
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bicyclic S-adenosylmethionine regeneration system applicable with different nucleosides or nucleotides as cofactor building blocks†
Désirée
Popadić
 a,
Dipali
Mhaindarkar
a,
Mike H. N.
Dang Thai
a,
Helen C.
Hailes
b,
Silja
Mordhorst‡
a and
Jennifer N.
Andexer
*a
a,
Dipali
Mhaindarkar
a,
Mike H. N.
Dang Thai
a,
Helen C.
Hailes
b,
Silja
Mordhorst‡
a and
Jennifer N.
Andexer
*a
aInstitute of Pharmaceutical Sciences, University of Freiburg, Albertstr. 25, 79104 Freiburg, Germany. E-mail: jennifer.andexer@pharmazie.uni-freiburg.de
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 22nd March 2021
Abstract
The ubiquitous cofactor S-adenosyl-L-methionine (SAM) is part of numerous biochemical reactions in metabolism, epigenetics, and cancer development. As methylation usually improves physiochemical properties of compounds relevant for pharmaceutical use, the sustainable use of SAM as a methyl donor in biotechnological applications is an important goal. SAM-dependent methyltransferases are consequently an emerging biocatalytic tool for environmentally friendly and selective alkylations. However, SAM shows undesirable characteristics such as degradation under mild conditions and its stoichiometric use is economically not reasonable. Here, we report an optimised biomimetic system for the regeneration of SAM and SAM analogues consisting of effective nucleoside triphosphate formation and an additional L-methionine regeneration cycle without by-product accumulation. The bicyclic system uses seven enzymes, S-methylmethionine as methyl donor and a surplus of inorganic polyphosphate, along with catalytic amounts of L-methionine and cofactor building block reaching conversions of up to 99% (up to 200 turnovers). We also show that the cycle can be run with cofactor building blocks containing different purine and pyrimidine nucleobases, which can be fed in at the nucleoside or nucleotide stage. These alternative cofactors are in turn converted to the corresponding SAM analogues, which are considered to be a key for the development of bioorthogonal systems. In addition to purified enzymes, the bicyclic system can also be used with crude lysates highlighting its broad biocatalytic applicability.
Introduction
S-Adenosyl-L-methionine (SAM, AdoMet, 1a) is an omnipresent and versatile cofactor needed for methylation, which is one of its most important functions (Fig. 1).1–3 It is featured in numerous biological processes such as DNA methylation, synthesis of secondary metabolites (e.g. that can act as neurotransmitters and antibiotics) as well as in epigenetic regulation, drug metabolism, and cancer development. SAM is formed in vivo by L-methionine adenosyltransferases (MATs, EC 2.5.1.6) from adenosine 5′-triphosphate (ATP, 2a) and L-methionine (L-Met, 3).4 Conventional SAM-dependent methyltransferases (MTs, EC 2.1.1§) transfer the methyl group in an SN2-type reaction in a regio- and stereoselective way onto e.g. O-, N-, S-, and C-atoms.5 Bioactive compounds have been shown to be improved in their physical and chemical properties by methylation (‘magic methyl effect’), demonstrating the importance of this modification for the pharmaceutical and biotechnological sectors.6,7 Commonly used chemical methylation agents such as methyl iodide or dimethyl sulfate are often carcinogenic, mutagenic, and harmful to the environment. The biocatalytic application of MTs, also using SAM analogues,8–12 are promising starting points for sustainable methods.13,14 While SAM analogues allowing to transfer larger alkyl chains have been widely employed for product diversification,15–25 the variation of the nucleobase has become a new focus, especially regarding the long-term goal to make bioorthogonal systems available.25–29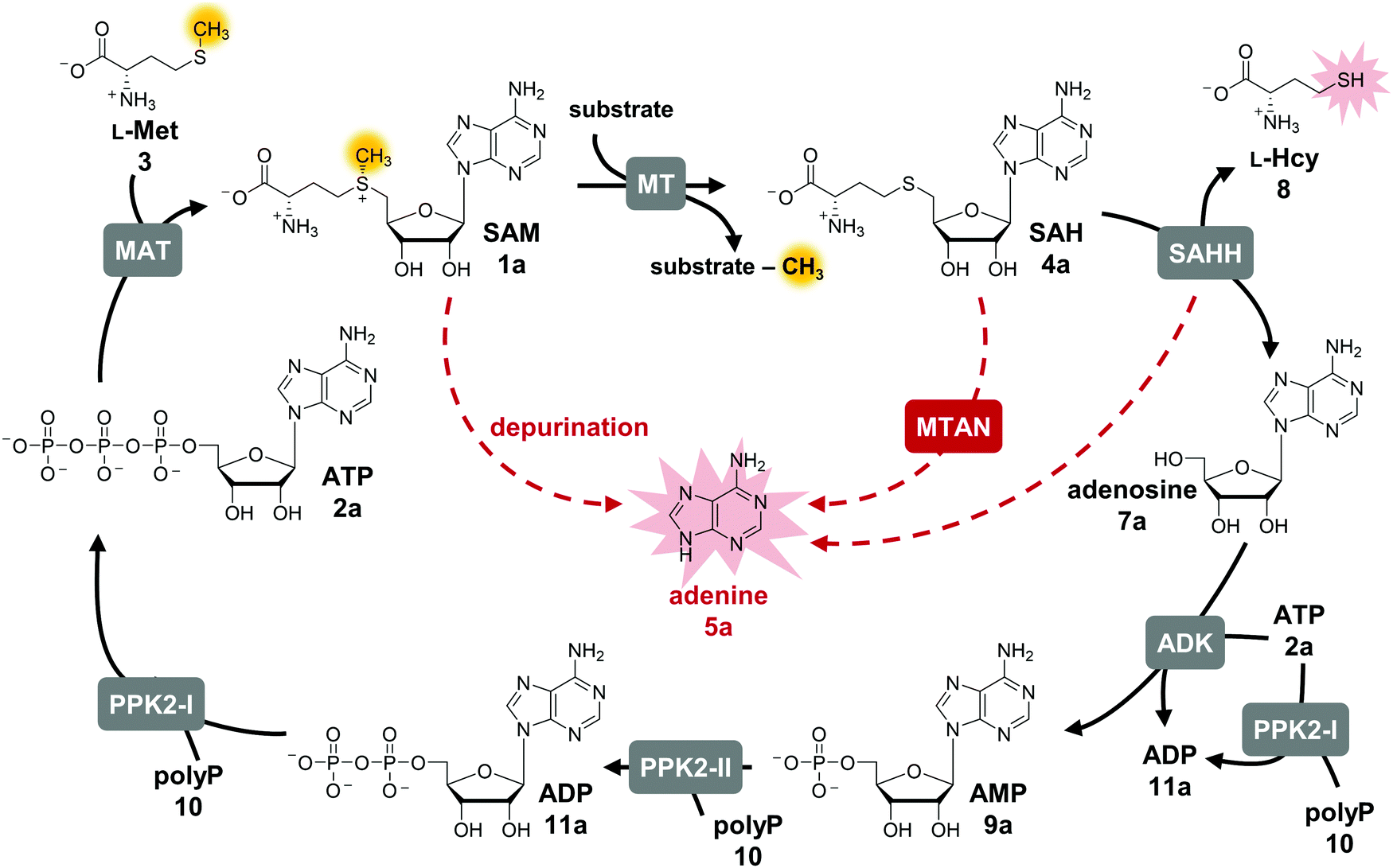 | ||
| Fig. 1 Cyclic SAM regeneration system and possible bottlenecks marked in red. MAT, L-methionine adenosyltransferase; MT, methyltransferase; MTAN, MTA/SAH nucleosidase; ADK, adenosine kinase; PPK2-I/II, polyphosphate kinases; SAHH, SAH hydrolase. | ||
The limitations of using SAM are its instability under physiological conditions, i.e. depurination and epimerisation at the sulfur centre, and the need of stoichiometric addition that is not economical on a technical scale.30,31 Possible SAM-derived degradation products include S-adenosyl-L-homocysteine (SAH, AdoHcy, 4a), adenine (5a), and methylthioadenosine (MTA). SAH is also the by-product of the enzymatic methylation reaction, and inhibits many MTs.32 Different approaches for regeneration systems have been reported to overcome these limitations (Table S1, ESI†).
