref. 114 with permission from the Royal Society of Chemistry. | ||
In 2018, Pokharia and co-workers reported on triorganotin(IV) ciprofloxacin complexes 15 and 16 (Fig. 9).115 Recently, a closely related diphenyltin(IV) derivative 17 has been reported by Hadjikakou and co-workers (Fig. 9).116
 | ||
| Fig. 9 Structures of compounds 15–17. | ||
Compounds 15 and 16 showed better antibacterial activity than ciprofloxacin against Gram-positive (S. aureus and E. faecalis), and Gram-negative (K. pneumoniae, E. coli, P. aeruginosa and P. mirabilis) strains. Likewise, diphenyltin complex 17 was more active than its constituents (ciprofloxacin and diphenyl dichloride) against S. aureus and S. epidermidis as well as against E. coli and P. aeruginosa strains. The MICs of 17 for these strains were in the nanomolar concentration range. Furthermore, complex 17 eradicated the biofilm of P. aeruginosa and S. aureus more effectively than ciprofloxacin did.
2.3. Derivatives of isoniazid, pyrazinamide and ethambutol
Rifampicin, isoniazid, pyrazinamide and ethambutol are four drugs used in treatment of the initial phase of tuberculosis.117 Isoniazid is a prodrug which upon transformation to the isonicotinoyl radical by the M. tuberculosis KatG enzyme and further reaction with NAD(P) inhibits mycobacterial cell wall formation.41,117 Smith and co-workers reported on ferrocenyl isoniazid (18–20), ferrocenyl pyrazinoic acid hydrazide (21–23) and half-sandwich derivatives 24–26 (Fig. 10).118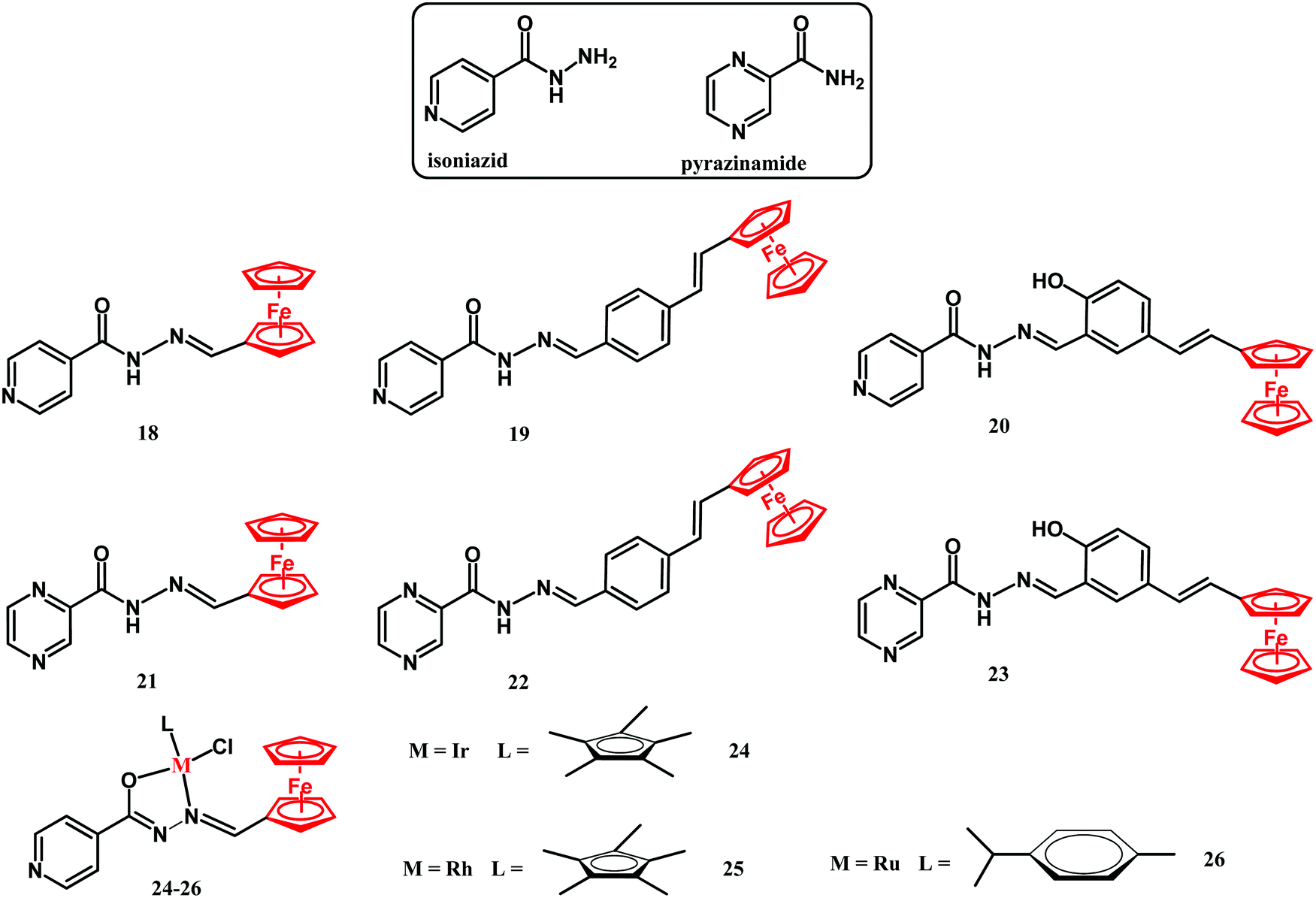 | ||
| Fig. 10 Structures of isoniazid, pyrazinamide and compounds 18–26. | ||
The activity of compounds 18–26 has been examined against the M. tuberculosis H37Rv strain in glycerol-based GAST-Fe and in glucose-based Middlebrook 7H9-ADC growth media. The compounds showed interesting growth medium-dependent M. tuberculosis eradication properties. Their antituberculosis activity in the glycerol-based GAST-Fe medium was noticeably higher than that in the glucose-based Middlebrook medium. The most active compound was the isoniazid derivative 18. The MIC90 value of 0.39 μM was comparable to that of the parental isoniazid. Incorporation of the second metal center gave no improvement in their activity, as the MICs of binuclear compounds 24–26 were in the 0.416 to 0.968 μM range. The authors hypothesized that the mechanisms of action of compound 18 might involve the disruption of the glycerol metabolism, resulting in the accumulation of toxic products in M. tuberculosis H37Rv cells.
In 2019, an interesting report on the cationic pentamethylcyclopentadienyl-Ir(III) ethambutol complex 27 (Fig. 11) was published by Merola and co-workers.119
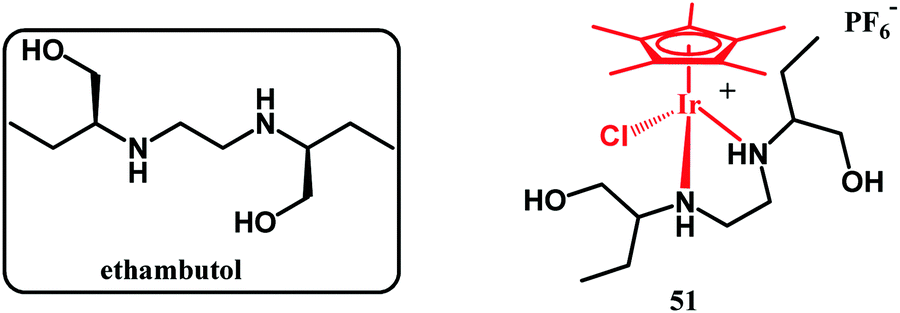 | ||
| Fig. 11 Structures of ethambutol and compound 27. | ||
This complex showed no activity against M. tuberculosis, which could be due to its ligand-dissociation inertness together with its positive charge, which impairs cellular uptake. On the other hand, 27 showed activity against S. aureus MSSA (MIC = 35 μg mL−1) and MRSA (MIC = 40 μg mL−1). Studies aiming to understand the mechanisms of antistaphylococci action are underway.
