
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent proteins of the EosFP clade: intriguing marker tools with multiple photoactivation modes for advanced microscopy
Karin
Nienhaus
 a and
Gerd Ulrich
Nienhaus
*abcd
a and
Gerd Ulrich
Nienhaus
*abcd
aInstitute of Applied Physics, Karlsruhe Institute of Technology, 76049 Karlsruhe, Germany. E-mail: uli@uiuc.edu
bInstitute of Nanotechnology, Karlsruhe Institute of Technology, 76021 Karlsruhe, Germany
cInstitute of Biological and Chemical Systems, Karlsruhe Institute of Technology, 76021 Karlsruhe, Germany
dDepartment of Physics, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
First published on 31st March 2021
Abstract
Optical fluorescence microscopy has taken center stage in the exploration of biological structure and dynamics, especially on live specimens, and super-resolution imaging methods continue to deliver exciting new insights into the molecular foundations of life. Progress in the field, however, crucially hinges on advances in fluorescent marker technology. Among these, fluorescent proteins (FPs) of the GFP family are advantageous because they are genetically encodable, so that live cells, tissues or organisms can produce these markers all by themselves. A subclass of them, photoactivatable FPs, allow for control of their fluorescence emission by light irradiation, enabling pulse-chase imaging and super-resolution microscopy. In this review, we discuss FP variants of the EosFP clade that have been optimized by amino acid sequence modification to serve as markers for various imaging techniques. In general, two different modes of photoactivation are found, reversible photoswitching between a fluorescent and a nonfluorescent state and irreversible green-to red photoconversion. First, we describe their basic structural and optical properties. We then summarize recent research aimed at elucidating the photochemical processes underlying photoactivation. Finally, we briefly introduce various advanced imaging methods facilitated by specific EosFP variants, and show some exciting sample applications.
 Karin Nienhaus | Dr Karin Nienhaus is a Research Associate at the Institute of Applied Physics, Karlsruhe Institute of Technology (KIT), Germany. She studied chemistry at the University of Münster, Germany. In 2003, she received her PhD in Chemistry at the University of Ulm, Germany. From 2000 to 2009, she worked as a Research Assistant at the Institute of Biophysics in Ulm, where she was mainly involved in spectroscopic studies of fluorescent proteins and heme proteins. Since 2009, she is employed at KIT. Her work focuses on investigating the molecular details of enzyme–substrate and receptor–ligand interactions and photoactivation processes in fluorescent proteins using various spectroscopic approaches. |
 Gerd Ulrich Nienhaus | Gerd Ulrich Nienhaus studied Physics and Physical Chemistry at the University of Münster, Germany and received his PhD degree in 1988. In 1990, he joined the Department of Physics at the University of Illinois at Urbana-Champaign, USA, where he is still appointed as an Adjunct Professor. In 1996, he became Professor and Director of the Institute of Biophysics at the University of Ulm, Germany. Since 2009, he is a Professor and Director of the Institute of Applied Physics at the Karlsruhe Institute of Technology (KIT), Germany. His interdisciplinary research ranges from the development of advanced optical microscopes via luminescence marker development to fundamental molecular and cellular biophysics. |
Introduction
Optical fluorescence microscopy has become a central experimental technique in the life sciences, allowing researchers to study biological structure and dynamics in specimens ranging from cells to tissues to entire organisms.1,2 This development was strongly driven by technological advances of microscopy components, including light sources (lasers), detectors (cameras, photodiodes), micromechanical and optical devices, electronics and computers. Nowadays, fluorescence microscopy with visible and near-infrared light is a highly sensitive, minimally invasive method to investigate live samples on time scales ranging from split seconds all the way up to many days and weeks. More recently, visualization of biological structures has become possible even at the molecular scale through the invention of super-resolution imaging techniques that bypass Abbe's diffraction limit in ingenious ways and offer theoretically unlimited image resolution.3–9 Despite all this progress, fluorescence microscopy could not have taken such a central role in the life sciences if it were not for parallel advances in an entirely different area, i.e., the development of luminescent marker tools. By selective and specific staining, biological molecules and structures become visible under the microscope and can be studied without interference from a myriad of other molecules in the specimen.A vast array of approaches have been accumulated to achieve specific labeling of samples for fluorescence microscopy.10,11 To accommodate the requirements of specific imaging experiments, an impressive variety of organic dyes and nanocrystals with intriguing photochemical properties have been synthesized, and intricate conjugation strategies have been devised for their selective attachment to target structures.11–13 In this account, we focus on fluorescent proteins (FPs) of the green fluorescent protein (GFP) family, which have become truly enabling tools for fluorescence imaging.14–17 Their key advantage over other labeling procedures is their genetic encodability. For example, the gene of a protein of interest can be extended to include the DNA code of a FP and introduced into the genome of a biological system (cell or organism). Subsequently, the sample produces the fluorescent fusion protein; no other preparative procedures are needed for labeling. While this approach is conceptually simple, it may take considerable effort to engineer a fusion protein in which the additional FP domain has minimal adverse effects on the biological function of the protein of interest, e.g., by designing appropriate amino acid linkers and selecting a suitable location for FP insertion. Notably, protein labeling via FP fusions has become even more powerful with the advent of CRISPR/Cas9 technology (recognized by the Nobel Prize in Chemistry 2020), which enables facile modification of target genes at their specific locus within the genome.18 Therefore, FP technology greatly facilitates the selective study of a protein in a cell or organism using fluorescence microscopy, requiring only established and straightforward genome modifications. Remarkably, although GFP was discovered as early as 1962 by Osamu Shimomura in tissue extracts of the jellyfish Aequorea victoria,19 it took more than three decades until the GFP gene20 was recombinantly expressed in Escherichia coli and Caenorhabditis elegans by Martin Chalfie and co-workers,21 and the advantage of genetic encoding of fluorescence markers was fully appreciated.
FP marker optimization by genetic engineering
All GFP-type proteins are made from polypeptide chains of ∼220–240 amino acids that are folded into 11 β-strands forming a barrel-like structure, which is capped by short helical sections and loops at either end, as shown for EosFP in Fig. 1a.22–24 A helix running along the central axis of the barrel is interrupted by the chromophore, 4-(p-hydroxybenzylidene)-5-imidazolinone (p-HBI) and variants thereof in yellow or red FPs, which forms autocatalytically from a tripeptide (His62-Tyr63-Gly64 in EosFP, Fig. 1b). The Gly and Tyr residues are strictly conserved in natural FP sequences, but the first amino acid of the triad may vary. Naturally occurring FPs have serious drawbacks for use as fluorescence markers, and tremendous efforts have been made to optimize them or to endow them with entirely new properties by genetic engineering. In the 1990s, Tsien and coworkers pioneered the engineering of advanced GFP variants, featuring bright fluorescence emission ranging from blue to yellow.25 Subsequently, homologs of GFP were discovered in anthozoa and other animals,26–31 so that a wide variety of FP sequences for development of FP markers became available. In some of these FPs, attractive new properties were discovered, notably, fluorescence emission in the red spectral region.32,33 Red emission is highly desirable for bioimaging, as cellular autofluorescence and light scattering are markedly reduced in the red spectral region. Other FPs displayed intriguing changes in their fluorescence properties in response to light exposure. This effect, known as photoactivation,34–41 is most attractive for fluorescence microscopy and affords new experimental approaches, e.g., pulse-chase imaging and super-resolution single molecule-based localization microscopy (SMLM).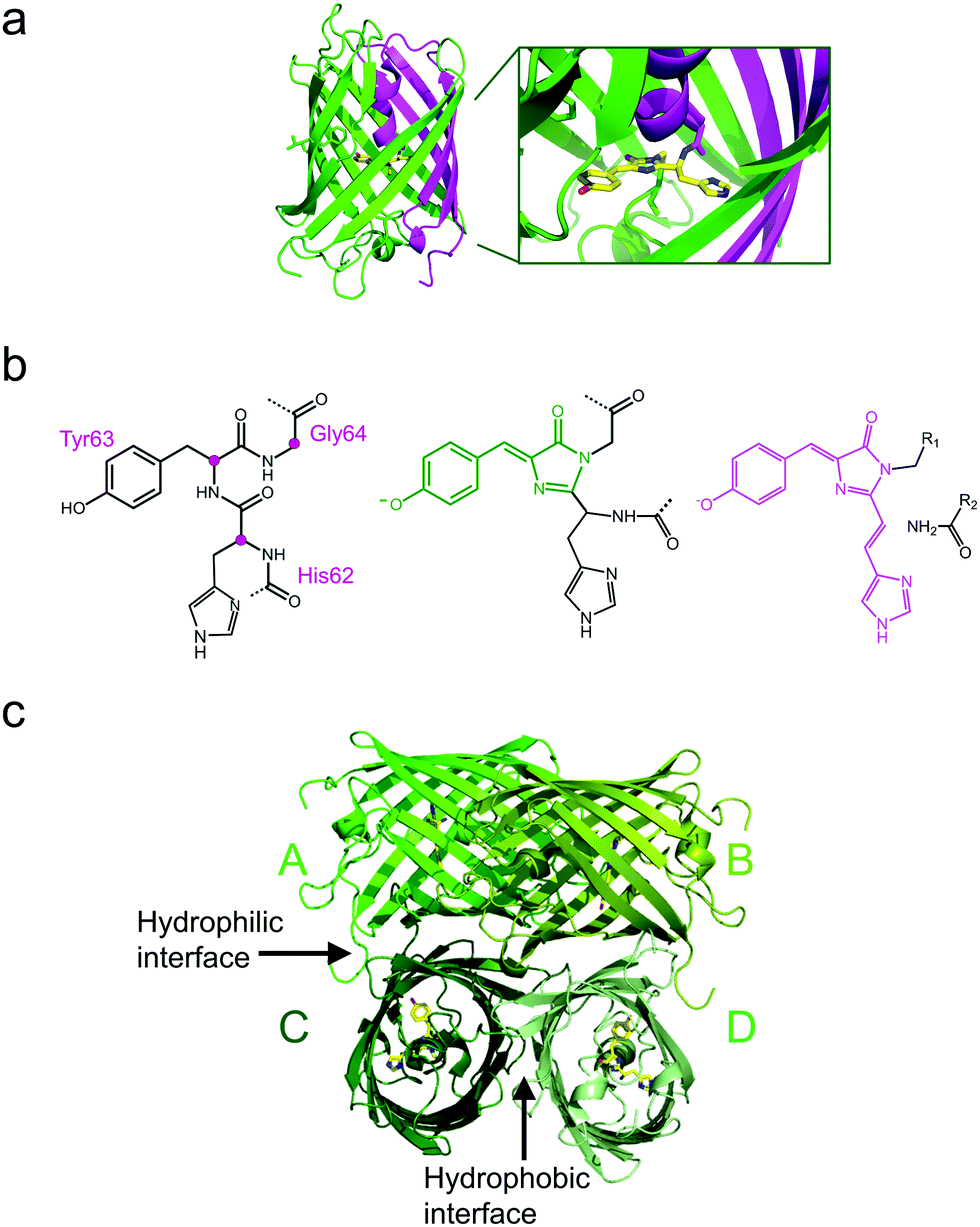 | ||
| Fig. 1 Overall structure of EosFP, a GFP-type protein. (a) Each monomer folds into an eleven-strand β-barrel, with the ends closed by loops and short α-helical structures. An α-helix running along its axis is interrupted by the chromophore generated from the tripeptide His62-Tyr63-Gly64. It is displayed in stick representation in the barrel and as a close-up. The N- and C-terminal fractions of the β-can that result from cleavage of the EosFP polypeptide chain upon green-to-red photoconversion are colored in magenta and green, respectively. (b) Chromophore-forming tripeptide (left, Cα atoms marked with magenta dots) and mature green and red chromophores (right, conjugated π-systems colored in green and red, respectively). (c) Tetrameric arrangement of protomers A–D, showing the hydrophilic AC and BD interfaces and the hydrophobic CD interface. | ||
Various biochemical and photochemical properties affect an FP's performance as a fluorescence marker.25,42 Biochemical properties that are targeted for FP marker optimization by amino acid modifications include the following: (1) DNA sequences are adjusted for optimal codon usage in cells chosen to express the FP marker. (2) Natural FPs typically form tetrameric (or in some cases dimeric) quaternary structures. Whereas this is not of concern in simple marker applications, e.g., highlighting cellular organelles, cells and tissues,43,44 the most prevalent application, i.e., selective protein labeling, requires monomeric FPs. If the FP marker has a tendency to oligomerize, this property is passed on to the fusion protein and typically causes severely compromised biological functions.45 Therefore, researchers aim to disrupt the binding interfaces between FP protomers by exchanging amino acids, e.g., by replacing hydrophobic amino acid residues with hydrophilic or even charged ones. Sometimes, a single mutation suffices to render a FP monomeric, as was the case for the dimeric natural Aequorea GFP.46 However, for anthozoan FPs, monomerization efforts have oftentimes proven extremely difficult.47–49 (3) After synthesis in the ribosome, only a fraction of polypeptide chains coding for a FP fold properly and form a functional fluorophore in the process, resulting in a maturation efficiency of less than one. Accordingly, the marker appears less bright (as judged from the overall sample staining), and the number of molecules is underestimated in quantitative fluorescence imaging studies. Therefore, it is highly desirable to optimize maturation by introducing suitable mutations.50 (4) Equally important is the temperature dependence of the maturation efficiency. It is typically high up to room temperature, in accordance with the environmental temperature of the organism from which the FP sequence was taken, but markedly declines above. Live-cell imaging of mammalian cell cultures require a temperature of 37 °C, however. Fortunately, the exchange of certain amino acids in the sequence has frequently been successful to generate thermostable variants expressing well at 37 °C.51,52 (5) Chromophore maturation may take from minutes to hours or even days for particular FPs,50,53 requiring molecular oxygen (O2) as the only further ingredient. In live imaging, fast maturation can be important, e.g., in developmental studies, so that marker production can keep up with the processes under study.