One of these is the incorporation of MTs in a linear enzyme cascade, in which SAM is synthesised in situ by MATs. In this cofactor supply system, stoichiometric amounts of ATP and L-methionine are used. Optionally, the system can be expanded with a third enzyme cleaving the inhibitory by-product SAH to shift the reaction equilibrium to the MT product. MTA/SAH nucleosidase (MTAN, EC 3.2.2.9) irreversibly cleaves SAH to adenine (5a) and S-ribosyl-L-homocysteine (6) and has therefore been the preferred enzyme for degrading SAH (Fig. S1A, ESI†).15,33,34 Alternatively, SAH hydrolase (SAHH, EC 3.3.1.1) hydrolyses SAH to adenosine (7a) and L-homocysteine (L-Hcy, 8). By adding a set of kinases, the linear cascade can be extended to start from adenosine; ATP is needed as phosphate donor for AMP (9a) synthesis from adenosine, while inorganic polyphosphate (polyP, 10) serves as phosphate donor to phosphorylate AMP to ADP (11a) and ADP to ATP (Fig. S1B, ESI†).35,36 As an alternative to MAT, adenosyl-chloride synthase (SalL, EC 2.5.1.94) can be used for SAM supply starting from 5′-chloro-5′-deoxyadenosine.16,37
In addition to linear supply cascades starting with MAT or chlorinase, a two-enzyme regeneration system based on the reverse reaction of halide MTs (HMTs, EC 2.1.1.165) has been reported (Fig. S1C, ESI†); it has been successfully used for the methylation of a range of different compounds, e.g. amino acids, inositol, and α-ketovaleric acid using methyl iodide as a stoichiometric methyl donor, with total turnover numbers (TTNs) up to 500.38,39 The other reported option for SAM regeneration is a biomimetic multi-enzyme system including the triple re-phosphorylation of adenosine (SAHH product) to ATP by employing adenosine kinase (ADK, EC 2.7.1.20) and family-2 polyphosphate kinases (PPK2, EC 2.7.4.1) (Fig. S1D, ESI†).35 Although the linear cascade starting from adenosine and the biomimetic cycle only differ in few parameters, the outcome regarding yield of MT product and number of cofactor regeneration cycles was significantly lower in the cyclic system. Here, we present a comprehensive study with the goal to identify possible bottlenecks, to optimise the system accordingly, and to explore its suitability for SAM nucleobase analogues.
Results and discussion
The exchange of MTAN (linear cascade) for SAHH (cycle) impacts the system in multiple ways: the MTAN-catalysed reaction is conveniently irreversible, while the equilibrium of the SAHH-catalysed reaction lies on the substrate side in vitro. In theory, this should not be an issue in the cycle as the following reaction (ADK) instantly converts adenosine to the mononucleotide. Nevertheless, the second product of SAHH, L-homocysteine, accumulates; it also contains a free thiol that might interfere with other cycle enzymes (Fig. 1). Further, accumulation of adenine was detected in the cyclic system leading to the loss of cofactor building block for regeneration of SAM (Fig. 1).35 Interestingly, during the development of the one-step regeneration system, Liao and Seebeck showed that adenine accumulates due to the presence of MTAN obtained as a contaminant during enzyme purification from E. coli BL21(DE3).38 Adenine has also been described to be a side product of the SAHH reaction.35,40 In addition, the formation of adenine is the result of non-enzymatic SAM depurination.27,31 All these factors are possible bottlenecks of the cyclic system that must be carefully addressed to evaluate their effect on the overall performance of the cofactor regeneration system.Starting point for optimisation
The previously published systems were tested with different O-, N- and C-MTs and used to alkylate numerous substrates with methyl and ethyl groups (Table S1, ESI†). The extended linear supply system achieved 75–99% of substrate conversion and over 103 ATP regeneration cycles. Swapping of MTAN for SAHH resulted in a true SAM regeneration cycle; albeit with decreased substrate conversions (up to 22%) and lower SAM regeneration turnovers (max. 11).35 Based on the previously stated limitations of the cyclic system, the starting point of this work was to improve the SAM regeneration cycle by (i) avoiding the formation of adenine and/or (ii) removing L-homocysteine from the system. As a first model, the catechol O-MT from Rattus norvegicus (RnCOMT) was used for the conversion of 3,4-dihydroxybenzoic acid (12) to vanillic acid (13) and isovanillic acid (14), with 13 being the main regioisomer formed (Fig. 2A).41,42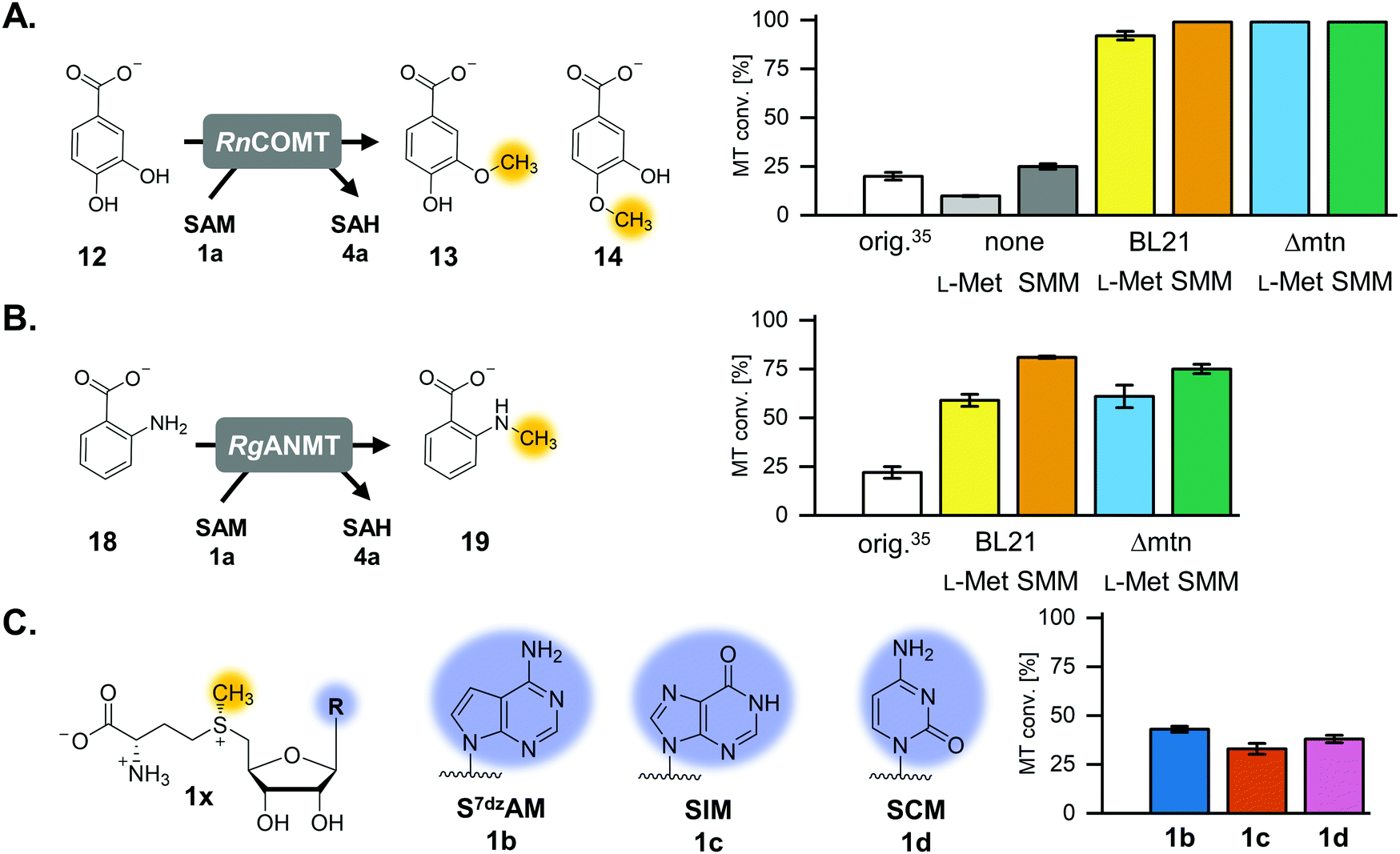 | ||