2.4. Derivatives of sulfonamides and trimethoprim
Sulfonamides are antimetabolites (antifolates) with bacteriostatic activity. Their mechanism of action stems from competitive binding of para-aminobenzoic acid (PABA) to dihydropteroate synthase (DHPS).41,117 DHPS plays a key role in dihydrofolic acid (DHF) biosynthesis. Similar to sulfonamides, trimethoprim also interferes with the folate biosynthesis pathway. It acts as an inhibitor of dihydrofolate reductase (DHFR), an enzyme which transforms DHF into tetrahydrofolic acid (THF). Folates are cofactors for nucleobase biosynthesis in bacteria. Thus, depletion of their cellular availability prevents DNA synthesis and impairs bacterial cell division and growth. In 2017, Aranciba and co-workers reported on ferrocenyl and cyrhetrenyl (Cyr) derivatives 28–33 (Fig. 12), all having a sulfonamide structural core and variable substituents in the phenyl para position.120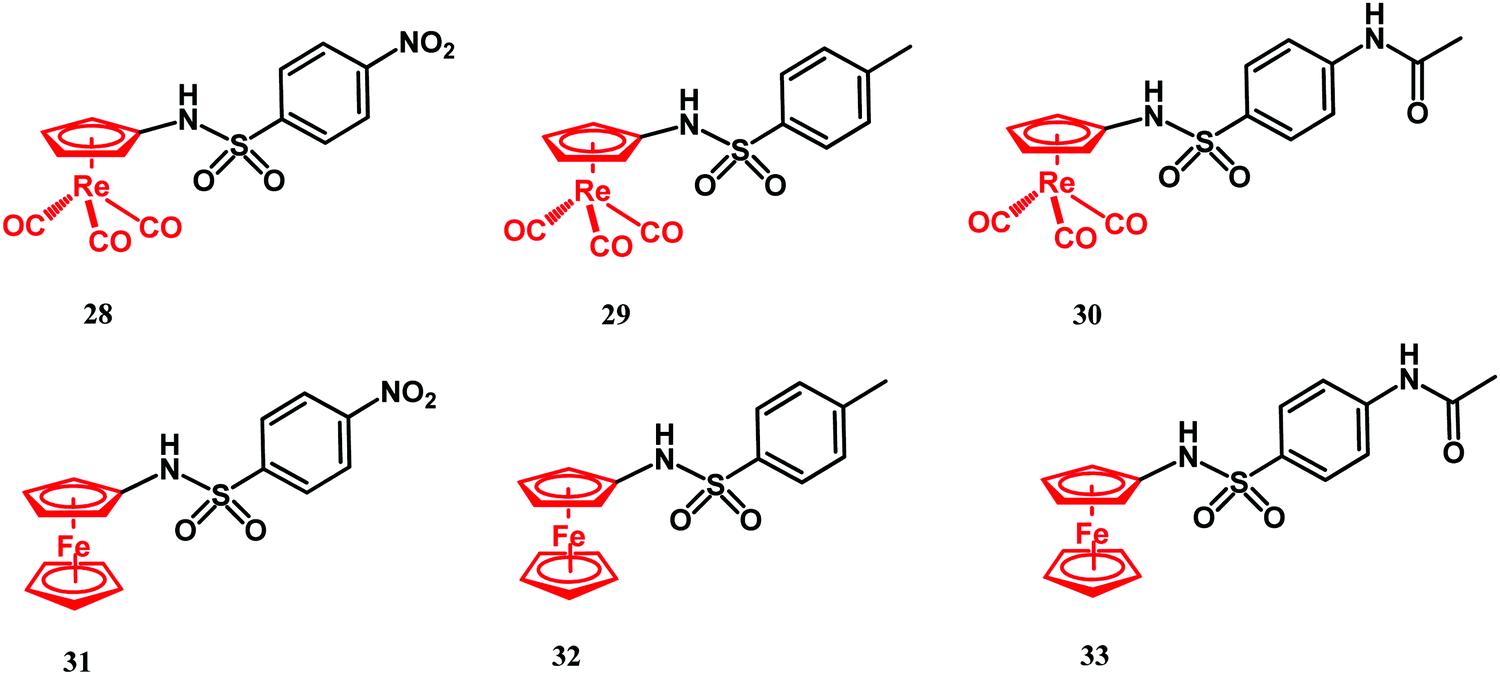 | ||
| Fig. 12 Structures of compounds 28–33. | ||
Compounds 28–33 were tested for antitubercular activity against the M. tuberculosis mc26230 model strain. Isoniazid was used as reference drug and had MIC99 = 0.4 μM. The antibacterial activity of Cyr derivatives 28–30 was higher than that of their ferrocenyl congeners 31–33 and ca. 465 times lower than isoniazid. The better antibacterial activity of the Cyr derivatives over the Fc compounds was explained by the electron-withdrawing vs. electron-donating properties of the former vs. the latter moiety.
In 2018, Sadler and co-workers reported on half-sandwich Ru(II), Rh(III) and Ir(III) complexes 34–47 (Fig. 13) containing sulfadoxine derived ligands.121 Sulfadoxine is a drug administered together with pyrimethamine in antimalarial therapy. They both block folate biosynthesis in the Plasmodium falciparum parasite by targeting DHPS and DHFR.
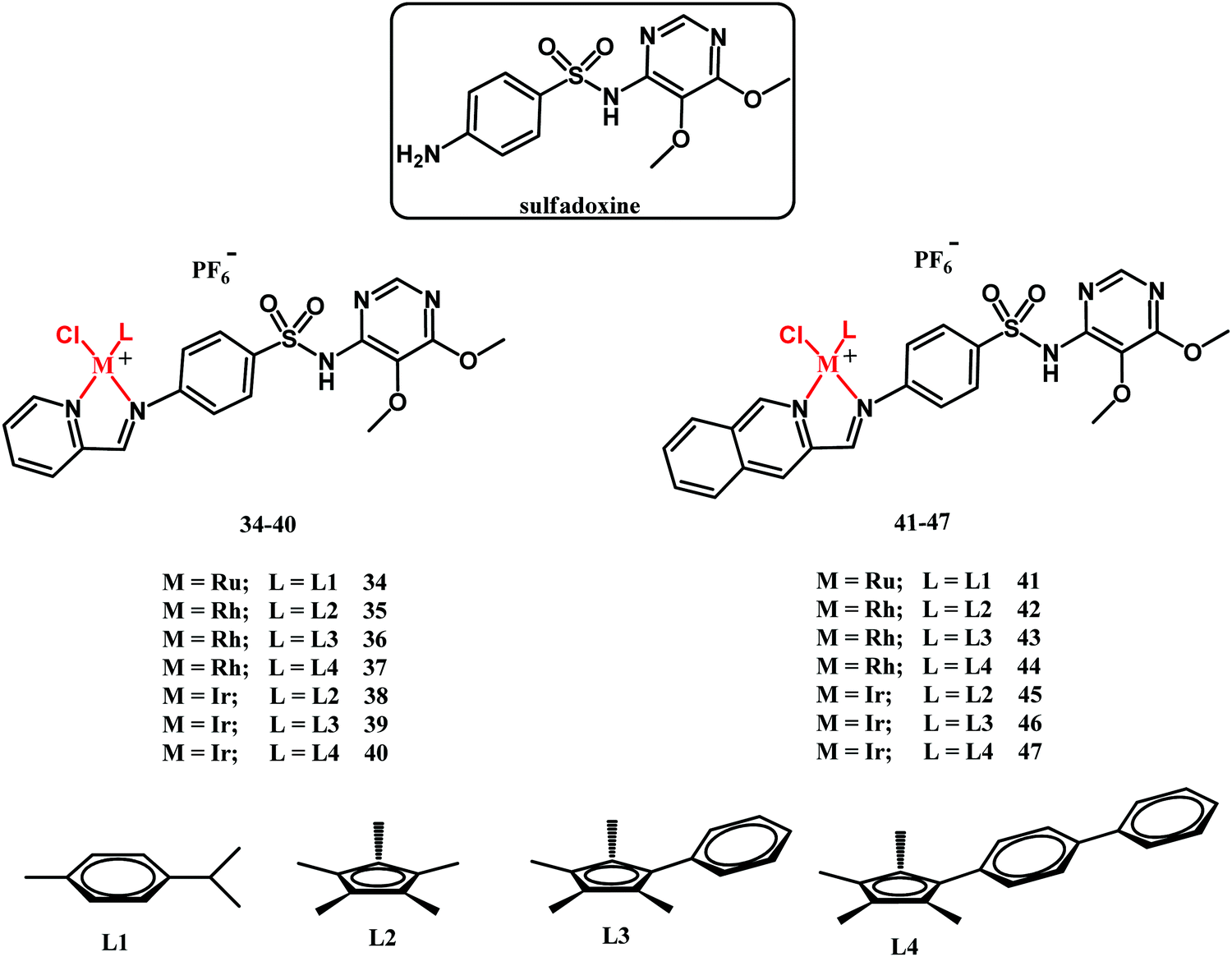 | ||
| Fig. 13 Structures of sulfadoxine and compounds 34–47. | ||
In vitro biological activity studies of 34–47 have been primarily directed toward the screening of their inhibitory activity against the P. falciparum 3D7 (chloroquine-sensitive), Dd2 (chloroquine-sensitive) and NG54 Late Stage Gametocyte (LSG) strains as well as against Trichomonas vaginalis parasite strain G3. Furthermore, the antibacterial potential of compounds 34–47 has been tested against the laboratory M. tuberculosis H37Rv strain. Ruthenium complexes 34 and 41 showed no antibacterial activity under the experimental conditions applied (MIC50 = 100 μM). The antituberculosis activity of pyridylimino-sulfadoxine rhodium complexes 35–37 was superior to the activity of their iridium counterparts 38–40. Interestingly, the reverse trend in activity has been observed for quinolylimino-sulfadoxine derivatives 42–47. In this respect, the most active compounds were iridium complexes 46 and 47 (both with MIC50 = 3.13 μM), whereas their rhodium counterparts 43 and 44 showed MIC50 = 6.25 and 50 μM, respectively. Compounds 35, 36, 43, 44, 46 and 47 exhibited antimycobacterial activity significantly higher than that of the parental sulfadoxine drug. On the other hand, none of the fourteen assayed complexes rivaled rifampicin and isoniazid antimycobacterial drugs in activity. The structure–activity relationship (SAR) for antimycobacterial activity is not straightforward and has not been provided in those studies. Compounds 34–47 showed no or limited activity against the T. vaginalis G3 strain. In respect of the antiplasmodial activity, the rhodium complexes were, in general, more active that the iridium compounds, whereas the ruthenium derivatives showed no activity. An interesting observation was that compounds 35–40 and 42–47 were active against sexual last stage gametocytes (LSG), whereas sulfadoxine, pyrimethamine and chloroquine were inactive. This feature clearly shows a beneficial role of the metal in the sulfadoxine activity improvement. SAR studies have shown that the quinolylimino-sulfadoxine compounds are more potent than their pyridyl-sulfadoxine analogues. Other activity-control factors are the size/lipophilicity of the Cp ligand and the rate of the chloride to water ligand exchange reaction.