Photochemical and photophysical properties of an FP marker can also be improved by genetic engineering, as the protein matrix modulates the chromophore environment in different ways and changes its properties as a marker dye. Accordingly, (1) color tuning of the absorption and emission bands can be achieved by amino acid replacements within (e.g., Tyr to Phe or His) and near the chromophore.25 An important role of the protein matrix is to constrain the dynamics of the p-HBI chromophore (or its derivatives) to keep it preferentially in a coplanar configuration.54 In fact, p-HBI freely diffusing in water is completely non-fluorescent.55 Amino acid exchanges that lead to a strengthening of the overall fold or chromophore interactions with the surrounding amino acids (and structural water molecules) can improve both (2) the absorption strength and (3) the fluorescence quantum yield (QY). (4) The photobleaching probability is also likely to depend on the rigidity of the FP scaffold and the enclosed fluorophore. As a case in point, oxygen plays an important role in photobleaching, and its diffusion to the fluorophore may be hindered in a more rigid environment. The photobleaching probability is a crucial performance parameter, as it represents the limited overall number of excitation/emission cycles of the fluorophore. Another important issue is (5) the protonation of the p-hydroxyphenyl moiety of the p-HBI chromophore, which affects the spectral properties in a major way. The neutral, protonated chromophore is usually non-fluorescent and its spectra are markedly blue-shifted from those of the anionic, deprotonated chromophore. In most (but not all) GFP-type proteins, its population varies with the pH of the protein environment. To ensure that the anionic chromophore is the predominant species at physiological pH, mutations can be introduced near the chromophore to lower its pK. Finally, we mention the (6) photoactivation capability, denoting photoinduced modifications of the FP's fluorescence properties. Two modes are being distinguished: Reversible photoactivation (photoswitching) typically arises from chromophore toggling between its trans and cis configurations, only one of which emits fluorescence with high yield. Irreversible photoactivation (photoconversion) refers to a permanent photochemical modification. Consequently, a non-fluorescent (dark) state may get photoactivated to a fluorescent (bright) state, or a bright state may be turned into another bright state with a different emission color (wavelength). A few FPs even combine reversible and irreversible modes. Photoactivation is a very attractive property, in fact, a prerequisite for some powerful imaging modalities. Consequently, considerable efforts have been made to optimize photoactivation properties of FPs, as we will discuss in detail below, using EosFP and its derivatives as an example.
Photoactivatable EosFP and its engineered descendants
Green-to-red photoconvertible FPs change their emission color in response to light irradiation, most effectively with violet light (wavelength ∼400 nm). Miyawaki and coworkers were the first to report this property for Kaede, a FP that they isolated from the stony coral Trachyphyllia geoffroyi.56 Soon thereafter, we introduced EosFP,22,57 originating from the scleractinian coral Lobophyllia hemprichii. Since then, the class of photoconvertible FPs has been greatly expanded with advanced EosFP variants,51,58–61 Dendra262,63 and variants,64 KikGr65 and its monomeric form mKikGr,66 mClavGR2,67 mMaple,68 pcDronpa,69 SAASoti,70,71 and a few other proteins.31,72,73 Among the latter, the “least evolved ancestor (LEA) protein” is an interesting example of an FP designed in the laboratory.73Early on, it was realized that photoconvertible FPs enable regional optical marking, which can be exploited for pulse-chase imaging.36,56,57,74,75 Selective irradiation tags FPs within a certain region of a specimen by converting them to red emitters, and then they are “chased” by imaging until they photobleach. In their Nobel prize-winning work in 2006, Betzig's group demonstrated single-molecule localization microscopy (SMLM) as a super-resolution imaging technique, using green-to-red photoconversion of a tandem-dimeric EosFP variant.5 Their work triggered efforts to engineer optimized photoconvertible FPs for super-resolution imaging. Remarkably, in EosFP, only a small number of amino acid exchanges sufficed to improve its marker properties and even to endow it with new properties beneficial for imaging. This likely explains its widespread use as a template for further FP development, as shown in the evolutionary tree in Fig. 2. There, we have limited ourselves to FP markers; sensors are not included.
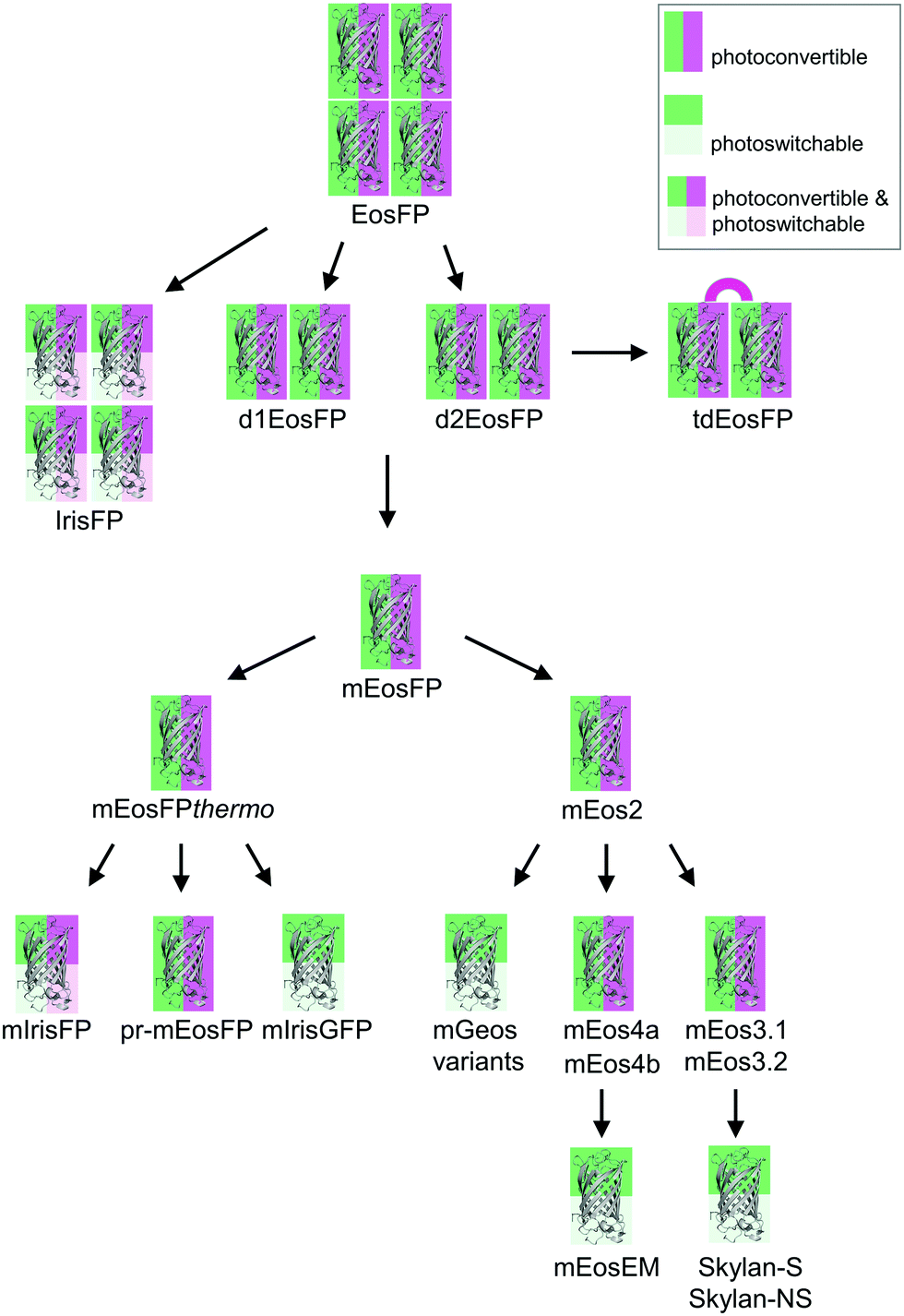 | ||
| Fig. 2 Evolutionary tree of photoactivatable EosFP. The oligomerization state is represented by the number of protein depictions, the background color encodes the photoactivation mode (photoconvertible, green/magenta; photoswitchable, green/light green; photoconvertible and photoswitchable, green/light green/magenta/light magenta). | ||
The four protomers A, B, C and D of natural EosFP associate via two types of interfaces, one with hydrophobic (AB, CD binding) and one with hydrophilic (AC, BD binding) character, to form a tightly associated tetramer (Fig. 1c).22 Single amino acid exchanges, Val123Thr and Thr158His, were sufficient to break up the AB and AC interfaces, respectively, resulting in dimeric variants, d1EosFP and d2EosFP (Fig. 2). Combining both mutations yielded a bright, monomeric variant, mEosFP.22 It has found widespread application in fluorescence imaging of plants,61 but not mammalian cells due to its low maturation efficiency above 30 °C. As an intermediate remedy, we designed a “pseudo-monomeric” marker by connecting two protomers of d2EosFP with a 12 amino-acid linker, which expresses well at 37 °C.74 Accordingly, this tandem-dimeric variant (tdEosFP) is twice as bright as the monomer and, therefore, attractive for imaging applications for which the label size is not of concern.5,76–79 Another immediate descendant of EosFP is its Phe173Ser mutant, denoted IrisFP,80 which combines photoconversion and photoswitching in the same FP, thereby enabling pulse-chase imaging with super-resolution via SMLM.
Subsequently, we found that a single point mutation in mEosFP, Ala69Val, provided an enhanced temperature tolerance, resulting in a bright marker for 37 °C expression, mEosFPthermo.58 As its other parameters remained unchanged within the experimental error, which is not surprising for such a tiny modification of a hydrophobic side chain, we never published its detailed characterization. So far, three further variants originated from mEosFPthermo (Fig. 2). Mutant Ala69Thr (pr-mEosFP) features “primed photoconversion”, i.e., photoactivation using combined blue-green (e.g., 488 nm) and far-red (600–850 nm) irradiation (vide infra) instead of 400 nm light.81 Monomeric IrisFP51 was also engineered from mEosFPthermo as well as a variant denoted mIrisGFP,82 showing reversible photoswitching but not green-to-red photoconversion. It is quite remarkable that such a diversity in photoactivation behavior can arise from a fairly small number of amino acid replacements (Fig. 3a).
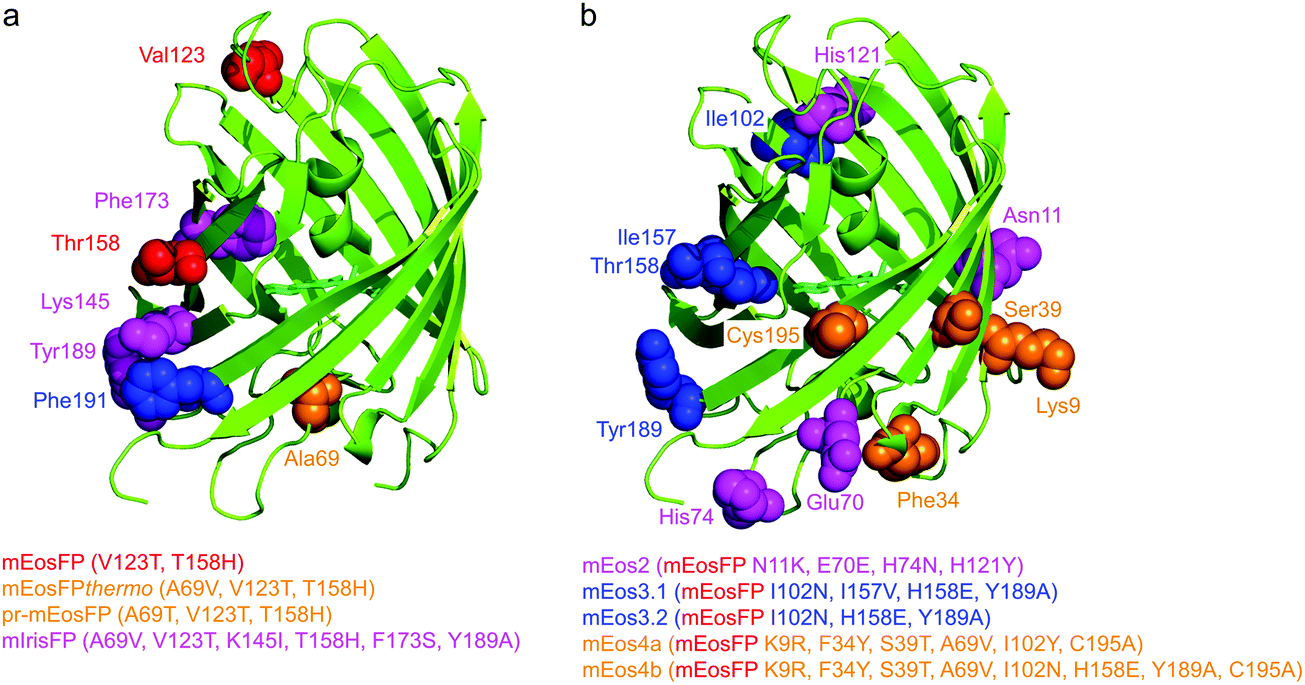 | ||
| Fig. 3 Genetic engineering of EosFP. Protomers are shown in cartoon representation; mutated residues are depicted in van der Waals representation, using the same colors as for the list of engineered variants below (with sequence changes specified in parentheses). (a) Modifications of EosFP to obtain monomeric mEosFP (red), the thermostable mEosFPthermo and the primable pr-mEosFP variants (orange), and mIrisFP (magenta). (b) Additional modifications of mEosFP to obtain mEos2 (magenta), mEos3.1 and mEos3.2 (blue) and mEos4a and mEos4b (orange). | ||
In parallel, McKinney et al.59 also pursued engineering of a monomeric EosFP expressing at 37 °C. Their variant, mEos2, however, had a higher dimerization tendency (KD = 20 μM) than its parent, mEosFP (233 μM), or mEosFPthermo (483 μM),60 which can be problematic at higher expression levels. This protein served as a starting point for further modifications. In an attempt to improve mEos2, mEos3.1 and mEos3.2 were introduced, reported to be truly monomeric, brighter and faster maturing than Eos2.60 Variants mEos4a and mEos4b were engineered that are able to fluoresce and photoconvert in heavily fixed (up to 1% OsO4), epoxy resin-embedded samples for correlative light and electron microscopy (CLEM).83 A further descendant of Eos4b, mEosEM, was recently reported to resist epon resin embedding after OsO4 treatment.84 Moreover, Chang et al.85 prepared a series of mutants by replacing His62, the first amino acid in the chromophore tripeptide, which is essential for green-to-red photoconversion, by other amino acids, yielding monomeric green EosFP (mGeos) variants with useful photoswitching capabilities. Amino acid modifications of Eos2 descendants are shown in Fig. 3b.