| Fig. 2 MTs and (co)substrates used in this study, along with the corresponding products and conversions for a maximum of 50 possible turnovers. (A) RnCOMT, catechol O-MT from Rattus norvegicus; (B) RgANMT, anthranilate N-MT from Ruta graveolens L. (C) Cofactor analogues with modified nucleobases and the conversions in the regeneration system using RnCOMT. | ||
Buffer screening
The reaction conditions for the assays were explored with respect to buffer and pH to improve MT conversion of RnCOMT with 12 as substrate. The highest substrate conversion was achieved in 50 mM Tris–HCl pH 8.0 at 37 °C and shaking of the reaction at 300 rpm (Fig. S3, details on the conditions tested are given in the ESI†). The optimised buffer conditions and preparation/incubation method resulted in a much improved conversion (92% vs. 20% for a maximum of 50 possible turnovers; Fig. 2A). Different amounts of AMP were added to the reaction mixture corresponding to 50, 100, or 200 possible TTN for the MT-catalysed reaction resulting in substrate conversions of 92% (46 TTN), 68% (68 TTN), and 47% (95 TTN), respectively (Table S5, ESI†). Without addition of AMP as cofactor building block, 9.9% MT conversion was detected; this is most likely due to cofactor molecules being co-purified with the enzymes (Table S5, ESI†). Small amounts of the undesired side products MTA and adenine, as well as SAH, were detected after 20 h of incubation (Fig. S4, ESI†).Isosteric adenosine analogues are not well tolerated by each cycle enzyme
One option to avoid the non-enzymatic depurination of SAM to adenine, is the use of isosteres such as S-7-deazaadenosyl-L-methionine (S7dzAM, 1b).27 As the monophosphorylated nucleoside of this reagent is not commercially available, 1 μM AMP was used in addition to 40 μM 7-deazaadenosine (tubercidin, 7b) to start the reaction (Fig. 3). Once phosphorylated 7-deazaadenosine is present, the cyclic system can work even if the small amount of AMP is degraded to adenine. The cofactor formed for methylation is S7dzAM. In comparison to the system with adenine as the backbone of the cofactor intermediates, the conversion dropped from 92% to 43% (Fig. 2). As expected, no adenine was detected in the reaction mixtures using 7-deazaadenosine (Fig. S5, ESI†). Depurination was avoided by the modified cofactor building block by prohibiting non-enzymatic SAM degradation under physiological conditions,27 as well as by averting the enzymatic degradation by possible MTAN contamination.34 Nevertheless, all enzymes have to accept the modified substrates; representatives of each enzyme family are described to accept (co)substrate analogues but mostly with lower conversions (detailed information in Table S2, ESI†).15,18,19,23,25,27,29,43–51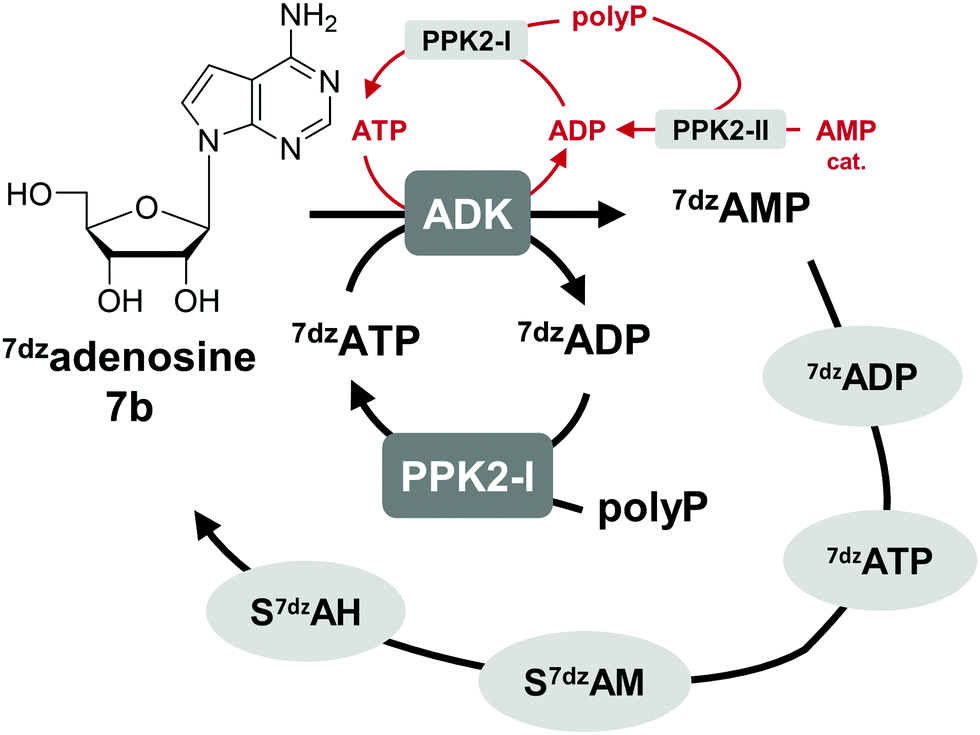 | ||
| Fig. 3 The use of 7dzadenosine as cofactor building block as a strategy to avoid depurination of SAM forming S7dzAM as cofactor. For the initial phosphorylation step catalysed by ADK, a catalytic amount of AMP is needed that is phosphorylated by PPK2s to ATP (depicted in red); in later cycles, 7dzATP can also be used. | ||
Besides EcMAT, another MAT originating from Thermococcus kodakarensis, TkMAT, which is likely to have a broader substrate spectrum like other archaeal MATs, was then tested in this set-up.44 Employing TkMAT decreased the MT conversion compared to EcMAT from 43% (22 TTN) to 23% (11 TTN, Table S6, ESI†). The reason for the decreased conversion might be the use of an enzyme from a hyperthermophile, whose optimal growth temperature is at 80 °C.44,52 Moreover, the substrate ranges of the involved enzymes have not been analysed in detail and other homologues of these enzymes involved could offer a better conversion with cofactor building block analogues. Theoretically, the use of isosteric adenosine analogues will be a good approach for more efficient methylation, if enzyme variants with an adapted substrate range can be identified.