A recent account on organometallic sulfonamide derivatives was published in 2019 by Metzler-Nolte and co-workers.122 They obtained sulfamethoxazole Ru(II) and Re(I) complexes 48–53 (shown in Fig. 14) and they tested them against four S. aureus strains (DSM 20231, methicillin-resistant ATCC 43300, BAA 976 and BAA977), as well as against Acinetobacter baumannii and Pseudomonas aeruginosa strains. Sulfamethoxazole is a DHPS inhibitor and is clinically used in combination with DHF inhibitor trimethoprim in a formulation known as cotrimoxazole (sulfamethoxazole:trimethoprim, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 formulation) to treat a wide range of bacterial infections.123,124
:1 formulation) to treat a wide range of bacterial infections.123,124
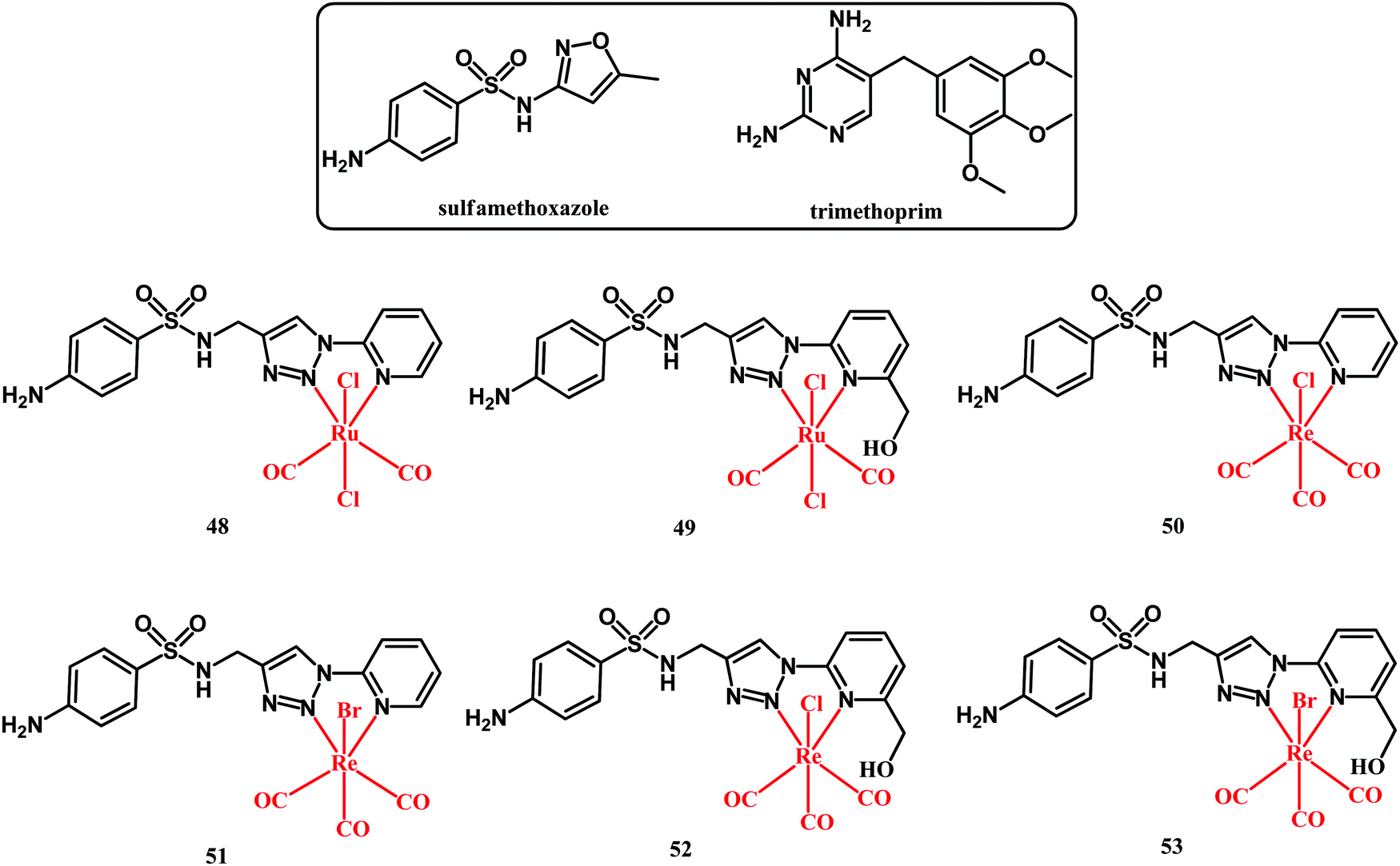 | ||
| Fig. 14 Structures of sulfamethoxazole, trimethoprim and compounds 48–53. | ||
Sulfamethoxazole alone was not active against the tested bacterial strains up to a concentration of 512 μg mL−1. On the contrary, trimethoprim alone was highly active against all strains except for Gram-negative A. baumannii and P. aeruginosa. For the majority of the strains, the cotrimoxazole activity was more potent on a molar basis than trimethoprim alone. Organometallic complexes 50, 51 and 52 showed medium activity against the S. aureus strains with MIC = 190–750 μM. This activity was lower than that of trimethoprim alone (MIC = 6.9–14.0 μM). Noticeably, an improvement in the activity was observed when a combination of complexes 50 and 51 with trimethoprim was tested. Like in the case of cotrimoxazole, a molar ratio of 20:1 (Re complex:trimethoprim) was formulated. Both Re-trimethoprim formulations showed similar activity toward all four S. aureus strains including the MRSA ATCC 43300 strain. In the latter case, cotrimoxazole had a MIC of 960:48 μM, whereas the MICs for the 50:trimethoprim and 51:trimethoprim formulations were 98:4.9 and 46:2.3 μM, respectively. This is an indication of a significant anti-MRSA activity increase. Although the mechanism behind such an activity increase was not given, the obtained results pinpoint the advantages offered by combined organometallic-organics for antibacterial therapy.
Recently Das, Ganeshpandian and co-workers reported on half-sandwich Ru(η6-p-cymene)-trimethoprim compound 54 (Fig. 15).125 The complex has been tested alone as well as after being loaded into polydiacetylene(PDA)-based liposomes against healthy human HEK-293, lung adenocarcinoma A549, breast carcinoma MCF-7 and liver carcinoma HepG2 cells. Compound 54 showed cytotoxicity toward healthy HEK-293 cells. Yet, this undesirable effect was diminished by the liposomal formulation of the drug. Neither 54 alone nor when encapsulated in liposomes showed activity against A549 and MCF-7 cells. On the contrary, compound 54 showed cytotoxic activity against liver carcinoma HepG2 cells. This activity was potentiated by the encapsulation of 54 into liposomal micelles. Insight into the action of compound 54 revealed that the Ru complex shows proapoptotic induction in HepG2 cells together with the ability of DNA scission. The antibacterial activity of 54 alone and when loading 54 into a liposomal carrier has been tested with the agar disk diffusion method against S. aureus and P. aeruginosa strains. The selection of these two strains was rationalized due to their prevalence as one of the most common pathogens which are responsible for nosocomial infections in patients with cancer.126 Rather unexpectedly, ruthenium trimethoprim complex 54 showed no improvement in antibacterial activity over the trimethoprim drug alone. The zone of inhibition for trimethoprim was 26 and 29 mm against the S. aureus and P. aeruginosa strains, respectively, at a 100 μM concentration. At the same high concentration, the zone of inhibition for 54 was 23 and 20 mm, respectively. Likewise, almost the same diameters of inhibition were obtained for 54 and trimethoprim at the lowest concentrations tested. For completeness, the liposome loaded 54 showed no antibacterial activity even at a 300 μM concentration. This can likely be explained by the hindered uptake of the liposome through the bacterial envelope. Although the mechanism of antibacterial activity of 54 was not examined, it can be predicted that the trimethoprim portion of the complex acts as a folate synthesis inhibitor, whereas the role of the Ru(η6-p-cymene) portion could be more speculative and may pertain to DNA/protein metalation.