Optical and structural properties of EosFP-type chromophores
Molecular brightness
Besides the photobleaching probability, molecular brightness is another important quantity that allows microscopists to assess a marker's performance in fluorescence imaging. Molecular brightness is the product of two photophysical quantities, the extinction coefficient, ε, measured at the peak of the absorption band, and the fluorescence QY, denoting the probability of photon emission after photon absorption. In Table 1, we have compiled literature values of these quantities for both green and red (anionic) chromophore species of EosFP variants. They are expected to differ somewhat between variants due to the altered structure and dynamics of the chromophore and its environment. Table 1 shows QY values ranging from 0.64–0.91 and 0.47–0.75 for the green and red chromophores, respectively. Interestingly, the two extreme values for the red fluorophore were obtained on the same variant, namely Eos2, by two different laboratories. Thus, we conclude that published QY values should be taken with a grain of salt, even though their determination relative to a standard dye involves a simple measurement. Likewise, ε values differ not only between variants, but also for the same variant measured by different laboratories (Table 1). A stunning example is mEosFPthermo, for which ε values of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 97000 M−1 cm−1 and 10000 and 41300 M−1 cm−1 have been reported for the green and red chromophore species, respectively. As we will briefly discuss in the next subsection, published ε values (Table 1) are only “apparent” parameters. Their determination is not trivial, and errors, assumptions and corrections involved in the process can lead to different results, but cannot explain discrepancies by as much as three- to fourfold. To conclude this subsection on a critical note, selecting a FP marker protein based on published parameters may not necessarily guide the user to the best choice.
000 and 97000 M−1 cm−1 and 10000 and 41300 M−1 cm−1 have been reported for the green and red chromophore species, respectively. As we will briefly discuss in the next subsection, published ε values (Table 1) are only “apparent” parameters. Their determination is not trivial, and errors, assumptions and corrections involved in the process can lead to different results, but cannot explain discrepancies by as much as three- to fourfold. To conclude this subsection on a critical note, selecting a FP marker protein based on published parameters may not necessarily guide the user to the best choice.
| EosFP variant | Green chromophore | Red chromophore | ||
|---|---|---|---|---|
| ε (M−1 cm−1) | QY | ε (M−1 cm−1) | QY | |
| EosFP | 7200057 |
0.7057 | 4100057 |
0.5557 |
| d1EosFP | 7480057 |
0.6857 | 4000057 |
0.6257 |
| d2EosFP | 5700059 |
0.6657 | 3300057 |
0.6057 |
| 8400057 |
4900059 |
|||
| tdEosFP | 6500059 |
0.9160 | 3200057 |
0.6260 |
| 8400057 |
4800059 |
|||
| mEosFP | 6720057 |
0.6457 | 3700057 |
0.6257 |
| 7200060 |
0.7060 | 4100060 |
0.5560 | |
| mEosFPthermo | 2700060 |
0.6764 | 1000060 |
0.6664 |
| 9720064 |
0.8460 | 4130064 |
0.6560 | |
| mEos2 | 5600059 |
0.8460 | 4130060 |
0.4760 |
| 75400 |
0.8483 | 4600059 |
0.7583 | |
| 8651083 |
5353083 |
|||
| 6142083 |
||||
| mEos3.1 | 8840060 |
0.8360 | 3350060 |
0.6260 |
| mEos3.2 | 6340060 |
0.8460 | 3220060 |
0.5560 |
| mEos4a | 8353083 |
0.8683 | 6100083 |
0.7183 |
| mEos4b | 7817083 |
0.8483 | 5550083 |
0.7183 |
Optical absorption bands of EosFP-type chromophores and their pH dependence
As a prototypical example, Fig. 4 presents optical absorption data of mEosFPthermo in the pH range 3–10. Spectra of the green form (Fig. 4a) show two absorption bands at 390 and 504 nm, denoted A and B,86,87 respectively. Their pH dependence reveals that the proton concentration in the solvent controls the fractions of neutral (phenol, A) and anionic (phenolate, B) green chromophore species residing in the protein interior.25 The variations of the A and B Band amplitudes with pH (Fig. 4c) deviate from a Hendersson–Hasselbalch relation, so a simple one-site protonation reaction does not accurately describe the data. Apparently, multiple protonation steps govern the neutral and anionic species populations, as seen before for other FPs.82,88,89 The best fit yields pK = 5.3 ± 0.1 (green lines in Fig. 4c) as the midpoint of the protonation transition. The low pK ensures that the majority of proteins is in the anionic, which is fluorescent form under physiological conditions. Green and yellow FPs usually have 5.0 < pK < 7.0,90 whereas blue and far-red variants often show pK < 4.90 Interestingly, there is an additional step-wise increase of the B band amplitude at pH > 8 in the data in Fig. 4c, reflecting a deprotonation in the chromophore vicinity that causes a further increase of ε by ca. 20%.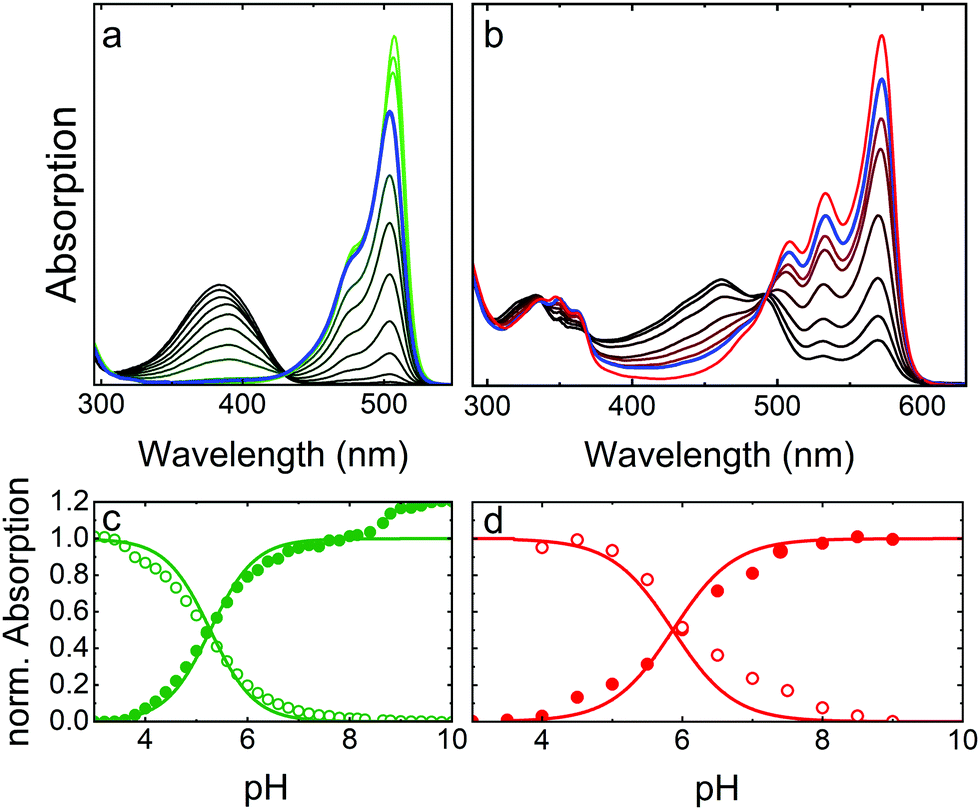 | ||
| Fig. 4 Optical absorption data of mEosFPthermo in the pH range 3–10. (a) Absorption spectra of the green chromophore; (b) absorption spectra of the red chromophore. The line colors in the two panels vary from (a) black to green and (b) black to red to indicate increasing sample pH. Spectra at pH 7.4 are plotted in blue. (c and d) Normalized peak absorption of the A (open symbols) and B (closed symbols) bands of the (c) green and (d) red chromophores. Fits with the Hendersson–Hasselbalch relation, yielding (c) pK = 5.3 and (d) pK = 5.8, are included as solid lines. | ||
Absorption spectra of red mEosFPthermo in the pH range 3–10 are shown in Fig. 4b. The absorption maxima of the A and B bands of mEosFPthermo are centered on 461 and 572 nm, respectively, and both possess a pronounced vibronic fine structure. Below 400 nm, S0–S2 transitions of the neutral and anionic red chromophores are apparent. As for the green form, the pH dependence of the A and B band absorbances (Fig. 4d) does not follow a simple one-site protonation model due to interactions between the chromophore and the protein matrix. Indeed, some FPs do not show a single-step chromophore protonation at all. Instead, there may be constant A and B band populations in a certain pH range89,91 or two distinct protonation steps,82,88 both bearing witness to the strong electrostatic coupling of the chromophore to the protein environment.92,93
To compare the absorption strengths of different FP species, absorption spectra and, specifically, the peak absorbances of the A and B bands must be converted to extinction coefficients, respectively, by using Beer's law. This requires knowledge of the concentration of our FP solution, which can be determined spectroscopically from the measured absorbance of our FP sample at 280 nm and the corresponding calculated extinction coefficient. We note in passing that mainly tryptophan (ε280 = 5500 M−1 cm−1) and tyrosine (ε280 = 1490 M−1 cm−1) residues contribute to the 280 nm protein band.94 If, however, our FP sample is not 100% pure, other types of proteins contribute to the 280 nm absorption as well. This results in an artificially inflated FP concentration and, therefore, a smaller extinction coefficient of the B band maximum. Moreover, since we aim for the ε value of the pure anionic (B band) species, we either have to choose a pH at which all chromophores are deprotonated, so that the A band fraction is zero, or we have to correct with the known pH dependence (Fig. 4). The just described approach is arguably most often used to determine ε values quoted in publications, yielding typical values of 70000–80000 M−1 cm−1 for the green EosFP chromophore. It disregards, however, the presence of non-mature (non-fluorescent) FP species, which can be viewed as an impurity in the determination of the protein concentration via the 280 nm band absorption. An alternative ε determination procedure involves taking absorption spectra before and after base denaturation of the FP sample. Using the peak extinction coefficient of the chromophore of completely denatured GFP at pH 13 (ε447 = 44000 M−1 cm−1)95 as a reference, the concentration of green chromophores in the denatured EosFP sample can be determined. This approach allows conversion of the peak absorbance of the B band (before denaturation) into an extinction coefficient, yielding values of ∼100000 M−1 cm−1 for the green EosFP chromophore (see mEosFPthermo in Table 1). Once the ε value of the anionic (B band) green species has been determined, the one of the red species results from the ratio of the B band peak absorbances before and after photoconversion. Notably, we convert only a small fraction of all chromophores to calculate this ratio, so that the error due to photobleaching is minimized. Finally, the maturation efficiency of the green FP (ca. 70–80%) can be calculated from the ratio of the apparent B band peak ε values obtained by using the 447 nm and the 280 nm peak extinction coefficients.
Isomerization and protonation states of the chromophore
The p-HBI moiety of the chromophore can adopt either a cis or a trans configuration, which poses a further complication in addition to protonation. The two configurations are shown for the green EosFP chromophore in Fig. 5a and b, respectively. The isolated p-HBI chromophore is known to be thermodynamically more stable in the cis form,96 but can easily rotate around the phenoxy (P) or imidazolinone (I) bonds of the methene bridge. Note that only rotations around the I-bond change the isomeric state. Bond rotations open non-radiative de-excitation pathways, making the free p-HBI dye completely non-fluorescent in solution.97–99 Therefore, to ensure a high fluorescence QY, the protein environment must stabilize the chromophore in one configuration or impose steric restrictions that prevent isomerization. Most natural FPs host a fluorescent cis chromophore, with the red-emitting eqFP611100 being a notable exception.101 Likewise, only a few engineered FPs have a fluorescent anionic trans chromophore.47,49,52,100–104 | ||
| Fig. 5 Isomerization and protonation states of the EosFP-type chromophore. (a) Neutral (left) and anionic (right) green trans chromophores. (b) Neutral (left, with arrows indicating isomerization around the phenoxy (P) and imidazolinone (I) bonds of the methene bridge) and anionic (right) green cis chromophores. Non-fluorescent and fluorescent species are depicted in grey and green, respectively. Absorption and emission spectra are shown below. Grey: A absorption bands of the neutral cis (and/or trans) chromophores; dark green: B absorption bands of the anionic (cis and/or trans) chromophores; bright green: emission spectrum of the anionic cis species. Cis–trans isomerization is reversible, as indicated by the grey-green double-headed arrow on the right. (c) Neutral (left) and anionic (right) red-emitting cis chromophores. Absorption and emission spectra are plotted below (grey: A absorption bands; red: B absorption bands; bright red: emission). Green-to-red photoconversion is irreversible, as indicated by the green-red arrow on the right. (d) Neutral (left) and anionic (right) red trans chromophores. Cis–trans isomerization is reversible, as indicated by the grey-red double-headed arrow on the right. Adapted from ref. 112 with permission from IOP Publishing, copyright 2016. | ||
In either isomeric state, the hydroxyphenyl moiety of the chromophore can, in principle, be protonated or deprotonated (Fig. 5). The corresponding A and B absorption bands of the cis and trans isomers overlap to a large extent, so it is not straightforward to unambiguously identify the isomerization state based on the absorption spectra.82,101,103 Fortunately, the cis configuration is predominant in EosFP in (dark) equilibrium, which is helpful in studies exploring the complicated photoactivation mechanisms.
Photoactivation mechanisms
Overview
Soon after the discovery of green-to-red photoconversion in Kaede in 2002,56 Mizuno et al.105 reported key structural changes resulting from this photoreaction: the polypeptide chain is cleaved between the Nα and Cα atoms of residue 62 in its chromophore tripeptide, His62-Tyr63-Gly64, and the conjugated π-electron system of the p-HBI chromophore is extended to include the imidazole sidechain of His62 (Fig. 5c, d), causing a red shift of its absorption and emission bands. The two chain fragments are tightly associated and stay together after photocleavage (Fig. 1a). These structural changes are characteristic of photoconverting FPs, as subsequently observed in X-ray structures of several green-to-red photoconverters including Kaede,106 EosFP,22 Dendra2,63 KikGR,107 LEA73 and pcDronpa.69 Whereas the end product of this photoreaction is undisputed, the detailed mechanisms by which it forms are still controversially debated.Photoconversion with light around 400 nm implies that the reaction is initiated by excitation of the neutral chromophore. Photoconversion via excitation of the anionic (green) chromophore with intense light (e.g., 488 nm) has also been reported for some FPs, albeit with low yield.62,108,109 Exposure with far-red light (600–850 nm) in addition to 488 nm light irradiation has been shown to greatly enhance the photoconversion yield in some cases.81,110,111 Moreover, photoswitching based on reversible cis–trans isomerization of the green and red chromophores was observed in several EosFP variants, e.g., in IrisFP.80 The involved chromophore states and excitation wavelengths are shown schematically in Fig. 6.112 In the following, we briefly summarize research aiming to shed light on the mechanisms underlying the different photoactivation modes.