Co-purified MTAN is not the only reason for adenine accumulation
Besides non-enzymatic SAM depurination, a possible reason for adenine accumulation is the contamination with MTAN from E. coli, which is not completely removed during purification. To eliminate the nucleosidase contamination, Liao and Seebeck generated an E. coli expression strain deficient in the gene encoding MTAN, E. coli Δmtn(DE3).38 In order to rule out a possible EcMTAN contamination, all enzymes were produced in the nucleosidase-deficient strain and purified as before. Protein yield from the mutant strain was lower compared to that from the E. coli BL21-Gold(DE3) strain, especially for MmSAHH, and some optimisation regarding the expression was carried out (ESI†). Using the optimised buffer conditions and the RnCOMT model system, the substrate conversion at 50, 100, and 200 possible turnovers was 99% (50 TTN, Fig. 2A), 85% (85 TTN), and 54% (108 TTN), respectively (Table S7, ESI†). Thus, the overall substrate conversion was higher compared to the system with enzymes purified from E. coli BL21-Gold(DE3). Interestingly, in addition to MTA and SAH, adenine was still detected in all samples after 20 h. This confirms that the postulated side reaction of SAHH is also responsible for adenine accumulation and not only EcMTAN contamination from enzyme production. This was double-checked by incubating MmSAHH purified from the nucleosidase-deficient strain, as well as MmSAHH purified from E. coli BL21-Gold(DE3) with SAH, which both showed the formation of adenine besides the hydrolysis to adenosine (Fig. S6, ESI†).Options for the removal of L-homocysteine
The second parameter for improving the regeneration system is the removal of the potentially inhibiting free thiol of L-homocysteine. This can be achieved either chemically or enzymatically. A chemical option is the use of a thiol scavenger, such as Ellman's reagent [5,5′-dithiobis(2-nitrobenzoic acid), DTNB]. However, the 2-nitro-5-thiobenzoic acid (TNB) formed is another free thiol, which could interfere with the enzymes and would accumulate during the reaction. Regarding enzymatic strategies to remove L-homocysteine, it would be ideal to re-methylate the compound yielding L-methionine that can be immediately re-used in the cycle. In nature, there are three main strategies for the synthesis of L-methionine from L-homocysteine: betaine-L-homocysteine S-methyltransferase (BHMT, EC 2.1.1.5), L-homocysteine S-methyltransferase (HSMT, EC 2.1.1.10) and L-methionine synthase (MS, EC 2.1.1.13) using the methyl donors betaine (15), S-methyl-L-methionine (SMM, 16) and 5-methyl-tetrahydrofolate (THF-CH3), respectively.53–55 Since SAH hydrolysis is postulated to be the bottleneck in the cyclic system, removal of L-homocysteine should also improve the substrate conversion by shifting the equilibrium to SAH hydrolysis.Methionine synthase in combination with THF–CH3 for methylation, which would yield tetrahydrofolate (THF) as a by-product, was not considered, as THF–CH3 is a complex molecule and would have to be provided stoichiometrically; nevertheless, a strategy aiming to boost SAM levels via THF–CH3 metabolism has been successfully used in vivo.56 Betaine and SMM are readily available. BHMT and HSMT (BHMT-2) are members of a family of zinc- and thiol/selenol-dependent MTs (Pfam 02574), and share a high degree of amino acid sequence identity (73% for the human homologues, Fig. 4).57–60
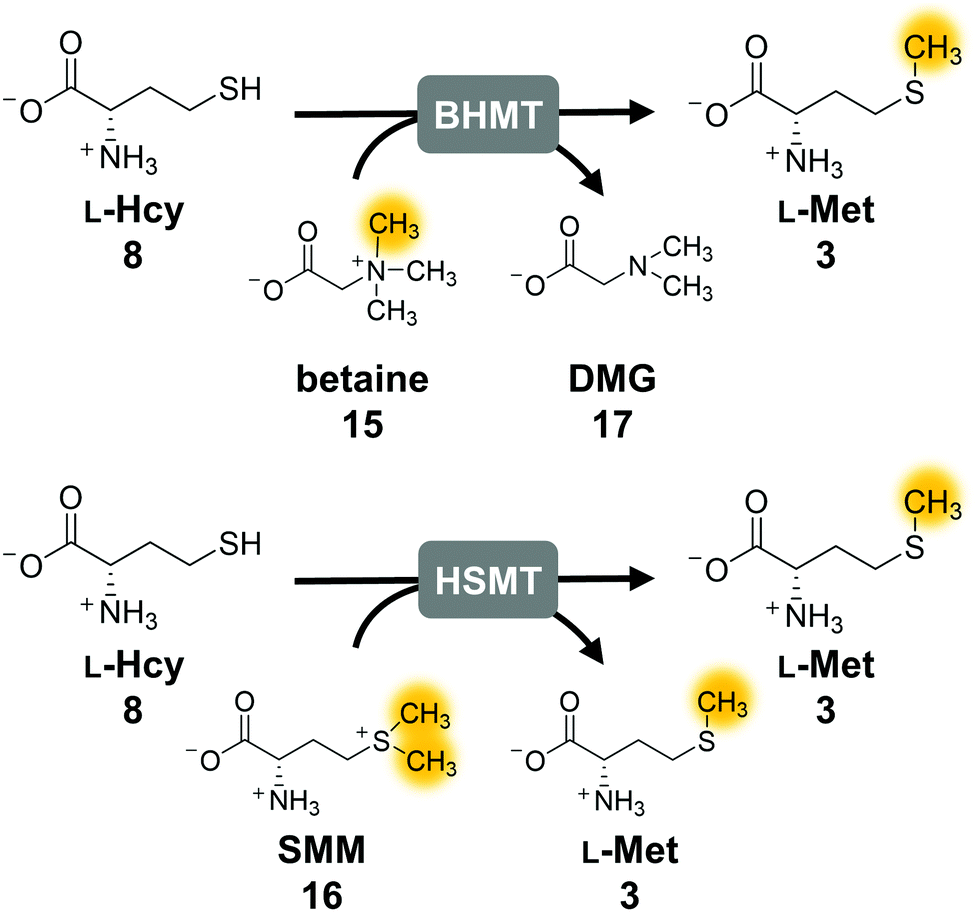 | ||
| Fig. 4 Enzymatic strategies to methylate L-homocysteine back to L-methionine using betaine or S-methylmethionine (SMM) as methyl donor. The orange marked methyl group is transferred to L-homocysteine. BHMT, betaine-L-homocysteine S-methyltransferase; HSMT; L-homocysteine S-methyltransferase. | ||
First experiments with BHMT from Rattus norvegicus (RnBHMT) did not lead to an improved performance of the system: substrate conversions decreased drastically to 19% (8 TTN), 13% (13 TTN) and 9% (17 TTN) for 50, 100, and 200 possible turnovers, respectively (Fig. S7 and Table S5, ESI†). As this might be mainly due to the reported inhibition of BHMT by various substrates and products such as dimethyl glycine (DMG, 17),61–63 we focused on HSMT using SMM as methyl donor.
The activity of ScHSMT from Saccharomyces cerevisiae was initially determined indirectly by incorporating this enzyme into the three-enzyme SAM supply cascade with RnCOMT and 12 as the MT substrate. Methylated products 13 and 14 were observed, and no substrate was left after 20 h confirming that ScHSMT catalyses the methylation of L-homocysteine to L-methionine, which was then used by EcMAT to synthesise SAM (Fig. S8, ESI†). In the next step, ScHSMT was integrated into the cyclic SAM regeneration system to regenerate L-methionine in addition to ATP.