 | ||
| Fig. 15 Structure of compound 54. | ||
3. Derivatives of non-antibacterial drugs (metformin, phenformin and aspirin)
Biguanides are a group of nitrogen-rich organic compounds with established pharmacological relevance. Belonging to this class are metformin and phenformin, two drugs used for diabetes treatment. In 2018, Sadler and co-workers reported on the synthesis and antimicrobial activity studies of a series of biguanide Ir(II) complexes.127 Within the studied series of compounds, metformin and phenformin complexes 55–57 and 58 (Fig. 16) were evaluated, respectively. The complexes were assayed against a wide panel of G-negative bacteria including E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii, and G-positive bacteria B. subtilis, S. pyogenes, E. faecalis, S. epidermidis and S. aureus (MSSA and MRSA), as well as against the pathogenic fungi Candida albicans and Cryptococcus neoformans. | ||
| Fig. 16 Structures of metformin, phenformin and compounds 55–58. | ||
The more hydrophilic complexes 55 and 56 were weakly active against the tested bacterial strains, with MICs over 54.3 μM. The more hydrophobic biphenyl derivative 57 showed instead superior antibacterial activity, with MICs in the range 3.2 to 12.6 μM against Gram-positive bacteria and 25 μM against an E. coli strain. The better activity of 57 can be explained by the increased uptake of the compound into bacterial cells. Likewise, even the more hydrophobic phenformin complex 58 showed superior antibacterial activity against Gram-positive bacteria with MICs in the range 0.17 to 2.7 μM. It also showed increased activity against the Gram-negative pathogens E. coli (MIC = 5.4 μM), K. pneumoniae (MIC = 21.6 μM), P. aeruginosa (MIC = 43.2 μM) and A. baumannii (MIC = 5.4 μM). Combined fluorescence microscopy and transmission electron microscopy (TEM) studies showed that the Ir(III) biguanide complexes do not disrupt the bacterial cell wall. Instead, Ir(III) biguanides can act as delivery systems to bacterial cells, where they dissociate to release free biguanidine ligands and reactive Ir(III) species. The latter can react with thiol-containing biomolecules, e.g., L-cysteine, to form dimers or other byproducts.127 On the other hand, the released binguanide can bind cellular metal ions and inhibit vital metabolic pathways in bacteria, which ultimately leads to the experimentally observed bactericidal effect. Importantly, as studied by Sadler, Ir(III) biguanide complexes showed overall low toxicity toward mammalian cells as well as low hemolytic activity. The Ir(III) biguanide complexes stand as an example of repurposing essentially non-antibacterial drugs into highly active antibacterial agents.
Another recent example of an organometallic derivative of a non-antibacterial drug which was examined for antibacterial activity is the aspirin Re(I) phenanthroline complex 59 (Fig. 17).128
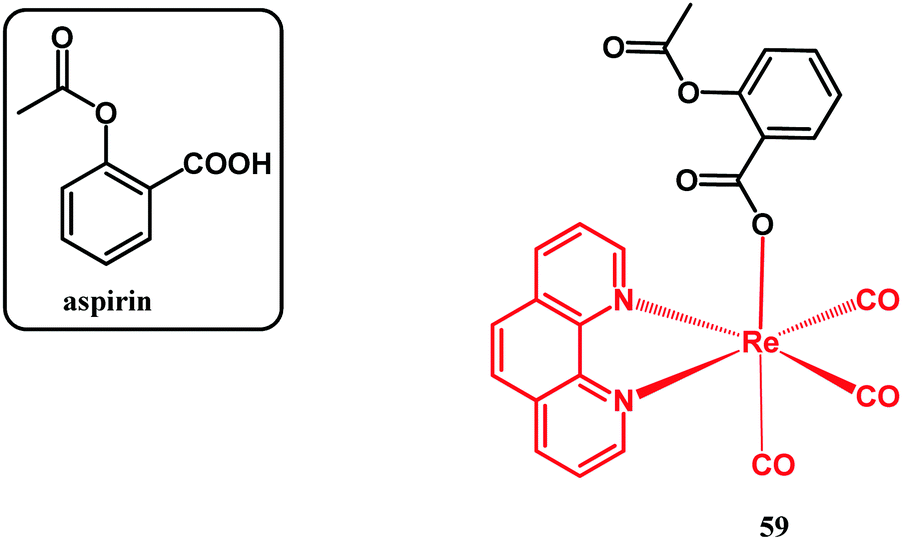 | ||
| Fig. 17 Structure of aspirin and compound 59. | ||
It has been reported by Kowalski and co-workers, with the main aim to study its optical properties, its accumulation in living mammalian cells by confocal microscopy and its anticancer activity. In connection with these major studies, the antibacterial activity of 59 was screened against S. aureus ATCC 6538 and ATCC 29213 and E. coli NCTC 8196 model strains. The examined complex showed activity with a MIC of 50.8 μM against all strains tested. This activity was better than that of nitrofurantoin and weaker than that of ampicillin, which were used as references in the course of the assays.
4. Metal-containing antibacterially active materials
Metals have been used as antimicrobial materials since antiquity, mainly for water disinfection, food preservation, crop protection, and health care. Their toxicity depends on several parameters including their physicochemical form (i.e., metal speciation), either in their elemental or oxidized states, their affinity to bind DNA, and their ability to disrupt membrane function, to bind donor ligands (including extra- and intracellular proteins and enzymes), to impair nutrient uptake, to alter signal transduction, and to induce oxidative stress, among others.129 In their elemental (zerovalent) form, metals can form nanoparticles under a controlled environment which confines crystal nucleation and growth. Those nanoparticles due to their large area per volume ratio are prone to rapid oxidation in physiological media and consequently their released ions are chemisorbed on the surface of the metal nanoparticles, the nanoparticle acting as a reservoir for the sustained release of oxidized species.130 Once in solution, their oxidation state can also change upon contact with cells. In this regard, Balfourier and co-workers demonstrated that gold nanoparticles are rapidly endocyted inside endosomes and biodegraded due to the acidic environment and the participation of NADPH oxidase as catalyst for the production of the highly oxidizing superoxide free radicals.131 Subsequently, metal-binding proteins (i.e., metallothioneins) are able to biomineralize dissolved species to form recrystallized nanoparticles.The oxidation state of metal nanoparticles is key to determine their antimicrobial action. In this regard, Xiu and co-workers demonstrated that under anaerobic incubation conditions the antimicrobial action of silver nanoparticles against E. coli strain K12 (ATCC 25404) was impaired due to the lack of ionic silver.132 Direct contact also plays a key role in the antimicrobial action. We demonstrated that silver and gold-loaded chitosan films were efficient in the elimination of Staphylococcus aureus ATTC 6538 and 9213 strains, whereas, under the same conditions, the exudates released from those films were unable to reduce bacterial contamination.133,134
The antimicrobial action also depends on the availability of the cytotoxic released ions. In 19th-century Europe, silver nitrate was used as eyedrops in newborns for the prevention of ophthalmia, reducing its incidence by a factor of 20–30.135 In this case, ionic silver was immediately available in aqueous solution, but, later on, sustained release systems have been developed using different materials as ion hosts to reduce the large cytotoxicity associated with uncontrolled release. Inorganic (e.g., zeolites, clays, etc.), inorganic–organic hybrid (MOFs, POMs, etc.) and organic (metal exchange resins) hosts are used to provide sustained release of antimicrobial ionic metals. Different application sectors have incorporated those materials to take advantage of their long-term antimicrobial action.
As we mentioned before, antimicrobial metals are commonly used in water disinfection, food preservation, crop protection, and health care. It is important to point out that not only the metal itself is responsible for its antimicrobial action but also its surface chemistry including ionic metallic species chemisorbed on the surface and also the presence of different ligands used during surface functionalization. In this section of the review we will focus only on health care applications including antimicrobial medical devices, surfaces and indoor air pollutant control with special emphasis on those studies which have been assessed in clinical settings.