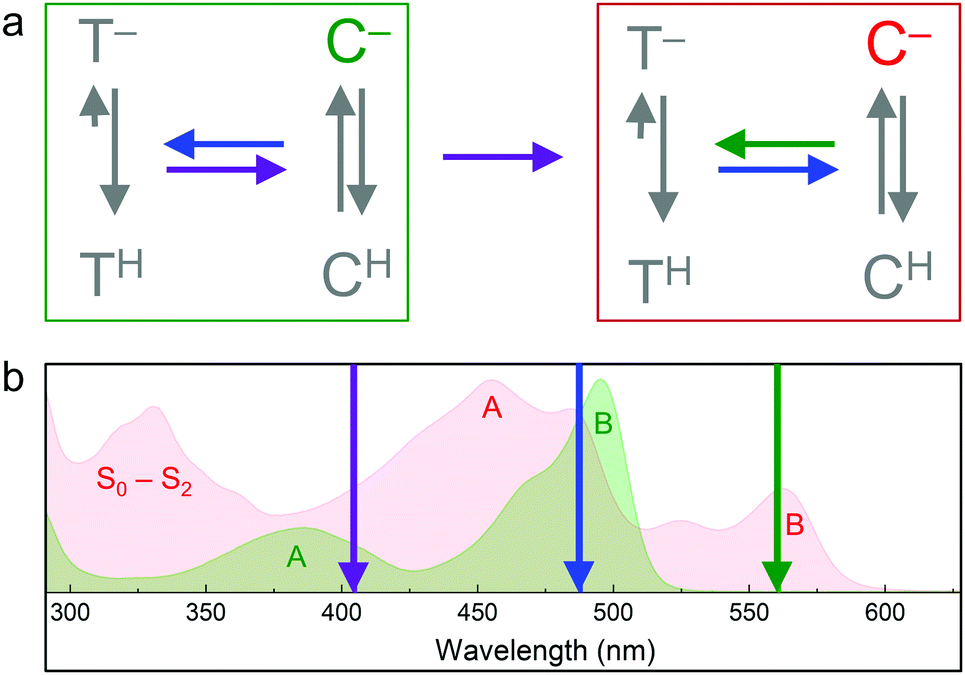 | ||
| Fig. 6 Transitions between EosFP-type chromophores. (a) Green (green box) and red (red box) chromophore states. C, cis. T, trans. Superscripts indicate the protonation state (H, protonated; −, deprotonated). Colored arrows represent light-induced transitions: 405 nm (violet), 473 or 488 nm (blue), 532 or 561 nm (green). Grey arrows indicate the pH-dependent ground state equilibria. (b) Absorption spectra of green and red pr-mEosFP, with absorption bands (A, B, S0–S2) labeled in the corresponding colors. Wavelengths of photoactivating laser irradiation are indicated by arrows (color-coding as in panel (a)). | ||
Green-to-red photoconversion via excitation of the neutral green cis chromophore
A variety of mechanisms have been invoked to explain peptide cleavage upon photoconversion, including light-induced β-elimination,105 unimolecular elimination (E1)107 and also bimolecular elimination (E2).22 To clarify the reaction mechanism, computational approaches can in general be quite helpful. Lelimousin et al.113 used excited-state hybrid quantum-chemical and molecular mechanics to gain more detailed insight into photoconversion in EosFP. Their results suggested that the electronically excited neutral green chromophore (S1 state) switches to the triplet state (T1) and back to the ground state singlet (S0) surface of the red species. Proton transfer from His62 to Phe61 was assumed to take place on the S1 surface, while backbone cleavage was found to occur on the T1 surface. By contrast, Li et al.114 proposed, based on quantum-chemical and molecular mechanics calculations using the ONIOM(B3LYP:AMBER) method, that photoconversion reactions (chromophore modifications, backbone cleavage) take place in the electronic ground state.A number of experimental studies also aimed at elucidating mechanistic details. Ultrafast spectroscopic studies revealed that the neutral green chromophore of Dendra2 undergoes excited-state proton transfer and becomes anionic within a few picoseconds.115 It remained unclear, however, if this species is involved in photoconversion. In a spectroscopic study of Kaede, backbone cleavage in the excited state was observed within 20 ps after excitation of the neutral green chromophore, generating the neutral red chromophore.116 This result is in support of earlier reports claiming that efficient photoconversion occurs via excitation of the neutral green chromophore.105,107,117 Notably, the picosecond timescale of the photoreaction may exclude the involvement of states with different spin multiplicity, which was proposed by Lelimousin et al.113 Based on time-resolved UV/vis and FTIR spectroscopy, Fare et al.118 recently suggested that photoconversion of EosFP is initiated by oxidation of the neutral green chromophore in the ground state via a tyrosyl radical, most likely Tyr177, that is transiently formed upon light-activation.
Wachter and coworkers119 performed structural studies on variants of a synthetic FP, denoted ALL-GFP, that was derived by statistical inference as the common ancestor of all FPs in the great star coral Montastraea cavernosa.120 They identified a least evolved ancestor, LEA, which harbors 13 mutations (including Gln62His in the chromophore triad), which was the minimum number necessary for efficient green-to-red photoconversion. A key finding was that the protein matrix around LEA's chromophore is more dynamic than the one of ALL-GFP, permitting substantial chromophore rearrangements in response to photoexcitation. Moreover, it was proposed that chromophore twisting is a pre-requisite for disruption of the electrostatic network and activation of a proton shuttling mechanism required for photoconversion.
From these computational and spectroscopic studies, it is evident that a great deal has been learned about photoconversion by excitation of the neutral species. However, our understanding of the detailed mechanism is still fragmented, and we expect further exciting and clarifying studies in the future.
Photoconversion via excitation of the anionic green chromophore
In addition to excitation of the neutral green chromophore with ∼400 nm light, excitation of the anionic green chromophore with 488 nm light was also found to induce photoconversion in Dendra2,62 albeit with markedly lower yield compared to excitation of the neutral form ref. 109. Pantazis and co-workers studied photoconversion on Dendra2 and observed that simultaneous exposure to 730 nm light in addition to 488 nm light for excitation of the green anionic chromophore markedly increased the photoconversion yield. They coined the phrase “primed conversion” for this process and proposed that the priming laser (∼488 nm) excites the green anionic chromophore and drives it into a primed (intermediate) state.121 In this state, absorption of a second photon of lower energy (730 nm) triggers the transition to the electronic ground state of the red species.Recently, Mohr et al.81 and Turkowyd et al.111 reported in parallel that the enormous, near-IR induced enhancement of green-to-red photoconversion via excitation of the anionic green chromophore is characteristic of green-to-red photoconvertible FPs carrying a threonine at sequence position 69. Amino acids in this position can regulate chromophore–protein interactions. In EosFP, Arg66 provides negative charge stabilization on the imidazolinone ring by hydrogen bonding of its guanidino moiety to the imidazolinone carbonyl oxygen.22 If there is a Thr69 instead of alanine, as in the natural Dendra2 sequence63 and mEos2 Ala69Thr (pr-mEos2),122 however, the Arg66 side chain hydrogen-bonds to the Thr69 hydroxyl, so that the chromophore becomes overall more flexible. Thus, the interaction of Thr69 with Arg66 is likely responsible for the appreciable photoconversion yield of Dendra2 upon excitation of the anionic green chromophore with 488 nm light in a confocal microscope,62 and the enhanced yield upon near-infrared light exposure.
To elucidate the conversion mechanism, we studied the yield of photoconversion as a function of the wavelength of the red light for Dendra2 and pr-mEosFP. Primed conversion is effective between 600 and 850 nm; the action spectra of the two FPs are similar in shape and show two slightly shifted peaks.81 Fluorescence quenching experiments revealed that the primed state is a triplet state with a lifetime of ∼5 ms.81 By using near-infrared transient absorption spectroscopy, Byrdin et al.123 identified a similar, long-lived triplet state after 488 nm excitation in IrisFP and enhanced GFP (eGFP), which do not have a threonine at sequence position 69 (EosFP numbering). They suggested that such a state may be common to all green p-HBI chromophores and involved in oxidative photochemical processes such as primed photoconversion or “oxidative redding”,124 the oxidant-mediated green-to-red photoconversion of eGFP. If that were the case, the crucial role of Thr69 would then be to facilitate formation of the red chromophore, presumably by enhancing the structural flexibility of the intermediate state. Further spectroscopic studies will likely shed light onto the details of this photoreaction.
Reversible cis–trans isomerization of the green and red chromophores
In 2008, Adam et al.80 reported photoswitching in IrisFP, the Phe173Ser mutant of tetrameric EosFP. By using X-ray crystallography and (in crystallo) optical spectroscopy, we showed that IrisFP displays reversible photoswitching between a bright green and a dark state via cis–trans isomerization of the chromophore, photoconverts irreversibly to the red-emitting state under violet light and exhibits reversible photoswitching also in the red form, again via cis–trans isomerization of the chromophore. Apparently, the significantly increased free volume in the chromophore pocket due to the amino acid exchange facilitates isomerization. Subsequently, monomeric FP variants were introduced that combine reversible and irreversible photoactivation, mIrisFP51 and NijiFP.64Reversible photoswitching of the green chromophore was analyzed in great detail in mIrisGFP (mEosFPthermo Phe173Ser) by using optical spectroscopy.82 This IrisFP variant was engineered to be monomeric but lost its green-to-red photoconversion capability in the process. Interestingly, it has His62 in the chromophore triad, which is seen as a necessary57 (but apparently not a sufficient) condition for effective photoconversion. We found that isomerization and protonation in mIrisGFP are strictly correlated because of very different acid–base environments that the chromophore's hydroxyphenyl group encounters in the two isomeric forms, so that pKcis ≈ 5.5 and pKtrans > 10. Consequently, excitation of the anionic cis chromophore (using 473 nm light) generates the neutral trans chromophore, and subsequent excitation of the neutral trans chromophore (using 405 nm light) induces back isomerization to the anionic cis state. Interestingly, cis–trans isomerization was much more efficient by exciting the neutral, non-fluorescent cis chromophore with 405 nm light rather than the anionic cis chromophore at 473 nm. This observation can be rationalized by quantum-chemical calculations by Olsen et al.,125 who found a barrierless energy surface for excited-state isomerization for the neutral but not for the anionic chromophore. Colletier et al.126 studied photoswitching of green IrisFP from the non-fluorescent trans to the fluorescent cis state by time-resolved absorption spectroscopy. They found evidence of three intermediate states, two with picosecond and one with microsecond lifetimes, associated with fast chromophore isomerization in the excited state and slow proton transfer in the ground state, respectively.
Evidence is mounting that reversible photoswitching is common to all EosFP variants, although with vastly different yields. For example, Thedie et al.108 showed that green mEos2 embedded in polyvinylalcohol can be switched off by 488 nm or strong 561 nm light. As the green-emitting state could readily be recovered by 405 nm light irradiation, the dark state was associated with a protonated chromophore either in a twisted or in a trans configuration. Green-to-red photoconversion of this dark state via 405 nm light exposure was not observed, however. A protonated and twisted chromophore was also generated by irradiation of the green anionic cis chromophore in the FP LEA.127 In this work, it was suggested that this state may be able to photoconvert upon 405 nm irradiation, but that photoswitching back to the anionic cis state is much more likely. For green mEos4b, a long-lived dark trans state was found upon 488 nm irradiation and characterized in detail.128 Based on kinetic crystallography, molecular dynamics, and Raman spectroscopy data, it was found that the relative stability of the cis and trans chromophores, judged from the number of hydrogen bonds holding it in place, is an important factor controlling the switching kinetics. Energetic issues of chromophore stabilization were also addressed in recent work by Boxer and co-workers,129 who modified the tyrosine moiety of the p-HBI chromophore in Dronpa2 using amber suppression. Their systematic analysis emphasizes the important role of the chromophore's charge distribution for modulating the energy surfaces of the singlet ground and excited states in response to I and P bond twisting.
Furthermore, there are also observations of photoswitching within the red form of photoconverting EosFP variants. Sun et al.130 investigated the effect of formaldehyde fixation on the photophysical properties of mEos3.2. From their experiments, they concluded that a fraction of red-emitting mEos3.2 molecules enter a long-lived dark state upon 405 nm irradiation from which they could be recovered by 561 nm irradiation. Recently, Bourgeois and coworkers131 also identified a long-lived dark state in the fluorescence emission from individual, red-converted mEos4b molecules. X-ray crystallography revealed that the dark-state chromophore is highly dynamic and adopts a “frustrated” trans conformation, with its hydroxyphenyl group bent out of plane to avoid steric clashes with a neighboring residue (Ile157). The absorption maximum at ∼475 nm and the finding that re-isomerization occurs upon 405-, 473- and 488 nm light illumination suggested that the trans chromophore is protonated (see Fig. 6b).
Our mechanistic understanding of the photochemical processes in EosFP variants is still incomplete, as shown in this section. Nevertheless, their heuristic optimization for specific imaging experiments has been enormously successful, as shown in the following section.
Advanced optical imaging with EosFP variants
Pulse chase imaging with conventional fluorescence microscopy
Optical pulse-chase imaging is a powerful technique to observe protein dynamics in live cells, tissues, and organisms labeled with a protein of interest fused to a photoconvertible FP. After imaging by excitation of the initially green FP, a subpopulation of molecules is selected and photoconverted by focused light irradiation to their red-emitting state. Subsequently, migration of these “tagged” molecules is followed in the red color channel without interference from newly expressed green-emitting proteins. 405 nm photoconversion light (typically from a GaN diode laser) is well separated in wavelength from photoexcitation light of green (e.g., 473 or 488 nm) or red (e.g., 532 or 561 nm) FP fluorophores (see Fig. 6b), so unintentional photoconversion by fluorescence excitation is not an issue.Fig. 7 shows examples of optical pulse-chase experiments. In Fig. 7a, the mRNA of tetrameric EosFP was microinjected into a Xenopus laevis oocyte. At developmental stage 3, one of the four cells was irradiated with 405 nm light to selectively photoconvert markers in this cell to the red state; the descendant cells were subsequently tracked over time. For organelle tracking, mitochondria in rabbit kidney cells were labeled with tdEosFP fused to a mitochondrial targeting signal. After photoconversion of individual mitochondria, migration as well as fusion and fission of mitochondria could be observed (Fig. 7b). Fig. 7c shows protein tracking in live cells at 37 °C using the tubulin-binding protein RITA132 fused to mEosFPthermo. After photoconversion in the region marked by the circle, red-emitting RITA fusion proteins appeared within 20 s in the region marked by the square in the leftmost image (see close-ups at 0, 20 and 40 s on the right).