The integration of ScHSMT into the SAM regeneration system along with the corresponding methyl donor D/L-SMM introduces new variables to the complex reaction system (Fig. 5A). Using each one molecule of L-SMM and L-homocysteine, HSMT produces two molecules of L-methionine; one molecule of L-SMM can be ultimately used to methylate two molecules of MT substrate. The bicyclic regeneration assay set-up contained 3 mM racemic SMM (or 1.5 mM) L-SMM, consequently producing 3 mM of L-methionine (for the methylation of 2 mM MT substrate). No degradation products of the methyl donor were detected after 20 h under the reaction conditions (Fig. S9, ESI†).
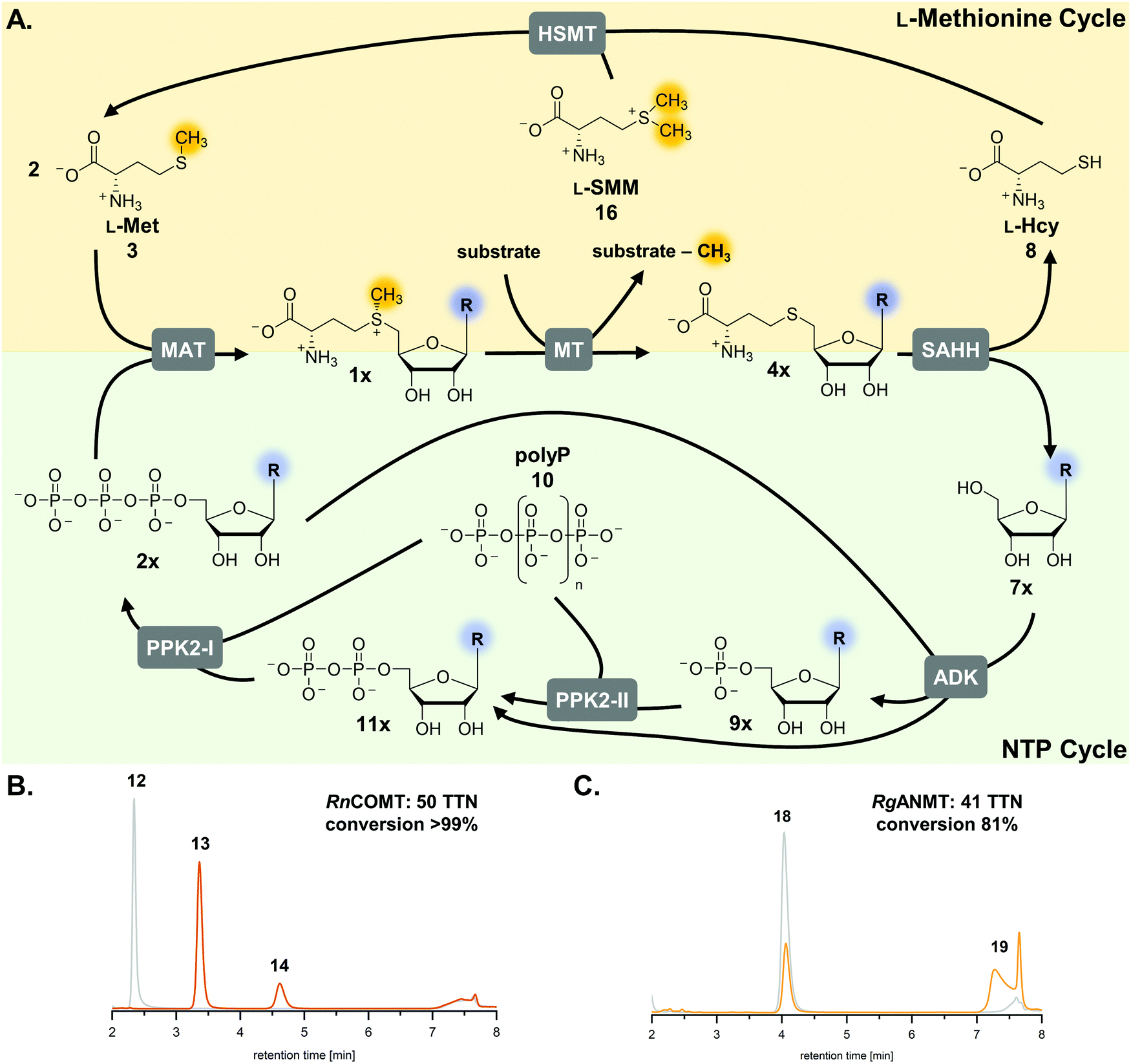 | ||
| Fig. 5 (A) Optimised cofactor regeneration system for SAM and its analogues. General scheme of the two parallel cyclic systems for L-methionine (yellow shading) and ATP (green shading) resulting in a bicyclic SAM regeneration system. Stoichiometric amounts of MT substrate, SMM, and polyP, and catalytic amounts of cofactor building block (9×) are needed. (B and C) Exemplary HPLC chromatograms for cyclic SAM regeneration assays with RnCOMT and RgANMT with the additional L-methionine regeneration with a maximum of 50 possible turnovers (orange: enzyme assay; grey: control without enzymes). MAT, L-methionine adenosyltransferase; MT, methyltransferase; ADK, adenosine kinase; PPK2-I/II, polyphosphate kinases; SAHH, SAH hydrolase; HSMT, L-homocysteine S-methyltransferase; TTN, total turnover number. | ||
Control reactions were set up to test if ScHSMT catalyses the methylation of other intermediates from the cyclic SAM regeneration system and if the other cycle enzymes utilise D/L-SMM as methyl donor. Besides L-homocysteine, the possible methyl acceptors for ScHSMT include the MT substrate 12 and SAH; however, no methylated products were observed in the corresponding control reactions (Fig. S10 and S11, ESI†). Furthermore, it was investigated whether EcMAT accepts D/L-SMM directly as an L-methionine substitute together with ATP to generate a SAM analogue. For easier detection, RnCOMT was coupled to the reaction and the assays were analysed for the conversion of MT substrate 12. The absence of methylated products of 12 indicated that either EcMAT did not accept D/L-SMM as a methyl donor or in the unlikely case of formed cofactor analogue that RnCOMT did not accept this (Fig. S12, ESI†). Similar results were observed for directly using D/L-SMM for methylation of the nucleophilic substrate 12 with RnCOMT (Fig. S13, ESI†). All control experiments were negative for RnBHMT and betaine as well (Fig. S10–S13, ESI†).
The L-methionine concentration was decreased to a catalytic amount of 40 μM, analogous to AMP. With this set-up, the substrate conversion was >99% for AMP concentrations of 40, 20, and 10 μM resulting in turnovers up to 200 (Fig. 2A, 5B and Fig. S14, Table S5, ESI†). When the assays were carried out using the enzymes purified from the nucleosidase-deficient E. coli strain in combination with L-methionine regeneration, the substrate conversion was the same for 50 and 100 possible turnovers, but strangely decreased to 64% for 200 possible turnovers compared to the E. coli BL21-Gold(DE3) strain produced enzymes (Table S7, ESI†); so far, we do not have an explanation for this effect. As observed earlier, at least some enzymes seem to co-purify with their corresponding cofactor building blocks resulting in a basal MT conversion of 9.9%; for the system with ScHSMT this background activity increased to 25% conversion for the RnCOMT reaction (Fig. 2A).
Reactions including ScHSMT did not show a build-up of SAH compared to those without L-homocysteine removal (Fig. S14 and S15, ESI†), supporting the positive effect of L-homocysteine removal on the SAHH reaction. Nevertheless, MTA and adenine were detected in all set-ups (Fig. S4, S14 and S15, ESI†); however, the loss of cofactor building block does not seem to have a major impact on the overall conversion. Closer inspection of the chromatograms revealed that the side products only occurred after the MT had completely converted the substrate; consequently, SAM was no longer required as a cofactor (Fig. S16, ESI†).
The purity of D/L-SMM is specified by the manufacturer to be >99.0%. When analysed by 1H NMR spectroscopy, L-methionine was detected as an impurity (Fig. S17 and S18, ESI†). Hence, another assay was carried out without any additional L-methionine. In this case, the MT substrate conversion was at >99% at 50 and 100 possible turnovers, however decreasing the cofactor building block further to 200 possible turnovers led to a drop of MT conversion to 83% (164 TTN) (Table S5 and Fig. S15, ESI†). This is a similar effect as observed for the different AMP amounts and is most likely due to the KM values of the different enzymes.
N-Methylation also improved by the optimised system with L-methionine regeneration
The optimised regeneration systems were also used for the methylation of anthranilic acid (18) with RgANMT (anthranilate N-methyltransferase from Ruta graveolens L.). In previously published experiments, we used RgANMT in the extended linear supply cascade as well as in the SAM (analogue) regeneration cycle for production of N-methylanthranilic acid (19) and N-ethylanthranilic acid (Table S1, ESI†). The results obtained regarding conversion and TTN were similar to O-MTs conversions in the cyclic system with a 22% yield (11 TTN) for methylation; however, in the linear cascade RgANMT did not reach full conversion as opposed to the O-MTs.35 This suggested a lower general activity of the enzyme, which in turn enables a better assessment of the impact of individual improvements. Indeed, using the improved six-enzyme system boosted conversion up to 59% (30 TTN), and adding the L-methionine regeneration system with ScHSMT further improved conversion up to 81% (41 TTN) for 50 possible turnovers (Fig. 2B, 5C and Fig. S19, S20, Table S5, ESI†).Cyclic SAM regeneration with L-methionine regeneration is more efficient than without
In order to provide further insights into the time course of the reaction, the assays for the SAM regeneration system with and without the L-methionine regeneration system were carried out with 40 μM cofactor building block (50 possible turnovers). The conversions with RnCOMT and RgANMT were monitored over 24 h (Fig. S21 and S22, ESI†). Both reactions showed a rapid increase in substrate conversion during the first 120 min of incubation. For the RnCOMT reaction, the conversion was nearly complete for both reaction set-ups after 240–360 min. As observed previously, a small residual amount of the substrate was present in the samples without L-methionine regeneration after 24 h. The same pattern emerged with RgANMT, but due to the lower activity of the enzyme, the differences again were more pronounced: the system with L-methionine regeneration reached a higher relative conversion after 240 min (93%) compared to the system without HSMT (82%). Consequently, removing both products of SAHH therefore might help the cyclic regeneration system to work more efficiently. The incubation time can be reduced to 480 min for the seven-enzyme system with this MT, however, this will have to be verified for systems that feature other enzyme variants or substrate analogues.Regeneration of SAM analogues is possible with nucleobase modifications
Using a cofactor for methylation or alkylation with a different nucleobase is interesting for the development of bioorthogonal systems with tailored variants of the enzymes that are selective for the altered cofactor and discriminate it compared to the natural one. Examples for such analogues are also found in nature. DNA is comprised of different nucleobases, and the inosine analogue of SAH, S-inosyl-L-homocysteine (SIH, 4c) has been found in the archaeon Methanocaldococcus jannaschii and the bacterium Streptomyces flocculus.45,64,65 Indeed, SIH seems to be part of an alternative SAM and L-methionine recycling pathway in archaea.45,66The experiment using 7-deazaadenosine described above already showed that, in principle, cofactor building blocks with altered nucleobases can be fed in the cycle. In order to gain more information about the performance of the regeneration system with nucleobase analogues, one purine as well as one pyrimidine nucleobase were used in the optimised cyclic system (Fig. 2C and 5A). TkMAT was used for the regeneration systems featuring cofactor analogues; initial experiments with different MATs showed that this enzyme accepts a range of nucleobases, whereas EcMAT did not accept inosine 5′-triphosphate (ITP, 2c) and cytidine 5′-triphosphate (CTP, 2d).44 Using TkMAT in a system with AMP as a cofactor building block and 50 possible turnovers, the substrate conversion dropped to 65% (32 TTN) compared to using EcMAT (Table S6, ESI†). This could be the case because TkMAT is from a hyperthermophile as discussed above. Adding the L-methionine regeneration system using ScHSMT increased the conversion to 83% (41 TTN) for 50 possible turnovers (Table S6, ESI†). Moreover, changing the nucleobase to hypoxanthine (5c) or cytosine (5d) showed that all enzymes accept the alternative nucleobases yielding methylated products. The cofactors formed are S-inosyl-L-methionine (SIM, 1c) and S-cytidyl-L-methionine (SCM, 1d), respectively (Fig. 2C). In combination with the L-methionine regeneration system, conversions increased from 23% to 33% (16 TTN) for SIM and from 20% to 38% (19 TTN) for SCM (Fig. 2C and Fig. S23, Table S6, ESI†). In comparison with no added cofactor building block, a significant increase of MT conversion was detected with the nucleotide analogues as cofactor building blocks supporting that the modification was accepted by the enzymes (Table S6, ESI†).
SAM analogues with extended alkyl chains at the sulfonium have been chemically and enzymatically synthesised and used for a range of purposes.15–20,22,23,25,43 When using different alkyl groups, the regeneration system for L-methionine cannot be applied, as the corresponding alkyl donor for the alkylation of L-homocysteine is not easily available. The cyclic SAM regeneration system has previously been published with L-ethionine as an alkyl donor forming S-adenosyl-L-ethionine (SAE) yielding the RgANMT product N-ethylanthranilic acid.35 In this case, using the optimised buffer conditions and enzyme concentrations did not improve the MT conversion (Fig. S24 and Table S8, ESI†). This suggests that the MAT and/or MT (the enzymes having to take altered substrates) do not cope well with the decreased amount of cofactor building block present compared to the extended linear cascade. Using L-ethionine in the extended linear SAE supply cascade showed >75% conversion for the RgANMT reaction previously,35 however ATP, and consequently the synthesised SAE, were present in a much higher concentration (up to 3 mM theoretically possible). In contrast, only 40 μM cofactor building block was present in the cyclic system, spread across all six intermediates possible (adenosine, AMP, ADP, ATP, SAE, SAH).
Increased substrate loading
To further test the limits of the system, the MT substrate loading was increased from 2 to 5 mM for 12 with RnCOMT. L-Methionine or D/L-SMM was increased to 7.5 mM to guarantee sufficient amount of methyl building block was present. The MT substrate conversion dropped to 37% (46 TTN), 23% (76 TTN), and 18% (90 TTN) for 125, 250, and 500 possible turnovers, respectively. Increasing AMP to 100 μM, making a maximum of 50 turnovers possible, led to an increase in conversion to 63% with 32 TTN. The addition of ScHSMT for L-methionine regeneration led to an increase in conversion of approximately 10% for all set-ups (Table S9, ESI†). Substrate or product inhibition might be a reason for the decreased conversion and is currently under investigation in our laboratory.The regeneration system can be used with crude lysates
In biotechnological applications, crude lysates are preferred enzyme preparations to circumvent the time-consuming purification of numerous enzymes, as well as to lower system costs.67 Enzymes were produced in E. coli BL21-Gold(DE3), cell pellets were collected, lysed by sonication and the cleared lysate was used for activity assays (for details ESI†). As a negative control, a strain carrying an empty vector was used. Quantification was not as straightforward as more peaks were present in the HPLC chromatogram (Fig. S25, ESI†); however, based on the individual quantification of substrate 12, conversions were 45% (22 TTN) for the six-enzyme system and 88% (44 TTN) for the bicyclic system (Table S10, ESI†). With further optimisation regarding the crude lysate used and MT substrate loading, as well as sample size for up-scaling, this is a promising launch point for the biocatalytic application of this SAM regeneration system.Conclusions
The strategy to remove L-homocysteine by re-methylating it to L-methionine using ScHSMT, greatly improves the performance of the SAM (analogue) regeneration cycle, especially for cofactor analogues where for example for SCM a 2-fold increase was observed. The methyl donor D/L-SMM can be efficiently used to remove L-homocysteine from the system, without generation of additional by-products. We assume that removing L-homocysteine drives the reaction of SAHH forward, as SAH was detected in assays without the L-methionine regeneration system but not when the L-methionine regeneration system was in place. Only when both adenosine and L-homocysteine are removed, the TTN for all cofactor regeneration systems improved notably, reaching full conversion for several reactions. The free thiol L-homocysteine might also interfere with other enzymes of the regeneration system. The SAM regeneration system was fully operational with only 10 μM cofactor building block distributed across six intermediates (on average 1.7 μM AMP, ADP, ATP, SAM, SAH, and adenosine).Interestingly, the cyclic SAM regeneration system can be implemented with different SAM analogues including S7dzAM, SIM, and SCM starting from either nucleoside or monophosphorylated nucleoside analogues. These are often more easily available compared to triphosphorylated nucleosides (for the three-enzyme supply cascade with MAT) or alkyl iodides (for halide MT supply cascade). These results demonstrate the ability of the system to be used as a toolbox with wide variation in the enzymes (besides MATs and MTs), MT substrates, as well as cofactor building blocks to access desired products that are difficult to synthesise chemically. Furthermore, the regeneration system is a starting point for application in bioorthogonal systems.5,19,24,26,29
Especially when using lysates, it will be possible to optimise parameters such as the enzyme/crude lysate loading and the ratio of substrate to cofactor building block for a specific enzyme-substrate combination in order to achieve the highest conversion possible with an adequate TTN. In combination with an efficient purification strategy, selectively methylated products could also be sustainably produced on a large biocatalytic scale.
Author contributions
D. P. (lead), D. M. (lead) and M. H. N. D. T. (supporting) investigated and curated data. S. M. (supporting), H. C. H. (supporting) and J. N. A. (lead) conceptualised and supervised the work. J. N. A. managed funding. D. P. and J. N. A. wrote the manuscript; all authors checked and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 235777276/RTG1976, the EU/BMBF (ERACo Biotech, BioDiMet) and the European Research Council (ERC project 716966). We thank Sascha Ferlaino for technical assistance with NMR analysis and Emina Čokljat and Katharina Strack for assistance with protein production, as well as Tabea Pappert for help with initial experiments with ScHSMT, and Simone Fiderer for help with initial experiments with TkMAT. We also thank Prof. Andrea Rentmeister (University of Münster) and Prof. Florian Seebeck (University of Basel) for many SAM-related discussions and Prof. Florian Seebeck for the donation of the E. coli Δmtn(DE3) strain.Notes and references
- M. Fontecave, M. Atta and E. Mulliez, Trends Biochem. Sci., 2004, 29, 243–249 CrossRef CAS PubMed
.
- W. M. Loenen, Biochem. Soc. Trans., 2006, 34, 330–333 CrossRef CAS
- M. Richter, Nat. Prod. Rep., 2013, 30, 1324–1345 RSC
- G. L. Cantoni, J. Biol. Chem., 1953, 204, 403–416 CrossRef CAS
- A.-W. Struck, M. L. Thompson, L. S. Wong and J. Micklefield, ChemBioChem, 2012, 13, 2642–2655 CrossRef CAS
- E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed
- I. J. W. McKean, P. A. Hoskisson and G. A. Burley, ChemBioChem, 2020, 21, 2890–2897 CrossRef CAS
- T. D. Huber, B. R. Johnson, J. Zhang and J. S. Thorson, Curr. Opin. Biotechnol., 2016, 42, 189–197 CrossRef CAS
- J. Zhang and Y. G. Zheng, ACS Chem. Biol., 2016, 11, 583–597 CrossRef CAS PubMed
- J. Deen, C. Vranken, V. Leen, R. K. Neely, K. P. F. Janssen and J. Hofkens, Angew. Chem., Int. Ed., 2017, 56, 5182–5200 CrossRef CAS PubMed
- Q. Tang, C. W. Grathwol, A. S. Aslan-Üzel, S. Wu, A. Link, I. V. Pavlidis, C. P. S. Badenhorst and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 1524–1527 CrossRef PubMed
- M. R. Bennett, S. A. Shepherd, V. A. Cronin and J. Micklefield, Curr. Opin. Chem. Biol., 2017, 37, 97–106 CrossRef CAS PubMed
- U. T. Bornscheuer, Philos. Trans. R. Soc., A, 2018, 376, 20170063 CrossRef PubMed
- S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem., Int. Ed., 2014, 53, 3965–3969 CrossRef CAS PubMed
- J. M. Lipson, M. Thomsen, B. S. Moore, R. P. Clausen, J. J. La Clair and M. D. Burkart, ChemBioChem, 2013, 14, 950–953 CrossRef CAS PubMed
- M. Thomsen, S. B. Vogensen, J. Buchardt, M. D. Burkart and R. P. Clausen, Org. Biomol. Chem., 2013, 11, 7606–7610 RSC
- B. J. C. Law, A.-W. Struck, M. R. Bennett, B. Wilkinson and J. Micklefield, Chem. Sci., 2015, 6, 2885–2892 RSC
- F. Muttach, F. Mäsing, A. Studer and A. Rentmeister, Chem. – Eur. J., 2017, 23, 5988–5993 CrossRef CAS PubMed
- C. Sommer-Kamann, A. Fries, S. Mordhorst, J. N. Andexer and M. Müller, Angew. Chem., Int. Ed., 2017, 56, 4033–4036 CrossRef CAS PubMed
- J. C. Sadler, L. D. Humphreys, R. Snajdrova and G. A. Burley, ChemBioChem, 2017, 18, 992–995 CrossRef CAS PubMed
-
T. D. Davis, S. Kunakom, M. D. Burkart and A. S. Eustaquio, in Methods in Enzymology, ed. B. S. Moore, Academic Press, 2018, vol. 604, pp. 367–388 Search PubMed
- I. J. W. McKean, J. C. Sadler, A. Cuetos, A. Frese, L. D. Humphreys, G. Grogan, P. A. Hoskisson and G. A. Burley, Angew. Chem., Int. Ed., 2019, 58, 17583–17588 CrossRef CAS PubMed
- A. J. Herbert, S. A. Shepherd, V. A. Cronin, M. R. Bennett, R. Sung and J. Micklefield, Angew. Chem., Int. Ed., 2020, 59, 14950–14956 CrossRef CAS PubMed
- N. V. Cornelissen, F. Michailidou, F. Muttach, K. Rau and A. Rentmeister, Chem. Commun., 2020, 56, 2115–2118 RSC
- J. M. Holstein, D. Stummer and A. Rentmeister, Chem. Sci., 2015, 6, 1362–1369 RSC
- T. D. Huber, F. Wang, S. Singh, B. R. Johnson, J. Zhang, M. Sunkara, S. G. Van Lanen, A. J. Morris, G. N. Phillips and J. S. Thorson, ACS Chem. Biol., 2016, 11, 2484–2491 CrossRef CAS
- C. Vranken, A. Fin, P. Tufar, J. Hofkens, M. D. Burkart and Y. Tor, Org. Biomol. Chem., 2016, 14, 6189–6192 RSC
- T. D. Huber, J. A. Clinger, Y. Liu, W. Xu, M. D. Miller, G. N. Phillips and J. S. Thorson, ACS Chem. Biol., 2020, 15, 695–705 CrossRef CAS PubMed
- L. W. Parks and F. Schlenk, J. Biol. Chem., 1958, 230, 295–305 CrossRef CAS
- J. L. Hoffman, Biochemistry, 1986, 25, 4444–4449 CrossRef CAS PubMed
- H. K. Chenault, E. S. Simon and G. M. Whitesides, Biotechnol. Genet. Eng. Rev., 1988, 6, 221–270 CrossRef CAS
- J. Siegrist, S. Aschwanden, S. Mordhorst, L. Thöny-Meyer, M. Richter and J. N. Andexer, ChemBioChem, 2015, 16, 2576–2579 CrossRef CAS
- J. E. Lee, K. A. Cornell, M. K. Riscoe and P. L. Howell, J. Biol. Chem., 2003, 278, 8761–8770 CrossRef CAS PubMed
- S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Angew. Chem., Int. Ed., 2017, 56, 4037–4041 CrossRef CAS PubMed
- J. N. Andexer and M. Richter, ChemBioChem, 2015, 16, 380–386 CrossRef CAS PubMed
- A. S. Eustáquio, F. Pojer, J. P. Noel and B. S. Moore, Nat. Chem. Biol., 2008, 4, 69–74 CrossRef
- C. Liao and F. P. Seebeck, Nat. Catal., 2019, 2, 696–701 CrossRef CAS
- C. Liao and F. P. Seebeck, Angew. Chem., Int. Ed., 2020, 59, 7184–7187 CrossRef CAS PubMed
- P. M. Ueland and J. Saebø, Biochim. Biophys. Acta, 1979, 585, 512–526 CrossRef CAS
- J. Vidgren, L. A. Svensson and A. Liljas, Nature, 1994, 368, 354–358 CrossRef CAS PubMed
- C. R. Creveling, N. Morris, H. Shimizu, H. H. Ong and J. Daly, Mol. Pharmacol., 1972, 8, 398–409 CAS
- C. Dalhoff, G. Lukinavičius, S. Klima
![[s with combining breve]](https://www.rsc.org/images/entities/char_0073_0306.gif) auskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed
auskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed - Z. J. Lu and G. D. Markham, J. Biol. Chem., 2002, 277, 16624–16631 CrossRef CAS PubMed
- D. Miller, H. Xu and R. H. White, J. Bacteriol., 2015, 197, 2284–2291 CrossRef CAS PubMed
- S. Shimizu, S. Shiozaki, T. Ohshiro and H. Yamada, Eur. J. Biochem., 1984, 141, 385–392 CrossRef CAS PubMed
- M. C. Long, S. C. Shaddix, O. Moukha-Chafiq, J. A. Maddry, L. Nagy and W. B. Parker, Biochem. Pharmacol., 2008, 75, 1588–1600 CrossRef CAS PubMed
- B. Nocek, S. Kochinyan, M. Proudfoot, G. Brown, E. Evdokimova, J. Osipiuk, A. M. Edwards, A. Savchenko, A. Joachimiak and A. F. Yakunin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17730–17735 CrossRef CAS PubMed
- L. E. Batten, A. E. Parnell, N. J. Wells, A. L. Murch, P. C. F. Oyston and P. L. Roach, Biosci. Rep., 2016, 36, e00294 CrossRef PubMed
- J. Nahálka and V. Pätoprstý, Org. Biomol. Chem., 2009, 7, 1778–1780 RSC
- B. P. Nocek, A. N. Khusnutdinova, M. Ruszkowski, R. Flick, M. Burda, K. Batyrova, G. Brown, A. Mucha, A. Joachimiak, Ł. Berlicki and A. F. Yakunin, ACS Catal., 2018, 8, 10746–10760 CrossRef CAS
- H. Atomi, T. Fukui, T. Kanai, M. Morikawa and T. Imanaka, Archaea, 2004, 1, 263–267 CrossRef CAS PubMed
- S. K. Shapiro and D. A. Yphantis, Biochim. Biophys. Acta, 1959, 36, 241–244 CrossRef CAS
- M. H. Stipanuk, Annu. Rev. Nutr., 1986, 6, 179–209 CrossRef CAS
- M. Thanbichler, B. Neuhierl and A. Böck, J. Bacteriol., 1999, 181, 662–665 CrossRef CAS PubMed
- S. Y. Chan and D. R. Appling, J. Biol. Chem., 2003, 278, 43051–43059 CrossRef CAS PubMed
- N. S. Millian and T. A. Garrow, Arch. Biochem. Biophys., 1998, 356, 93–98 CrossRef CAS PubMed
- J. C. Evans, D. P. Huddler, J. Jiracek, C. Castro, N. S. Millian, T. A. Garrow and M. L. Ludwig, Structure, 2002, 10, 1159–1171 CrossRef CAS
- K. Li, G. Li, L. M. T. Bradbury, A. D. Hanson and S. D. Bruner, Biochem. J., 2016, 473, 277–284 CrossRef CAS
- P. Ranocha, F. Bourgis, M. J. Ziemak, D. Rhodes, D. A. Gage and A. D. Hanson, J. Biol. Chem., 2000, 275, 15962–15968 CrossRef CAS PubMed
- S. S. Szegedi, C. C. Castro, M. Koutmos and T. A. Garrow, J. Biol. Chem., 2008, 283, 8939–8945 CrossRef CAS PubMed
- C. Castro, N. S. Millian and T. A. Garrow, Arch. Biochem. Biophys., 2008, 472, 26–33 CrossRef CAS PubMed
- R. Waditee and A. Incharoensakdi, Curr. Microbiol., 2001, 43, 107–111 CrossRef CAS PubMed
- M. K. Speedie, J. J. Zulty and P. Brothers, J. Bacteriol., 1988, 170, 4376–4378 CrossRef CAS
- J. J. Zulty and M. K. Speedie, J. Bacteriol., 1989, 171, 6840–6844 CrossRef CAS
- D. V. Miller, B. J. Rauch, K. Harich, H. Xu, J. J. Perona and R. H. White, Microbiology, 2018, 164, 969–981 CrossRef CAS
-
K. Faber, Biotransformations in Organic Chemistry: A Textbook, Springer, Berlin, 2018 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental information, as well as supplementary figures and tables are available. See DOI: 10.1039/d1cb00033k |
| ‡ Present address: Institute of Microbiology, ETH Zürich, Vladimir-Prelog-Weg 1-5/10, 8093 Zurich, Switzerland. |
| § Currently (03/2021), 363 MT-catalysed reactions (2.1.1.1–2.1.1.372) are classified. |
| This journal is © The Royal Society of Chemistry 2021 |