4.1. Antibiofouling medical surfaces and indoor air pollutant control
Medical surfaces can be coated with metal containing paints as a prophylactic measure to prevent biofouling. Zinc oxide and titanium oxide are the most frequent semiconductor metal oxides used as nanofillers in paints to prevent healthcare associated nosocomial infections in healthcare facilities. Those nanofillers are added to modify the rheological properties of the paints, promoting fast drying and avoiding paint drips, and also to take advantage of their photocatalytic action against pathogenic microorganisms. Recent advances focus on the development of highly active visible-light induced photocatalytic coatings based on those materials instead of the traditional UV-activated ones. In this regard, Krumdieck and co-workers developed intricate titanium dioxide nanostructures containing carbon and prepared by chemical vapor deposition on stainless steel doors and faucet handles, rendering a 100 times larger surface area and enhanced antimicrobial action compared to conventional titania coatings and powders.136 However, Hu and co-workers have recently shown that non harmful bacteria can become harmful (i.e., spore forming bacteria) when using antimicrobial paints and they state that we should be judicious in the use of antimicrobial products.137 Most antimicrobial paints are validated using model Gram + (e.g., S. aureus) and Gram – bacteria (e.g., E. coli), but it is important to recall that a large amount of known bacteria can remain in a viable but nonculturable state. In this regard, Robben and co-workers demonstrated that commercially available household cleaners in combination with inorganic salts can induce a viable but non-culturable state in five human pathogens.138Besides Ti and Zn, several other metals have been reported as antimicrobials on medical wards. Copper nanoparticles and copper-zeolite nanocomposites have been used as nanofillers within polymeric paints to render antimicrobial coatings on plastic waiting room chairs and on metal hospital IV pools, respectively, reducing the total viable microorganisms present, regardless of the microorganism tested.139 A randomized control trial between 2010 and 2011 in the ICUs of 3 hospitals was carried out by distributing patients admitted in those units in rooms with or without copper alloy surfaces, and the rates of incident acquired infections and/or colonization with MRSA or vancomycin-resistant Enterococcus (VRE) in each type of room were compared.140 Patients cared for in rooms having copper alloy surfaces (i.e., bed rails, overbed tables, IV poles, and arms of the visitor's chair) had a significantly lower rate of incident infection and/or colonization than patients treated in standard rooms. A prospective cohort study involving 621 patients hospitalized in a medical intensive care unit having titania-based photocatalysts coated on high touch surfaces and walls also revealed that the MRSA acquisition rate was significantly reduced.141
Commercial formulations of silver including AgION Technology's AgION™ (i.e., silver/zinc ions contained in an LTA type zeolite) have been demonstrated as efficient antimicrobial coatings.142 Silver exchanged Y-type zeolites have not only been used on surfaces but also as filters to remove airborne pathogens (bacteria and fungi) in medical facilities.143 Titania-coated cordierite foams have also been used to photo-catalytically degrade gaseous acetaldehyde, which is associated with sick building syndrome, and airborne or droplet-based infectious pathogens: E. coli, P. aeruginosa, L. pneumophila, K. pneumoniae, and MRSA.144
4.2. Antimicrobial medical devices and metal-based treatments
The extensive medicinal use of metals was displaced with the discovery of antibiotics, although there are still several medical devices in use which incorporate metals for the treatment of pathogenic microorganisms.Silver-based catheters (e.g., AcryMed's SilvaGard™) are widely used in clinical settings. A 12-month randomized crossover trial with 27878 patients compared rates of nosocomial catheter-associated urinary tract infection in patients with silver-coated and uncoated catheters, revealing a decrease in the risk of infection by 21% among study wards randomized to silver-coated catheters and by 32% among patients in whom silver-coated catheters were used on the wards.145 A meta-analysis of 117 reports and eight trials with a total of 2355 patients satisfying the inclusion criteria revealed that silver alloy catheters are significantly more effective in preventing urinary tract infections than silver oxide catheters are.146 Silver-containing catheters are recommended for short term use only and their benefit for patients with long-term catheters remains unclear.
Endotracheal tubes (ETTs) used in mechanically ventilated patients can be colonized by biofilm forming bacteria and thus contribute to the development of ventilator-associated pneumonia. Thorarinsdottir and co-workers reported that compared to uncoated PVC-based ETTs, the use of noble-metal-coated (containing silver, gold, and palladium (Bactiguard® AB, Sweden)) PVC-based ETTs was associated with reduced high-grade biofilm formation, although no significant difference was observed between silicon-coated ETTs and noble-metal-coated ETTs.147 ETTs containing silver sulfadiazine in the interior of their lumen showed the lack of bacterial biofilm in 23 patients intubated in intensive care units following a phase I–II randomized clinical trial.148
Silver coated tumor endoprostheses have been associated with a lower rate of early periprosthetic infection in 85 patients and also debridement with antibiotic treatment and retention of the implant appeared to be more successful with those silver-coated implants.149 Implantcast Ltd's Mutars® (i.e., silver coated bone replacements) showed a reduced rate of infection in 51 sarcoma patients receiving those megaprostheses compared to those receiving uncoated titanium controls.150
Silver sulfadiazine, as a topical antiseptic used in partial and full thickness burns to prevent infection, promotes regeneration and reduces inflammation. Several common formulations include silver, copper and bismuth in their formulation and are commercialized as topical antiseptics (e.g., Silvadene®, Xeroform®, etc.).151 Chronic wounds are difficult to treat and frequently polymicrobial populations are colonizing those and tissue debridement and antibiotic therapy fail in their management. Antimicrobial wound dressings have been designed to release antimicrobial compounds and some of them include metals in their formulation. To date, more studies are needed to corroborate that antimicrobial-releasing dressings are more clinically and cost efficient than conventional wound dressings in the management of chronic infected wounds.152 For instance, after the analysis of 12 randomized controlled trials reporting 13 comparisons, O'Meara and co-workers concluded that current evidence does not support the use of silver-based products in the management of venous leg ulcers.153
Metal ions are also used in the treatment of eye infections and in periodontal and peri-implant diseases. Eye drops, mouthwashes, dentifrices, dental implants and delivery devices have incorporated metallic ions (i.e., silver, copper, zinc, etc.) to prevent infection, but special attention should be paid to their potential toxicological effects.154
Not only are metals incorporated into devices but also they can be applied systemically. In this regard, a phase 1 clinical trial in individuals with cystic fibrosis and chronic P. aeruginosa airway infections intravenously treated with gallium (as a disruptor of bacterial iron metabolism) revealed improved lung function inhibiting P. aeruginosa growth.155 The use of iron chelators (such as gallium) and other metals as siderophore synthesis inhibitors opens new avenues in the management of pathogenic bacteria.
4.3. Photodynamic and photothermal antimicrobial therapy based on metal nanoparticles
PDT uses light and tissue oxygen to generate ROS with the aid of organic photosensitizers. Those organic molecules (e.g., porphyrins, chlorophylls and dyes) are prone to photobleaching and, consequently, metal nanoparticles have been introduced in this field to avoid such a limitation. Upconversion nanoparticles (i.e., rare-earth based lanthanide- or actinide-doped transition metals) and large band-gap semiconductor nanoparticles (e.g., zinc oxide, titanium oxide, copper oxide, quantum dots, etc.) have been used in the photoinactivation of pathogenic bacteria usually colonizing infected topical wounds due to the inherent limitation in the light penetration depth achievable. For instance, rapid sterilization and accelerated wound healing have been reported by using Zn2+ and graphene oxide in the management of bacteria-infected wounds using preclinical models.156 Mao and co-workers recently reported the use of MOFs for the in vivo labelling of bacteria and simultaneous photodynamic treatment and therapy guided observation by using fluorescence imaging.157 PEGylated W18O49 nanosheets have been used for the multi-modal imaging of the gastrointestinal tract and imaging-guided photothermal (PTT) sterilization in vivo using externally applied near infrared light.158 The combination of both PDT and PTT is also possible by combining photothermal MoS2 films and an organic photosensitizer (IR780) assisted by glutathione oxidation accelerated by NIR light, rendering synergistic and rapid killing of Staphylococcus aureus biofilms in vivo.159Despite all those successful approaches, future research should be focused on the synthesis of metal nanoparticles having reduced toxicity and fast biodegradability in order to compete with conventional organic photosensitizers.
6. Conclusion and outlook
This review primarily focuses on organometallic drug derivatives and metal-containing materials having antibacterial activity. In addition, this antibacterial action is also reported for non-antibacterial drugs like metformin, phenformin and aspirin. The data discussed herein show that the combination of an organometallic moiety with an organic pharmacophore (drug) in many cases results in derivatives which are able to circumvent drug resistance in bacteria. This activity is achieved by the bimodal (or multimodal) mode of action of organometallic–drug conjugates. In such cases, the chances of developing antibiotic resistance are reduced because simultaneous bacterial mutations against several mechanisms of action are improbable. On the other hand, the impaired uptake of some organometallic–drug derivatives and their potential toxicity against mammalian cells hinder the full exploration of their antibacterial activity in the clinic. This review also shows that metals and metal nanoparticles can benefit traumatology, orthopedic surgery, wound management, and ocular, periodontal and respiratory diseases thanks to their prophylactic and bactericidal use; however, despite all recent advances and even having several metal-based products on the market, more multicenter prospective clinical trials are needed to validate the large amount of scientific literature encompassing metal based antimicrobial materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from University of Łódź, Poland is gratefully acknowledged.References
- N. J. Planavsky, D. Asael, A. Hofmann, C. T. Reinhard, S. V. Lalonde, A. Knudsen, X. Wang, F. O. Ossa, E. Pecoits, A. J. B. Smith, N. J. Beukes, A. Bekker, T. M. Johnson, K. O. Konhauser, T. W. Lyons and O. J. Rouxel, Nat. Geosci., 2014, 7, 283–286 CrossRef CAS.
- C. Thomazo, M. Ader and P. Philippot, Geobiology, 2011, 9, 107–120 CrossRef CAS.
- K. Lepot, Earth-Sci. Rev., 2020, 209, 103296 CrossRef CAS.
- K. Lepot, A. Addad, A. H. Knoll, J. Wang, D. Troadec, A. Béché and E. J. Javaux, Nat. Commun., 2017, 8, 14890–14900 CrossRef CAS.
- H. W. Jannasch and M. J. Mottl, Science, 1985, 229, 717–725 CrossRef CAS.
- E. Spieck, M. Spohn, K. Wendt, E. Bock, J. Shively, J. Frank, D. Indenbirken, M. Alawi, S. Lücker and J. Hüpeden, ISME J., 2020, 14, 364–379 CrossRef CAS.