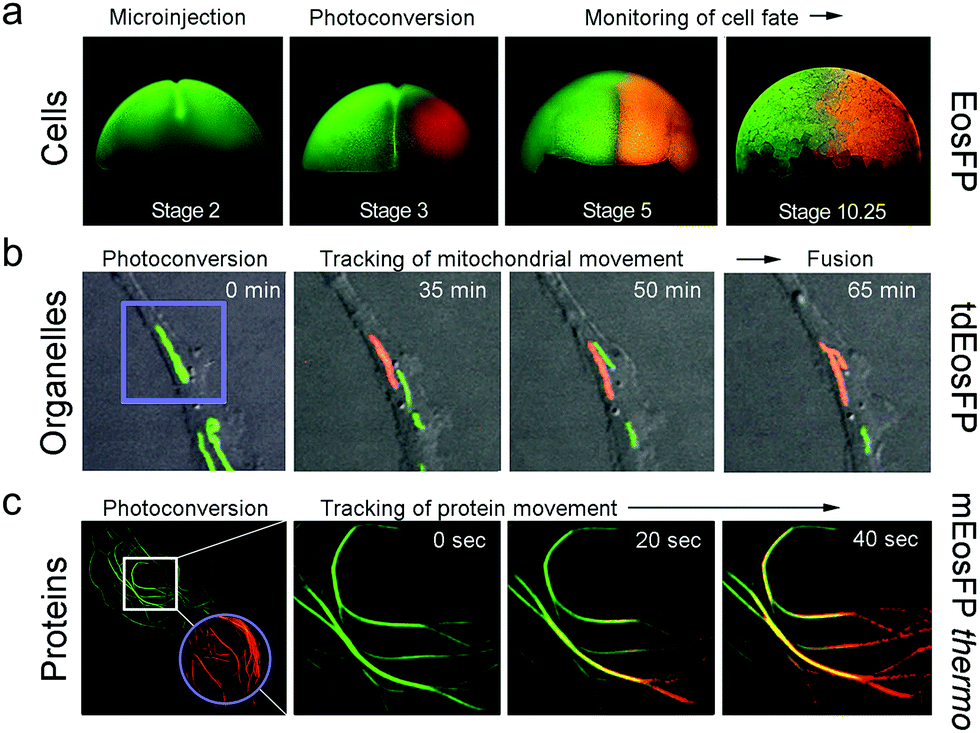 | ||
| Fig. 7 Application of EosFP and variants in pulse-chase experiments. (a) Cell tracking. The mRNA of tetrameric EosFP was microinjected into a Xenopus laevis oocyte, so that all descendant cells express the marker as shown for stage 2 (two cells). At stage 3 (four cells), EosFP molecules in one cell were selectively photoconverted to the red state and the descendant cells were tracked over time. (b) Organelle tracking. Mitochondria in rabbit kidney cells were labeled with tdEosFP fused to a mitochondrial targeting signal. After photoconversion in the region marked by the violet square by focused 405 nm irradiation, the red-labeled markers were followed over time. (c) Protein tracking. Parts of the cytoskeleton of a HeLa cell are highlighted by RITA-mEosFPthermo. After photoconversion of the FPs in the region marked by the violet circle at time zero, red-emitting RITA proteins appeared within 20 s in the region marked by the white square in the leftmost image (see close-ups at 0, 20 and 40 s on the right). Adapted from ref. 58 with permission from Wiley-VCH, copyright 2011. | ||
SMLM and super-resolution pulse chase imaging
Super-resolution SMLM is a camera-based widefield microscopy technique that relies on the localization of individual fluorophores.5–7 Oftentimes, SMLM is carried out with photoactivatable FPs, using either green-to-red photoconversion or reversible photoswitching. Suitable light irradiation ensures that only a small number of fluorophores are in the emitting state in a single camera frame (dwell time of milliseconds) and imaged until they cease to emit, either due to photobleaching or entering a transient, long-lived dark state. In each image, the spots from only a few, randomly activated fluorophores are not likely to overlap spatially. Therefore, the center of gravity of all photon positions measured for an individual fluorophore before photobleaching can be precisely computed and taken as an estimate of the fluorophore location. The final image is reconstructed by superimposing all measured fluorophore locations from a large number (typically 102–104) of camera frames (Fig. 8).133,134 | ||
| Fig. 8 Schematic depictions of SMLM. The fluorescence emission properties of the inactive (“off”) and active (“on”) chromophore species are indicated by the color code of the lettering. Grey: dark, non-emitting; green: green-emitting; magenta: red-emitting. (a) SMLM based on irreversible green-to-red photoconversion by using 405 nm light activation. (b) SMLM based on photoconversion by using dual-color (488 nm/∼700 nm), i.e., primed light activation. (c) SMLM based on reversible photoswitching (shown for the green chromophore). | ||
For SMLM with photoconvertible EosFP variants, we irradiate the sample with low-intensity 405 nm (Fig. 8a) or dual-color (primed) photoconverting light (Fig. 8b). Therefore, only a small number of green fluorophores turn red within each camera frame and are subsequently localized by imaging with, e.g., 532- or 561 nm excitation light in the red color channel. Photoswitchable EosFP variants are so-called “negative” photoswitchers,38,135–137i.e., upon excitation of their anionic fluorescent species (with 473- or 488 nm light for green-emitting FPs) off-switching competes with fluorescence. Thus, imaging with simultaneous low-intensity 405 nm activation light switches only a small number of fluorophores to the on-state within each frame so that they can be localized individually (Fig. 8c).
A FP marker that combines both photoactivation modes such as mIrisFP permits pulse-chase imaging experiments with sub-diffraction resolution in live cells by using dual-color SMLM.51 Green-to-red photoconversion allows for photo-induced tagging and chasing of a subset of proteins in the cell, as discussed in the previous subsection, and the photoswitching capability of the green and red fluorophore species can be used for SMLM (Fig. 8c). Notably, this imaging scheme requires that the probability of photoconversion is less than the one of photoswitching the green chromophore with 405 nm light.
We first demonstrated super-resolution pulse-chase imaging in experiments aimed to visualize the dynamics of focal adhesion complexes, which are known to continuously assemble and disassemble during cell migration. For imaging, we used live HeLa cells expressing a paxillin–mIrisFP fusion protein after transfection. Paxillin is a multidomain adaptor protein and a crucial component of focal adhesions, as shown in the diffraction-limited image obtained by total internal reflection (TIRF) microscopy (Fig. 9a). First, a SMLM image of focal adhesion points was taken, using photoswitching of green mIrisFP (Fig. 9b). Then, mIrisFP molecules in the region marked by the violet oval in Fig. 9b were photoconverted to the red form. After 75 s, the SMLM image taken using photoswitching of red mIrisFP reveals that a significant fraction of the tagged molecules appeared in locations outside the irradiated region (Fig. 9c). A patch of focal adhesions (marked with a circle in Fig. 9b–d) was already present while the green image was taken (Fig. 9b) and grew in strength over the following 600 s by recruiting red-converted paxillin–mIrisFP from the photoconversion region (Fig. 9c and d).
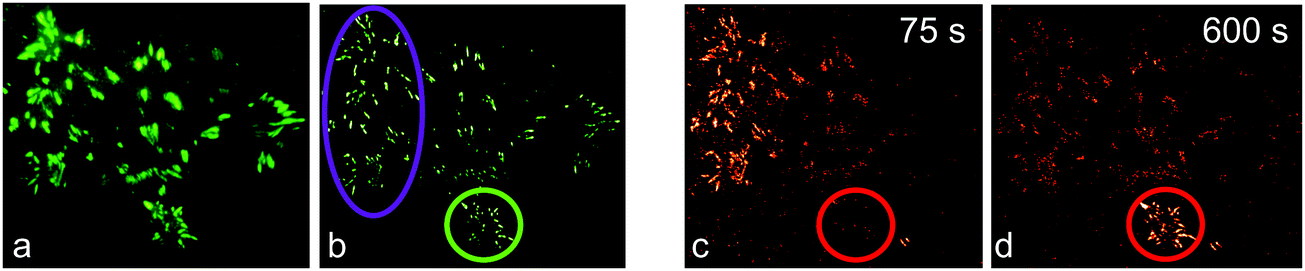 | ||
| Fig. 9 Dual-color SMLM combined with pulse-chase imaging on a live HeLa cell. (a) TIRF microscopy image of green paxillin–mIrisFP located at focal adhesions. (b) SMLM reconstruction using photoswitching of green mIrisFP. Afterwards, mIrisFP molecules in the region marked by the violet oval were irradiated for 30 s with 405 nm light for green-to-red photoconversion. (c and d) SMLM images of red paxillin–mIrisFP (c) 75 s and (d) 600 s after photoconversion. | ||
Dual-channel SMLM
Oftentimes, simultaneous imaging of two target proteins with sub-diffraction resolution is desirable, e.g., to analyze their mutual interaction. Dual-channel SMLM can be realized by using fluorescent markers with different photoactivation mechanisms, e.g., mEosFPthermo and mIrisGFP. The following imaging strategy ensures complete decoupling of the two channels: First, a SMLM image of mEosFPthermo is captured in the red channel using 405 nm photoconversion and 561 nm excitation light. Once all green mEosFPthermo molecules are depleted by photoconversion and subsequent bleaching, a SMLM image of mIrisGFP is acquired in the green channel using 473 nm (off-switching) excitation and 405 nm (on-switching) light.We have employed this approach to study desmin, a muscle-specific intermediate filament protein, in live cells. Desmin mutations have been implicated in various muscle diseases and are known to cause formation of protein aggregates. To mimic the clinically relevant heterozygous situation, we transfected SW-13 (adrenal cortex carcinoma) cells with equal amounts of plasmid for co-expression of mIrisGFP fused to wild-type desmin and mEosFPthermo fused to desmin mutant Asn116Ser (and other variants).138 Notably, SW-13 cells do not express desmin on their own. Dual-channel SMLM revealed that filaments as well as aggregates existed within the same cells, both of them containing wild-type and Asn116Ser mutant desmin (Fig. 10). By contrast, homozygous transient expression revealed that wild-type desmin assembled into filamentous networks, whereas the Asn116Ser mutant formed cytoplasmic aggregates. Interestingly, the localization patterns were found to vary from mutant to mutant.138
 | ||
| Fig. 10 Dual-channel SMLM of SW-13 cells co-expressing wild-type desmin fused to mIrisGFP and desmin mutant Asn116Ser fused to mEosFPthermo. (a and b) SMLM images of (a) the wild-type desmin fusion protein using photoswitching of mIrisFP and (b) the mutant fusion protein using 405 nm photoconversion of mEosFPthermo. (c) Dual-color (overlay) SMLM image. Scale bar, 1 μm. Adapted from ref. 138 with permission from American Society for Biochemistry and Molecular Biology, copyright 2012. | ||
Primed conversion offers an alternative approach to dual-channel super-resolution SMLM, requiring only a single wavelength for fluorescence excitation. To this end, we use two photoconverting FP markers, one capable of regular 405 nm photoconversion and the other one of primed photoconversion. We can then distinguish the two FPs, which have essentially identical spectral properties, solely on the basis of their susceptibility to the different photoconversion methods.
To demonstrate this approach, we fused (405 nm converting) mEos2 to E-MAP-115139 and (primeable) pr-mEos2 to Lifeact140 to visualize microtubules and actin fibers, respectively. In the regular wide-field image, the green-emitting mEos2 and pr-mEos2 are indeed indistinguishable (Fig. 11a). Then, we collected a SMLM image of the red-emitting LifeAct-pr-mEos2 using primed photoconversion (Fig. 11b). Subsequently, we imaged the remaining E-MAP-115-mEos2 markers, which do not convert under primed conversion illumination, by SMLM using 405 nm photoconversion (Fig. 11c). The overlay image of the two channels is shown in Fig. 11d.
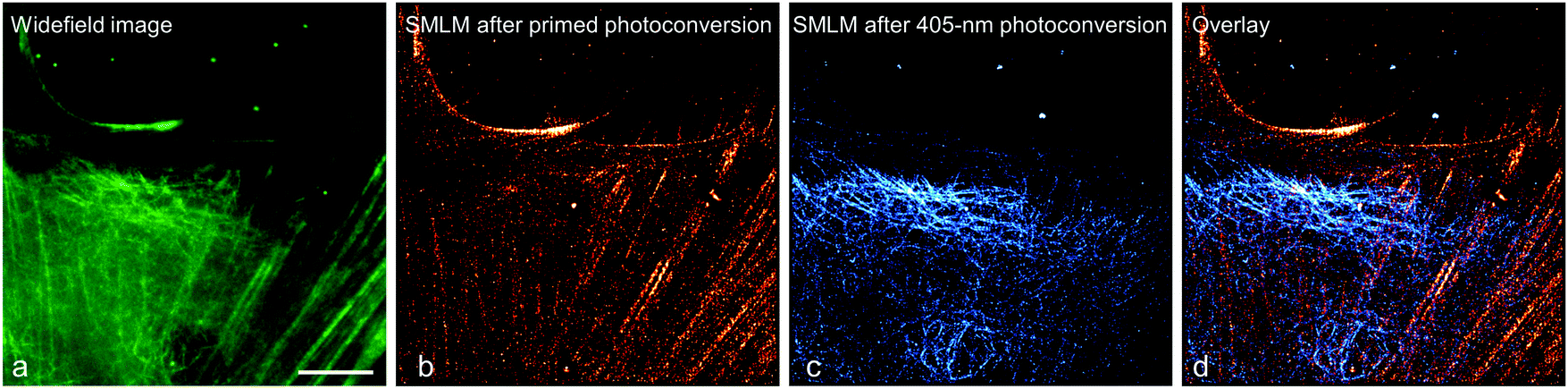 | ||
| Fig. 11 Sequential dual-channel SMLM using EosFP variants with orthogonal green-to-red photoconversion modes. (a) Widefield image of a HeLa cell, with actin filaments and microtubules labeled with LifeAct-pr-mEos2 and E-MAP-115-mEos2, respectively. The green emission from the two markers is indistinguishable. (b) SMLM of red-converted LifeAct-pr-mEos2 using primed photoconversion (488/642 nm). (c) SMLM of red-converted E-MAP-115-mEos2 using 405 nm photoconversion, with the fluorescence of red-emitting mEos2 depicted in blue color to distinguish the two markers in the overlay shown in (d). Scale bar, 10 μm. Adapted from ref. 81 with permission from John Wiley and Sons, copyright 2017. | ||
Quantitative SMLM approaches
Quantitative SMLM goes beyond mere imaging and can provide insight into biomolecular dynamics, clustering and oligomerization of proteins in biological systems.133 SMLM lends itself to protein quantification, as photons are collected from each individual photoactivated FP until, in the ideal case, its fluorophore photobleaches. However, FPs typically have long-lived, non-emissive states from which they can recover thermally or by light-activation.141 Thus, they undergo multiple, temporally separated activation-deactivation cycles, leading to overcounting the FPs or FP fusion constructs. By contrast, undercounting can result from incomplete maturation and photoconversion. For accurate counting, it is therefore important to characterize the photophysical properties of the FP label and include this information in the analysis.142–144 By taking the number of photophysical (bright and dark) states, the kinetics of interconversion, and the fraction of active fluorophores into account, the number of molecules can be determined from the blinking statistics. Hummer et al.145 used this approach to extract oligomerization states of membrane-bound CD86-mEos2, VSVG-mEos2, and CD80-mEos2 from SMLM data.146 Furthermore, Krueger et al.147 used quantitative SMLM to determine the oligomerization state of the toll-like receptor TLR4 fused to mEos2 in HEK 293 cells in the presence and absence of the co-receptors MD2 and CD14 and in response to different lipopolysaccharide ligands.Manley et al.78 combined SMLM with single-particle tracking (SPT). By taking successive SMLM image frames of the membrane proteins Gag and VSVG fused to tdEosFP and linking the signals of the same molecule in consecutive frames according to their proximity, they reconstructed short single-molecule trajectories. Here, sequential photoactivation of a small set of FP molecules in each image facilitates linking the signals associated with a particular FP over successive images and allows tracking at a much higher fluorophore density than in traditional SPT, where all fluorescent labels are simultaneously active.
In our lab, we have studied the transport of vesicles to and from the hyphal tip of live Aspergillus nidulans cells by combining SMLM with high-speed pulse-chase imaging.148 We found that the chitin synthase ChsB (fused to mEosFPthermo) was transported from the cell body to the hyphal tip and vice versa at two different speeds. By using motor protein deletion mutants, the fast movements (7–10 μm s−1) were assigned to transport of secretory vesicles loaded with ChsB by kinesin-1, and the slower ones (2–7 μm s−1) to transport of early endosomes loaded with ChsB by kinesin-3.
Correlative light and electron microscopy (CLEM)
Multi-channel imaging can also be performed by using two entirely different modalities, such as electron microscopy (EM), optical microscopy, Raman microscopy or magnetic resonance imaging. Such correlative approaches can provide complementary information, as they are generally based on different mechanisms for contrast generation. Correlative light and electron microscopy (CLEM) combines EM for ultrastructural analysis with optical microscopy, most often, fluorescence microscopy for visualizing proteins or organelles that are specifically tagged by fluorescent markers. This approach has become much more powerful with the advent of super-resolution optical microscopy, which closes the resolution gap between EM and optical microscopy.Before EM data collection, samples are typically fixed, stained with osmium tetroxide (OsO4), a widely used contrast agent that binds to lipid membranes, and embedded in a resin. Unfortunately, most FPs respond to OsO4 staining with a loss of fluorescence, precluding their use in CLEM. To combine transmission EM (TEM) and SMLM based on labeling with photoconverting EosFP-type markers, Paez-Segala et al.83 engineered and characterized a variety of mEos2 mutants. Two of them, mEos4a and mEos4b, were much more resistant to heavy staining (1%) with OsO4 than mEos2, mEos3.1 and mEos3.2, both in the green and, after photoconversion, in the red forms, thus enabling correlative SMLM and high-quality TEM. Very recently, Fu et al.84 introduced three more mutations into mEos4b to obtain variant mEosEM, for which they reported an improved performance after sample embedding in epon, an epoxy resin that is harder than the low-polymerization-temperature hydrophilic resin, glycidyl methacrylate, used by Paez-Segala et al.83 and better preserves the ultrastructure of the specimens. We note that, after epon embedding, mEosEM did no longer photoconvert but showed reversible photoswitching in the green form, which can be exploited for SMLM.
Nonlinear structured illumination microscopy
Structured illumination microscopy (SIM) is a linear fluorescence imaging technique, offering up to two-fold resolution enhancement by applying stripe-like periodically patterned illumination.149 Its nonlinear extension, saturated structured illumination microscopy (SSIM),150 enables super-resolution imaging by using high excitation power to drive fluorophores into saturation. A non-linear excitation/emission response, which is the origin of the additional resolution enhancement of SSIM,151 can also be generated with photoswitchable FP markers, using orders of magnitude less laser power, however. This imaging modality was termed patterned activation nonlinear structured illumination microscopy (PA NL-SIM).152 Patterned activation of the FP markers is followed by patterned excitation and readout of the emission. More specifically, a set of raw wide field images is recorded by shifting and rotating the activation/illumination pattern. The super-resolution image is reconstructed from the raw images based on the known illumination pattern. Different from SMLM, which favors photoactivatable FPs emitting all their photons in a single burst and featuring a high on/off contrast ratio for localization, PA NL-SIM can tolerate photoswitchable FP markers with lower contrast ratio. It demands, however, that its photon budget, i.e., all photons emitted prior to photobleaching, can be expended over up to hundreds of on–off cycles without a significant decay in photon emission per cycle and, for live imaging, without adding any toxic anti-bleaching chemicals.153,154 Zhang et al.155 engineered an FP starting from mEos3.1 with optimal properties for PA NL-SIM, which they denoted Skylan-NS. Using PA NL-SIM with total internal reflection microscopy (TIRF) excitation, Betzig and coworkers152 demonstrated the excellent performance of Skylan-NS. They imaged caveolae, flask-shaped invaginations of the plasma membrane with a diameter of 50–100 nm (Fig. 12), formed by oligomerization of the caveolin protein, using Skylan-NS fused to caveolin. | ||
| Fig. 12 Images of caveolae on the plasma membrane of a live COS-7 cell expressing Skylan-NS-caveolin at 23 °C. TIRF images (a) with deconvolution and combined with (b) SIM, (c) PA NL-SIM, and (d) saturated PA NL-SIM (45-nm resolution) are shown. Insets in (a–d) show individual caveolae. (e) Diversity of caveolae ring diameters as visualized by PA NL-SIM. (f) Larger rings may represent surface-docked vesicles. (g) Clusters of caveolae reminiscent of clathrin plaques. Scale bars, 100 nm. Adapted from ref. 152 with permission from American Association for the Advancement of Science, copyright 2015. | ||
Super-resolution optical fluctuation imaging (SOFI)
Unlike SMLM, which uses the emission intensity to localize many individual fluorophores for reconstruction of super-resolved images, SOFI relies on temporal fluctuations of the intensity of individual emitters contributing to each pixel of the image.156 Therefore, SOFI requires fluorophores with flickering emission intensity. To produce a super-resolved SOFI image, a time series of tens to hundreds of images is acquired, and the intensity fluctuations of each individual pixel (of sub-resolution size) are analyzed by calculating nth-order cumulants, which are functions related to nth-order correlation functions.156 An image generated from the nth-order cumulant yields an n-fold enhanced resolution over the regular (intensity) image.157 The super-resolved image is background-free and the pixel amplitudes represent the number of fluorescent labels if they exhibit the same emission properties, including the intensity levels of the dark (off) and bright (on) states and the photoswitching kinetics.Photoswitchable FPs have been successfully employed for SOFI, exploiting their ability to repeatedly switch between a bright on-state and a dark off-state.158 Zhang et al.159 developed Skylan-S from mEos2 and reported beneficial properties especially for SOFI, including a high intensity in the on-state and an enhanced contrast ratio and photobleaching stability. To demonstrate that Skylan-S is an excellent probe for SOFI, they imaged clathrin-coated pits (CCPs) in living U2OS cells (Fig. 13), using a fusion protein of clathrin and Skylan-S. CCPs are cell membrane invaginations with diameters of 100–200 nm mediating endocytosis. They grow over the course of several ten seconds, and finally bud off to form intracellular vesicles.160
 | ||
| Fig. 13 Widefield and SOFI images of CCPs in live U2OS cells, using clathrin fused with Skylan-S for labeling. (a–c) TIRFM images. (d–f) SOFI images from second-order cumulants. (g–i) SOFI images from third-order cumulants. (b, e and h) Close-up views of the boxed regions in panels a, d, and g, respectively. (c, f and i) Close-up views of the boxed regions in panels b, e, and h, respectively. Scale bars, 10 μm (a, d and g), 3 μm (b, e and h), and 500 nm (c, f and i). Adapted from ref. 159 with permission from American Chemical Society, Copyright 2015. | ||
Conclusions and outlook
Optical fluorescence microscopy, especially super-resolution imaging, has been established as one of the most powerful and versatile tools for studying the molecular basis of life. The method crucially relies on our ability to label biomolecules selectively with fluorescent markers, which is an unavoidable perturbation of the system. For live-cell imaging with super-resolution, FPs will be indispensable due to their genetic encodability, which ensures simple labeling procedures. Recent advances in gene editing, notably, CRISPR/Cas9, have made powerful tools available for targeted gene modification, e.g., to introduce a FP domain, at a specific locus, making the FP labeling approach even more powerful. Gene editing ensures that expression levels are more native-like and artifacts due to transient overexpression can be avoided. However, the smaller numbers of expressed labels (with respect to overexpression via transient transfection) make imaging experiments more demanding and call for highly sensitive microscopes and the brightest (photoactivatable) labels. Accordingly, the quest for FPs with enhanced brightness, suitable photoactivation properties, and a large photon budget will continue.In this review, we have focused on photoactivatable FPs of the EosFP clade and shown that only a few modifications of the natural amino acid sequence resulted in many FP variants with improved and sometimes surprising new properties advantageous for certain imaging techniques. We anticipate that the usual trial-and-error or random approaches for identifying suitable sequence modifications will be increasingly substituted by rational engineering, based on better understanding of the structure–function relationships in FPs, giving guidance for the development of further improved EosFP variants and other GFP-type proteins, e.g., with respect to the intriguing array of light-tunable optical properties. In fact, whereas all EosFP variants included in this review have shown their usefulness in certain applications, they are by no means optimized, easy-to-use devices. For example, apart from regular photoactivation, FPs exhibit intermittency of their fluorescence emission due to transient (and often long-lived) population of non-emissive states. This can be a blessing in some applications (e.g., shelving in dark states can reduce photobleaching effects137) but a curse in others (e.g., an already registered FP may be mistaken for a different one upon recovery from a long-lived dark state in quantitative SMLM). Often, carefully adjusted sample illumination with multiple laser wavelengths can help release FPs from these states for best imaging performance.51,108,130 In view of the continuous appearance of new variants even after almost 20 years of EosFP engineering, we are looking forward to further advanced variants, optimized for specific applications.
Notably, there is a large pool of FP sequences in a variety of animals that has not been systematically explored. Screening campaigns may reveal FPs with new and advanced properties that can be exploited for biological imaging. Therefore, we envision that there are still considerable opportunities for advances in FP marker technology. Likewise, we expect progress with other genetically encoded labeling techniques not covered in this review, e.g., chemical tags such as Halo or SNAP for protein labeling161 or light-up aptamers for RNA labeling.162–164 These approaches employ synthetic dyes with excellent fluorescence properties, superior to those of FP-based labels, but require an external supply of fluorophores, limiting their versatility and ease of use. In view of the central role of optical fluorescence microscopy in the life sciences, there can be no doubt that the quest for bright, selective and sophisticated optical fluorescence markers for the imaging of biological structures will continue to be a hot research topic for years to come.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge many researchers for their collaboration in the development and characterization of FP markers, specifically, EosFP, for more than 20 years. Special thanks are due to Jörg Wiedenmann, who brought us into this exciting research field. He discovered EosFP and many other FPs in anthozoa and optimized them to serve as fluorescence markers. In the early years, Franz Ostwald's molecular and cellular biology expertise was essential for testing our FPs in cells and model organisms. We also thank Dominique Bourgeois and Periklis (Laki) Pantazis and their groups for working with us on the characterization of dual-mode photoactivation of IrisFP and the mechanistic basis of primed conversion in pr-mEosFP and other FPs, respectively. The Nienhaus lab was funded by the Helmholtz Association, program Materials Systems Engineering (MSE), the Karlsruhe School of Optics and Photonics (KSOP) and the Deutsche Forschungsgemeinschaft (SFB 1324 (project Z02) and GRK 2039).References
- Fluorescence Microscopy: From Principles to Biological Applications, ed. U. Kubitscheck, Wiley-VCH GmbH, Weinheim, Germany, 2017 Search PubMed.
- J. C. Stockert and A. Blazquez-Castro, Fluorescence Microscopy in Life Sciences, ed. U. A. E. Sharjah, Bentham Science Publishers, 2017 Search PubMed.
- Far-Field Optical Nanoscopy, ed. P. Tinnefeld, C. Eggeling and S. W. Hell, Springer Berlin Heidelberg, 2015 Search PubMed.
- S. W. Hell, Science, 2007, 316, 1153–1158 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- S. T. Hess, T. P. Girirajan and M. D. Mason, Biophys. J., 2006, 91, 4258–4272 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- L. Schermelleh, R. Heintzmann and H. Leonhardt, J. Cell Biol., 2010, 190, 165–175 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, J. Mol. Biol., 2016, 428, 308–322 CrossRef CAS PubMed.
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS PubMed.
- R. P. Haugland, The Handbook—A Guide to Fluorescent Probes and Labeling Technologies, ed. O. R. Eugene, Molecular Probes, 11th edn, 2005 Search PubMed.
- K. Nienhaus and G. U. Nienhaus, in Fluorescence Microscopy: From Principles to Biological Applications, ed. U. Kubitscheck, Wiley-VCH GmbH, 2017, pp. 143–174 Search PubMed.
- J. Lippincott-Schwartz and G. H. Patterson, Science, 2003, 300, 87–91 CrossRef CAS PubMed.
- E. A. Rodriguez, R. E. Campbell, J. Y. Lin, M. Z. Lin, A. Miyawaki, A. E. Palmer, X. Shu, J. Zhang and R. Y. Tsien, Trends Biochem. Sci., 2017, 42, 111–129 CrossRef CAS PubMed.
- R. N. Day and M. W. Davidson, Chem. Soc. Rev., 2009, 38, 2887–2921 RSC.
- G. U. Nienhaus, Angew. Chem., Int. Ed., 2008, 47, 8992–8994 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- O. Shimomura, F. H. Johnson and Y. Saiga, J. Cell. Physiol., 1962, 59, 223–239 CrossRef CAS PubMed.
- D. C. Prasher, V. K. Eckenrode, W. W. Ward, F. G. Prendergast and M. J. Cormier, Gene, 1992, 111, 229–233 CrossRef CAS PubMed.
- M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Science, 1994, 263, 802–805 CrossRef CAS PubMed.
- K. Nienhaus, G. U. Nienhaus, J. Wiedenmann and H. Nar, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9156–9159 CrossRef CAS PubMed.
- M. Ormö, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392–1395 CrossRef PubMed.
- F. Yang, L. G. Moss and G. N. Phillips, Jr., Nat. Biotechnol., 1996, 14, 1246–1251 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- M. V. Matz, A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov and S. A. Lukyanov, Nat. Biotechnol., 1999, 17, 969–973 CrossRef CAS PubMed.
- J. Wiedenmann, C. Elke, K. D. Spindler and W. Funke, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14091–14096 CrossRef CAS PubMed.
- A. Miyawaki, Cell Struct. Funct., 2002, 27, 343–347 CrossRef CAS PubMed.
- G. S. Baird, D. A. Zacharias and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11984–11989 CrossRef CAS PubMed.
- N. C. Shaner, R. E. Campbell, P. A. Steinbach, B. N. Giepmans, A. E. Palmer and R. Y. Tsien, Nat. Biotechnol., 2004, 22, 1567–1572 CrossRef CAS PubMed.
- F. Oswald, F. Schmitt, A. Leutenegger, S. Ivanchenko, C. D'Angelo, A. Salih, S. Maslakova, M. Bulina, R. Schirmbeck, G. U. Nienhaus, M. V. Matz and J. Wiedenmann, FEBS J., 2007, 274, 1102–1109 CrossRef CAS PubMed.
- J. Wiedenmann, S. Ivanchenko, F. Oswald and G. U. Nienhaus, Mar. Biotechnol., 2004, 6, 270–277 CrossRef CAS PubMed.
- V. V. Verkhusha and K. A. Lukyanov, Nat. Biotechnol., 2004, 22, 289–296 CrossRef CAS PubMed.
- Y. Ishitsuka, K. Nienhaus and G. U. Nienhaus, Methods Mol. Biol., 2014, 1148, 239–260 CrossRef CAS PubMed.
- J. Lippincott-Schwartz and G. H. Patterson, Trends Cell Biol., 2009, 19, 555–565 CrossRef CAS PubMed.
- K. A. Lukyanov, D. M. Chudakov, S. Lukyanov and V. V. Verkhusha, Nat. Rev. Mol. Cell Biol., 2005, 6, 885–891 CrossRef CAS PubMed.
- K. D. Piatkevich and V. V. Verkhusha, Curr. Opin. Chem. Biol., 2010, 14, 23–29 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, Chem. Soc. Rev., 2014, 43, 1088–1106 RSC.
- D. M. Shcherbakova, P. Sengupta, J. Lippincott-Schwartz and V. V. Verkhusha, Annu. Rev. Biophys., 2014, 43, 303–329 CrossRef CAS PubMed.
- A. Acharya, A. M. Bogdanov, B. L. Grigorenko, K. B. Bravaya, A. V. Nemukhin, K. A. Lukyanov and A. I. Krylov, Chem. Rev., 2017, 117, 758–795 CrossRef CAS PubMed.
- D. Bourgeois and V. Adam, IUBMB Life, 2012, 64, 482–491 CrossRef CAS PubMed.
- G. U. Nienhaus, K. Nienhaus and J. Wiedenmann, in Fluorescent Proteins I: From Understanding to Design, ed. G. Jung, Springer, 2012, vol. 11, pp. 241–263 Search PubMed.
- D. Shcherbo, E. M. Merzlyak, T. V. Chepurnykh, A. F. Fradkov, G. V. Ermakova, E. A. Solovieva, K. A. Lukyanov, E. A. Bogdanova, A. G. Zaraisky, S. Lukyanov and D. M. Chudakov, Nat. Methods, 2007, 4, 741–746 CrossRef CAS PubMed.
- M. Z. Lin, M. R. McKeown, H. L. Ng, T. A. Aguilera, N. C. Shaner, R. E. Campbell, S. R. Adams, L. A. Gross, W. Ma, T. Alber and R. Y. Tsien, Chem. Biol., 2009, 16, 1169–1179 CrossRef CAS PubMed.
- J. Wiedenmann, F. Oswald and G. U. Nienhaus, IUBMB Life, 2009, 61, 1029–1042 CrossRef CAS PubMed.
- D. A. Zacharias, J. D. Violin, A. C. Newton and R. Y. Tsien, Science, 2002, 296, 913–916 CrossRef CAS PubMed.
- S. Kredel, F. Oswald, K. Nienhaus, K. Deuschle, C. Röcker, M. Wolff, R. Heilker, G. U. Nienhaus and J. Wiedenmann, PLoS One, 2009, 4, e4391 CrossRef PubMed.
- R. E. Campbell, O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7877–7882 CrossRef CAS PubMed.
- J. Wiedenmann, B. Vallone, F. Renzi, K. Nienhaus, S. Ivanchenko, C. Röcker and G. U. Nienhaus, J. Biomed. Opt., 2005, 10, 14003 CrossRef PubMed.
- E. Balleza, J. M. Kim and P. Cluzel, Nat. Methods, 2018, 15, 47–51 CrossRef CAS PubMed.
- J. Fuchs, S. Böhme, F. Oswald, P. N. Hedde, M. Krause, J. Wiedenmann and G. U. Nienhaus, Nat. Methods, 2010, 7, 627–630 CrossRef CAS PubMed.
- S. Kredel, K. Nienhaus, M. Wolff, F. Oswald, S. Ivanchenko, F. Cymer, A. Jeromin, F. J. Michel, K.-D. Spindler, R. Heilker, G. U. Nienhaus and J. Wiedenmann, Chem. Biol., 2008, 15, 224–233 CrossRef CAS PubMed.
- T. D. Craggs, Chem. Soc. Rev., 2009, 38, 2865–2875 RSC.
- J. W. Park and Y. M. Rhee, J. Am. Chem. Soc., 2016, 138, 13619–13629 CrossRef CAS PubMed.
- S. R. Meech, Chem. Soc. Rev., 2009, 38, 2922–2934 RSC.
- R. Ando, H. Hama, M. Yamamoto-Hino, H. Mizuno and A. Miyawaki, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12651–12656 CrossRef CAS PubMed.
- J. Wiedenmann, S. Ivanchenko, F. Oswald, F. Schmitt, C. Röcker, A. Salih, K. D. Spindler and G. U. Nienhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15905–15910 CrossRef CAS PubMed.
- J. Wiedenmann, S. Gayda, V. Adam, F. Oswald, K. Nienhaus, D. Bourgeois and G. U. Nienhaus, J. Biophoton., 2011, 4, 377–390 CrossRef CAS PubMed.
- S. A. McKinney, C. S. Murphy, K. L. Hazelwood, M. W. Davidson and L. L. Looger, Nat. Methods, 2009, 6, 131–133 CrossRef CAS PubMed.
- M. Zhang, H. Chang, Y. Zhang, J. Yu, L. Wu, W. Ji, J. Chen, B. Liu, J. Lu, Y. Liu, J. Zhang, P. Xu and T. Xu, Nat. Methods, 2012, 9, 727–729 CrossRef CAS PubMed.
- J. Mathur, R. Radhamony, A. M. Sinclair, A. Donoso, N. Dunn, E. Roach, D. Radford, P. S. Mohaghegh, D. C. Logan, K. Kokolic and N. Mathur, Plant Physiol., 2010, 154, 1573–1587 CrossRef CAS PubMed.
- N. G. Gurskaya, V. V. Verkhusha, A. S. Shcheglov, D. B. Staroverov, T. V. Chepurnykh, A. F. Fradkov, S. Lukyanov and K. A. Lukyanov, Nat. Biotechnol., 2006, 24, 461–465 CrossRef CAS PubMed.
- V. Adam, K. Nienhaus, D. Bourgeois and G. U. Nienhaus, Biochemistry, 2009, 48, 4905–4915 CrossRef CAS PubMed.
- V. Adam, B. Moeyaert, C. C. David, H. Mizuno, M. Lelimousin, P. Dedecker, R. Ando, A. Miyawaki, J. Michiels, Y. Engelborghs and J. Hofkens, Chem. Biol., 2011, 18, 1241–1251 CrossRef CAS PubMed.
- H. Tsutsui, S. Karasawa, H. Shimizu, N. Nukina and A. Miyawaki, EMBO Rep., 2005, 6, 233–238 CrossRef CAS PubMed.
- S. Habuchi, H. Tsutsui, A. B. Kochaniak, A. Miyawaki and A. M. van Oijen, PLoS One, 2008, 3, e3944 CrossRef PubMed.
- H. Hoi, N. C. Shaner, M. W. Davidson, C. W. Cairo, J. Wang and R. E. Campbell, J. Mol. Biol., 2010, 401, 776–791 CrossRef CAS PubMed.
- A. L. McEvoy, H. Hoi, M. Bates, E. Platonova, P. J. Cranfill, M. A. Baird, M. W. Davidson, H. Ewers, J. Liphardt and R. E. Campbell, PLoS One, 2012, 7, e51314 CrossRef PubMed.
- B. Moeyaert, N. Nguyen Bich, E. De Zitter, S. Rocha, K. Clays, H. Mizuno, L. van Meervelt, J. Hofkens and P. Dedecker, ACS Nano, 2014, 8, 1664–1673 CrossRef CAS PubMed.
- G. Lapshin, A. Salih, P. Kolosov, M. Golovkina, Y. Zavorotnyi, T. Ivashina, L. Vinokurov, V. Bagratashvili and A. Savitsky, J. Innovative Opt. Health Sci., 2015, 8, 1550028 CrossRef.
- I. D. Solovyev, A. V. Gavshina, A. S. Katti, A. I. Chizhik, L. M. Vinokurov, G. D. Lapshin, T. V. Ivashina, M. G. Khrenova, I. I. Kireev, I. Gregor, J. Enderlein and A. P. Savitsky, Sci. Rep., 2018, 8, 15542 CrossRef PubMed.
- Y. A. Labas, N. G. Gurskaya, Y. G. Yanushevich, A. F. Fradkov, K. A. Lukyanov, S. A. Lukyanov and M. V. Matz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4256–4261 CrossRef CAS PubMed.
- H. Kim, T. J. Grunkemeyer, C. Modi, L. Chen, R. Fromme, M. V. Matz and R. M. Wachter, Biochemistry, 2013, 52, 8048–8059 CrossRef CAS PubMed.
- G. U. Nienhaus, K. Nienhaus, A. Hölzle, S. Ivanchenko, F. Renzi, F. Oswald, M. Wolff, F. Schmitt, C. Röcker, B. Vallone, W. Weidemann, R. Heilker, H. Nar and J. Wiedenmann, Photochem. Photobiol., 2006, 82, 351–358 CrossRef CAS PubMed.
- K. L. Litvinenko, N. M. Webber and S. R. Meech, J. Phys. Chem. A, 2003, 107, 2616–2623 CrossRef CAS.
- L. Zhou, T. Obhof, K. Schneider, M. Feldbrugge, G. U. Nienhaus and J. Kamper, Sci. Rep., 2018, 8, 3611 CrossRef PubMed.
- H. Shroff, C. G. Galbraith, J. A. Galbraith and E. Betzig, Nat. Methods, 2008, 5, 417–423 CrossRef CAS PubMed.
- S. Manley, J. M. Gillette, G. H. Patterson, H. Shroff, H. F. Hess, E. Betzig and J. Lippincott-Schwartz, Nat. Methods, 2008, 5, 155–157 CrossRef CAS PubMed.
- A. Miyawaki, Nat. Rev. Mol. Cell Biol., 2011, 12, 656–668 CrossRef CAS PubMed.
- V. Adam, M. Lelimousin, S. Boehme, G. Desfonds, K. Nienhaus, M. J. Field, J. Wiedenmann, S. McSweeney, G. U. Nienhaus and D. Bourgeois, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18343–18348 CrossRef CAS PubMed.
- M. A. Mohr, A. Y. Kobitski, L. R. Sabater, K. Nienhaus, C. J. Obara, J. Lippincott-Schwartz, G. U. Nienhaus and P. Pantazis, Angew. Chem., Int. Ed., 2017, 56, 11628–11633 CrossRef CAS PubMed.
- S. Gayda, K. Nienhaus and G. U. Nienhaus, Biophys. J., 2012, 103, 2521–2531 CrossRef CAS PubMed.
- M. G. Paez-Segala, M. G. Sun, G. Shtengel, S. Viswanathan, M. A. Baird, J. J. Macklin, R. Patel, J. R. Allen, E. S. Howe, G. Piszczek, H. F. Hess, M. W. Davidson, Y. Wang and L. L. Looger, Nat. Methods, 2015, 12, 215–218 CrossRef CAS PubMed.
- Z. Fu, D. Peng, M. Zhang, F. Xue, R. Zhang, W. He, T. Xu and P. Xu, Nat. Methods, 2020, 17, 55–58 CrossRef CAS PubMed.
- H. Chang, M. Zhang, W. Ji, J. Chen, Y. Zhang, B. Liu, J. Lu, J. Zhang, P. Xu and T. Xu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4455–4460 CrossRef CAS PubMed.
- M. A. Elsliger, R. M. Wachter, G. T. Hanson, K. Kallio and S. J. Remington, Biochemistry, 1999, 38, 5296–5301 CrossRef CAS PubMed.
- M. Chattoraj, B. A. King, G. U. Bublitz and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8362–8367 CrossRef CAS PubMed.
- K. Nienhaus, F. Renzi, B. Vallone, J. Wiedenmann and G. U. Nienhaus, Biochemistry, 2006, 45, 12942–12953 CrossRef CAS PubMed.
- K. Nienhaus, F. Renzi, B. Vallone, J. Wiedenmann and G. U. Nienhaus, Biophys. J., 2006, 91, 4210–4220 CrossRef CAS PubMed.
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS PubMed.
- W. W. Ward, H. J. Prentice, A. F. Roth, C. W. Cody and S. C. Reeves, Photochem. Photobiol., 1982, 35, 803–808 CrossRef CAS.
- C. Scharnagl, R. Raupp-Kossmann and S. F. Fischer, Biophys. J., 1999, 77, 1839–1857 CrossRef CAS.
- R. Bizzarri, R. Nifosi, S. Abbruzzetti, W. Rocchia, S. Guidi, D. Arosio, G. Garau, B. Campanini, E. Grandi, F. Ricci, C. Viappiani and F. Beltram, Biochemistry, 2007, 46, 5494–5504 CrossRef CAS PubMed.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
- W. W. Ward, Methods Biochem. Anal., 2006, 47, 39–65 Search PubMed.
- X. He, A. F. Bell and P. J. Tonge, FEBS Lett., 2003, 549, 35–38 CrossRef CAS PubMed.
- W. Weber, V. Helms, J. A. McCammon and P. W. Langhoff, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6177–6182 CrossRef CAS PubMed.
- A. A. Voityuk, M. E. Michel-Beyerle and N. Rösch, Chem. Phys., 1998, 231, 13–25 CrossRef CAS.
- A. D. Kummer, C. Kompa, H. Niwa, T. Hirano, S. Kojima and M. E. Michel-Beyerle, J. Phys. Chem. B, 2002, 106, 7554–7559 CrossRef CAS.
- J. Wiedenmann, A. Schenk, C. Röcker, A. Girod, K. D. Spindler and G. U. Nienhaus, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11646–11651 CrossRef CAS PubMed.
- K. Nienhaus, H. Nar, R. Heilker, J. Wiedenmann and G. U. Nienhaus, J. Am. Chem. Soc., 2008, 130, 12578–12579 CrossRef CAS PubMed.
- F. V. Subach, G. H. Patterson, S. Manley, J. M. Gillette, J. Lippincott-Schwartz and V. V. Verkhusha, Nat. Methods, 2009, 6, 153–159 CrossRef CAS PubMed.
- D. C. Loos, S. Habuchi, C. Flors, J. Hotta, J. Wiedenmann, G. U. Nienhaus and J. Hofkens, J. Am. Chem. Soc., 2006, 128, 6270–6271 CrossRef CAS PubMed.
- H. Shinoda, K. Lu, R. Nakashima, T. Wazawa, K. Noguchi, T. Matsuda and T. Nagai, Cell Chem. Biol., 2019, 26, 1469–1479 CrossRef CAS PubMed.
- H. Mizuno, T. K. Mal, K. I. Tong, R. Ando, T. Furuta, M. Ikura and A. Miyawaki, Mol. Cell, 2003, 12, 1051–1058 CrossRef CAS PubMed.
- I. Hayashi, H. Mizuno, K. I. Tong, T. Furuta, F. Tanaka, M. Yoshimura, A. Miyawaki and M. Ikura, J. Mol. Biol., 2007, 372, 918–926 CrossRef CAS PubMed.
- H. Tsutsui, H. Shimizu, H. Mizuno, N. Nukina, T. Furuta and A. Miyawaki, Chem. Biol., 2009, 16, 1140–1147 CrossRef CAS PubMed.
- D. Thedie, R. Berardozzi, V. Adam and D. Bourgeois, J. Phys. Chem. Lett., 2017, 8, 4424–4430 CrossRef CAS PubMed.
- N. S. Makarov, C. Cirloganu, J. W. Perry, K. A. Lukyanov and K. M. Solntsev, J. Photochem. Photobiol., A, 2014, 280, 5–13 CrossRef CAS.
- N. V. Klementieva, K. A. Lukyanov, N. M. Markina, S. A. Lukyanov, E. V. Zagaynova and A. S. Mishin, Chem. Commun., 2016, 52, 13144–13146 RSC.
- B. Turkowyd, A. Balinovic, D. Virant, H. G. G. Carnero, F. Caldana, M. Endesfelder, D. Bourgeois and U. Endesfelder, Angew. Chem., Int. Ed., 2017, 56, 11634–11639 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, J. Phys.: Condens. Matter, 2016, 28, 443001 CrossRef PubMed.
- M. Lelimousin, V. Adam, G. U. Nienhaus, D. Bourgeois and M. J. Field, J. Am. Chem. Soc., 2009, 131, 16814–16823 CrossRef CAS PubMed.
- X. Li, L. W. Chung, H. Mizuno, A. Miyawaki and K. Morokuma, J. Phys. Chem. B, 2010, 114, 16666–16675 CrossRef CAS PubMed.
- E. Fron, M. Van der Auweraer, B. Moeyaert, J. Michiels, H. Mizuno, J. Hofkens and V. Adam, J. Phys. Chem. B, 2013, 117, 2300–2313 CrossRef CAS PubMed.
- E. Fron, M. Sliwa, V. Adam, J. Michiels, S. Rocha, P. Dedecker, J. Hofkens and H. Mizuno, Photochem. Photobiol. Sci., 2014, 13, 867–874 CrossRef CAS PubMed.
- J. Wiedenmann and G. U. Nienhaus, Expert Rev. Proteomics, 2006, 3, 361–374 CrossRef CAS PubMed.
- C. Fare, L. Yuan, V. Cordon-Preciado, J. J. Michels, M. J. Bearpark, P. Rich and J. J. van Thor, J. Phys. Chem. B, 2020, 124, 7765–7778 CrossRef CAS PubMed.
- H. Kim, T. Zou, C. Modi, K. Dorner, T. J. Grunkemeyer, L. Chen, R. Fromme, M. V. Matz, S. B. Ozkan and R. M. Wachter, Structure, 2015, 23, 34–43 CrossRef CAS PubMed.
- J. A. Ugalde, B. S. Chang and M. V. Matz, Science, 2004, 305, 1433 CrossRef CAS PubMed.
- W. P. Dempsey, L. Georgieva, P. M. Helbling, A. Y. Sonay, T. V. Truong, M. Haffner and P. Pantazis, Nat. Methods, 2015, 12, 645–648 CrossRef CAS PubMed.
- R. Berardozzi, V. Adam, A. Martins and D. Bourgeois, J. Am. Chem. Soc., 2016, 138, 558–565 CrossRef CAS PubMed.
- M. Byrdin, C. Duan, D. Bourgeois and K. Brettel, J. Am. Chem. Soc., 2018, 140, 2897–2905 CrossRef CAS PubMed.
- A. M. Bogdanov, A. S. Mishin, I. V. Yampolsky, V. V. Belousov, D. M. Chudakov, F. V. Subach, V. V. Verkhusha, S. Lukyanov and K. A. Lukyanov, Nat. Chem. Biol., 2009, 5, 459–461 CrossRef CAS PubMed.
- S. Olsen, K. Lamothe and T. J. Martinez, J. Am. Chem. Soc., 2010, 132, 1192–1193 CrossRef CAS PubMed.
- J. P. Colletier, M. Sliwa, F. X. Gallat, M. Sugahara, V. Guillon, G. Schiro, N. Coquelle, J. Woodhouse, L. Roux, G. Gotthard, A. Royant, L. M. Uriarte, C. Ruckebusch, Y. Joti, M. Byrdin, E. Mizohata, E. Nango, T. Tanaka, K. Tono, M. Yabashi, V. Adam, M. Cammarata, I. Schlichting, D. Bourgeois and M. Weik, J. Phys. Chem. Lett., 2016, 7, 882–887 CrossRef CAS PubMed.
- T. D. Krueger, L. Tang, L. Zhu, I. L. Breen, R. M. Wachter and C. Fang, Angew. Chem., Int. Ed., 2020, 59, 1644–1652 CrossRef CAS PubMed.
- E. De Zitter, J. Ridard, D. Thedie, V. Adam, B. Levy, M. Byrdin, G. Gotthard, L. Van Meervelt, P. Dedecker, I. Demachy and D. Bourgeois, J. Am. Chem. Soc., 2020, 142, 10978–10988 CrossRef CAS PubMed.
- M. G. Romei, C. Y. Lin, I. I. Mathews and S. G. Boxer, Science, 2020, 367, 76–79 CrossRef CAS PubMed.
- M. Sun, K. Hu, J. Bewersdorf and T. D. Pollard, Biophys. J., 2021, 120, 21–34 CrossRef CAS PubMed.
- E. De Zitter, D. Thedie, V. Monkemoller, S. Hugelier, J. Beaudouin, V. Adam, M. Byrdin, L. Van Meervelt, P. Dedecker and D. Bourgeois, Nat. Methods, 2019, 16, 707–710 CrossRef CAS PubMed.
- S. A. Wacker, C. Alvarado, G. von Wichert, U. Knippschild, J. Wiedenmann, K. Clauss, G. U. Nienhaus, H. Hameister, B. Baumann, T. Borggrefe, W. Knöchel and F. Oswald, EMBO J., 2011, 30, 43–56 CrossRef CAS PubMed.
- P. Sengupta, S. B. van Engelenburg and J. Lippincott-Schwartz, Chem. Rev., 2014, 114, 3189–3202 CrossRef CAS PubMed.
- P. N. Hedde and G. U. Nienhaus, Protoplasma, 2014, 251, 349–362 CrossRef CAS PubMed.
- A. C. Stiel, M. Andresen, H. Bock, M. Hilbert, J. Schilde, A. Schönle, C. Eggeling, A. Egner, S. W. Hell and S. Jakobs, Biophys. J., 2008, 95, 2989–2997 CrossRef CAS PubMed.
- S. Gayda, P. N. Hedde, K. Nienhaus and G. U. Nienhaus, in Far-Field Optical Nanoscopy, ed. P. Tinnefeld, C. Eggeling and S. W. Hell, Springer-Verlag Berlin Heidelberg, 2011 Search PubMed.
- K. Nienhaus and G. U. Nienhaus, ACS Nano, 2016, 10, 9104–9108 CrossRef CAS PubMed.
- A. Brodehl, P. N. Hedde, M. Dieding, A. Fatima, V. Walhorn, S. Gayda, T. Saric, B. Klauke, J. Gummert, D. Anselmetti, M. Heilemann, G. U. Nienhaus and H. Milting, J. Biol. Chem., 2012, 287, 16047–16057 CrossRef CAS PubMed.
- K. Faire, C. M. Waterman-Storer, D. Gruber, D. Masson, E. D. Salmon and J. C. Bulinski, J. Cell Sci., 1999, 112, 4243–4255 CrossRef CAS.
- J. Riedl, A. H. Crevenna, K. Kessenbrock, J. H. Yu, D. Neukirchen, M. Bista, F. Bradke, D. Jenne, T. A. Holak, Z. Werb, M. Sixt and R. Wedlich-Söldner, Nat. Methods, 2008, 5, 605–607 CrossRef CAS PubMed.
- N. Durisic, L. Laparra-Cuervo, A. Sandoval-Alvarez, J. S. Borbely and M. Lakadamyali, Nat. Methods, 2014, 11, 156–162 CrossRef CAS PubMed.
- P. Annibale, S. Vanni, M. Scarselli, U. Rothlisberger and A. Radenovic, PLoS One, 2011, 6, e22678 CrossRef CAS PubMed.
- N. Durisic, L. L. Cuervo and M. Lakadamyali, Curr. Opin. Chem. Biol., 2014, 20, 22–28 CrossRef CAS PubMed.
- A. Lee, K. Tsekouras, C. Calderon, C. Bustamante and S. Presse, Chem. Rev., 2017, 117, 7276–7330 CrossRef CAS PubMed.
- G. Hummer, F. Fricke and M. Heilemann, Mol. Biol. Cell, 2016, 27, 3637–3644 CrossRef CAS PubMed.
- F. Fricke, J. Beaudouin, R. Eils and M. Heilemann, Sci. Rep., 2015, 5, 14072 CrossRef PubMed.
- C. L. Krüger, M. T. Zeuner, G. S. Cottrell, D. Widera and M. Heilemann, Sci. Signaling, 2017, 10, eaan1308 CrossRef PubMed.
- L. Zhou, M. Evangelinos, V. Wernet, A. F. Eckert, Y. Ishitsuka, R. Fischer, G. U. Nienhaus and N. Takeshita, Sci. Adv., 2018, 4, e1701798 CrossRef PubMed.
- M. G. Gustafsson, J. Microsc., 2000, 198, 82–87 CrossRef CAS PubMed.
- M. G. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13081–13086 CrossRef CAS PubMed.
- R. Heintzmann and T. Huser, Chem. Rev., 2017, 117, 13890–13908 CrossRef CAS PubMed.
- D. Li, L. Shao, B. C. Chen, X. Zhang, M. Zhang, B. Moses, D. E. Milkie, J. R. Beach, J. A. Hammer, 3rd, M. Pasham, T. Kirchhausen, M. A. Baird, M. W. Davidson, P. Xu and E. Betzig, Science, 2015, 349, aab3500 CrossRef PubMed.
- E. H. Rego, L. Shao, J. J. Macklin, L. Winoto, G. A. Johansson, N. Kamps-Hughes, M. W. Davidson and M. G. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E135–143 CrossRef CAS PubMed.
- Y. Wu and H. Shroff, Nat. Methods, 2018, 15, 1011–1019 CrossRef CAS PubMed.
- X. Zhang, M. Zhang, D. Li, W. He, J. Peng, E. Betzig and P. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10364–10369 CrossRef CAS PubMed.
- T. Dertinger, R. Colyer, G. Iyer, S. Weiss and J. Enderlein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22287–22292 CrossRef CAS PubMed.
- T. Dertinger, R. Colyer, R. Vogel, J. Enderlein and S. Weiss, Opt. Express, 2010, 18, 18875–18885 CrossRef PubMed.
- P. Dedecker, G. C. Mo, T. Dertinger and J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10909–10914 CrossRef CAS PubMed.
- X. Zhang, X. Chen, Z. Zeng, M. Zhang, Y. Sun, P. Xi, J. Peng and P. Xu, ACS Nano, 2015, 9, 2659–2667 CrossRef CAS PubMed.
- Y. Li, L. Shang and G. U. Nienhaus, Nanoscale, 2016, 8, 7423–7429 RSC.
- C. A. Hoelzel and X. Zhang, ChemBioChem, 2020, 21, 1935–1946 CrossRef CAS PubMed.
- S. Neubacher and S. Hennig, Angew. Chem., Int. Ed., 2019, 58, 1266–1279 CrossRef CAS PubMed.
- R. Wirth, P. Gao, G. U. Nienhaus, M. Sunbul and A. Jäschke, J. Am. Chem. Soc., 2019, 141, 7562–7571 CrossRef CAS PubMed.
- M. Sunbul, J. Lackner, A. Martin, D. Englert, B. Hacene, K. Nienhaus, G. U. Nienhaus and A. Jäschke, Nat. Biotechnol., 2021 DOI:10.1038/s41587-020-00794-3.
| This journal is © The Royal Society of Chemistry 2021 |