- A. Schippers, L. N. Neretin, J. Kallmeyer, T. G. Ferdelman, B. A. Cragg, R. J. Parkes and B. B. Jørgensen, Nature, 2005, 433, 861–864 CrossRef CAS.
- K. R. Redeker, J. P. J. Chong, A. Aguion, A. Hodson and D. A. Pearce, J. R. Soc., Interface, 2017, 14, 20170729 CrossRef.
- D. A. Cowan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19749–19750 CrossRef CAS.
- J. K. Fredrickson, J. M. Zachara, D. L. Balkwill, D. Kennedy, S. W. Li, H. M. Kostandarithes, M. J. Daly, M. F. Romine and F. J. Brockman, Appl. Environ. Microbiol., 2004, 70, 4230–4241 CrossRef CAS.
- R. Conrad and A. V. Vlassov, Med. Sci. Rev., 2015, 2, 92–103 CrossRef.
- H. G. Groot, Y. J. van de Vegte, N. Verweij, E. Lipsic, J. C. Karper and P. van der Harst, Sci. Rep., 2020, 10, 14771–14779 CrossRef CAS.
- C. Pleguezuelos-Manazo, J. Puschhof, A. R. Huber, A. van Hoeck, H. M. Wood, J. Nomburg, C. Gurjao, F. Manders, G. Dalmasso, P. B. Stege, F. L. Paganelli, M. H. Geurts, J. Beumer, T. Mizutani, Y. Miao, R. van der Linden and S. van der Elst, Genomics England Research Consortium, K. C. Garcia, J. Top, R. J. L. Willems, M. Giannakis, R. Bonnet, P. Quirke, M. Meyerson, E. Cuppen, R. van Boxtel and H. Clevers, Nature, 2020, 580, 269–273 CrossRef.
- M. A. Spyrou, K. I. Bos, A. Herbig and J. Krause, Nat. Rev. Genetics, 2019, 20, 323–340 CrossRef CAS.
- A. Fleming, Rev. Infect. Dis., 1929, 2, 129–139 CrossRef.
- M. Barber, Lancet, 1948, 2, 641–644 CrossRef CAS.
- A. Schatz, E. Bugie and S. A. Waksman, Exp. Biol. Med., 1944, 55, 66–69 CrossRef CAS.
- J. Cotton and D. A. Mitchison, Br. Med. J., 1948, 2, 1009–1015 CrossRef.
- M. P. Jevons, Br. Med. J., 1961, 1, 124–125 CrossRef.
- M. T. Parker and J. H. Hewitt, Lancet, 1970, 295, 800–804 CrossRef.
- L. Elwell, M. Roberts, L. Mayer and S. Falkow, Antimicrob. Agents Chemother., 1977, 11, 528–533 CrossRef CAS.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS.
- B. Marshall, M. Roberts, A. Smith and S. B. Levy, J. Infect. Dis., 1984, 149, 1028–1029 CrossRef CAS.
- M. D. Iseman, N. Eng. J. Med., 1993, 329, 784–791 CrossRef CAS.
- D. M. P. De Oliveira, B. M. Forde, T. J. Kidd, P. N. A. Harris, M. A. Schembri, S. A. Beatson, D. L. Paterson and M. J. Walker, Clin. Microbiol. Rev., 2020, 33, e00181 CrossRef CAS.
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, 122–129 CrossRef.
- WHO. WHO's global report on antimicrobial resistance 2019, https://www.who.int/antimicrobial-resistance/interagency-coordination-group/final-report/en/.
- P. Wiggins, Exp. Opin. Invest. Drugs, 2004, 13, 889–902 Search PubMed.
- D. J. Hoban, D. J. Biedenbach, A. H. Mutnick and R. N. Jones, Microbiol. Infect. Dis., 2003, 45, 279–285 CrossRef CAS.
- D. M. Morens, G. K. Folkers and A. S. Fauci, Nature, 2004, 430, 242–249 CrossRef CAS.
- S. Kumari and V. J. Ram, Drugs Today, 2004, 40, 487–500 CrossRef CAS.
- D. A. Mitchison, Am. J. Respir. Crit. Care Med., 2005, 171, 699–706 CrossRef.
- S. B. Levy, Sci. Am., 1998, 278, 46–53 CrossRef CAS.
- K. Bush, Clin. Infect. Dis., 2001, 32, 1085–1089 CrossRef CAS.
- D. L. Paterso, W.-C. Ko, A. Von Gottberg, S. Mohapatra, J. M. Casellas, H. Goossens, L. Mulazimoglu, G. Trenholme, K. P. Klugman, R. A. Bonomo, L. B. Rice, M. M. Wagener, J. G. McCormack and V. L. Yu, Ann. Intern. Med., 2004, 140, 26–32 CrossRef.
- P. A. Bradford, Clin. Microbiol. Rev., 2001, 14, 933–951 CrossRef CAS.
- J. F. Barrett, Exp. Opin. Ther. Targets, 2004, 8, 515–519 CrossRef.
- C. D. Salgado, B. M. Farr and D. P. Calfee, Clin. Infect. Dis., 2003, 36, 131–139 CrossRef.
- C. G. Whitney, M. M. Farley, J. Hadler, L. H. Harrison, C. Lexau, A. Reingold, L. Lefkowitz, P. R. Cieslak, M. Cetron, E. R. Zell, J. H. Jorgensen and A. Schuchat, N. Engl. J. Med., 2000, 343, 1917–1924 CrossRef CAS.
- K. A. Gordon, D. J. Bidenbach and R. N. Jones, Diagn. Microbiol. Infect. Dis., 2003, 46, 285–289 CrossRef CAS.
- R. J. Anderson, P. W. Groundwater, A. Todd and A. J. Worsley, Antibacterial Agents. Chemistry, Mode of Action, Mechanisms of Resistance and Clinical Applications, Wiley and Sons, Ltd, Chichester, West Sussex, UK, 2012 Search PubMed.
- S. Levy, The Antibiotic Paradox: How Misuse of Antibiotics Destroys their Curative Powers, Perseus, Cambridge, 2002 Search PubMed.
- F. von Nussbaum, M. Brands, B. Hinzen, S. Weigand and D. Häbich, Angew. Chem., Int. Ed., 2006, 45, 5072–5129 CrossRef CAS.
- C. Wetzel, M. Lonneman and C. Wu, Eur. J. Med. Chem., 2021, 209, 112931 CrossRef CAS.
- T. T. Tran, D. Panesso, N. N. Mishra, E. Mileykovskaya, Z. Guan, J. M. Munita, J. Reyes, L. Diaz, G. M. Weinstock, B. E. Murray, Y. Shamoo, W. Dowhan, A. S. Bayer and C. A. Arias, mBio–Am. Soc. Microbiol, 2013, 4, e00281–13 Search PubMed.
- V. G. Meka and H. S. Gold, Clin. Infect. Dis., 2004, 39, 1010–1015 CrossRef CAS.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O’Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D’Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS.
- S. Gibbons, Nat. Prod. Rep., 2013, 30, 988–1027 RSC.
- K. M. G. O’Connell, J. T. Hodgkinson, H. F. Sore, M. Welch, G. P. C. Salmond and D. R. Spring, Angew. Chem., Int. Ed., 2013, 52, 10706–10733 CrossRef.
- J. T. Staley and A. Konopka, Annu. Rev. Microbiol., 1985, 39, 321–346 CrossRef CAS.
- F. Cieplik, D. Deng, W. Crielaard, W. Buchalla, E. Hellwig, A. Al-Ahmad and T. Maisch, Crit. Rev. Microbiol., 2018, 44, 571–589 CrossRef CAS.
- N. A. Smith, P. Zhang, S. E. Greenough, M. D. Horbury, G. J. Clarkson, D. McFeely, A. Habtemariam, L. Salassa, V. G. Stavros, C. G. Dowson and P. J. Sadler, Chem. Sci., 2017, 8, 395–404 RSC.
- E. Sauvageot, M. Elie, S. Gaillard, R. Daniellou, P. Fechter, I. J. Schalk, V. Gasser, J.-L. Renaud and G. L. A. Mislin, Metallomics, 2017, 9, 1820–1827 RSC.
- S. Wu, A. Li, X. Zhao, C. Zhang, B. Yu, N. Zhao and F.-J. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 17177–17183 CrossRef CAS.
- M. Tinajero-Trejo, N. Rana, C. Nagel, H. E. Jesse, T. W. Smith, L. K. Wareham, M. Hippler, U. Schatzschneider and R. K. Poole, Antioxid. Redox Signaling, 2016, 24, 765–780 CrossRef CAS.
- J. Betts, C. Nagel, U. Schatzschneider, R. Poole and R. M. la Ragione, PLoS One, 2017, 12, e0186359 CrossRef.
- N. Rana, H. Jesse, M. Tinajero-Trejo, J. Butler, M. L. von und zur Mühlen, C. Nagel, U. Schatzschneider and R. K. Poole, Microbiology, 2017, 163, 1477–1489 CrossRef CAS.
- A. Regiel-Futyra, J. M. Dąbrowski, O. Mazuryk, K. Śpiewak, A. Kyzioł, B. Pucelik, M. Brindell and G. Stochel, Coord. Chem. Rev., 2017, 351, 76–117 CrossRef CAS.
- A. Frei, Antibiotics, 2020, 9, 90 CrossRef CAS.
- M. B. Harbut, C. Vilchèze, X. Luo, M. E. Hensler, H. Guo, B. Yang, A. K. Chatterjee, V. Nizet, W. R. Jacobs Jr., P. G. Schultz and F. Wang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4453–4458 CrossRef CAS.
- F. Li, J. G. Collins and F. R. Keene, Chem. Soc. Rev., 2015, 44, 2529–2542 RSC.
- M. Patra, G. Gasser and N. Metzler-Nolte, Dalton Trans., 2012, 41, 6350–6358 RSC.
- M. A. Sierra, L. Casarrubios and M. C. de la Torre, Chem. – Eur. J., 2019, 25, 7232–7242 CrossRef CAS.
- U. Schatzschneider, Antimicrobial activity of organometal compounds: Past, present, and future prospects, in Advances in Bioorganometallic Chemistry, ed. T. Hirao and T. Moriuchi, Elsevier, Amsterdam, 2018, ISBN 978-0-12-814197-7 Search PubMed.
- B. S. Ludwig, J. D. G. Correia and F. E. Kühn, Coord. Chem. Rev., 2019, 396, 22–48 CrossRef CAS.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627–2639 RSC.
- B. Albada and N. Metzler-Nolte, Acc. Chem. Res., 2017, 50, 2510–2518 CrossRef CAS.
- P. Güntzel, C. Nagel, J. Weigelt, J. W. Betts, C. A. Pattrick, H. M. Southam, R. M. La Ragione, R. K. Poole and U. Schatzschneider, Metallomics, 2019, 11, 2033–2042 RSC.
- Z. Ude, I. Romero-Canelón, B. Twamley, D. F. Hughes, P. J. Sadler and C. J. Marmion, J. Inorg. Biochem., 2016, 160, 210–217 CrossRef CAS.
- C. Schmidt, L. Albrecht, S. Balaupramaniam, R. Misgeld, B. Karge, M. Brönstrup, A. Prokop, K. Baumann, S. Reichl and I. Ott, Metallomics, 2019, 11, 533–545 RSC.
- C. Schmidt, B. Karge, R. Misgeld, A. Prokop, R. Franke, M. Brönstrup and I. Ott, Chem. – Eur. J., 2017, 23, 1869–1880 CrossRef CAS.
- F. Chen, J. Moat, D. McFeely, G. Clarkson, I. J. Hands-Portman, J. P. Furner-Pardoe, F. Harrison, C. G. Dowson and P. J. Sadler, J. Med. Chem., 2018, 61, 7330–7344 CrossRef CAS.
- Q. Laurent, L. K. Batchelor and P. J. Dyson, Organometallics, 2018, 37, 915–923 CrossRef CAS.
- A. Frei, M. Amado, M. A. Cooper and M. A. T. Blaskovich, Chem. – Eur. J., 2020, 26, 2852–2858 CrossRef CAS.
- A. Mansour and K. Radacki, Dalton Trans., 2020, 49, 4491–4501 RSC.
- A. Gupta, P. Prasad, S. Gupta and P. K. Sasmal, ACS Appl. Mater. Interfaces, 2020, 12, 35967–35976 CrossRef CAS.
- Y. Zheng, W. Liu, Y. Chen, H. Jiang, H. Yan, I. Kosenko, L. Chekulaeva, I. Sivaev, V. Bregadze and X. Wang, Organometallics, 2017, 36, 3484–3490 CrossRef CAS.
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- G. Palermo, A. Magistrato, T. Riedel, T. von Erlach, C. A. Davey, P. J. Dyson and U. Rothlisberger, ChemMedChem, 2016, 11, 1199–1210 CrossRef CAS.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 1549–1560 CrossRef CAS.
- S. R. Martínez, A. M. Durantini, M. C. Becerra and G. Cosa, ACS Infect. Dis., 2020, 6, 2468–2477 CrossRef.
- P. Belenky, J. D. Ye, C. B. M. Porter, N. R. Cohen, M. A. Lobritz, T. Ferrante, S. Jain, B. J. Korry, E. G. Schwarz, G. C. Walker and J. J. Collins, Cell Rep., 2015, 13, 968–980 CrossRef CAS.
- J. J. Foti, B. Devadoss, J. A. Winkler, J. J. Collins and G. C. Walker, Science, 2012, 336, 315–319 CrossRef CAS.
- R. Rubbiani, B. Wahrig and I. Ott, J. Biol. Inorg. Chem., 2014, 19, 961–965 CrossRef CAS.
- R. Koch, Dtsch. Med. Wochenstr, 1890, 16, 756–757 Search PubMed.
- N. C. Lloyd, H. W. Morgan, B. K. Nicholson and R. S. Ronimus, Angew. Chem., Int. Ed., 2005, 44, 941–944 CrossRef CAS.
- B. Rosenberg, L. van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632–635 RSC.
- G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS.
- E. O. Fischer and W. Pfab, Z. Naturforsch., B: J. Chem. Sci., 1952, 7, 377–379 Search PubMed.
- Ferrocenes: Ligands, Materials, and Biomolecules, ed. P. Štěpnička, Wiley-VCH, Chichester, 2008 Search PubMed.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS.
- K. Kowalski, Coord. Chem. Rev., 2018, 366, 91–108 CrossRef CAS.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2020, 104, 5931–5986 CrossRef.
- M. Wenzel, M. Patra, C. H. R. Senges, I. Ott, J. J. Stepanek, A. Pinto, P. Prochnow, C. Vuong, S. Langklotz, N. Metzler-Nolte and J. E. Bandow, ACS Chem. Biol., 2013, 8, 1442–1450 CrossRef CAS.
- P. Macheboeuf, C. Contreras-Martel, V. Job, O. Dideberg and A. Dessen, FEMS Microbiol. Rev., 2006, 30, 673–691 CrossRef CAS.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32, 234–258 CrossRef CAS.
- L. I. Llarrull, S. A. Testero, J. F. Fisher and S. Mobashery, Curr. Opin. Microbiol., 2010, 13, 551–557 CrossRef CAS.
- Z. Yao, D. Kahne and R. Kishony, Mol. Cell, 2012, 48, 705–712 CrossRef CAS.
- K. Bush and J. F. Fisher, Annu. Rev. Microbiol., 2011, 65, 455–478 CrossRef CAS.
- D. M. Livermore, Clin. Microbiol. Rev., 1995, 8, 557–584 CrossRef CAS.
- E. I. Edwards, R. Epton and G. Marr, J. Organomet. Chem., 1976, 107, 351–357 CrossRef CAS.
- E. M. Lewandowski, Ł. Szczupak, S. Wong, J. Skiba, A. Guśpiel, J. Solecka, V. Vrček, K. Kowalski and Y. Chen, Organometallics, 2017, 36, 1673–1676 CrossRef CAS.
- J. D. Smith, M. Kumarasiri, W. Zhang, D. Hesek, M. Lee, M. Toth, S. Vakulenko, J. F. Fisher, S. Mobashery and Y. Chen, Antimicrob. Agents Chemother., 2013, 57, 3137–3146 CrossRef CAS.
- C. J. Adamski, A. M. Cardenas, N. G. Brown, L. B. Horton, B. Sankaran, B. V. V. Prasad, H. F. Gilbert and T. Palzkill, Biochemistry, 2015, 54, 447–457 CrossRef CAS.
- E. M. Lewandowski, Ł. Szczupak, A. Kowalczyk, G. Mendoza, M. Arruebo, L. M. C. Jacobs, P. Stączek, Y. Chen and K. Kowalski, ChemBioChem, 2020, 21, 2187–2195 CrossRef CAS.
- M. Michaut, A. Steffen, J.-M. Contreras, C. Morice, A. Paulen, I. J. Schalk, P. Plésiat and G. L. A. Mislin, Bioorg. Med. Chem. Lett., 2020, 30, 127098 CrossRef CAS.
- L. A. Mitscher, Chem. Rev., 2005, 105, 559–592 CrossRef CAS.
- D. J. Dwyer, M. Kohanski, B. Hayete and J. J. Collins, Mol. Syst. Biol., 2007, 3, 91 CrossRef.
- Ł. Szczupak, A. Kowalczyk, D. Trzybiński, K. Woźniak, G. Mendoza, M. Arruebo, D. Steverding, P. Stączek and K. Kowalski, Dalton Trans., 2020, 49, 1403–1415 RSC.
- R. Joshi, S. K. Yadav, H. Mishra, N. Pandey, R. Tilak and S. Pokharia, Heteroatom Chem., 2018, 29, 21433–21446 CrossRef.
- M. P. Chrysouli, C. N. Banti, N. Kourkoumelis, E. E. Moushi, A. J. Tasiopoulos, A. Douvalis, C. Papachristodoulou, A. G. Hatzidimitriou, T. Bakas and S. K. Hadjikakou, Dalton Trans., 2020, 49, 11522–11535 RSC.
- R. W. Lacey, J. Antimicrob. Chemother., 1979, 5(suppl. B), 75–83 CrossRef CAS.
- T. Stringer, R. Seldon, N. liu, D. F. Warner, C. Tam, L. W. Cheng, K. M. Land, P. J. Smith, K. Chibale and G. S. Smith, Dalton Trans., 2017, 46, 9875–9885 RSC.
- C. M. DuChane, G. W. Karpin, M. Ehrich, J. O. Falkinham III and J. S. Merola, Med. Chem. Commun., 2019, 10, 1391–1398 RSC.
- C. Quintana, G. Silva, A. H. Klahn, V. Artigas, M. Fuentealba, C. Biot, I. Halloum, L. Kremer, N. Novoa and R. Arancibia, Polyhedron, 2017, 134, 166–172 CrossRef CAS.
- P. Chellan, V. M. Avery, S. Duffy, J. A. triccas, G. Nagalingam, C. Tam, L. W. Cheng, J. Liu, K. M. Land, G. J. Clarkson, I. Romero-Canelón and P. J. Sadler, Chem. – Eur. J., 2018, 24, 10078–10090 CrossRef CAS.
- R. G. Miller, M. Vázquez-Hernández, P. Prochnow, J. E. Bandow and N. Metzler-Nolte, Inorg. Chem., 2019, 58, 9404–9413 CrossRef CAS.
- R. Raz, B. Chazan, Y. Kennes, R. Colodner, E. Rottensterich, M. Dan, I. Lavi and W. Stamm, Clin. Infect. Dis., 2002, 34, 1165–1169 CrossRef CAS.
- M. A. Foltzer and R. E. Reese, Med. Clin. North Am., 1987, 71, 1177–1194 CrossRef CAS.
- D. Gopalakrishnan, C. Sumithaa, A. M. Kumar, N. S. P. Bhuvanesh, S. Ghorai, P. Das and M. Ganeshpandian, New J. Chem., 2020, 44, 20047–20059 RSC.
- M. Kamboj and K. A. Sepkowitz, Lancet Oncol., 2009, 10, 589–597 CrossRef.
- F. Chen, J. Moat, D. McFeely, G. Clarkson, I. J. Hands-Portman, J. P. Furner-Pardoe, F. Harrison, C. G. Dowson and P. J. Sadler, J. Med. Chem., 2018, 61, 7330–7344 CrossRef CAS.
- J. Skiba, A. Kowalczyk, P. Stączek, T. Bernaś, D. Trzybiński, K. Woźniak, U. Schatzschneider, R. Czerwieniec and K. Kowalski, New J. Chem., 2019, 43, 573–583 RSC.
- J. Lemire, J. Harrison and R. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS.
- A. Henglein, Chem. Mater., 1998, 10, 444–450 CrossRef CAS.
- A. Balfourier, N. Luciani, G. Wang, G. Lelong, O. Ersen, A. Khelfa, D. Alloyeau, F. Gazeau and F. Carn, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 103–113 CrossRef CAS.
- Z. Xiu, Q. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS.
- A. Regiel, S. Irusta, A. Kyzioł, M. Arruebo and J. Santamaria, Nanotechnology, 2012, 24, 015101 CrossRef.
- A. Regiel-Futyra, M. Kus-Liśkiewicz, V. Sebastian, S. Irusta, M. Arruebo, G. Stochel and A. Kyzioł, ACS Appl. Mater. Interfaces, 2015, 7, 1087–1099 CrossRef CAS.
- C. S. F. Crede, Die verhütung der augenentzündung der neugeborenen, Arch. Gynakol., 1881, 17, 50–53 CrossRef.
- S. P. Krumdieck, R. Boichot, R. Gorthy, J. G. Land, S. Lay, A. J. Gardecka, M. I. J. Polson, A. Wasa, J. E. Aitken, J. A. Heinemann, G. Renou, G. Berthome, F. Charlot, T. Encinas, M. Braccini and C. M. Bishop, Sci. Rep., 2019, 9, 1883 CrossRef.
- J. Hu, S. B. Maamar, A. J. Glawe, N. Gottel, J. A. Gilbert and E. M. Hartmann, Indoor Air, 2019, 29, 551–562 CrossRef.
- C. Robben, S. Fister, A. K. Witte, D. Schoder, P. Rossmanith and P. Mester, Sci. Rep., 2018, 8, 15132 CrossRef.
- H. Palza, M. Nuñez, R. Bastías and K. Delgado, Int. J. Antimicrob. Agents, 2018, 51, 912–917 CrossRef CAS.
- C. D. Salgado, K. A. Sepkowitz, J. F. John, J. R. Cantey, H. H. Attaway, K. D. Freeman, P. A. Sharpe, H. T. Michels and M. G. Schmidt, Infect. Control Hosp. Epidemiol., 2013, 34, 479–486 CrossRef.
- M. H. Kim, S. G. Lee, K. S. Kim, Y. J. Heo, J. E. Oh and S. J. Jeong, BMC Infect. Dis., 2018, 18, 610 CrossRef CAS.
- M. M. Cowan, K. Z. Abshire, S. L. Houk and S. M. Evans, J. Ind. Microbiol. Biotechnol., 2003, 30, 102–106 CrossRef CAS.
- J. H. Shen, Y. S. Wang, J. P. Lin, S. H. Wu and J. J. Horng, J. Air Waste Manag. Assoc., 2013, 64, 13–18 CrossRef.
- Y. Yao, T. Ochiai, H. Ishiguro, R. Nakano and Y. Kubota, Appl. Catal., B, 2011, 106, 592–599 CrossRef CAS.
- T. B. Karchmer, E. T. Giannetta, C. A. Muto, B. A. Strain and B. M. Farr, Arch. Intern. Med., 2000, 160, 3294–3298 CrossRef CAS.
- S. Saint, J. G. Elmore, S. D. Sullivan, S. S. Emerson and T. D. Koepsell, Am. J. Med., 1998, 105, 236–241 CrossRef CAS.
- H. R. Thorarinsdottir, T. Kander, A. Holmberg, S. Petronis and B. Klarin, Crit. Care, 2020, 24, 382 CrossRef.
- L. Berra, T. Kolobow, P. Laquerriere, B. Pitts, S. Bramati, J. Pohlmann, C. Marelli, M. Panzeri, P. Brambillasca, F. Villa, A. Baccarelli, S. Bouthors, H. T. Stelfox, L. M. Bigatello, J. Moss and A. Pesenti, Intensive Care Med., 2008, 34, 1030–1037 CrossRef CAS.
- H. Wafa, R. J. Grimer, K. Reddy, L. Jeys, A. Abudu, S. R. Carter and R. M. Tillman, Bone Joint J., 2015, 97-B, 252–257 CrossRef CAS.
- J. Hardes, C. von Eiff, A. Streitbuerger, M. Balke, T. Budny, M. P. Henrichs, G. Hauschild and H. Ahrens, J. Surg. Oncol., 2010, 101, 389–395 CrossRef.
- J. Cambiaso-Daniel, S. Boukovalas, G. H. Bitz, L. K. Branski, D. N. Herndon and D. M. Culnan, Ann. Plast. Surg., 2018, 1 Search PubMed.
- G. Norman, J. Christie, Z. Liu, M. J. Westby, J. M. Jefferies, T. Hudson, J. Edwards, D. P. Mohapatra, I. A. Hassan and J. C. Dumville, Cochrane Database Syst. Rev., 2017, 7, CD011821 Search PubMed.
- S. O’Meara, D. Al-Kurdi, Y. Ologun, L. G. Ovington, M. Martyn-St James and R. Richardson, Cochrane Database Syst. Rev., 2014, 10, CD003557 Search PubMed.
- R. P. Allaker, J. Dent. Res., 2010, 89, 1175–1186 CrossRef CAS.
- C. H. Goss, Y. Kaneko, L. Khuu, G. D. Anderson, S. Ravishankar, M. L. Aitken, N. Lechtzin, G. Zhou, D. M. Czyz, K. McLean, O. Olakanmi, H. A. Shuman, M. Teresi, E. Wilhelm, E. Caldwell, S. J. Salipante, D. B. Hornick, R. J. Siehnel, L. Becker, B. E. Britigan and P. K. Singh, Sci. Transl. Med., 2018, 10, eaat7520 CrossRef.
- Y. Li, X. Liu, L. Tan, Z. Cui, X. Yang, Y. Zheng, K. W. K. Yeung, P. K. Chu and S. Wu, Adv. Funct. Mater., 2018, 28, 1800299 CrossRef.
- D. Mao, F. Hu, Kenry, S. Ji, W. Wu, D. Ding, D. Kong and B. Liu, Adv. Mater., 2018, 30, 1706831 CrossRef.
- Z. Liu, J. Liu, R. Wang, Y. Du, J. Ren and X. Qu, Biomaterials, 2015, 56, 206–218 CrossRef CAS.
- M. Li, L. Li, K. Su, X. Liu, T. Zhang, Y. Liang, D. Jing, X. Yang, D. Zheng, Z. Cui, Z. Li, S. Zhu, K. W. K. Yeung, Y. Zheng, X. Wang and S. Wu, Adv. Sci., 2019, 6, 1900599 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |