
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic strategies for asymmetric synthesis
Mélanie
Hall
 ab
ab
aInstitute of Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: melanie.hall@uni-graz.at
bField of Excellence BioHealth – University of Graz, Austria
First published on 1st June 2021
Abstract
Enzymes, at the turn of the 21st century, are gaining a momentum. Especially in the field of synthetic organic chemistry, a broad variety of biocatalysts are being applied in an increasing number of processes running at up to industrial scale. In addition to the advantages of employing enzymes under environmentally friendly reaction conditions, synthetic chemists are recognizing the value of enzymes connected to the exquisite selectivity of these natural (or engineered) catalysts. The use of hydrolases in enantioselective protocols paved the way to the application of enzymes in asymmetric synthesis, in particular in the context of biocatalytic (dynamic) kinetic resolutions. After two decades of impressive development, the field is now mature to propose a panel of catalytically diverse enzymes for (i) stereoselective reactions with prochiral compounds, such as double bond reduction and bond forming reactions, (ii) formal enantioselective replacement of one of two enantiotopic groups of prochiral substrates, as well as (iii) atroposelective reactions with noncentrally chiral compounds. In this review, the major enzymatic strategies broadly applicable in the asymmetric synthesis of optically pure chiral compounds are presented, with a focus on the reactions developed within the past decade.
 Mélanie Hall | Mélanie Hall studied chemistry at the École Nationale Supérieure de Chimie de Rennes (ENSCR), France, and received her PhD in chemistry from the University of Graz, Austria, under Prof. Kurt Faber (2007). After a postdoctoral research stay with Prof. Andreas Bommarius at the Georgia Institute of Technology in Atlanta, USA, she returned to the University of Graz, where she obtained her habilitation in organic chemistry (venia docendi) in 2016. She is currently assistant professor for sustainable bioorganic synthetic chemistry at the Institute of Chemistry. Her research is dedicated to the field of biocatalysis, with particular focus on asymmetric synthesis and enzymatic synthetic technologies. |
Introduction
Biocatalysis, which can be broadly defined as the use of enzymes to catalyze chemical reactions, is found in diverse fields of application. While its historical use by ancient civilizations in brewery, bakery and wine making was not conscious, advances mostly in the 20th century in neighboring disciplines such as biochemistry, enzymology, molecular biology, and more recently in protein crystallography, genetic engineering and computational tools, contributed to a global and molecular understanding of enzymes and their modes of action. At the same time, scientists could finally produce, manipulate and modify enzymes on demand, which participated in the establishment of modern biocatalysis.1,2 The use of enzymes in chemistry has a long tradition, such as in the food, textile and detergent industry,3,4 however biocatalysis started to dramatically impact the fine chemical and pharmaceutical industry only recently at the turn of the 21st century. Besides the strong motivation to develop environmentally more acceptable chemical processes, the faster access to proteins and the ability to modify them largely contributed to a mindset shift in industry.5,6 Enzymes can compete with traditional chemical (catalytic) methods for a broad range of chemical transformations, including those long considered nonnatural. Exemplary is the identification of the catalytic activity of aldoxime dehydratases in the Kemp elimination reaction7 – a base-catalyzed deprotonation of a benzisoxazole ring – for which, besides chemical bases, only de novo designed proteins were known to be active catalysts.8Baeyer–Villiger monooxygenases (BMVOs) represent another beautiful example of how nature designs efficient enzymatic machineries to catalyze difficult chemical reactions. Chemists employ peracids or peroxides as oxidant in the eponymic reaction for the insertion of oxygen between two carbon atoms. After attack of the carbonyl group of the substrate by the oxidant, the reaction proceeds through formation of the so-called Criegee intermediate. The collapse of this intermediate leads to migration of one of the alkyl substituents of the substrate carbonyl to the oxygen atom derived from the oxidant, and eventually results in the formation of the ester or lactone product. BVMOs simply use molecular oxygen as oxidant in the enzymatic version of the reaction and rely on a reduced flavin cofactor to activate the oxygen. The resulting peroxyflavin is a powerful nucleophile that can attack carbonyls, leading to a biological Criegee intermediate. The rest of the reaction can be considered analogous to the chemical oxidation.9 A major advantage of the biological catalyst is the benign and nontoxic environment required for the oxygenation reaction: air, importantly seconded by high regio- and enantioselectivity.
The interest in using enzymes in asymmetric synthesis is motivated by the exquisite chemo, regio- and stereoselectivity of enzymes. Concepts underlying enzymatic strategies in asymmetric reactions are similar to those found in organic synthesis:
– A prochiral compound can be transformed stereoselectively into a chiral product in enantiopure form. Usually, such reactions involve nucleophilic attack onto sp2 hybridized carbon atoms embedded in a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-, CO-, or CN-double bond, and imply face recognition of the substrate molecular plane by the enzymes.
C-, CO-, or CN-double bond, and imply face recognition of the substrate molecular plane by the enzymes.
– A prochiral sp3 hybridized atom can be converted into a chiral center by the formal enantioselective replacement of one of the two enantiotopic groups, such as hydrogen atoms on carbon or electron lone pairs on sulfur.
– In case the chiral information is already present, a racemic substrate can be converted enantioselectively according to the principles of kinetic resolution.
– Finally, the case of atroposelective reactions deserves attention. The chiral information is contained not in a center but in an axis that can be either formed or, if already existing, resolved.
In this article, the major strategies relying on enzymes for the key step of asymmetric synthesis are reviewed and classified in one of the following categories: (i) stereoselective reactions involving transformations of sp2 hybridized carbon atoms, (ii) enantioselective reactions and (iii) atroposelective reactions. For each category, the focus was intentionally put on most recent literature where relevant, while each strategy is introduced in the light of the underlying chemical concept.
Nonnatural asymmetric reactions catalyzed by P450 monooxygenases and pioneered by Frances Arnold are out of the scope of this review. Excellent and comprehensive articles can be recommended for the interested reader.10–14 The enzymes selected for this review were chosen due to a certain level of generality and broad applicability in synthesis, usually indicated by a large substrate scope. In selected cases, (industrial) examples that largely contributed to the acceptance of biocatalysis in asymmetric synthesis15 are presented in details. The broad topic covering the use of hydrolases in enantioselective reactions was not included,16–18 except on special occasions. Finally, special techniques leading to optically pure chiral compounds, such as desymmetrization, stereoinversion, cyclic deracemization and enantioconvergent processes19–26 were intentionally not broadly covered.
1. Stereoselective reactions involving transformations of sp2 carbons
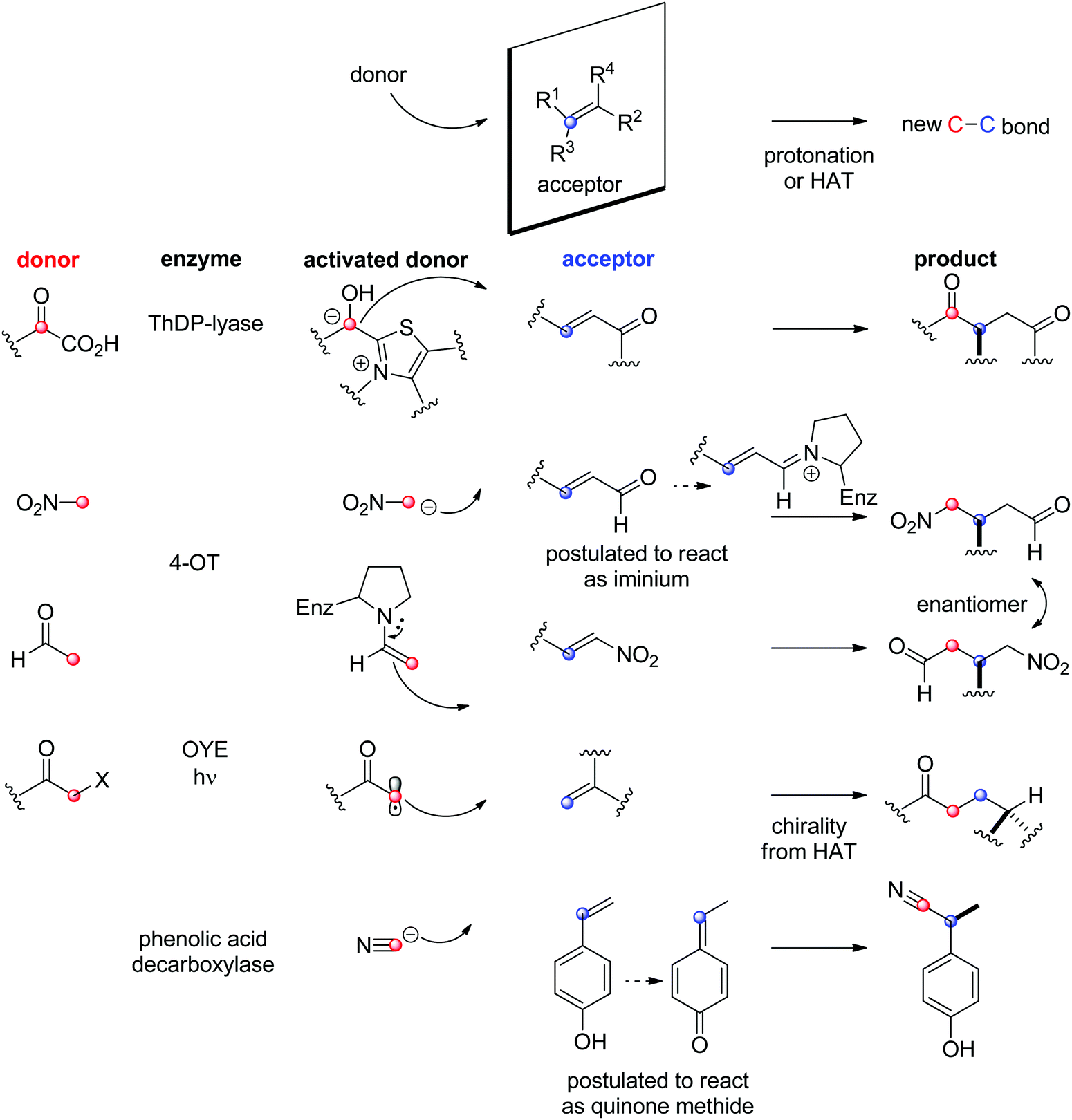
The biocatalytic stereoselective conversion of prochiral molecules into chiral products often involves substrates bearing at least one sp2 hybridized carbon atom embedded in a double bond, such as CC-, CN- or CO-bond. Transformations of these double bonds include reduction reactions (stereoselective C–H bond formation), transamination and hydroamination (stereoselective C–N-bond formation), epoxidation (stereoselective C–O-bond formation), and addition of nucleophilic carbon (stereoselective C–C-bond formation). All these reactions proceed through selective substrate face recognition by the enzyme of interest and may lead to the formation of up to two chiral centers (Scheme 1).
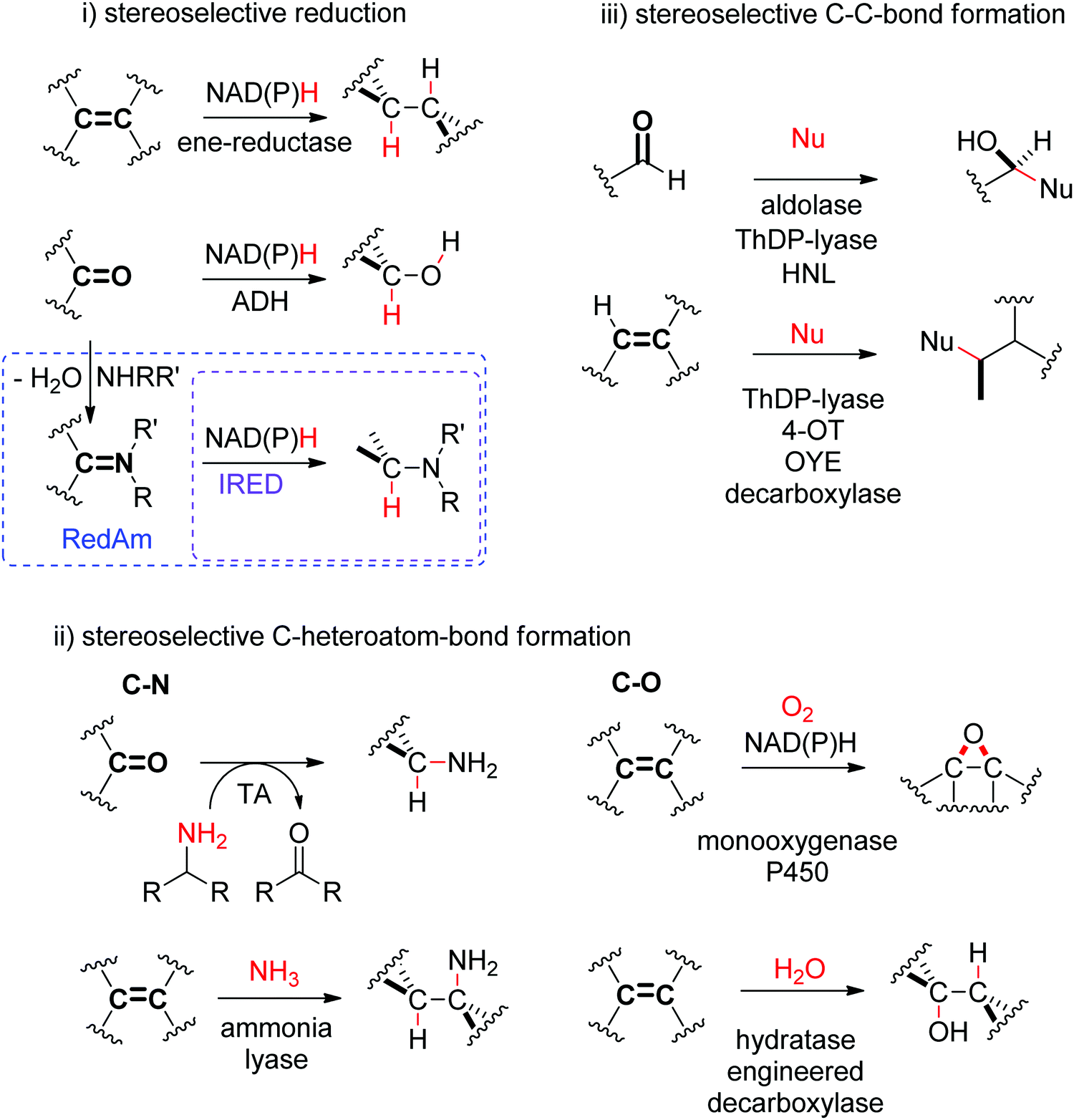 | ||
| Scheme 1 Enzymatic asymmetric strategies relying on stereoselective reactions: (i) reduction, (ii) C–heteroatom-bond formation and (iii) C–C-bond formation by addition of nucleophilic carbon (4-OT: 4-oxalocrotonate tautomerase, ADH: alcohol dehydrogenase, HNL: hydroxynitrile lyase, IRED: imine reductase, OYE: Old Yellow Enzyme, RedAm: reductive aminase, TA: transaminase, ThDP: thiamine diphosphate, Nu: nucleophile). | ||
1.1. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C-, CO-, CN-double bond reduction
C-, CO-, CN-double bond reduction
In biology, the reduction of double bonds is typically a formal addition of [2H], which proceeds via a hydride addition/protonation sequence. The hydride added stereoselectively on the most electron-deficient (carbon) atom is derived from a molecule of nicotinamide cofactor NAD(P)H,27 and usually, the final product is obtained by protonation – often catalyzed by an acidic amino acid – through the reaction medium.
C-double bonds.28 Ene-reductases from the flavoprotein family of Old Yellow Enzymes (OYEs) have become the golden standard for this reaction in biocatalysis owing to a broad substrate scope and large protein diversity.29,30 Bacterial enoate reductases from obligate anaerobes are specific for α,β-unsaturated carboxylic acids (enoates). Due to their dependence on an [Fe–S]-cluster, these enzymes are oxygen-sensitive and therefore less suitable for practical applications in synthesis. Nicotinamide-dependent reductases from the NADP-dependent leukotriene B4 dehydrogenase subfamily of medium chain dehydrogenases/reductases (MDR) are flavin-independent enzymes found in plants and mammals, which have not yet demonstrated generality through high stereoselectivity on a large panel of substrates. Some of these alkene reductases have however displayed sufficient stereoselectivity and could be applied in the synthesis of a few chiral molecules.31–33
Ene-reductases from the flavin mononucleotide (FMN)-dependent OYE family34 accept a broad range of α,β-unsaturated compounds activated on Cα by an electron-withdrawing group, and that include nitro, carbonyl, nitrile, imide and carboxylic acid derivatives (Scheme 2).30 Strongly activated oximes can also be reduced.35 Based on detailed structural and mechanistic studies, the reaction is well understood and proceeds according to a ping-pong bi–bi-mechanism. In a first step (the reductive half-reaction), FMN is reduced by NAD(P)H. Following exit of the oxidized cofactor from the active site, binding of the activated substrate by two conserved residues (His/His or His/Asn) via H-bonding with the electron-withdrawing group further contributes to reduce the electron density of the CC-double bond and favors attack by the reduced flavin. The transfer of the hydride from N5 onto Cβ (the oxidative half-reaction) is followed by protonation at Cα from a conserved Tyr residue located on the other side of the CC-double bond, ensuring overall trans-specific addition of 2H (Scheme 3).36,37 This is in contrast to the cis-specific homogeneous hydrogenation methods. In case of mixed binding modes38 of the substrate, the stereoselectivity may be affected and the ee values of the product become moderate to poor.39–41 Homologues have been discovered in all kingdoms of life and many crystal structures of OYEs are now available, thereby fueling protein engineering campaigns toward improved catalytic properties.42,43
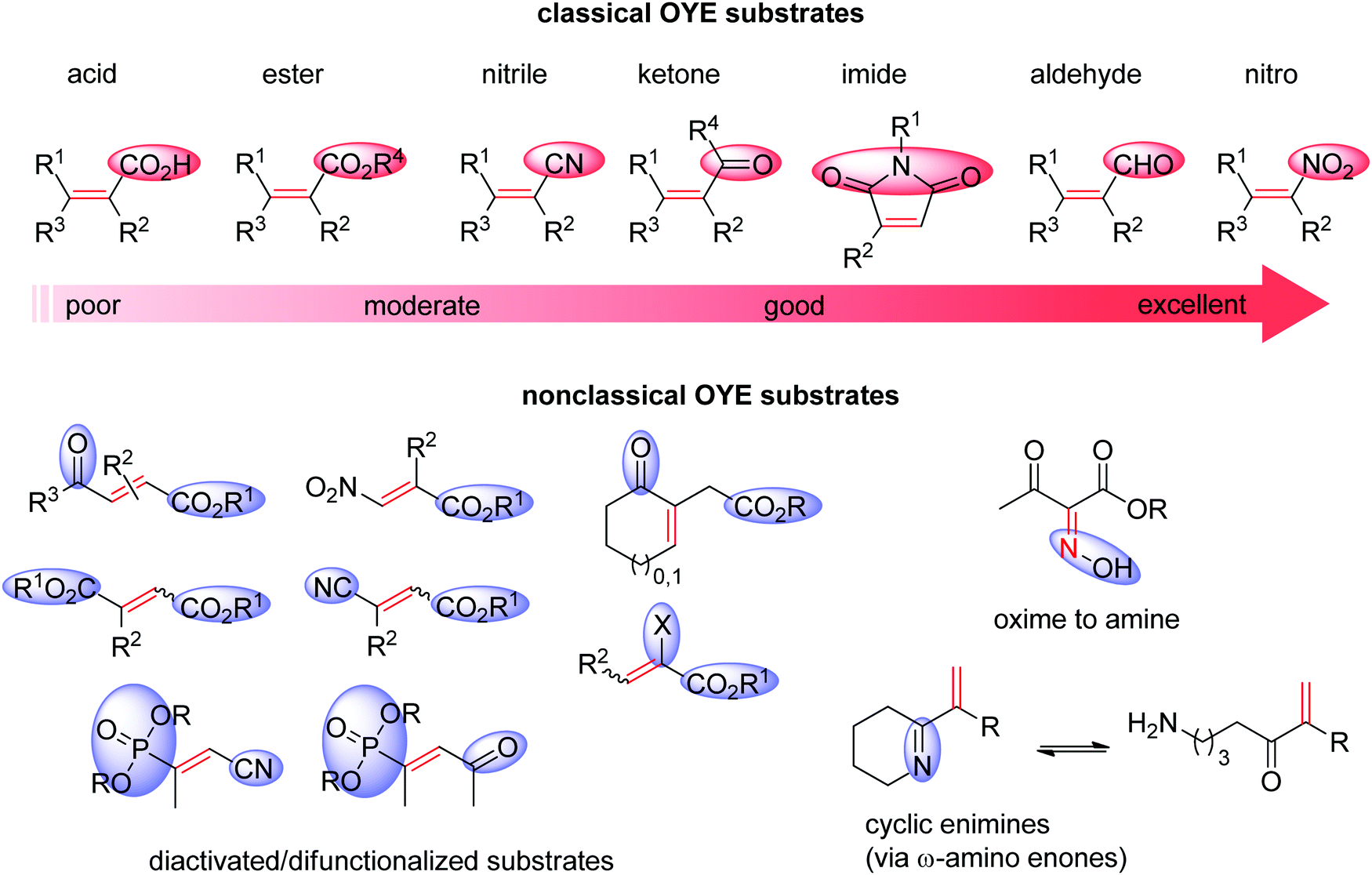 | ||
| Scheme 2 Classical substrates for Old Yellow Enzymes (OYEs) listed according to reactivity, and nonclassical substrates. | ||
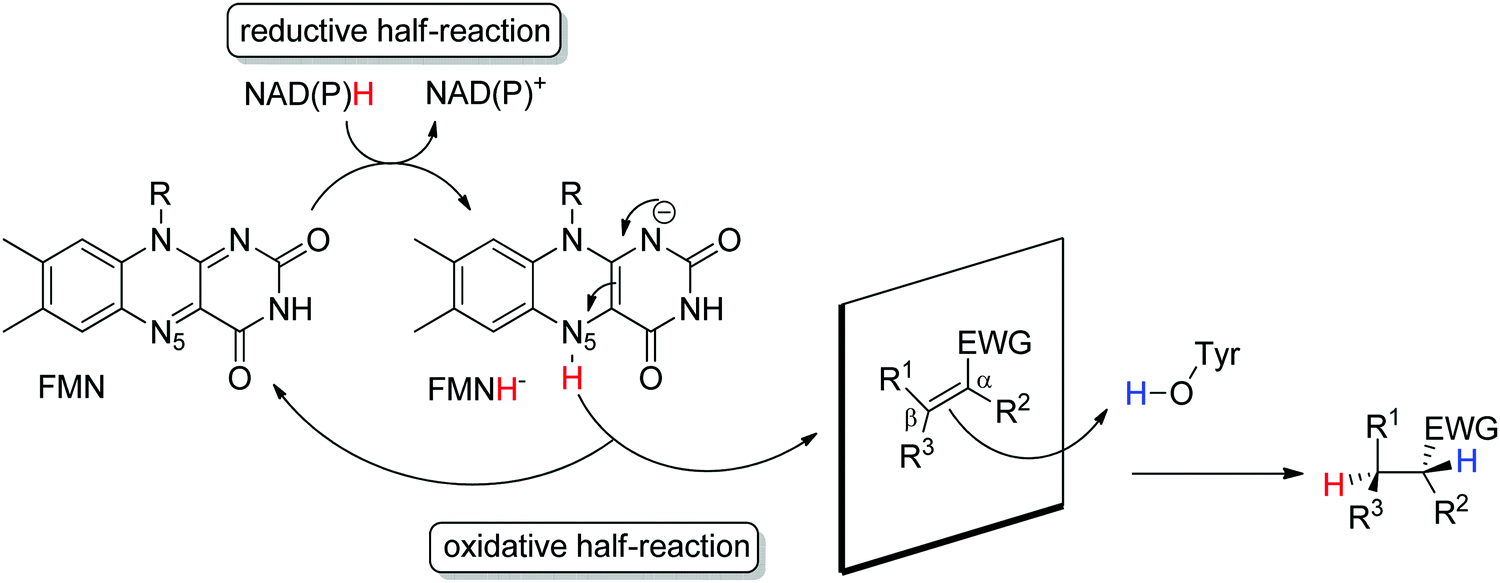 | ||
| Scheme 3 Stereoselective reduction of activated alkenes by FMN-dependent Old Yellow Enzymes (EWG: electron-withdrawing group, FMN: flavin mononucleotide). | ||
OYEs have been exploited in a number of reactions in order to generate chiral molecules in enantiopure form, many of which find application as small building blocks in synthesis. For instance, (R)-levodione,44 (S)-citronellal45,46 and (R)-helional47 were obtained by OYE-catalyzed reduction reactions. Similarly, this biocatalytic platform was employed to access (R)-derivatives of the Roche ester,48 the (R)-nitro precursor of a β-amino acid49 and the precursor of (R)-2-methylsuccinate (Fig. 1).50
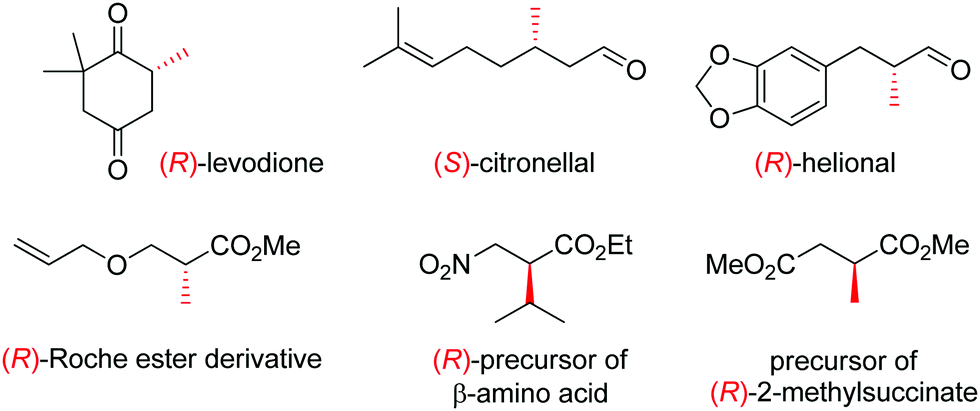 | ||
| Fig. 1 Important chiral building blocks obtained by stereoselective bioreduction catalyzed by ene-reductases of the OYE family.44–50 | ||
An important advantage of this biocatalytic reductive platform is the availability of two main strategies to control the stereochemical outcome of the reactions51–56 (Scheme 4): (i) in substrate-based methods, modulation of the alkene configuration or the type and size of the substituents impacts the absolute configuration of the product. (ii) In enzyme-based methods, advantage is taken of the large protein diversity – and the resulting variation in enzyme active site configuration – to identify stereocomplementary enzymes. In addition, protein engineering can be employed to modify on purpose the active site toward altered substrate binding and thus influence the stereoselectivity of the reaction.
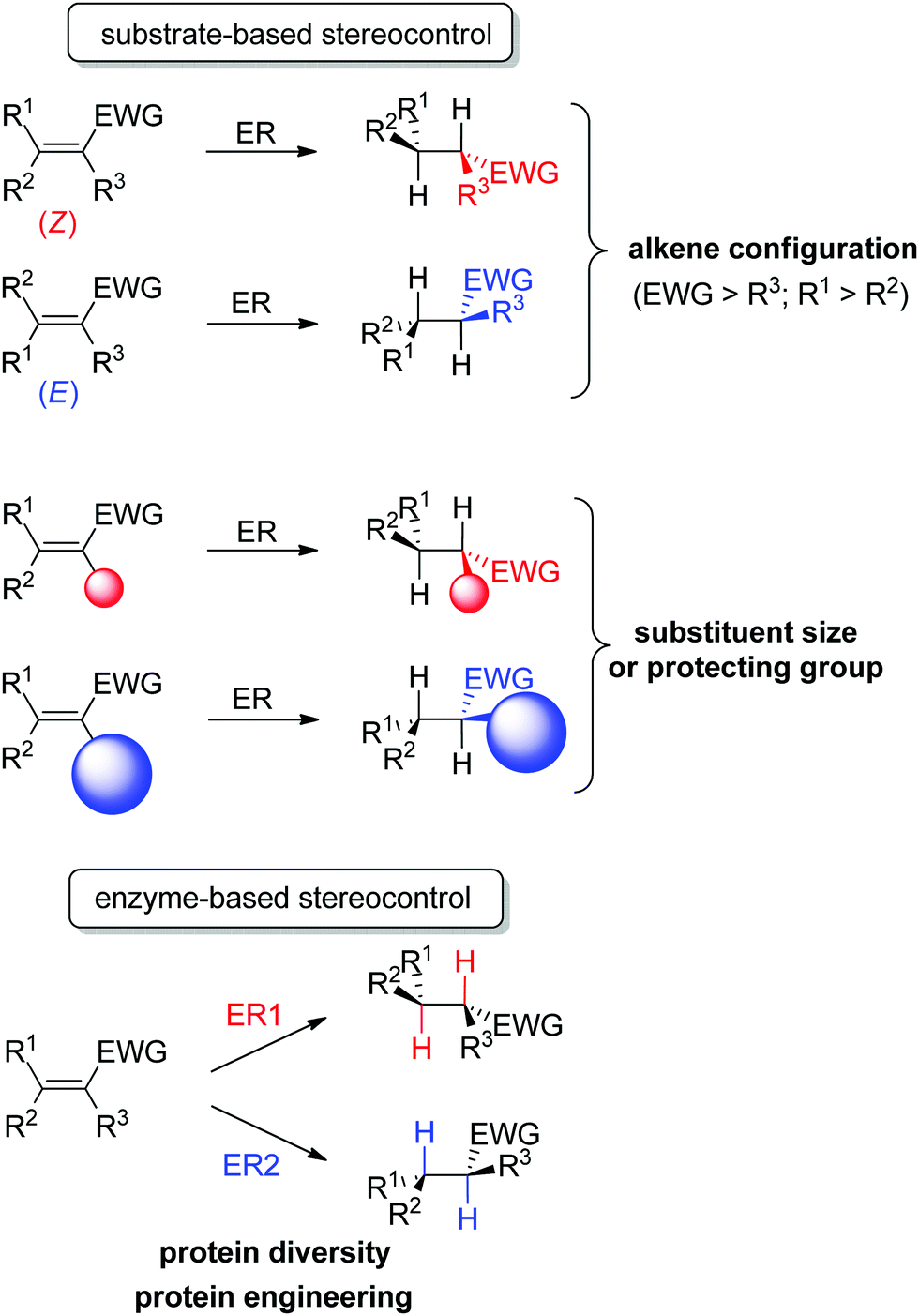 | ||
| Scheme 4 Stereocontrol strategies with ene-reductases based on substrate or enzyme (ER: ene-reductase, EWG: electron-withdrawing group). | ||
Some examples can be highlighted (Scheme 5): the use of YqjM to access both enantiomers of methyl 2-chloropentanoate by using either (Z)- or (E)-configurated methyl 2-chloropent-2-enoate as starting material;57 the conversion by OYE3 of α,β-dehydroamino acids bearing a benzoyl- or phenylacetyl-protected amino group into the (S)- or (R)-product, respectively;58 the use of either OPR1 or OPR3, both isoenzymes from Lycopersicon esculentum, to generate (R)- or (S)-1-nitro-2-phenyl-propane, respectively;45 the reduction of (S)-carvone to the cis-(2R,5S)- or trans-(2S,5S)-dihydrocarvone product by wild-type OYE1 or its variant W116I, respectively.42 Both stereocontrol strategies can be efficiently merged, as was shown in the reduction of cyclohexenone substrates bearing an enol ether moiety in the α-position toward the corresponding acyloin derivatives. The pair methoxy group/OYE3 led to the formation of the (R)-enantiomer in 97% ee while the pair benzyloxy/XenB yielded the (S)-enantiomer with 96% ee.59
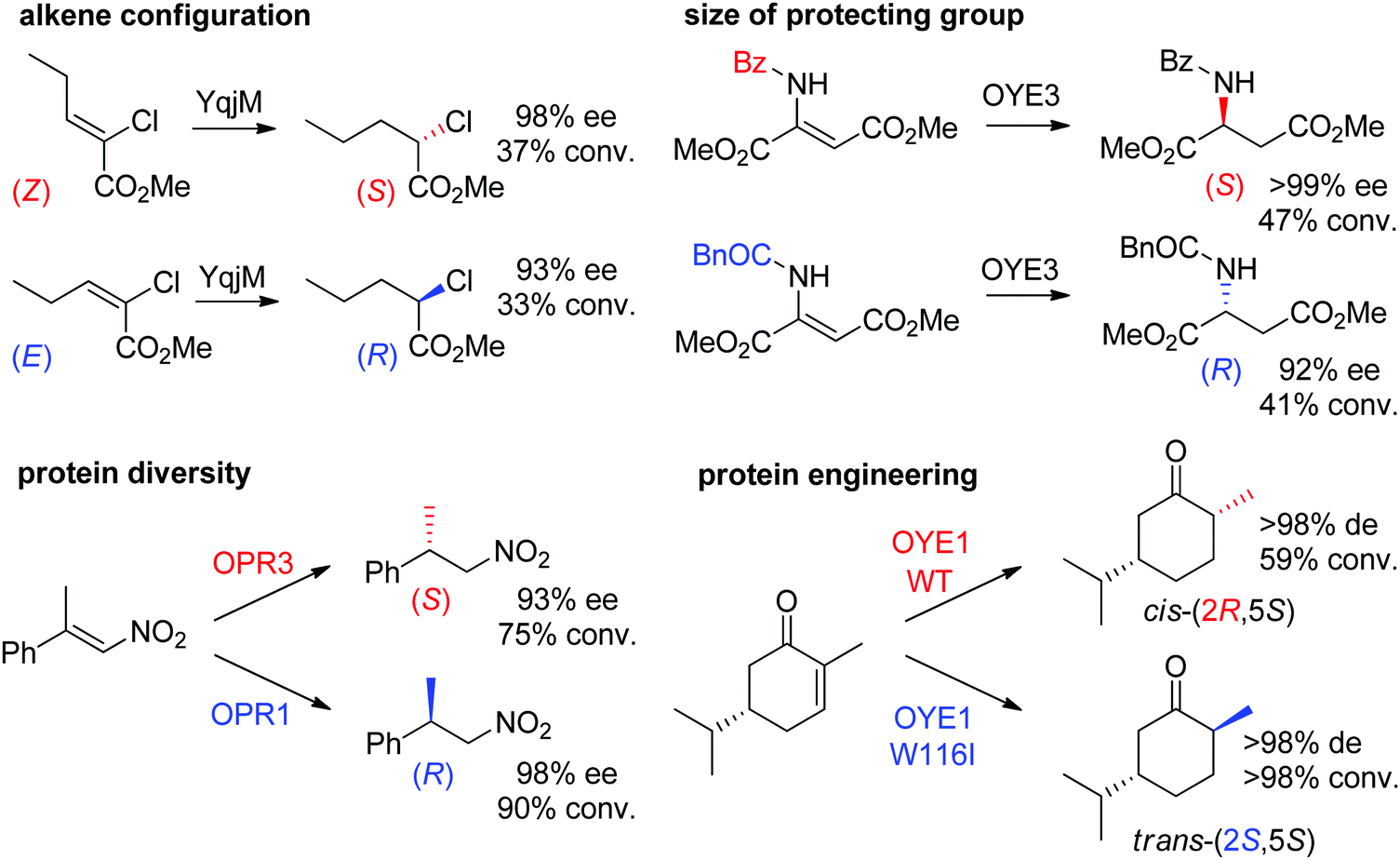 | ||
| Scheme 5 Examples of stereocontrol strategies using ene-reductases.42,45,57,58 Reprinted from ref. 252 Copyright 2020, with permission from Elsevier. | ||
In theory, the stereoselective reduction of tetrasubstituted alkenes by OYEs can generate up to two chiral centers. In practice, such highly substituted compounds are challenging to obtain in highly optically pure form, often due to the racemization of the labile α-center and the steric constraints posed by the substrate substitution in the enzyme active site. In a few published examples, the corresponding products were obtained with high diastereoselectivity (Scheme 6).60,61 In both reported cases, the resulting products were further reduced by alcohol dehydrogenases, thereby granting access to γ-butyrolactones with two, and halohydrins with three contiguous stereocenters, respectively, with excellent ee and de values (>99%).
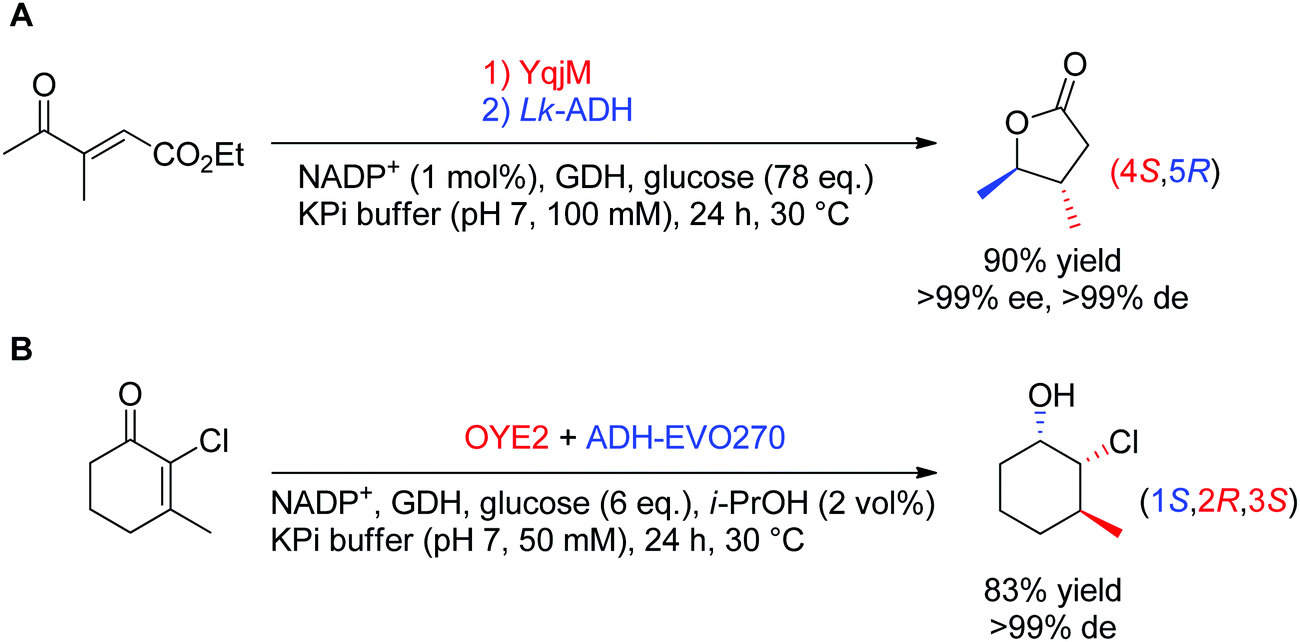 | ||
| Scheme 6 Access to two (A) or three (B) chiral centers via bi-enzymatic reductive cascades in (A) one-pot two-step60 and (B) one-pot one-step.61 A recycling system was used in both cases to regenerate NADPH (ADH-EVO270: commercial alcohol dehydrogenase, GDH: glucose dehydrogenase, Lk-ADH: ADH from Lactobacillus kefir). | ||
Further recent developments in the field62 include reductive (dynamic) kinetic resolution of racemic substituted lactones through the stereorecognition of a distant γ-chiral center,63 and reduction of chemically rich molecules (with more than one functional group). In the latter case, several difunctionalized products could be obtained and reduction was particularly favored in the case of substrates undergoing double activation (Scheme 2).41,49,58,64
Despite the large spectrum of applications in synthesis at the laboratory scale, ene-reductases from the OYE family are still underrepresented in industrial biocatalytic processes.50 As a consequence of the enzyme typically low substrate concentration tolerance (<20 mM), efforts in process engineering are required to render OYE-catalyzed reactions compatible with large scale process conditions. Immobilization of ene-reductases may increase process robustness. Lately, co-immobilization with a cofactor recycling enzyme partner showed encouraging results.65–68
Even more challenging remains the difficult predictability in the substrate acceptance and stereopreference, and currently, identification of a biocatalyst suitable for a desired reaction is inevitably tied up with large screening efforts.
Lately, ene-reductases could be successfully employed in the CC-bond reduction of cyclic enimines (Scheme 2).69 The reaction was postulated to proceed via spontaneous hydrolytic ring opening of the cyclic imines, thus liberating a carbonyl functionality that could act as an electron-withdrawing activating group for the CC-double bond (enone-type). The reaction was coupled to an imine reductive step catalyzed by imine reductase (see Section 1.1.3). In the case where two chiral centers were generated consecutively, conversion to heterocyclic amines through a one-pot cascade set-up was high, however diastereoselectivities remain moderate.
Mechanistically derived from the initial reductive step targeting the CC-double bond of γ-halogenated α,β-unsaturated aldehydes and ketones, a reductive carbocyclization reaction could be selectively achieved using OYEs in which the catalytic proton donor had been silenced (mutation of tyrosine to phenylalanine).70 Advantage was taken of the intrinsic reactivity of the intermediate enolate generated, of the proximity of the electrophilic γ-center, and of the slow nonenzymatic protonation ‘quenching’ step, which all contributed to an intramolecular nucleophilic substitution leading to cyclization (Scheme 7). The reaction formed chiral 1,2-disubstituted cyclopropanes and diastereoselectivity was particularly high with the YqjM mutant Y169F.
 | ||
| Scheme 7 Reductive carbocyclization catalyzed by a tyrosine variant of YqjM.70 | ||
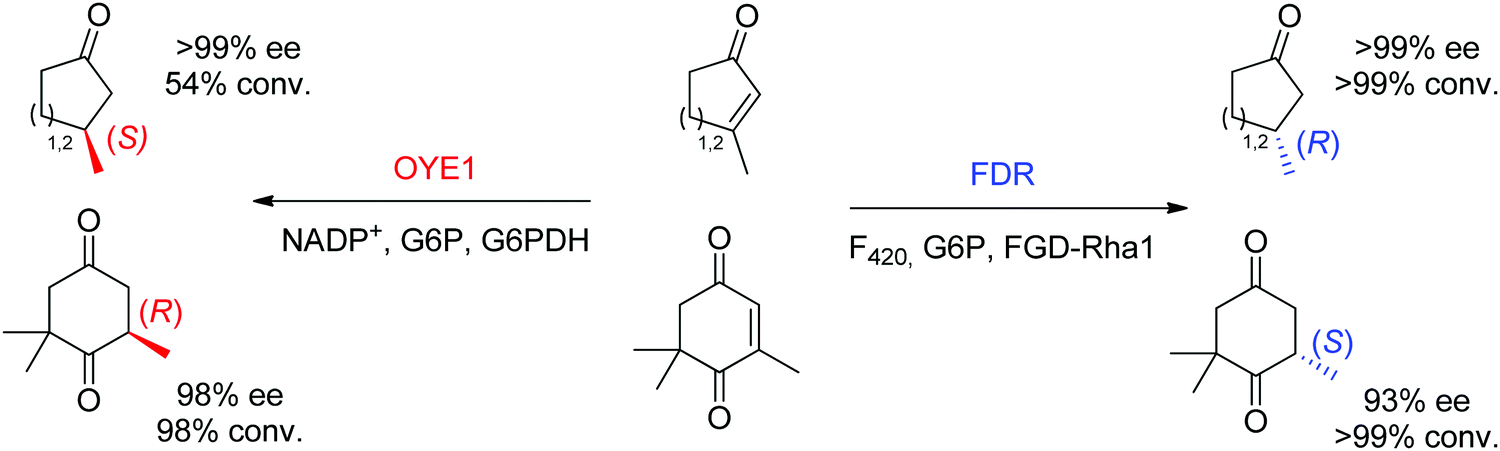
Finally, additional biocatalysts suitable for CC-double bond reductions have been identified with nicotinamide-independent F420H2-dependent reductases (FDRs) from the superfamily of flavin/deazaflavin oxidoreductases (FDORs).62,71 While initially applied to the reduction of quinones, coumarins and pyrones to nonchiral compounds,72 reductases from Mycobacterium hassiacum and Rhodococcus jostii RHA1 lately showed excellent stereoselectivity in the reduction of β-substituted α,β-unsaturated aldehydes and cyclic enones.73 Despite overall low catalytic rates, these enzymes may turn relevant as stereocomplementary catalysts to OYEs in the reduction of particular substrates. For instance, ketoisophorone, invariably reduced to (R)-levodione by OYEs, was transformed into the (S)-enantiomer by FDRs. Importantly, the cofactor F420 can be recycled by a F420-dependent glucose 6-phosphate dehydrogenase at the expense of glucose 6-phosphate. As recently demonstrated, these enzymes also proceed via a trans-addition of [2H] and could reduce tetrasubstituted cyclic enones with high stereo- and diastereoselectivity.61 In selected cases, including the reduction of ketoisophorone and 3-methylcycloalk-2-enone, FDR appeared stereocomplementary to OYE1 (Scheme 8).74
While aldehydes are readily reduced by several ADHs, no chiral center is generated in the reaction and corresponding reactions will not be treated here. However, dynamic kinetic resolutions of racemic α-substituted aldehydes were successfully attained and are briefly discussed in Section 2.2. Since the reaction is reversible, ADHs are frequently used in oxidation reaction and can be implemented in enantioselective processes with chiral alcohols, such as in (dynamic) kinetic resolution, stereoinversion or cyclic deracemization.83,84
A major reason for the broad acceptance of ADHs in the organic chemistry laboratory85 is the possibility to predict the stereochemical outcome of the reaction based on the choice of the enzyme and the configuration of the substrate (Scheme 9). So-called Prelog ADHs follow the Prelog rule,86 that is the carbonyl is attacked on the Re-face by the hydride of NAD(P)H and furnishes the (S)-alcohol, as long as the smallest substituent has the lowest priority according to the Cahn-Ingold-Prelog (CIP) sequence rule (used to name stereoisomers with fixed absolute configuration). Stereocomplementary enzymes denoted as anti-Prelog ADHs deliver, in the contrary and under same considerations, (R)-alcohols after attack on the Si-face.87
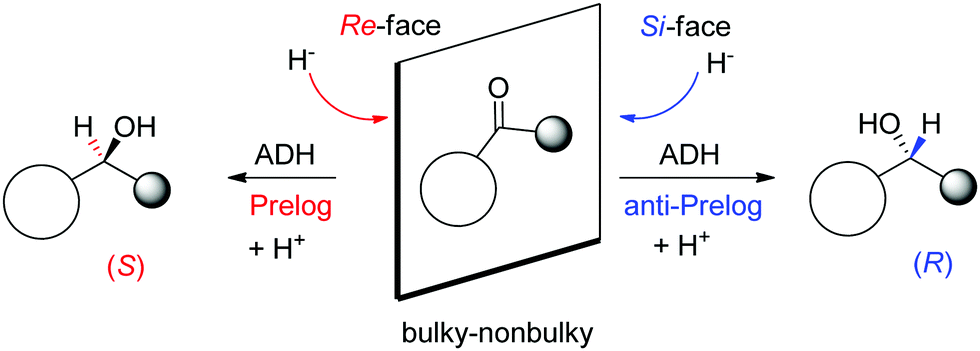 | ||
| Scheme 9 Prelog and anti-Prelog rule applied to the reduction of carbonyl compounds by alcohol dehydrogenases (ADH). H− denotes the hydride of NAD(P)H, not shown for clarity. Republished from ref. 38 with permission of John Wiley & Sons – Books, copyright 2021; permission conveyed through Copyright Clearance Center, Inc. | ||
Owing to a fantastic protein diversity, ADHs display stereopreference on various substrate types. Not only bulky–nonbulky substrates with major size differences between the two substituents, but also bulky–bulky substrates such as diaryl ketones can be reduced to enantiopure alcohols.80,88–92
For particularly challenging substrates, such as large compounds or multifunctionalized molecules, the implementation of protein engineering methods can tailor a particular ADH for the targeted application.93–96 Such approach has been regularly employed under industrial setting and the resulting catalysts often displayed, in addition to perfect stereoselectivity, high catalytic activity as well as increased robustness toward process conditions.97–99
The statin side chain, with its two asymmetric hydroxylated centers, can be conveniently prepared by an ADH-catalyzed reductive strategy. In one synthetic approach to atorvastatin – a blockbuster drug used in the prevention of cardiovascular disease and the treatment of high levels of cholesterol – an enzymatic cascade was developed to access the key chiral precursor of the final active pharmaceutical ingredient (API). An ADH engineered by Codexis was applied to the reduction of 4-chloroacetoacetate into (S)-4-chloro-3-hydroxybutyrate, while the reduced nicotinamide cofactor was regenerated by glucose/glucose dehydrogenase (Scheme 10). The reaction was performed at pH 7 and 25 °C and yielded (S)-4-chloro-3-hydroxybutyrate in 96% yield and >99.5% ee. This intermediate was then further converted in a sequence of enzymatic dehalogenation–epoxidation catalyzed by a halohydrin dehalogenase and chemical cyanolysis to deliver (R)-4-cyano-3-hydroxybutyrate, which could be incorporated into the final API.100,101
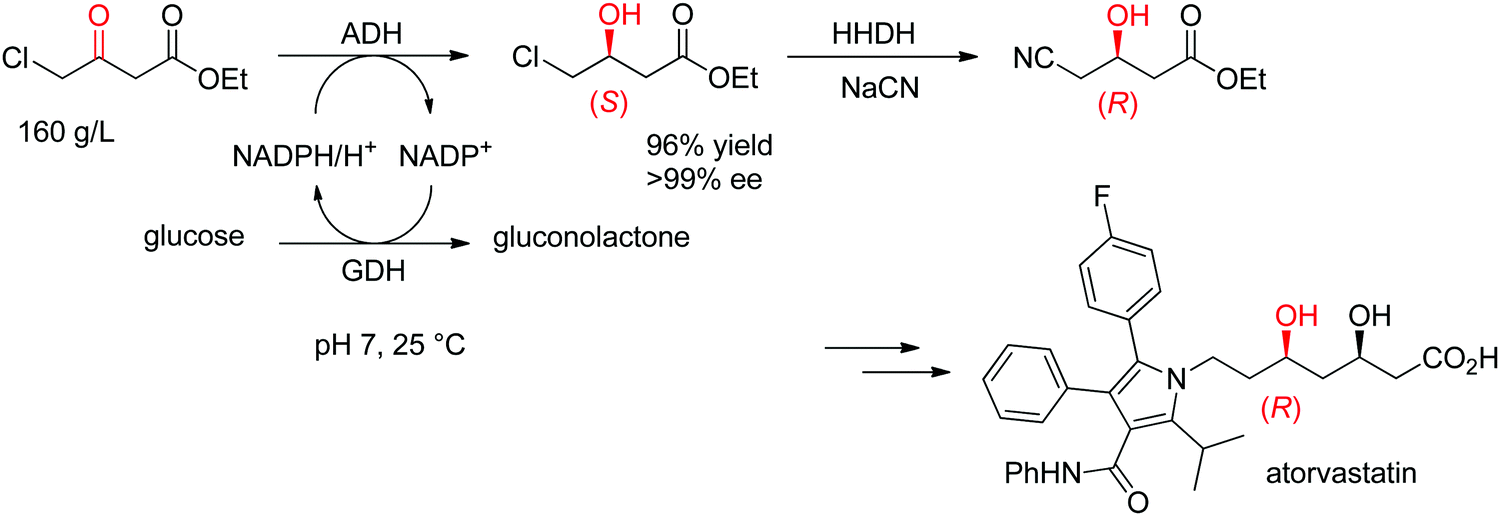 | ||
| Scheme 10 Enzymatic cascade for the asymmetric synthesis of the chiral precursor to atorvastatin based on an ADH-catalyzed step (GDH: glucose dehydrogenase, HHDH: halohydrin dehalogenase).100 Republished from ref. 38 with permission of John Wiley & Sons – Books, copyright 2021; permission conveyed through Copyright Clearance Center, Inc. | ||
A chemoenzymatic process has been developed by Merck & Co, Inc. for the synthesis of montelukast, an anti-asthmatic agent. The key step of asymmetric biocatalytic reduction replaced the chemical synthesis that was based on a chiral reducing agent and could provide the crucial (S)-alcohol intermediate en route to montelukast in enantiopure form. An ADH was engineered by Codexis in several rounds of directed evolution to accept the bulky poly-functionalized substrate [(E)-methyl 2-(3-(3-(2-(7-chloroquinolin-2-yl)vinyl)phenyl)-3-oxopropyl)benzoate] under challenging reaction conditions (Scheme 11). The engineered biocatalyst was finally employed in 67 vol% organic solvent in form of a mixture of isopropanol and toluene over 40–45 h reaction time at up to 45 °C on 230 kg scale at 100 g L−1 substrate, whereby isopropanol served as hydride source via the coupled-substrate cofactor recycling approach.102
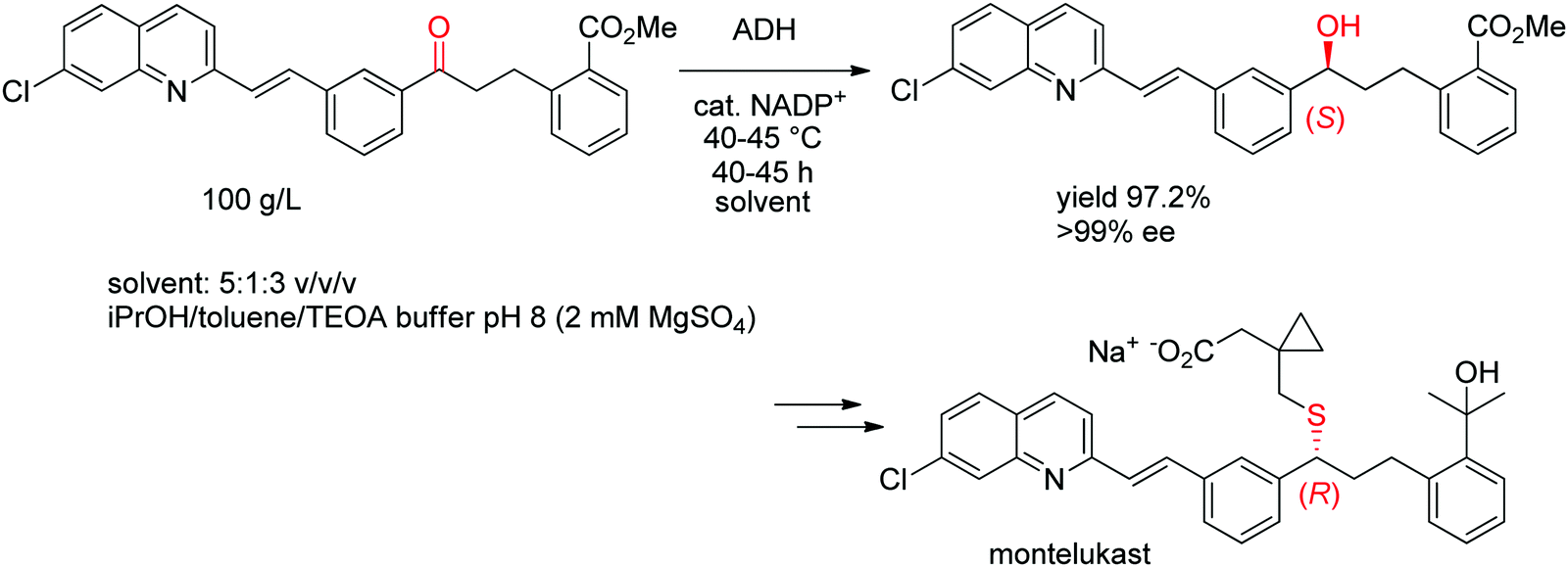 | ||
| Scheme 11 Asymmetric synthesis of the (S)-precursor to montelukast by ADH-catalyzed reduction (ADH: engineered alcohol dehydrogenase, TEOA: triethanolamine).102 Republished from ref. 38 with permission of John Wiley & Sons – Books, copyright 2021; permission conveyed through Copyright Clearance Center, Inc. | ||
Recently, promiscuous reductive activity was found in the Baeyer–Villiger monooxygenase cyclohexanone monooxygenase (CHMO) from Acinetobacter sp. NCIM 9871. Initially, the wild-type enzyme showed only poor activity in the reduction of an aromatic α-keto ester substrate under anaerobic conditions. Rational design based on site-directed mutagenesis was therefore applied and led to the generation of a highly efficient and selective biocatalyst that followed the anti-Prelog rule on a range of aromatic α-keto esters. Importantly, in the case of this artificial ketoreductase, the hydride delivery was suggested to proceed through the reduced FAD, akin to the mechanism of Old Yellow Enzymes in CC-double bond reduction reactions (see Section 1.1.1).103
N reduction in presence of NADPH are imine reductases.105,106 The field of enzymatic imine reduction is a prime example of how rapidly the field of biocatalysis can evolve.107 Within the last decade, the portfolio of imine reductases (often denoted IREDs) grew from a few examples known from specific biosynthetic pathways with little application potential in biocatalysis – most prominent being the case of dihydrofolate reductase involved in the synthesis of tetrahydrofolic acid – to large collections of stereocomplementary enzymes, for which specific sequence motifs could be identified.108 The stereoselectivity has been assigned to two distinct stereocomplementary superfamilies.109 New imine-reducing enzymes can also be obtained by introducing mutations in closely related enzyme classes, as was shown with β-hydroxyacid dehydrogenases. An engineered enzyme bearing two mutations was found highly stereoselective in the reduction of 6-phenyl-2,3,4,5-tetrahydropyridine, yielding the corresponding (S)-product with 97% ee, thereby outperforming wild-type IREDs.110
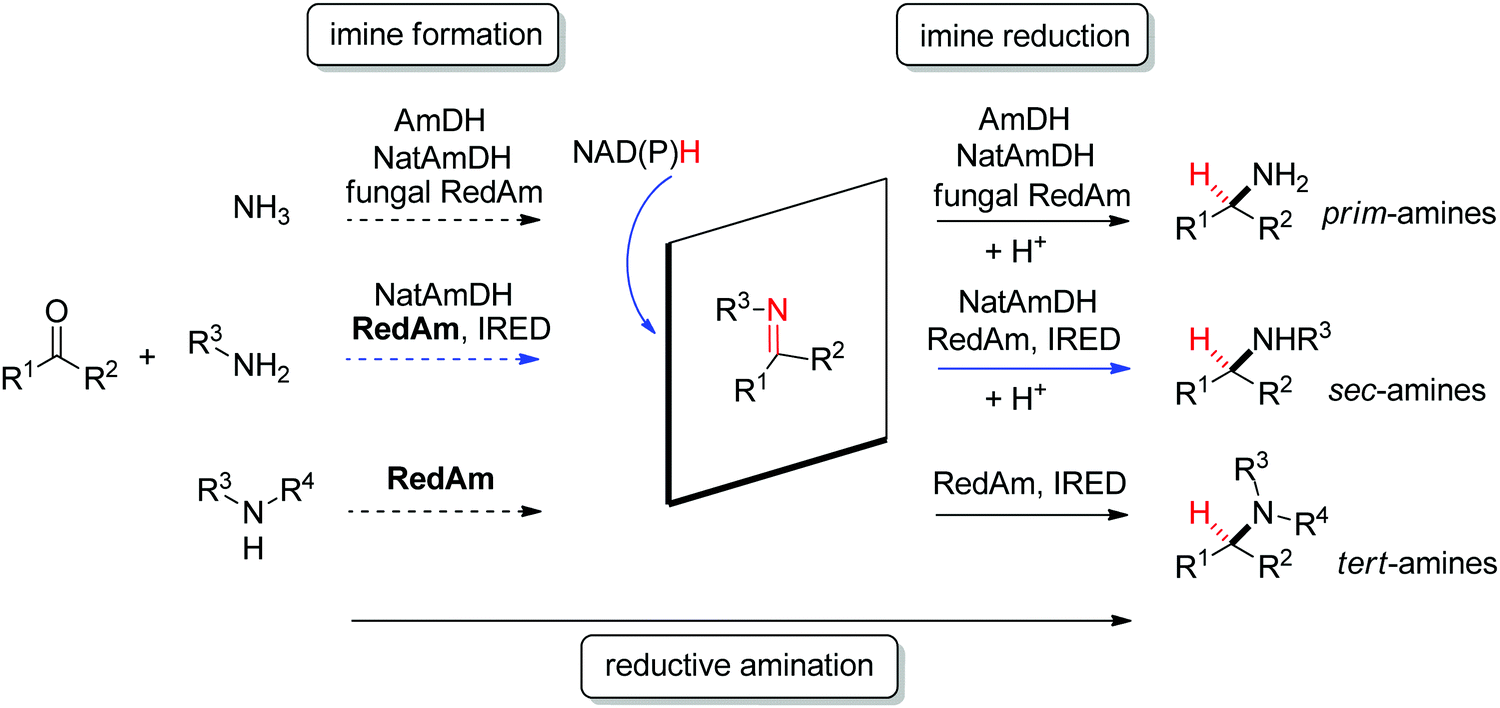 | ||
| Scheme 12 Stereoselective enzymatic strategies in imine reduction to access primary, secondary and tertiary chiral amines. An enzyme-catalyzed imine formation may be included, rendering a reductive amination. Hydride attack of the imine intermediate depicted arbitrarily for the case of primary amine (blue arrow) as amine substrate. Dashed arrows indicate that the reaction can be enzymatically catalyzed (IRED: imine reductase, (Nat)AmDH: (native) amine dehydrogenase, RedAm: reductive aminase). | ||
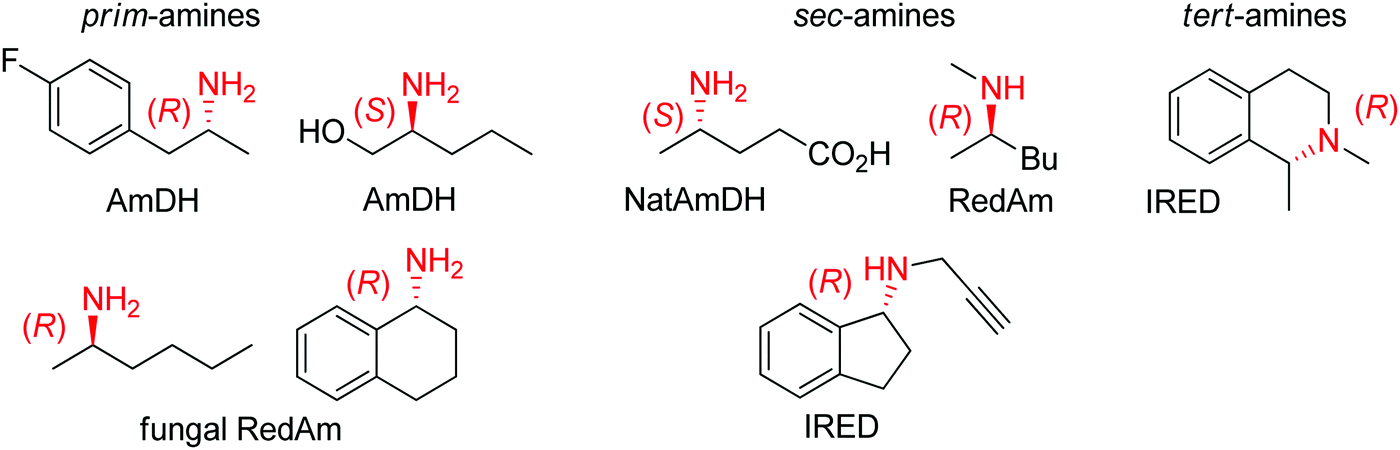 | ||
| Fig. 2 Sampling of products obtained by biocatalytic reductive amination, except tertiary amines, which were obtained by enzymatic iminium reduction only (IRED: imine reductase, (Nat)AmDH: (native) amine dehydrogenase, RedAm: reductive aminase).114,115,119–123 | ||
Imine reductases have been preferentially applied to the reduction of cyclic imines,111–113 which present a higher stability in aqueous reaction media. By including in situ formation of the imine substrates in the reaction scheme, exocyclic and acyclic imines can also be converted to chiral secondary amines. To favor the reversible formation of the imine in aqueous media, a large excess of the amine in comparison to the carbonyl compound is employed. Since both imine and iminium ion are accepted, IREDs can be employed for the formation of secondary and tertiary amines (Fig. 2).114–116
In addition, the CN reduction reaction may be enzymatically coupled to preceding catalyzed imine formation. The resulting enzymatic reductive amination of carbonyl compounds with primary amines could be observed with some imine reductases.117,118 However, to reach synthetically relevant conversion levels, a large excess of the amine substrate is necessary, poising both the atom economy of the reaction and downstream processing. Since chemoselectivity is high and the carbonyl substrate cannot be depleted by alternative reductive pathways, this approach is still very attractive and advantageous compared to some general chemical approaches.
A subclass of imine reductases called reductive aminases (RedAms) was later shown capable of catalyzing the imine formation more efficiently, rendering a biocatalytic reductive amination of carbonyl compounds with primary and secondary amines.122,124 This system does not require an excess of amine and offers a more attractive synthesis of secondary and tertiary amines enabled by the use of near-stoichiometric carbonyl/amine equivalents. Although ammonia was not well accepted by the first identified reductive aminases, the discovery of fungal RedAms accepting the simplest amine donor considerably enlarged the possibility to obtain primary amines by enzymatic reductive amination, a synthetic advantage that was further strengthened by increased thermostability of the proteins.123 Recently, a large set of imine reductases were shown to operate at a close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry (amine/carbonyl), with only a few enzymes being categorized as reductive aminases. This highlights the difficulty in assigning activity in reductive amination at low stoichiometric excess to particular imine reductases according to their structure and sequence similarities. In this work, the authors importantly identified anilines and other heteroaromatic amines as amine partners.125
:1 stoichiometry (amine/carbonyl), with only a few enzymes being categorized as reductive aminases. This highlights the difficulty in assigning activity in reductive amination at low stoichiometric excess to particular imine reductases according to their structure and sequence similarities. In this work, the authors importantly identified anilines and other heteroaromatic amines as amine partners.125
Overall, sequence-activity relationships with imine reductases are highly substrate-dependent and no general rule could be delineated to anticipate the outcome of a particular reaction or from specific pairs of substrate/enzyme.126
IREDs can be further creatively combined with chemical steps, as shown by combining an enzymatic imine reduction with a base-mediated rearrangement to generate chiral 2,2-disubstituted azepanes and benzazepines.127
An example of application of IREDs in industry was reported by GSK for the synthesis of a molecule currently in clinical trials for use in the treatment of leukemia. Here, the chiral information was already present in the amine substrate, which was used as racemic starting material and coupled to an aldehyde by enzymatic reductive amination (Scheme 13). After three rounds of directed evolution, excellent catalytic and process performance was obtained for the selected imine reductase. This kinetic resolution was performed with the engineered enzyme on 20 L scale at acidic pH (4.6) and delivered the chiral secondary amine product with 99.7% ee in almost 85% isolated yield.128 Importantly, the green metrics of the process could be considerably improved compared to previous chemical approach. Finally, the reaction was elegantly installed in a redox-neutral cascade,129 in which the aldehyde was obtained by oxidation of the corresponding alcohol precursor by an alcohol dehydrogenase, thus rendering a formal amination of primary alcohol130,131 in presence of catalytic amounts of the nicotinamide cofactor and at equimolar amine loading.
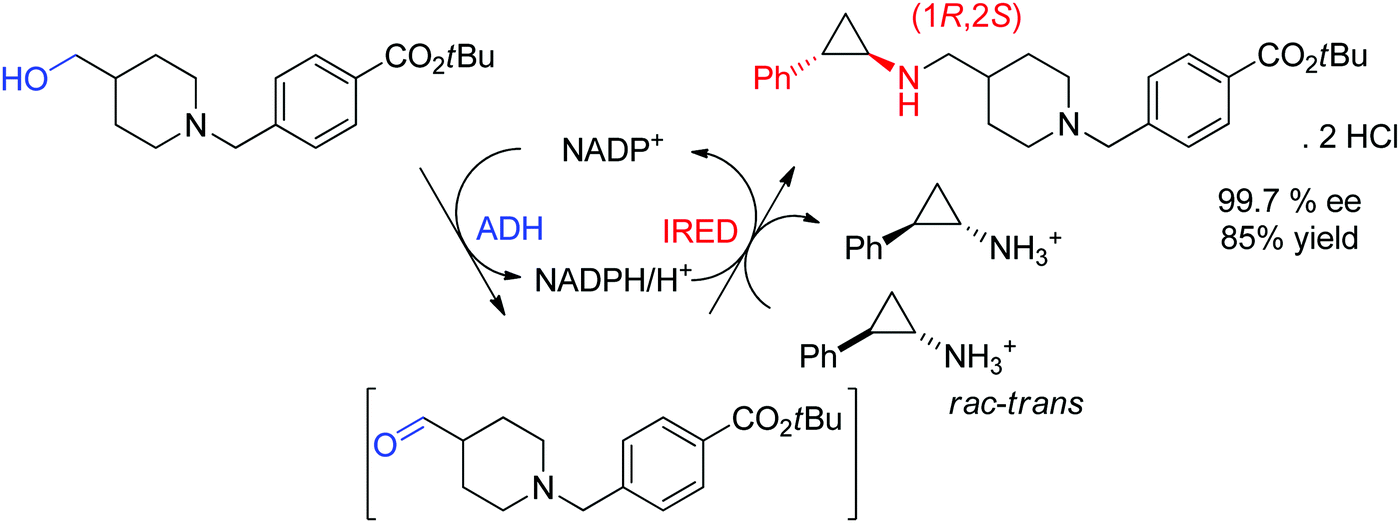 | ||
| Scheme 13 Application of imine reductase (IRED) in the kinetic resolution of a racemic chiral amine substrate via reductive amination in an enzymatic cascade for formal amination of a primary alcohol.128 | ||
Other success stories have contributed to extend the list of biocatalysts used in the reductive amination of highly diverse ketone substrates. The first breakthrough came from the engineering of amino acid dehydrogenases, which naturally catalyze the reversible reductive amination of α-ketoacids with ammonia and have a long history in the asymmetric synthesis of amino acids.132 During a protein engineering campaign, and by targeting few crucial residues responsible especially for binding of the amino acid substrate, Bommarius and coworkers could abolish the requirement of leucine dehydrogenase for the substrate carboxylic acid moiety used as anchor. This provided the first example of engineered amine dehydrogenases (AmDHs) accepting ‘simple’ ketones for the synthesis of primary amines.133,134 Soon after, other leucine dehydrogenases and phenylalanine dehydrogenases were mutated in a comparable manner and provided a range of efficient biocatalysts to access chiral primary amines in enantiopure form.135
Conceptually related was the engineering of an ε-deaminating L-lysine dehydrogenase into an AmDH. Through introduction of only one mutation, the requirement for the terminal α-amino acid functionality, although not so strict with the native enzyme, was lost and the new biocatalyst could efficiently accept a range of ketones in asymmetric reductive amination reaction with ammonia, including aromatic substrates at up to 100 mM concentration and 50 °C. Rather astonishing here was that the native enzyme accepts an aldehyde as natural carbonyl substrate and therefore did not 'need' to display stereoselectivity in the first place. The enzyme was successfully applied to the reductive amination of acetophenone on 600 mg scale using ammonium formate, and the final (R)-amine could be obtained in perfect enantiopurity and 85% isolated yield. A formate dehydrogenase was employed to regenerate the nicotinamide cofactor.136
The scope of biocatalytic reductive amination was finally recently enlarged through the discovery of a family of stereoselective native amine dehydrogenases for the synthesis of primary and secondary amines. As in the case of engineered AmDHs, a large excess of ammonia is employed, typically in form of ammonium formate, while the necessary nicotinamide cofactor is used in catalytic amounts and recycled in situ by a regeneration system such as glucose/glucose dehydrogenase.121,137
As opposed to imine reductases and reductive aminases, engineered AmDHs are specific for NADH, while native AmDHs show slight preference for NADPH.
Finally, the case of opine dehydrogenase deserves mention. The native enzyme accepts α-keto acids and couples them with α-amino acids.138 Codexis evolved this enzyme toward an active biocatalyst for the reductive amination of ketones with a range of secondary amines. In many cases, chiral tertiary amines could be obtained with excellent stereoselectivity.139
1.2. C–Heteroatom-bond formation
1.2.1.1. Transamination of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) O-bond.
Transaminases catalyze the formal swap of functional groups via amino-transfer between a carbonyl acceptor and an amino donor, which means that for amine synthesis, a ketone is formed as by-product. These aminotransferases are pyridoxal-5′-phosphate (PLP)-dependent enzymes and use the cofactor as amine shuttle between the donor and the acceptor. In the amination direction, PLP first gets aminated by the amine donor to form pyridoxamine-5′-phosphate (PMP), which then transfers the amino group to the carbonyl substrate (Scheme 14).140 For applications in biocatalysis, so-called ω-transaminases have been lately dominating (later shortly denoted transaminases). These enzymes accept aliphatic ketones as substrates, without requirement for other functional groups.141–143
O-bond.
Transaminases catalyze the formal swap of functional groups via amino-transfer between a carbonyl acceptor and an amino donor, which means that for amine synthesis, a ketone is formed as by-product. These aminotransferases are pyridoxal-5′-phosphate (PLP)-dependent enzymes and use the cofactor as amine shuttle between the donor and the acceptor. In the amination direction, PLP first gets aminated by the amine donor to form pyridoxamine-5′-phosphate (PMP), which then transfers the amino group to the carbonyl substrate (Scheme 14).140 For applications in biocatalysis, so-called ω-transaminases have been lately dominating (later shortly denoted transaminases). These enzymes accept aliphatic ketones as substrates, without requirement for other functional groups.141–143
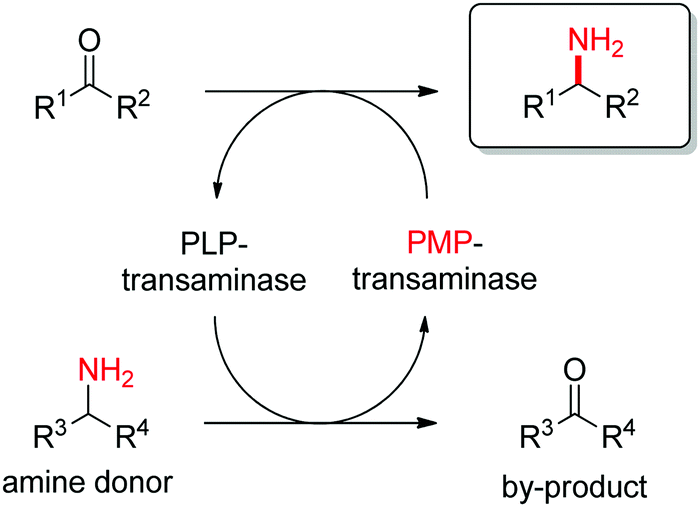 | ||
| Scheme 14 Overall concept of amino-transfer using PLP-dependent transaminase (PLP: pyridoxal-5′-phosphate, PMP: pyridoxamine-5′-phosphate). | ||
Prochiral ketones are converted stereoselectively to primary amines. Mechanistically, the face recognition occurs at the stage of the planar quinonoid ‘imine-type’ intermediate generated between the substrate carbonyl and the aminated cofactor (PMP), which locks the substrate in a defined position; the change from prochiral to chiral molecule occurs through stereoselective protonation of this intermediate by a conserved lysine (Scheme 15). Both (R)- and (S)-selective transaminases have been identified144 and while the protein folds differ, the stereocomplementarity can be simply explained by the different positioning of the lysine in relation to the cofactor binding pocket: the key catalytic residue is situated on the Si-face of the PLP in (S)-transaminases and on the Re-face in (R)-transaminases.145–148 Determinant for the stereo-outcome of the reaction is thus the reaction of the carbonyl with the aminated cofactor and generation of the prochiral imine intermediate.
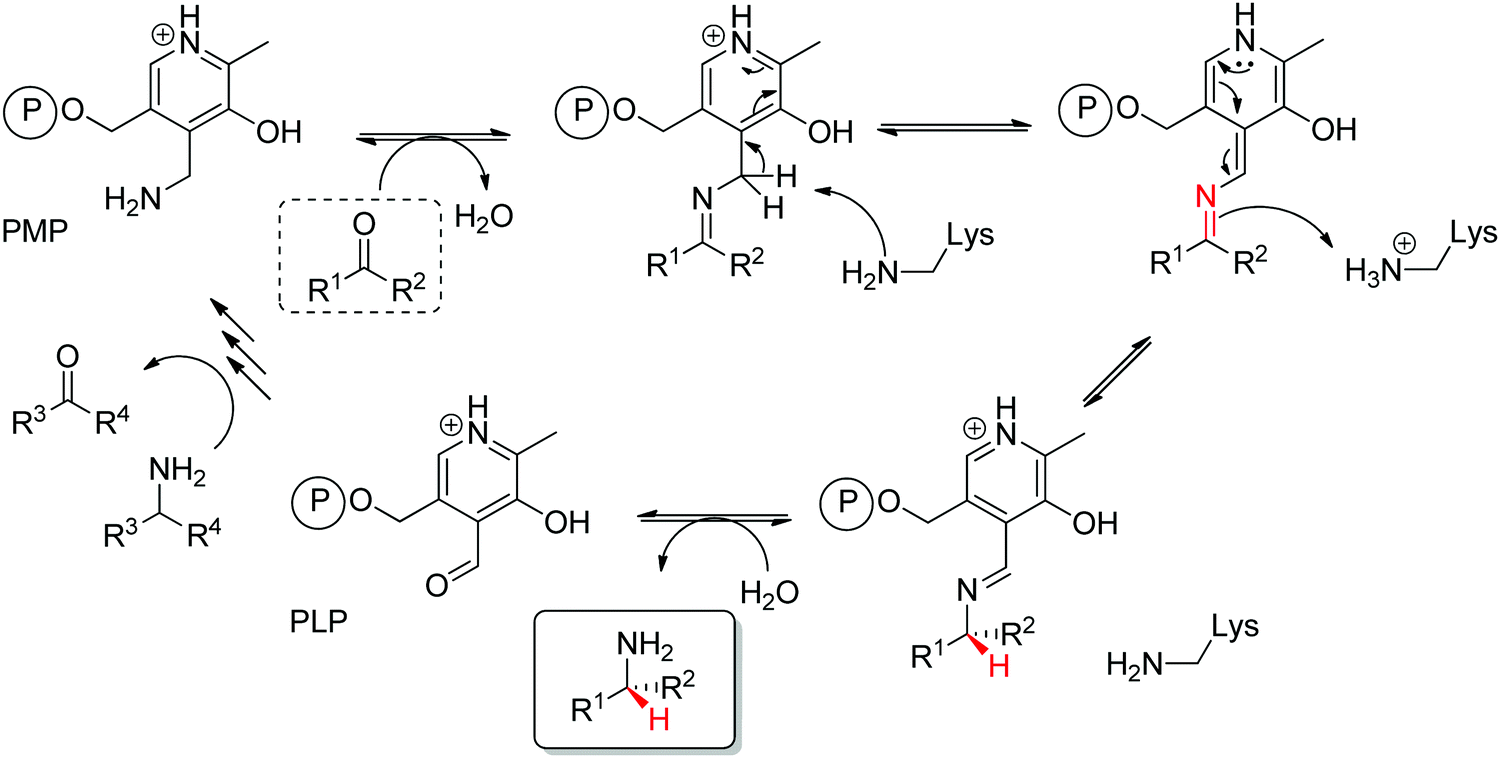 | ||
| Scheme 15 Mechanism of transaminase for the amination of a carbonyl compound (PLP: pyridoxal-5′-phosphate, PMP: pyridoxamine-5′-phosphate). The (first) steps of amination of PLP to PMP are not detailed for clarity (e.g., PLP bound to the enzyme as an internal aldimine via a lysine residue before formation of the external aldimine with the amine donor en route to PMP formation). | ||
While considered only a decade ago ideal biocatalysts for the preparation of chiral amines, the field has developed in such an incredible manner over the last few years that other options that do not suffer from equilibrium limitations typical for transfer reactions and from low atom economy due to inherent generation of by-products are rapidly becoming prevalent in the laboratory (e.g., amine dehydrogenases and reductive aminases, see Section 1.1.3). Importantly, major efforts in the field have delivered suitable solutions to render transaminase-catalyzed reactions attractive in synthesis.149 To favor the amination reaction, strategies involve for instance the (enzymatic) removal of the ketone co-product, the use of 'smart' amine donors (e.g., 1,2-diamines that dimerize after one amine transfer reaction150), or the regioselective conversion of diketones to yield imines followed by reduction to the corresponding cyclic amines.151,152
The industrial biocatalytic synthesis of the antidiabetic drug sitagliptin by Merck & Co, Inc. is a testimony of the strong synthetic value of transaminases for accessing primary chiral amines. The ketone precursor is a bulky multifunctionalized molecule. Protein engineering was necessary to reach both high level of activity and stereoselectivity and render the transaminase amenable to application under process conditions (Scheme 16). The engineered biocatalyst displayed 27 mutations and could convert the ketone at high substrate concentration (0.5 M and 1 M of isopropylamine as amino donor) in 50% DMSO at 45 °C with perfect stereoselectivity (>99% ee), furnishing the final product in high yield (>90%). This example quickly became a benchmark for industrial biotransformations.153,154
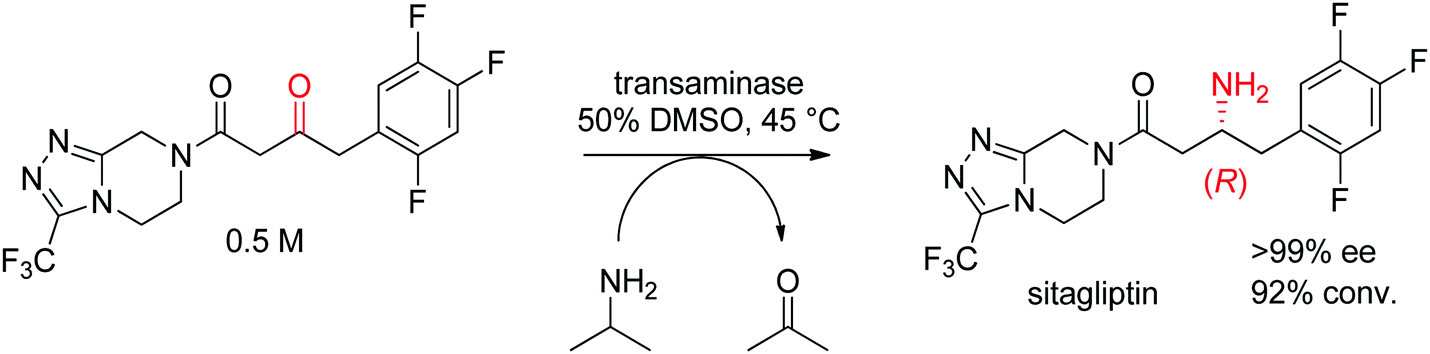 | ||
| Scheme 16 Transaminase-catalyzed amine transfer developed by Merck & Co, Inc. to access (R)-sitagliptin in optically pure form.153 | ||
1.2.1.2. Hydroamination of C
C-bond.
Enzymatic hydroaminations of alkenes are performed by ammonia lyases. The enzymes catalyze the reversible addition of ammonia across CC-double bonds of α,β-unsaturated carboxylic acids. The reaction is stereoselective and leads to the formation of chiral α-amino acids, however, to favor the synthetic direction, large excess of ammonia is necessary.155 The case of amino mutases will not be discussed here. These mechanistically diverse isomerases transfer the amino group of α-amino acids to the β-position via a formal 1,2-shift, yielding chiral β-amino acids; while the reaction is stereoselective, the chiral information is not necessarily retained.156
Prevalent ammonia lyases include aromatic ammonia lyases phenylalanine and tyrosine ammonia lyases (PAL and TAL), and (methyl)aspartate ammonia lyases. Several of these enzymes can accept a range of unsaturated (di)carboxylic acids beyond their natural substrate.157,158
PAL has been employed for instance for the preparation of a range of (nonnatural) aromatic amino acids starting from 3-arylacrylates with various substitution. For instance, several (S)-halophenylalanines could be obtained with excellent enantiopurity and high space time yields (up to >200 g L−1 d−1) by employing a cyanobacterial PAL with ammonium carbamate (4 M) as reaction buffer at high substrate loadings (>0.3 M).159
D-Arylalanines cannot be obtained with high stereoselectivity by PAL. The nonperfect stereoselectivity of a newly identified wild-type enzyme could be exploited to access D-arylalanines with high enantiopurity. The concept (Scheme 17) relied on the low selectivity in the hydroamination reaction (step 1), combined with a kinetic resolution of the nonenantiopure amino acids through enantioselective oxidation to the corresponding imino acid by an L-amino acid deaminase (step 2), followed by nonselective chemical reduction back to the amino acid (step 3). Steps 2–3 allowed enrichment of the D-amino acid products through a cyclic deracemization, finally furnishing high ee values after multiple cycles. Reactions performed on 100 mL allowed access to 50–60 mg of isolated products.160
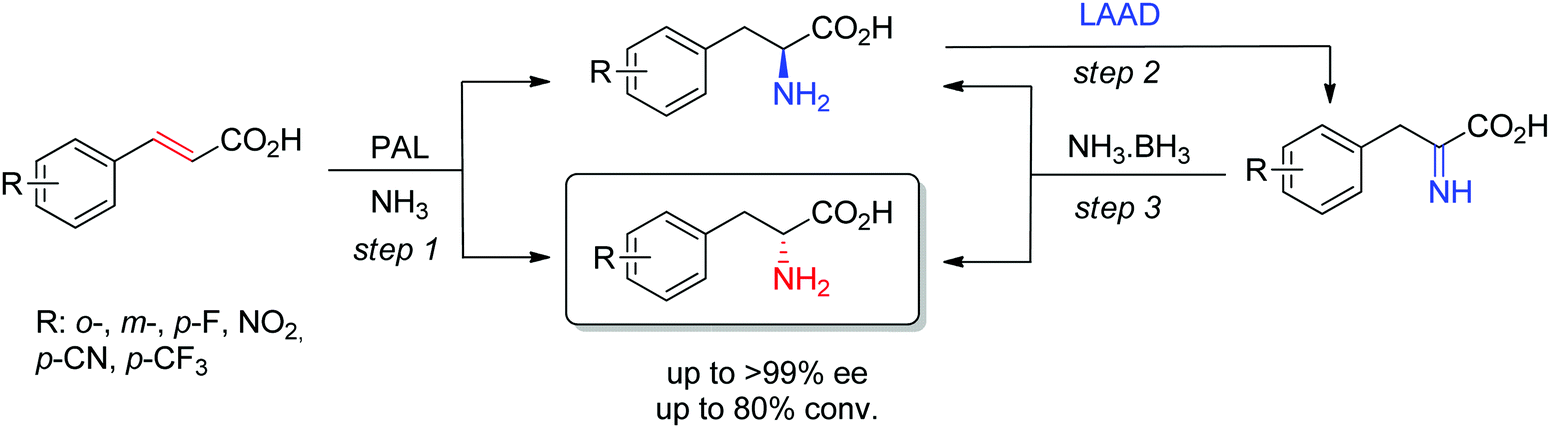 | ||
| Scheme 17 Access to D-arylalanines in a cascade through enzymatic nonselective hydroamination and cyclic deracemization combining enantioselective oxidation and chemical reduction (LAAD: L-selective amino acid deaminase, PAL: phenylalanine ammonia lyase).160 | ||
In selected cases, primary amines are also accepted in place of ammonia, as was shown in the synthesis of N-substituted L-aspartic acids using for instance mutated methylaspartate ammonia lyase on fumarate.161–163
Asymmetric direct β-hydroamination of α,β-unsaturated carboxylic acids by ammonia lyases was made possible through computational redesign of aspartate ammonia lyase AspB from Bacillus sp. YM55-1. The natural enzyme was highly specific for the dicarboxylic acid substrate fumarate, respectively for aspartate in the deamination direction. By employing the algorithm Rosetta Enzyme Design initially developed for de novo protein design,164 the requirement for a second carboxylic acid moiety could be abolished. The best resulting variants successfully transformed a range of substituted acrylates into the corresponding aliphatic and aromatic β-amino acids with high stereoselectivity. The reaction was run on a kg-scale at 300 g L−1 concentration of crotonic acid at pH 9 and 55 °C and selectively yielded (R)-β-aminobutanoic acid with 99% conversion and >99% ee (Scheme 18).165
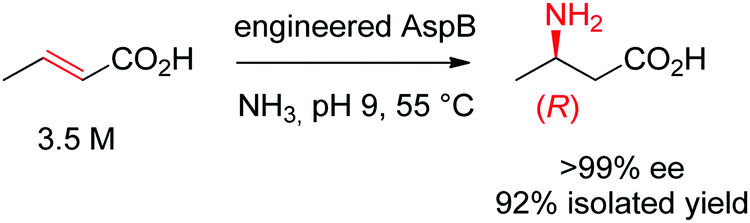 | ||
| Scheme 18 Hydroamination of crotonic acid to (R)-β-aminobutanoic acid by an engineered aspartate ammonia lyase AspB from Bacillus sp. YM55-1.165 | ||
By combining several enzymes in a one-pot cascade, a formal hydroamination of nonactivated alkenes was developed. The sequence epoxidation–isomerization–transamination was achieved with α-methyl styrene by the action of styrene monooxygenase, styrene oxide isomerase and transaminase. The three enzymes, plus alanine dehydrogenase used to drive the amination reaction, were co-expressed heterologously in E. coli, and the resulting biocatalyst employed as whole cells could catalyze the formal anti-Markovnikov selective formation of (S)-2-phenylpropan-1-amine in up to 97% ee.166 This elegant approach demonstrates the maturity of the field, since formal reactions can now be achieved in one pot by smartly combining individual enzymes in cascade reactions.
1.2.2.1. Alkene epoxidation. A range of enzymes are available to catalyze the asymmetric cis-epoxidation of C
C-double bonds via formal insertion of oxygen (Scheme 19). The reaction is particularly valuable as the oxirane ring may be opened through various (nucleophilic) substitution reactions, including enzymatic routes, thereby offering access to hydroxy-functionalized compounds with up to two chiral centers. The stereoselective attack on one face of the CC-bond requires oxygen activation, and this is achieved by diverse cofactor-dependent enzymes, such as flavin-dependent monooxygenases, iron-dependent nonheme monooxygenases and iron-heme dependent monooxygenases (P450 enzymes).167 Exception are heme-thiolate (fungal) peroxygenases, which use hydrogen peroxide directly as oxidant.168 The substrate spectrum depends heavily on the type of biocatalysts.
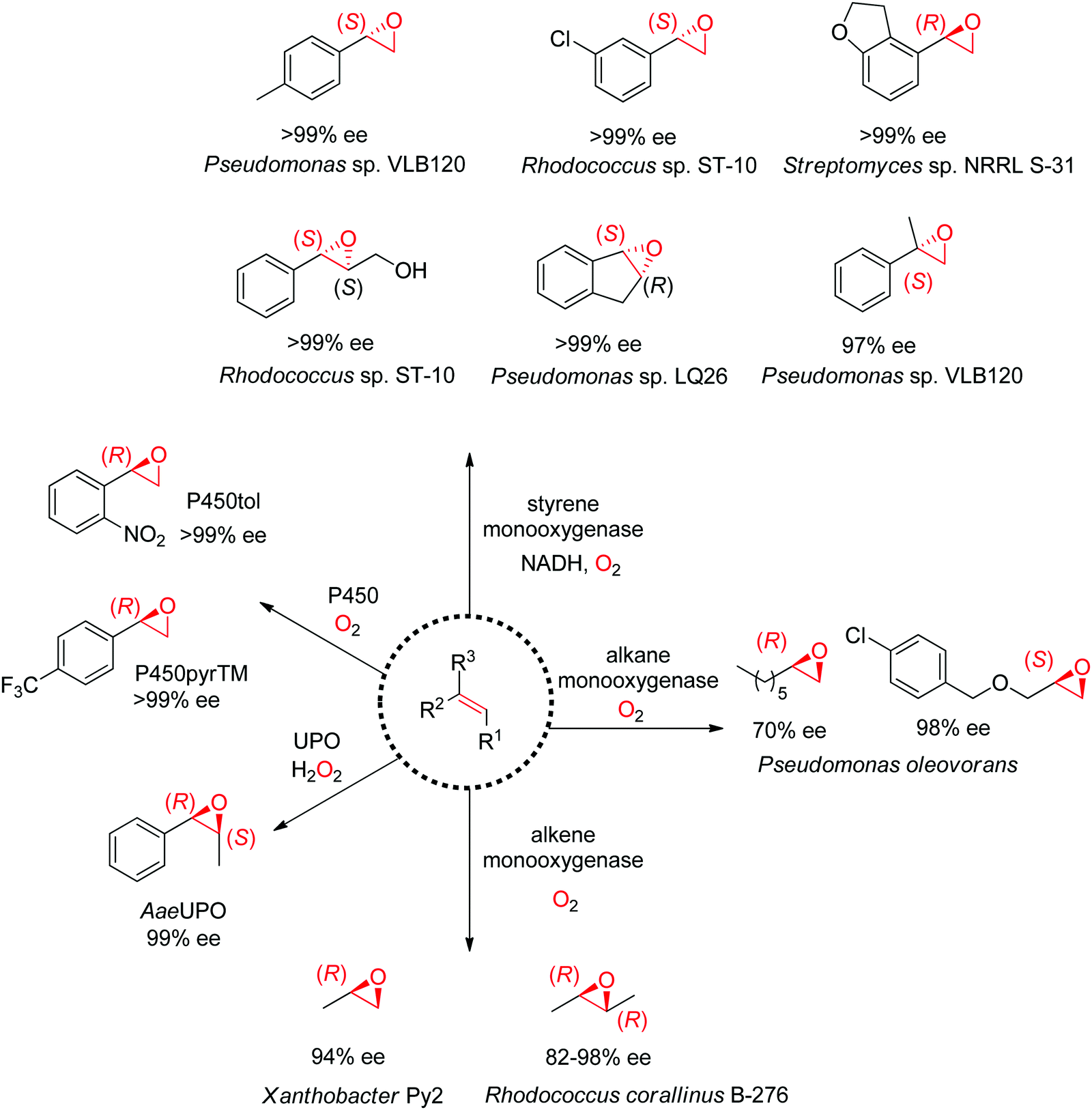 | ||
| Scheme 19 Enzymatic platform for the stereoselective epoxidation of alkenes and examples of products formed (UPO: unspecific peroxygenase). P450pyrTM: triple mutant of P450 monooxygenase from Sphingomonas sp. HXN-200, P450tol: P450 monooxygenase from Rhodococcus coprophilus TC-2, AaeUPO: UPO from Agrocybe aegerita. | ||
Despite being some of the most widely applied systems in enzymatic epoxidation reactions, styrene monooxygenases are not simple biocatalysts. Considered a prototype enzyme of this class of flavoenzymes, styrene monooxygenase from Pseudomonas sp. is composed of two components: the NADH-dependent flavin reductase StyB catalyzes the necessary activation of the flavin of the FAD-dependent epoxidase StyA through reduction with nicotinamide. The reduced FAD then transfers to StyA, where it reacts with molecular oxygen to generate a C(4a)-peroxyflavin intermediate, which can attack the alkene substrate, usually in a highly stereoselective manner.169 To simplify the reaction set-up, whole cells of E. coli recombinantly expressing the two components are employed and provide sufficient amount of NADH for the initial reductive step, while air serves as the source of oxidant. Poorly functionalized small molecules are best accepted, a clear advantage compared to traditional chemical methods, and typically yield (S)-epoxides. A range of α- and β-substituted styrenes, as well as terminal and cyclic alkenes, have been successfully epoxidized with the StyA/StyB system. In addition to substituted styrenes, 1-phenylethenes are good substrates for styrene monooxygenases (Scheme 19).170–172 Exquisite enantioselectivity has been observed on racemic substituted allylic alcohols, which, combined to the perfect stereoselectivity of the epoxidation, led to a kinetic resolution toward the formation of oxirane-containing secondary alcohols with excellent de values. E values were in several cases >200 (Scheme 20).173
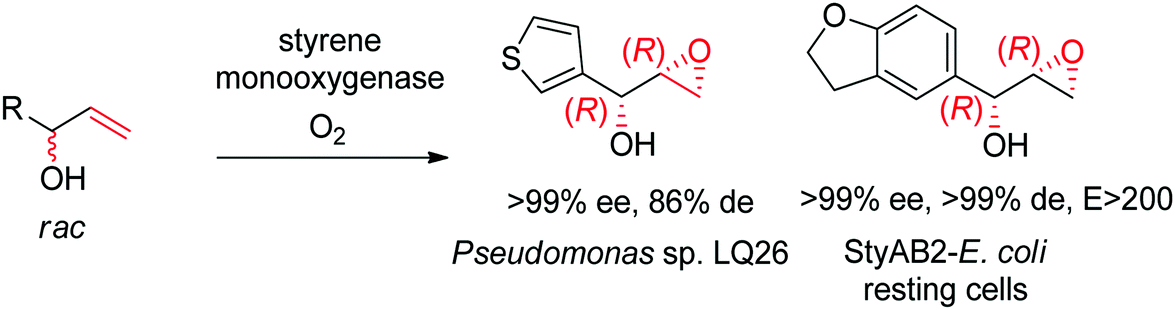 | ||
| Scheme 20 Kinetic resolution of racemic allylic alcohols by styrene monooxygenase-catalyzed epoxidation.173 | ||
Biocatalytic cascades initiated by styrene monooxygenase-catalyzed epoxidation reaction have allowed the functionalization of styrenes beyond epoxidation and generated in one pot chiral α-hydroxy acids and α- and β-amino acids.174,175
An exception to the (S)-selective styrene monooxygenases was recently identified with the characterization of an (R)-selective styrene monooxygenase from Streptomyces sp. Only the oxygenase component A could be identified through genome mining, and when coupled to the reductase component B from Pseudomonas sp., it showed activity on a few styrene derivatives, which were all converted to the corresponding (R)-epoxides with ee values up to >99%.176 The primary sequence of this protein was then used to perform searches in protein databases and led to the identification of eight (R)-selective styrene monooxygenases with enlarged substrate scope.177 Notably, from a phylogeny perspective, the (R)-selective enzymes appear grouped in a clade away from the (S)-selective ones.
Nonheme monooxygenases that depend on iron include membrane-bound xylene monooxygenases178,179 – with activity pattern comparable but not superior to that of styrene monooxygenases – and alkane and alkene monooxygenases (Scheme 19). Alkane monooxygenase from Pseudomonas oleovorans, which also catalyzes the hydroxylation of alkanes, contains three components – the nonheme iron ω-hydroxylase and the two-component electron-transport chain system – and is thus practically employed as whole-cell biocatalyst. The enzyme works well with linear terminal alkenes. (R)-Epoxides are usually obtained.180–182 Alkene monooxygenases are also multi-component biocatalysts and preferentially take short-chain alkenes as substrates.183,184
Catalytically versatile P450 monooxygenases rely on an iron(IV)-oxo porphyrin-cation-radical species (so-called ‘compound I’) for the oxygen atom transfer. The substrate spectrum is broad and includes diverse molecules such as terminal alkenes, unsaturated fatty acids and styrene derivatives. The major limitation is common to catalysis by P450s: the low substrate concentration tolerance limits their applications in preparative synthesis. Importantly, P450s are usually stereocomplementary to styrene monooxygenases and (R)-styrene epoxides are obtained. Several P450-based systems have been disclosed. For instance, natural P450tol showed exquisite stereoselectivity on a variety of styrenes,185 while P450pyr could be specifically engineered to deliver (R)-styrene epoxides with excellent enantiopurity (Scheme 19).186 The engineered triple mutant could also effectively convert ortho-substituted styrenes with (S)-selectivity, 1,1-disubstituted alkenes as well as cyclic alkenes. Lately, an engineered P450BM3 employed in the presence of a dual functional small molecule could produce (R)-styrene epoxide in the peroxygenase mode.187
Systems based on heme-dependent unspecific peroxygenases (UPOs) that utilize hydrogen peroxide as oxidant are currently emerging.188,189 A few of these enzymes mostly originating from fungi have been well characterized and include UPOs from Agrocybe aegerita (AaeUPO),190Coprinellus radians (CraUPO)191 and Marasmius rotula (MroUPO).192 A major attractive feature of these biocatalysts is their catalytic efficiency, translating into high TTN values (up to 110000), coupled with exquisite stereoselectivity, as seen in the cis-epoxidation of styrene derivatives (Scheme 19).193 As with P450 enzymes, the tolerance to high substrate concentration remains poor, which currently prevents their use in large scale applications. Finally, the heme center is poorly compatible with the oxidant; this problem however can be partly circumvented by in situ H2O2 production strategy.194
Lastly, the engineering of 4-oxalocrotonate tautomerase (4-OT) delivered remarkable catalytic promiscuity. The designed protein could accept different (organic) hydroperoxides to catalyze the epoxidation of a range of α,β-unsaturated aldehydes. As in the case of the Michael addition of nitromethane on α,β-unsaturated aldehydes catalyzed by 4-OT195 (see Section 1.3.2.1), the epoxidation reaction was suggested to proceed via an enzyme-bound iminium intermediate. Stereocomplementary versions using 4-OT displaying three mutations were obtained by changing the oxidant from t-BuOOH for attack on the Re-face, to hydrogen peroxide for attack on the Si-face of the alkene, and granted access to epoxides with ee values up to 98%; diastereoselectivity was in most cases good to very good (Scheme 21).196 In both cases, the reaction was scalable and was combined with a chemical reduction by NaBH4 to deliver the corresponding epoxy-alcohols.
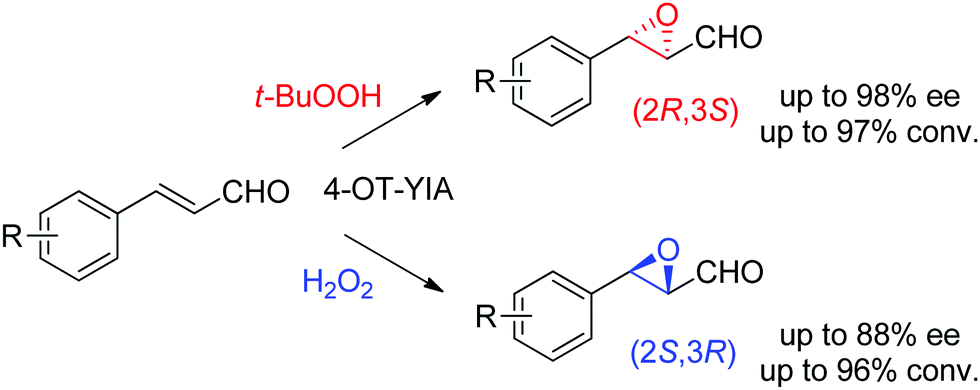 | ||
| Scheme 21 Stereocomplementary epoxidation of α,β-unsaturated aldehydes via switch of oxidant with a triple variant of 4-oxalocrotonate tautomerase (4-OT).196 | ||
1.2.2.2. Alkene hydration. A few enzymes can be employed to catalyze the asymmetric hydration of alkenes, a challenging reaction for chemists. Unactivated alkenes can be converted to the corresponding alcohols by linalool dehydratase isomerase, carotenoid-1,2-hydratases or fatty acid hydratases, however, many of these hydratases are mostly active on a small subset of (natural) substrates.197 The oleate hydratase from Elizabethkingia meningoseptica was engineered to catalyze the stereoselective hydration of terminal and internal alkenes devoid of a carboxylic acid group. In the presence of a carboxylic acid decoy molecule, a range of secondary (S)-alcohols were obtained with excellent stereoselectivity (up to 99% ee).198
Cofactor independent ferulic acid decarboxylase (FDC) is broadly defined as a phenolic acid decarboxylase. The enzyme catalyzes the reverse decarboxylation of phenolic acids, a feature which has been exploited in biocatalysis for carboxylation reactions of nonnatural substrates.199 Catalytic promiscuity of FDC from Enterobacter sp. could be identified in the asymmetric hydration of hydroxystyrene derivatives.200,201 A range of (S)-4-(1-hydroxyethyl)phenols were obtained with ee values up to 71% and conversion up to 82%.
1.3. C–C-Bond formation
Several enzymatic strategies are available for asymmetric C–C-bond forming reactions202,203 and involve attack of a reactive (nucleophilic or radical) intermediate species onto a sp2 hybridized carbon atom, either within a CO- or CC-double bond (Scheme 1). The nucleophiles are diverse and include enolate/enamine, umpoled carbonyl carbon, cyanide and nitroalkane. Examples with more specialized enzymes have been reviewed.204,205
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) O-bond.
The field of biocatalytic C–C-bond forming reactions using carbonyl as electrophile has long been dominated by lyases and aldolases for the synthesis of α- and β-hydroxy carbonyl derivatives, respectively (Scheme 22).206,207 Although both enzymes couple two carbonyl compounds together, they use very distinct catalytic machinery to that end, which explains the different nature of the products formed. Lyases rely on the thiamine diphosphate (ThDP) cofactor to perform an astonishing Umpolung reaction with the donor substrate, usually an aldehyde, but certain enzymes employ α-ketoacids such as pyruvate in decarboxylative carboligation reactions. The now nucleophilic carbonyl atom attacks the acceptor, (almost) invariably an aldehyde, yielding α-hydroxy ketones (acyloins). Aldolases, on the other hand, can activate the α-carbon of ketone donors via two distinct mechanisms:208,209 type I aldolases make use of a catalytic lysine to generate a reactive enamine intermediate, while type II aldolases require Zn2+ as a Lewis acid to activate the (often α-hydroxy functionalized) ketone donor via an enolate. The electrophilic acceptor was until recently always an aldehyde (vide infra).
O-bond.
The field of biocatalytic C–C-bond forming reactions using carbonyl as electrophile has long been dominated by lyases and aldolases for the synthesis of α- and β-hydroxy carbonyl derivatives, respectively (Scheme 22).206,207 Although both enzymes couple two carbonyl compounds together, they use very distinct catalytic machinery to that end, which explains the different nature of the products formed. Lyases rely on the thiamine diphosphate (ThDP) cofactor to perform an astonishing Umpolung reaction with the donor substrate, usually an aldehyde, but certain enzymes employ α-ketoacids such as pyruvate in decarboxylative carboligation reactions. The now nucleophilic carbonyl atom attacks the acceptor, (almost) invariably an aldehyde, yielding α-hydroxy ketones (acyloins). Aldolases, on the other hand, can activate the α-carbon of ketone donors via two distinct mechanisms:208,209 type I aldolases make use of a catalytic lysine to generate a reactive enamine intermediate, while type II aldolases require Zn2+ as a Lewis acid to activate the (often α-hydroxy functionalized) ketone donor via an enolate. The electrophilic acceptor was until recently always an aldehyde (vide infra).
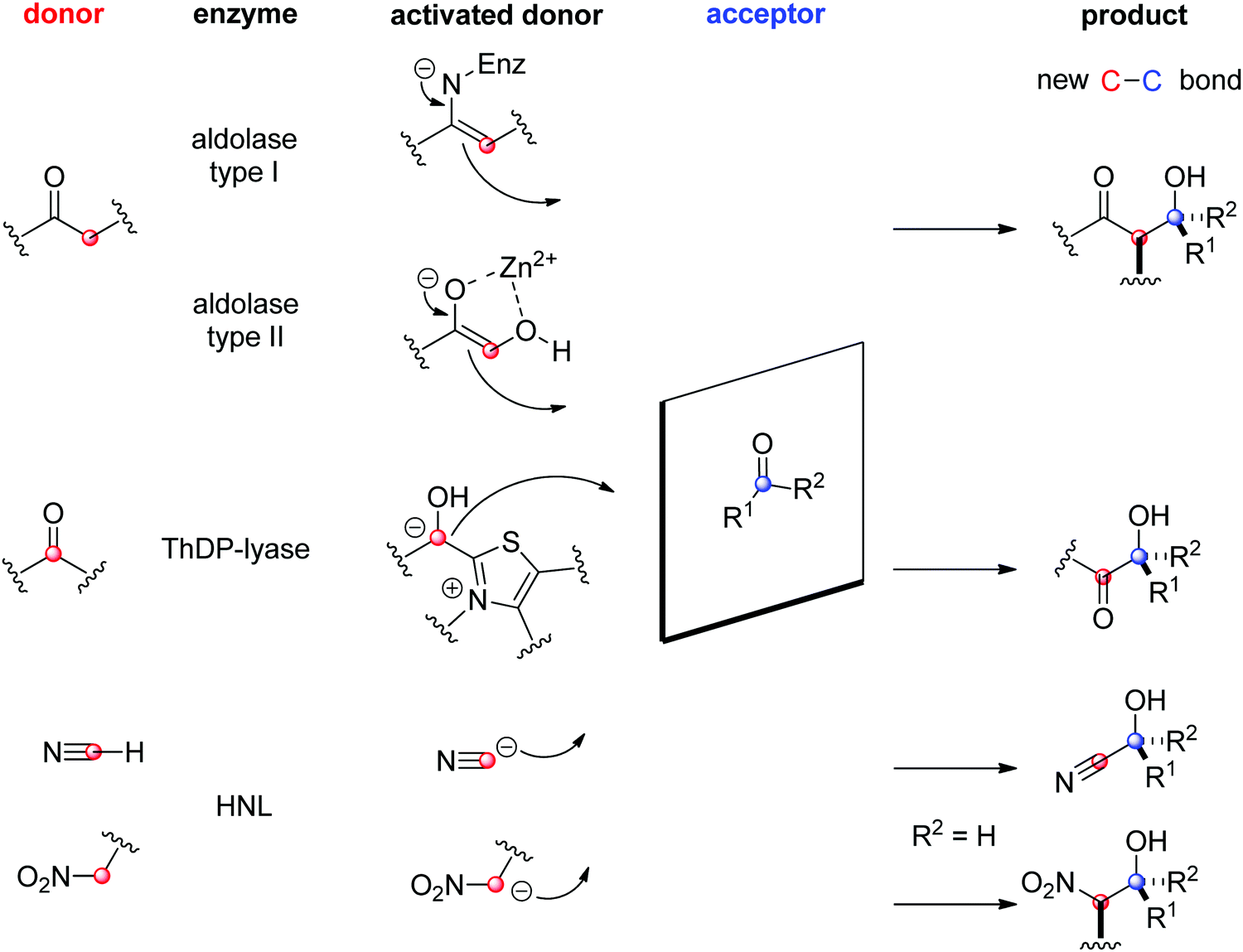 | ||
| Scheme 22 Enzymatic strategies for stereoselective C–C-bond formation with a carbonyl acceptor (HNL: hydroxynitrile lyase, ThDP: thiamine diphosphate). | ||
The asymmetric synthesis of tertiary alcohols through C–C-bond forming reactions appeared possible with the identification of a ThDP-dependent enzyme that could accept ketones as acceptors. YerE from Yersinia pseudotuberculosis showed high stereoselectivity in the reaction of pyruvate with cyclic and open-chain ketones as well as ketoesters.210 More recently, highly selective access to tertiary alcohols could be granted by aldolases. Rhamnulose-1-phosphate aldolase (RhuA) from Bacteroides thetaiotaomicron employed dihydroxyacetone phosphate as nucleophile. The reactions with hydroxy functionalized ketones proceeded with high stereo- and diastereoselectivity to form the final products with two chiral centers.211 Later, and following previous indications that such reaction was indeed possible,212,213 a pyruvate aldolase was confirmed to accept ketones as electrophile with high stereoselectivity.214
Recently, biocatalytic formation of quaternary stereocenters by aldol addition of 3,3-disubstituted 2-oxoacid derivatives to aldehydes was demonstrated using metal-dependent 3-methyl-2-oxobutanoate hydroxymethyltransferase (KPHMT) that functioned as a type II aldolase. With some chiral nucleophiles, the reaction was enantioselective. The authors suggested that the formation of the (E)-enolate intermediate reactive species was favored, while the attack of the aldehyde was stereoselective, eventually yielding stereopure quaternary carbons. In the case of aldehydes other than formaldehyde, an additional chiral center was formed.215
Hydroxynitrile lyases (HNLs) have been investigated for a long time as catalysts in cyanohydrin formation reactions. An aldehyde or a ketone is being stereoselectively attacked by cyanide as the nucleophile and forms chiral α-hydroxynitriles. Stereocomplementary enzymes exist: (R)-selective HNLs are predominantly found in cyanogenic plants, including several Prunus species (such as those popularly known as apricot, plum, cherry and almond),216,217 or (noncyanogenic) weed plant Arabidopsis thaliana.218 One of the most investigated (S)-selective counterparts was identified in the rubber tree (Hevea brasiliensis).219 The substrate scope is broad and highly dependent on the enzyme.220
A major addition to the reaction portfolio of HNLs was the development of a biological Henry reaction, in which nitroalkanes react as nonnatural nucleophiles with aldehydes to generate (S)-β-nitro alcohols (Scheme 23). The first example was demonstrated with nitromethane and high ee values for the products were obtained (92% ee in the case of benzaldehyde). Higher ee values could be obtained at acidic pH values (pH 5.5). With nitroethane, two chiral centers are generated, however despite high stereoselectivity in the nucleophilic attack of the CO-double bond (95% ee), the anti-diastereoselectivity on benzaldehyde was only moderate (80% de).221,222 An (R)-stereocomplementary homologue was identified with HNL from Arabidopsis thaliana, which accepted nitromethane as donor and a range of substituted aldehydes as acceptors.223
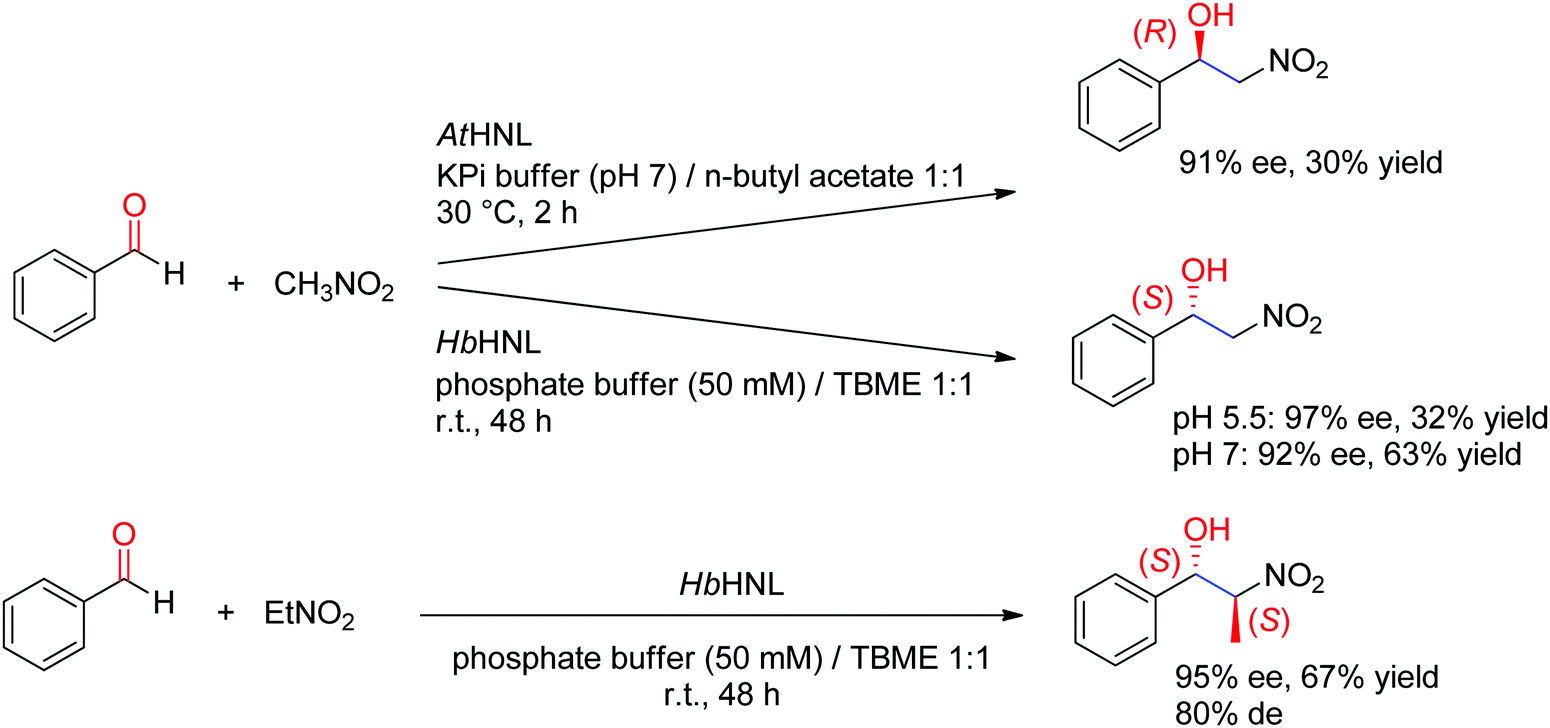 C-bond.
C-bond.
1.3.2.1. Activated alkenes (Michael addition). Enzymatic asymmetric versions of the so-called Michael addition – the nucleophilic attack of a C
C-double bond within an activated alkene – include different scenarios, depending on the nature of both the nucleophilic donor and the electrophilic acceptor (Scheme 24).
 | ||
| Scheme 24 Enzymatic strategies for stereoselective C–C-bond formation with an alkene acceptor (4-OT: 4-oxalocrotonate tautomerase, HAT: hydrogen atom transfer, OYE: Old Yellow Enzyme, ThDP: thiamine diphosphate). | ||
With ThDP-dependent lyases, an exception in the type of acceptor generally accepted (see Section 1.3.1) was discovered, and examples of a biological Stetter reaction were identified with the decarboxylative Michael addition of pyruvate onto α,β-unsaturated ketones. PigD from Serratia marcescens for instance catalyzed the 1,4-addition of pyruvate on a range of aliphatic and (hetero)aromatic α,β-unsaturated ketones with perfect chemo- and stereoselectivity to produce 2-substituted 1,4-diketones. Besides pyruvate, also 2-oxobutanoate was accepted as donor.224,225 An intramolecular nondecarboxylative version was developed with a benzaldehyde lyase and allowed the generation of chiral chroman-4-one derivatives with up to 98% ee.226
The chemically related Michael addition of acetaldehyde onto nitroalkenes was shown possible with 4-oxalocrotonate tautomerase (4-OT), an enzyme that relies on an N-terminal catalytic proline to activate the aldehyde (donor) substrate via enamine chemistry (Scheme 25). The products were obtained in high enantiopurity227 and the stereoselectivity could be controlled by substrate engineering, allowing access to both (R)- and (S)-γ-nitroaldehydes.228 Protein engineering also turned useful for the design of stereocomplementary biocatalysts.229 For donors other than acetaldehyde, a second chiral center was installed with high diastereoselectivity (up to 94% de). Finally, a smartly designed swap of functional groups along with a key mutation in 4-OT later allowed the reaction of nitroalkanes onto α,β-unsaturated aldehydes, and, in the case of nitromethane and cinnamaldehyde, elegantly provided stereocomplementary access to the γ-nitroaldehyde product (compared to the addition of acetaldehyde onto trans-β-nitrostyrene).195 Here, the unsaturated aldehyde acceptor was suggested to be activated by the proline via an iminium ion intermediate. Chemically analogous, a cupin loaded with copper was shown able to catalyze the asymmetric addition of nitromethane to azachalcones. Highest activity and stereoselectiviy were obtained after protein engineering and a single mutation was sufficient to obtain excellent values (up to 99% ee for the (S)-product with H52A).230
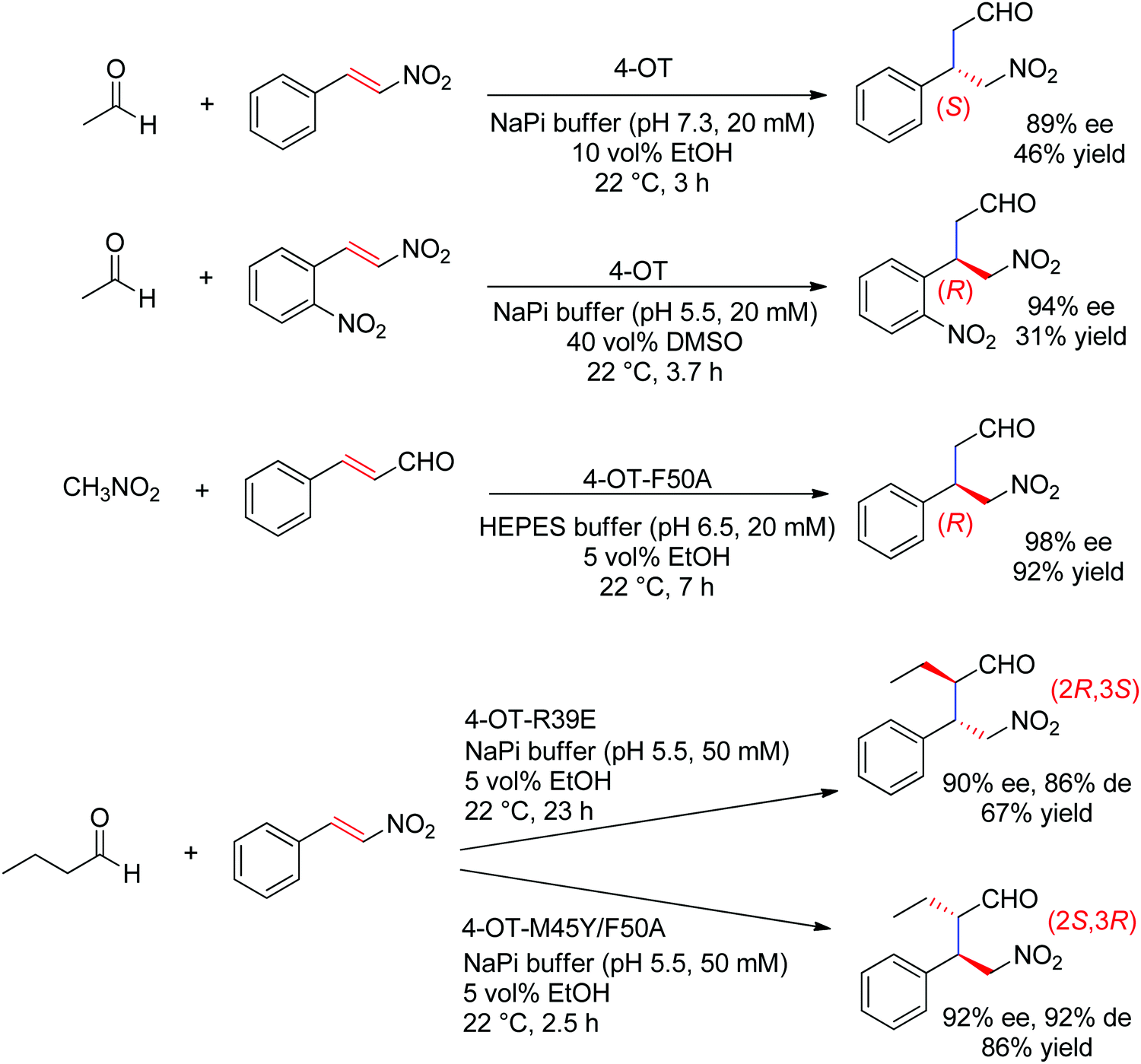 | ||
| Scheme 25 Examples of stereocomplementary strategies in 4-oxalocrotonate tautomerase (4-OT)-catalyzed Michael addition reactions.195,227–229 | ||
1.3.2.2. Nonactivated alkenes. A few cases of enzymatic asymmetric C–C-bond forming reactions with nonactivated prochiral alkenes as electrophile have been reported and involve the catalytic promiscuity of enzymes that otherwise catalyze completely different chemical reactions.
Following the discovery of catalytic promiscuity of the cofactor independent ferulic acid decarboxylase (FDC) from Enterobacter sp. in asymmetric hydration reactions (see Section 1.2.2.2),201 a range of diverse nucleophiles was tested and demonstrated the ability of FDC to also form new C–N-, C–S- and C–C-bonds. The reaction with 4-hydroxystyrene was shown to proceed via asymmetric 1,6-conjugate addition of the nucleophile. The synthetically relevant addition of cyanide onto 4-vinylphenol delivered (S)-configurated 2-(4-hydroxyphenyl)propanenitrile in good yield and with 85% ee. Other phenolic acid decarboxylases were active, but despite higher stereoselectivity (up to 91% ee), conversions remained modest.231
A few creative C–C-bond forming protocols involving Old Yellow Enzymes have been developed, in which, following reduction by NAD(P)H, a key flavin radical semiquinone was obtained by photoexcitation. Under light irradiation, α-chloroamide derivatives underwent a reaction similar to the dehalogenation of α-bromoesters catalyzed by OYEs (see Section 2.1.3). The structure of the substrates was chosen such that cyclization occurred before the hydrogen atom transfer, thanks to a reactive CC-bond within the molecule. A range of enantiopure lactams with diverse substitution pattern could be generated through this radical dehalogenation/stereoselective intramolecular C–C-bond forming protocol. Although up to two chiral centers could be formed, in that case diastereoselectivity was poor (<60% de).232
More recently, a case of stereoselective formal intermolecular hydroalkylation of nonactivated alkenes catalyzed by Old Yellow Enzymes was reported by the Zhao lab (Scheme 26).233 In that case too, photoexcitation was crucial to initiate the radical reaction with α-halogenated carbonyl compounds. Overall, the reaction can be seen as the intermolecular equivalent of the cyclization developed with α-chloroamides (see above). The Hyster group later reported the same reaction with α-chloroamides devoid of CC-double bonds, however stereoselectivity was on average less pronounced.234 In both cases, a γ-chiral center was generated and did not result from the radical C–C-bond forming step but from the final hydrogen atom transfer (HAT) from the flavin semiquinone.
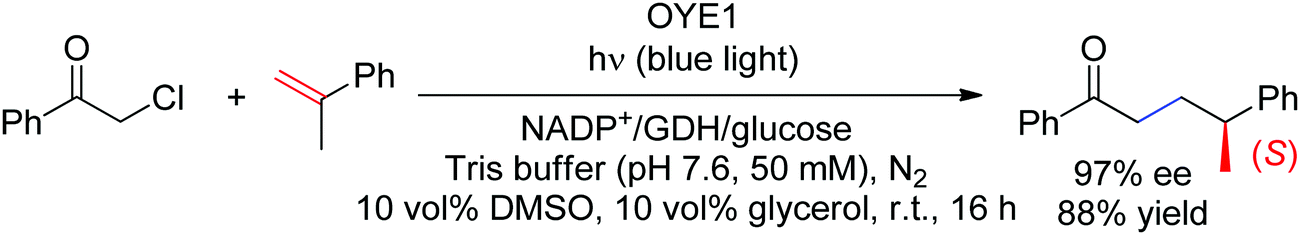 | ||
| Scheme 26 Asymmetric intermolecular hydroalkylation of nonactivated alkenes catalyzed by Old Yellow Enzyme under light irradiation (GDH: glucose dehydrogenase, OYE1: Old Yellow Enzyme from Saccharomyces pastorianus).233 | ||
Other asymmetric biocatalytic C–C-bond forming reactions exist, many derived from biosynthetic pathways. While these complementary methods are powerful approaches, they tend to be specific for particular substrate templates. They often involve a cyclization reaction (some are fully intramolecular) and will not be covered here.205,235
2. Enantioselective reactions
2.1. Conversion of prochiral carbons into chiral centers
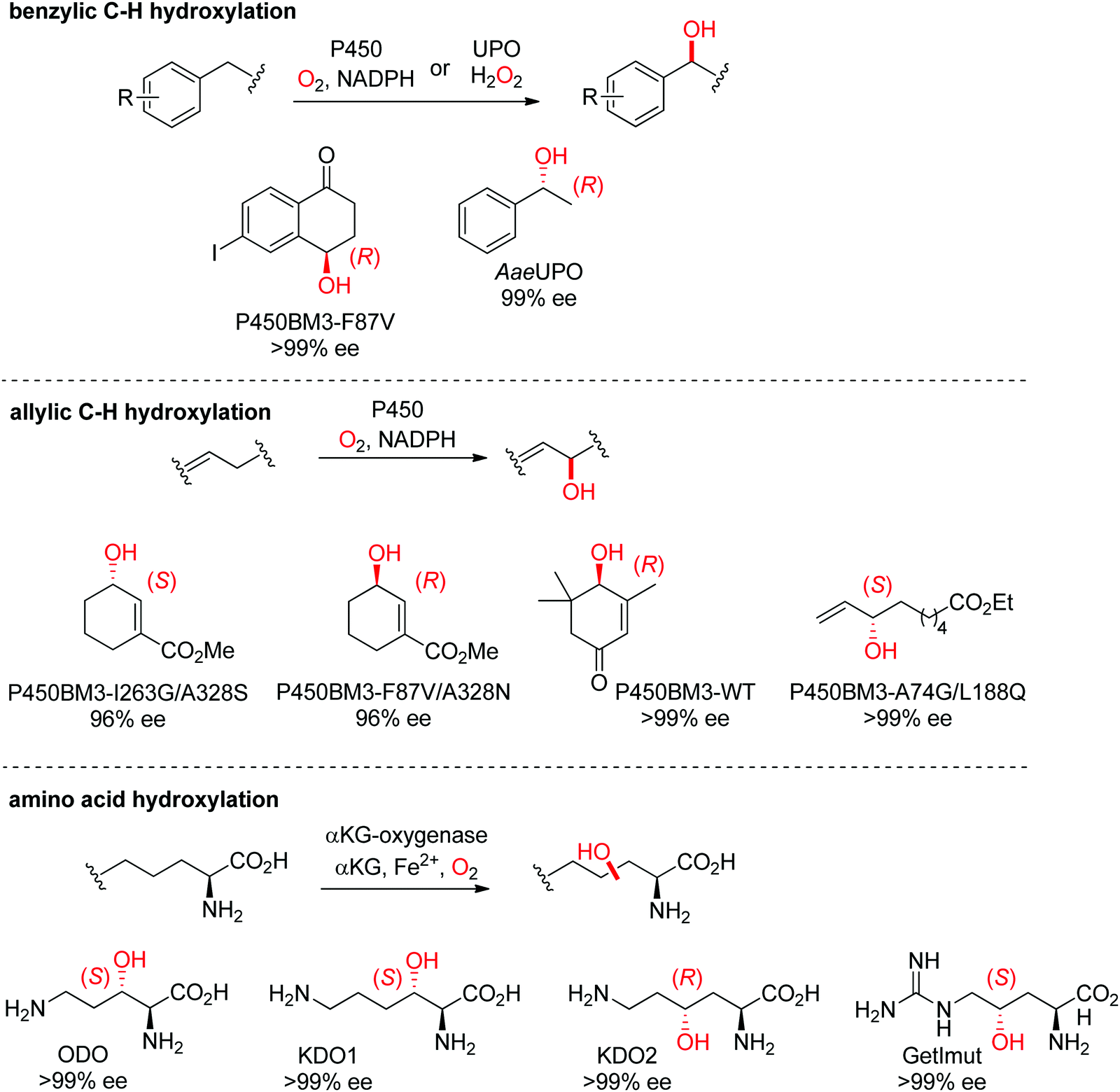 | ||
| Scheme 27 Range of hydroxylated products obtained by enantioselective C–H hydroxylation reactions catalyzed by cytochrome P450s, unspecific peroxygenases (UPOs) and α-ketoglutarate and nonheme iron-dependent oxygenases. α-KG: α-ketoglutarate, AaeUPO: UPO from Agrocybe aegerita,237 KDO1: L-lysine dioxygenase from Catenulispora acidiphila,239 KDO2: L-lysine dioxygenase from Chitinophaga pinensis,239 GetImut: engineered citrulline 4-hydroxylase from Streptomyces sp. L-49973,241 ODO: L-ornithine dioxygenase from Catenulispora acidiphila,239 P450BM3: P450 monooxygenase from Bacillus megaterium.242,244–246 | ||
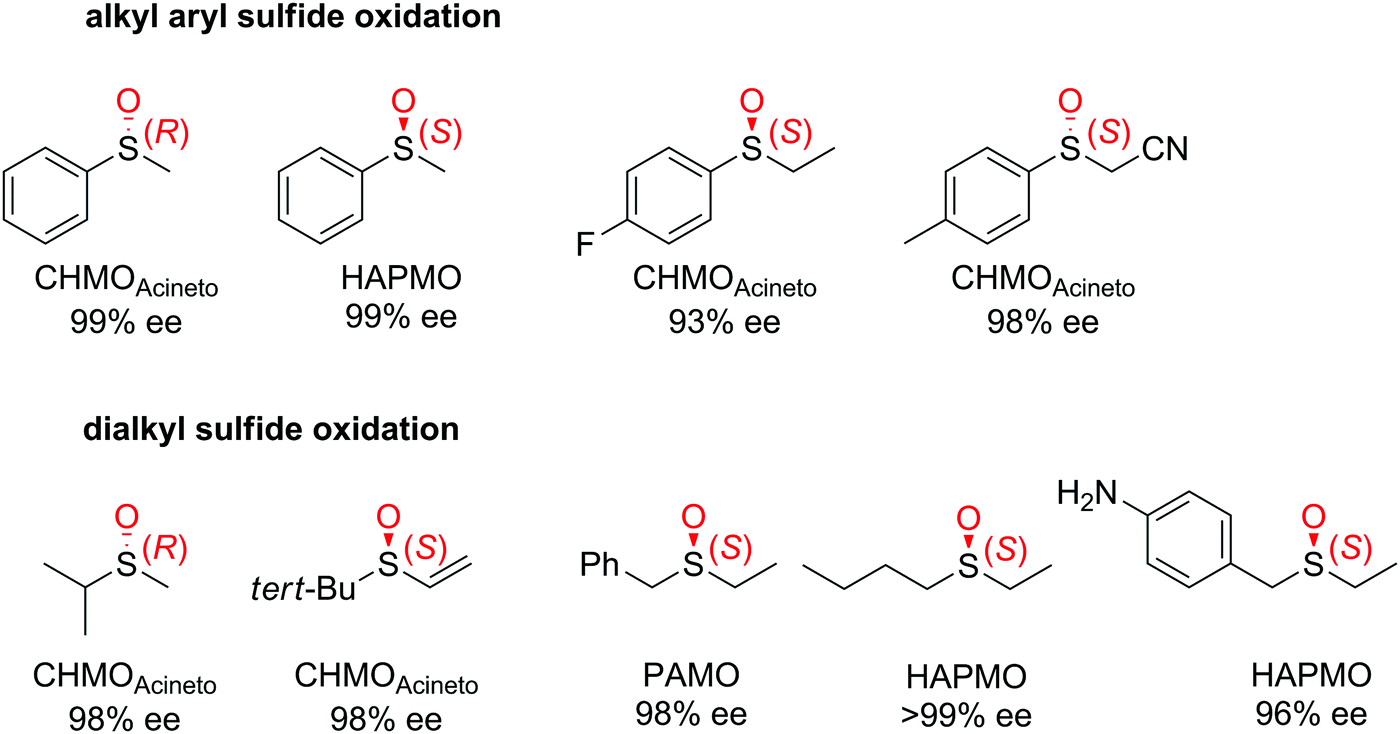 | ||
| Fig. 3 Range of chiral sulfoxides obtained by enantioselective sulfoxidation of alkyl aryl sulfides.262–264 CHMOAcineto: cyclohexanone monooxygenase from Acinetobacter calcoaceticus NCIMB 9871, HAPMO: 4-hydroxyacetophenone monooxygenase from Pseudomonas putida JD1, PAMO: phenylacetone monooxygenase from Thermobifida fusca. Reprinted from ref. 252 Copyright 2020, with permission from Elsevier. | ||
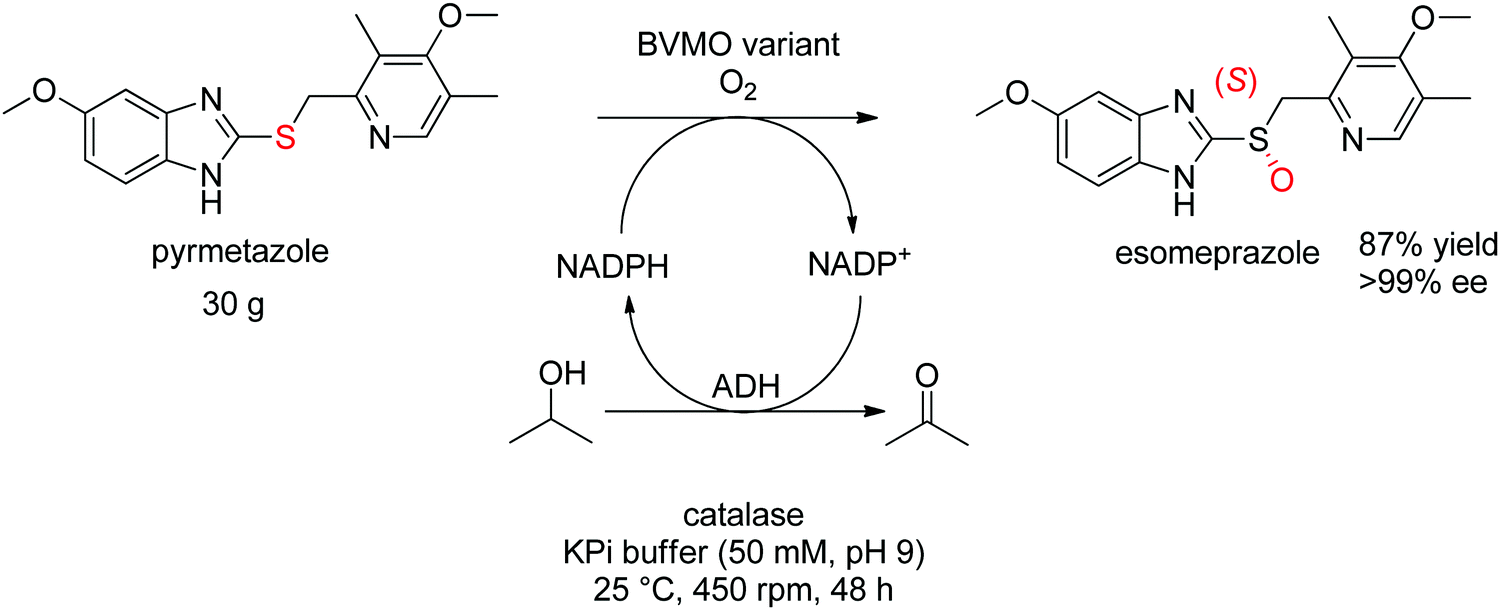
For more challenging substrates, directed evolution was successfully applied. An engineered BVMO developed by Codexis was employed in the asymmetric oxidation of pyrmetazole, a precursor to the drug esomeprazole used in the treatment of stomach acid reflux. The (S)-enantiomer of the target product could be obtained in 87% yield and in enantiopure form (>99% ee) from a reaction run on 30 g scale. The coupled-enzyme approach was selected for the regeneration of NADPH, and relied on isopropanol as reductant and an alcohol dehydrogenase (Scheme 28).
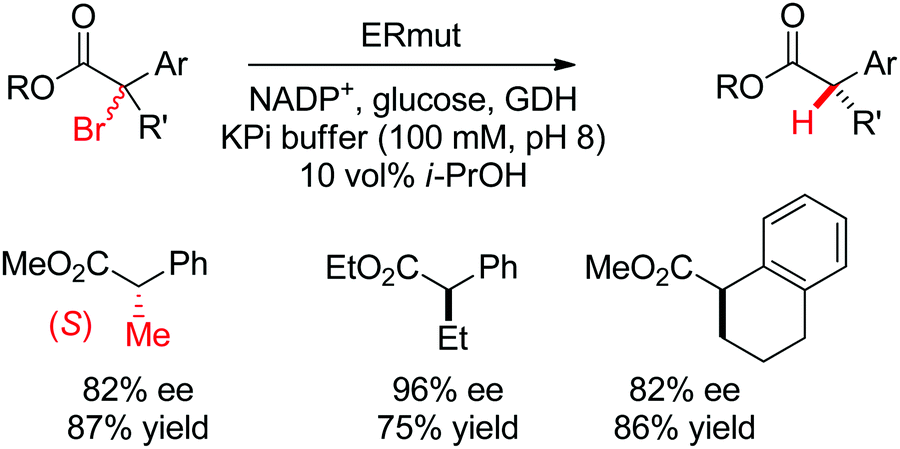 | ||
| Scheme 29 Enantioselective dehalogenation of α-bromoesters by a tyrosine mutant of ene-reductase from Gluconobacter oxydans (ERmut).266 GDH: glucose dehydrogenase. | ||
Despite recent and significant advances in the identification of enzymes active in halogenation reactions,267,268 the field of biocatalytic asymmetric halogenation remains highly underdeveloped.269 Recently, this area gathered particular attention as cases of enantioselective biohalogenation were reported.270,271 Most notably, the potential in biocatalysis of WelO5, a carrier-independent iron/α-ketoglutarate dependent halogenase reported to chlorinate regio- and enantioselectively 12-epi-fischerindole U, has been explored.272 Engineering of a highly similar homologue, WelO5*, led to variants with an enlarged substrate scope, albeit with low catalytic activity.273 In parallel, complementary engineering efforts on a homologous protein from Westiella intricata HT-29-1 by structure-guided directed evolution delivered enzymes with excellent chemo-, regio-, and diastereoselective chlorination activity on hapalindole derivatives.274 While promising, these enzymes will have to demonstrate generality to become potent tools in synthesis on a broad panel of nonnatural substrates at synthetically relevant concentration.275,276 In contrast, for the halogenation of aromatic molecules, several enzymatic protocols are available.269
2.2. (Dynamic) kinetic resolution
In cases where the chiral center is already in place in the starting material, the enantioselective transformation of racemic molecules into enantiopure compounds can be attained by several strategies that rely on enzymatic kinetic resolution.277 The underlying concept is that a biocatalyst, facing two enantiomers of the same substrate, will preferentially convert one enantiomer to the final product. In an ideal case, the reaction with the other enantiomer is so slow that it will not be observed on the timescale of the reaction and the final product is therefore obtained with high enantiopurity in a maximum 50% yield. 50% of the starting material is recovered in enantiopure form. When an in situ racemization step specific for the substrate is included, a maximum product yield of 100% can be reached since the nonreacting enantiomer is transformed through racemization into the preferred enantiomer that reacts with the biocatalyst of interest (dynamic kinetic resolution). The final product is obtained with high enantiopurity. Historical examples with lipases will not be treated here and the reader can consult the abundant literature for more information and examples.16–18,208,278,279 Instead, the focus will be briefly directed to recent examples of kinetic resolution and dynamic kinetic resolution with redox reactions, such as those involving α-substituted carbonyl compounds, which usually racemize spontaneously in (slightly basic) aqueous media.The kinetic resolution of secondary thiols was achieved by enantioselective oxidation using an engineered alcohol oxidase. While the wild-type enzyme was inactive, several variants of the enzyme HFMO (5-(hydroxymethyl)furfural oxidase) could catalyze the oxidation of (S)-1-arylethanethiols and in several cases, perfect kinetic resolution was obtained (E > 200), with conversions reaching max. theoretical yield of 50%. Enantiopure secondary (R)-thiols were obtained, along with acetophenone derivatives, which formed spontaneously by hydrolysis of the oxidized thioketone products (Scheme 30).280
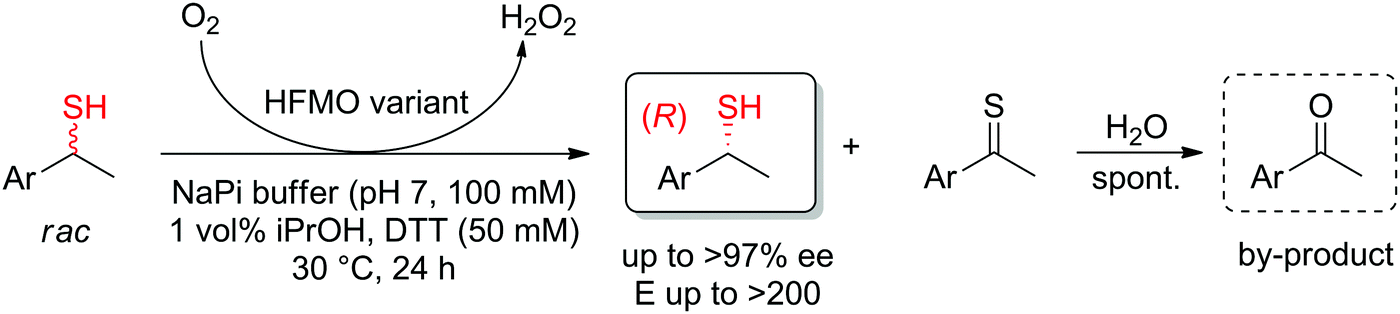 | ||
| Scheme 30 Kinetic resolution of sec-thiols through enantioselective oxidation catalyzed by variants of 5-(hydroxymethyl)furfural oxidase (HFMO).280 | ||
The light-driven kinetic resolution of α-amino and α-hydroxy carboxylic acids was made possible through (S)-selective decarboxylation catalyzed by the fatty acid photodecarboxylase from Chlorella variabilis NC64A (CvFAP). E values up to >200 could be achieved and highest turnover numbers (TON) were obtained with the single variant CvFAP-G462Y (up to ∼4000). The reaction conducted under irradiation at 450 nm at 20 °C was scaled up to 100 mg and allowed the isolation of (R)-2-hydroxyoctanoic acid in 45% yield and 99% ee, and of (R)-2-hydroxyhexanoic acid in 40% yield and 98% ee.281
Enantioselective Baeyer–Villiger monooxygenases (BVMOs) have been employed in the kinetic resolution of racemic α-substituted ketones and lactones.252,277 Owing to exquisite selectivity, E values >200 have been obtained with 2-substituted cyclopentanone and cyclohexanone derivatives, as well as with (aryl) aliphatic ketones.282 With particular compounds, cases of enantiocomplementary regiodivergent monooxygenation have been observed, in which the two enantiomers of a chiral substrate were converted to two regioisomeric products with high enantiopurity (Scheme 31A).283,284 The application of BVMOs under basic pH (9–10)285 or in combination with a weekly basic anionic exchange resin286 promoted the dynamic kinetic resolution of α-alkylated ketones through in situ racemization of the substrates (Scheme 31B).287
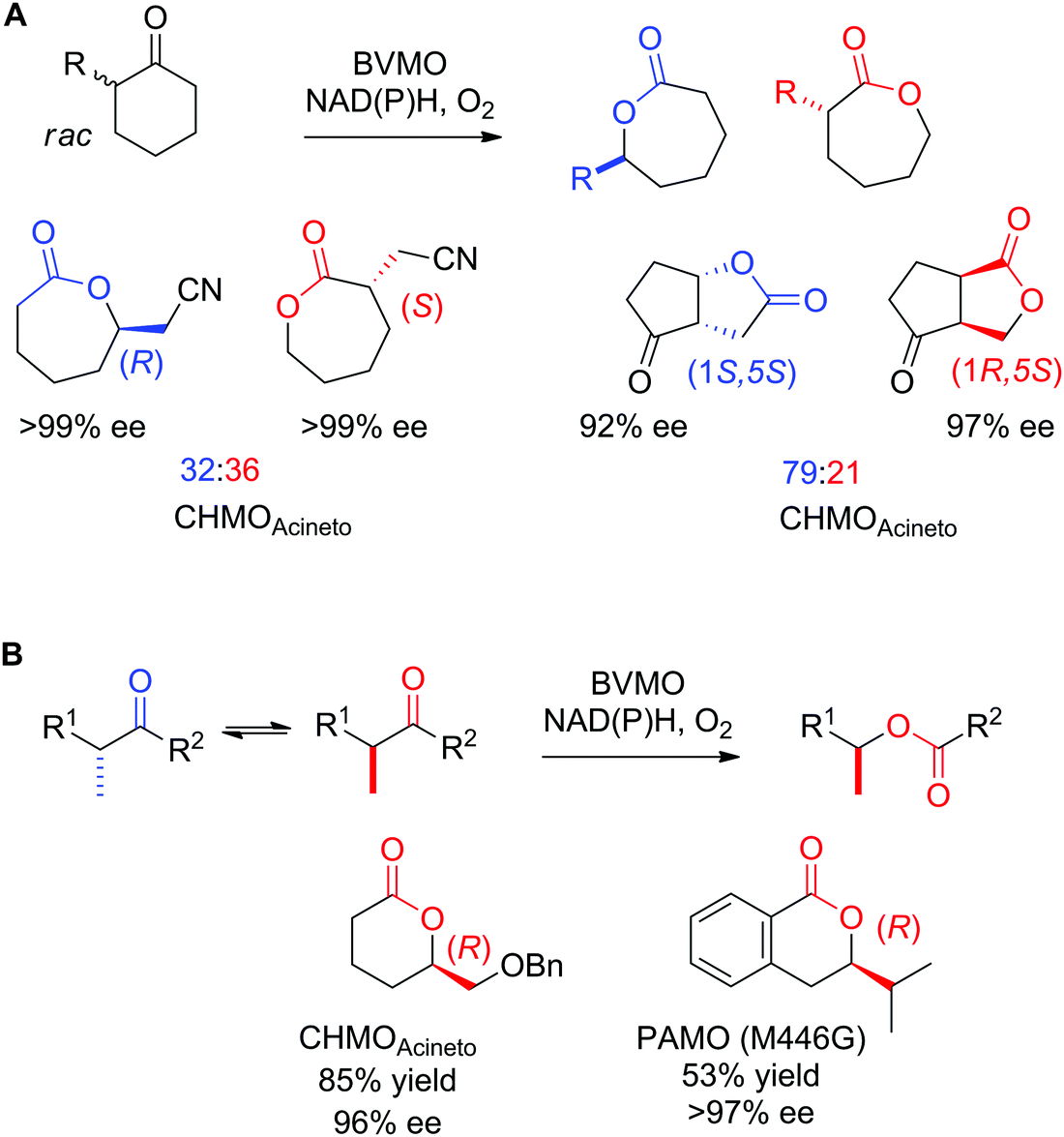 | ||
| Scheme 31 Applications of enantioselective Baeyer–Villiger monooxygenases in (A) enantiocomplementary regiodivergent process283,284 and (B) dynamic kinetic resolution of racemic substrates285–287 with examples of products obtained with high enantiopurity. CHMOAcineto: cyclohexanone monooxygenase from Acinetobacter calcoaceticus NCIMB 9871, PAMO (M446G): variant of phenylacetone monooxygenase from Thermobifida fusca. Reprinted from ref. 252 Copyright 2020, with permission from Elsevier. | ||
Highly stereo- and enantioselective ADHs haven been employed in dynamic kinetic resolution processes of α-substituted chiral carbonyl compounds toward formation of optically pure α-alkyl-β-hydroxy esters,288 α-alkyl-β-hydroxy ketones, α-amino-β-hydroxy esters, and variously substituted halohydrins.82,289,290 The stereoselective reduction reaction combined with the stereorecognition of the existing chiral center and in situ racemization of the substrate ensure that the final products possessing two chiral centers are obtained as one stereoisomer only of the four theoretically possible ones in high yields.
With α-substituted aldehydes, similar approach is possible and leads to enantiopure α-substituted primary alcohols. (S)-Profenols could be obtained in excellent enantiopurity (up to 99% ee) by applying a thermostable ADH from Sulfolobus sulfataricus to the enantioselective reduction of a series of racemic 2-arylpropionaldehydes at pH 9, including the precursor to naproxen (Scheme 32).291 The reduced nicotinamide was regenerated by the coupled-substrate approach employing ethanol as hydride source.
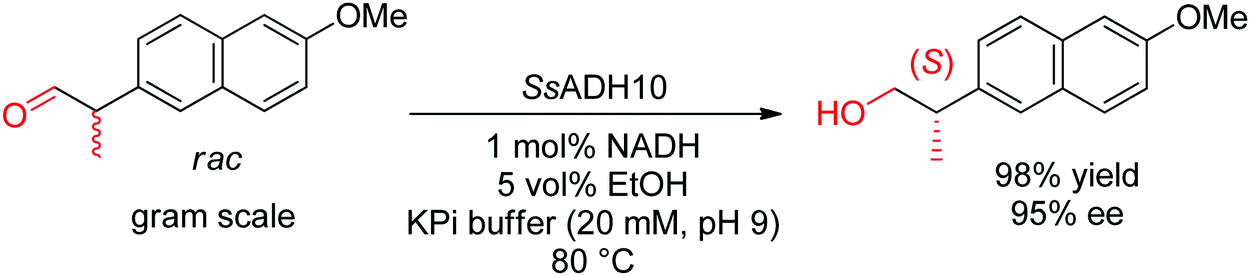 | ||
| Scheme 32 Reductive dynamic kinetic resolution of α-substituted aldehyde catalyzed by an alcohol dehydrogenase from Sulfolobus sulfataricus (SsADH10) to access precursor of (S)-naproxen.291 Republished from ref. 38 with permission of John Wiley & Sons – Books, copyright 2021; permission conveyed through Copyright Clearance Center, Inc. | ||
Under different reaction conditions, simultaneous access to (S)-profens and profenols was granted following the disproportionation of racemic 2-arylpropanals catalyzed by ADH from horse liver (HLADH). This parallel interconnected dynamic asymmetric transformation delivered a 1:1 mixture of corresponding alcohols and carboxylic acids, which were obtained with high enantiopurity (up to 99% ee) and high conversion levels (Scheme 33).292 Racemization of the substrate was observed at the reaction pH of 7.5. The system rendered a redox-neutral cascade and only catalytic amounts of the oxidized nicotinamide cofactor were necessary.
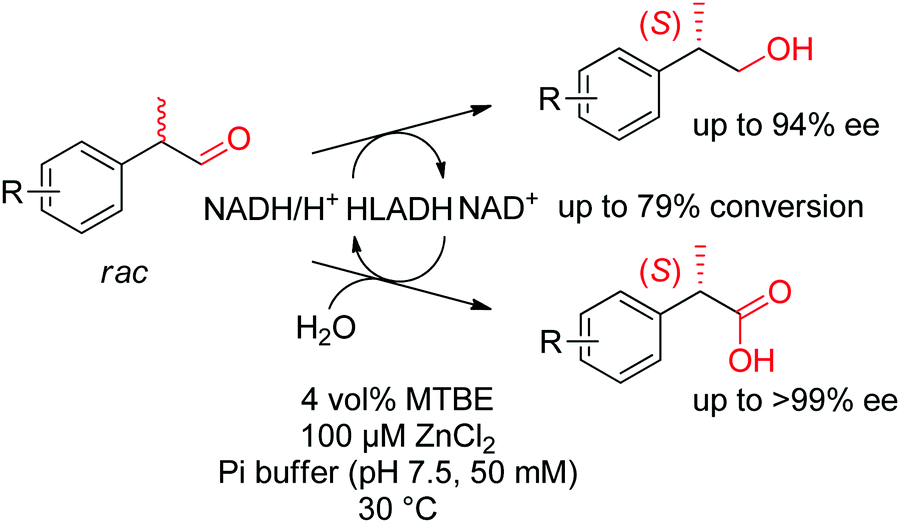 | ||
| Scheme 33 Asymmetric disproportionation of racemic 2-arylpropanals catalyzed by alcohol dehydrogenase from horse liver (HLADH) under redox-neutral conditions to form (S)-profens and profenols.292 Reproduced from ref. 129 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Complementary to the numerous enzymatic methods available to generate chiral amines (see Sections 1.1.3 and 1.2.1), kinetic resolution of racemic amines via oxidative protocols offers very efficient approaches that usually rely on flavin-dependent amine oxidases,293,294 in particular amino acid oxidases and most prominent monoamine oxidases (MAOs). From the latter class of enzymes, MAO from Aspergillus niger (MAO-N) has been employed in a wide range of deracemization protocols, due to its broad substrate scope and acceptance of primary and secondary amines. A large number of variants have been generated and allow the oxidation of many substrates, including tertiary amines.295,296 A ‘simple’ kinetic resolution leads in the case of perfect enantioselectivity to maximum 50% yield of the nonconverted enantiopure amine and generates 50% side product in form of an imine (or iminium ion) that usually hydrolyzes to the ketone. Combining (non)selective (chemical) reduction of the imine back to the (racemic) amine with the enzymatic oxidation step leads to a cyclic deracemization in which yields may reach 100% for the enantiopure desired chiral amine. A variant of MAO-N was identified during a directed evolution campaign that showed strong preference for the oxidation of (S)-α-methylbenzylamine. Combined with a nonselective reduction using the ammonia–borane complex, (R)-α-methylbenzylamine could be obtained with 93% ee and 77% yield.297 This process was later applied to the deracemization of key intermediates to active pharmaceutical ingredients. In the case of 1-phenyl-1,2,3,4-tetrahydroisoquinoline, a precursor to solifenacin used in the treatment of overactive bladder, the (S)-enantiomer could be obtained in 90% isolated yield and 98% ee after 48 h reaction time at the gram-scale (Scheme 34).295 The wild-type and variants of MAO-N have been applied in a number of synthetic sequences,298 either multi-enzymatic, such as for the synthesis of chiral 2,5-disubstituted pyrrolidines triggered by a transaminase-catalyzed step,299 or chemoenzymatic, as seen in the synthesis of a chiral precursor to boceprevir, a drug developed by Merck & Co, Inc. for the treatment of hepatitis C.300
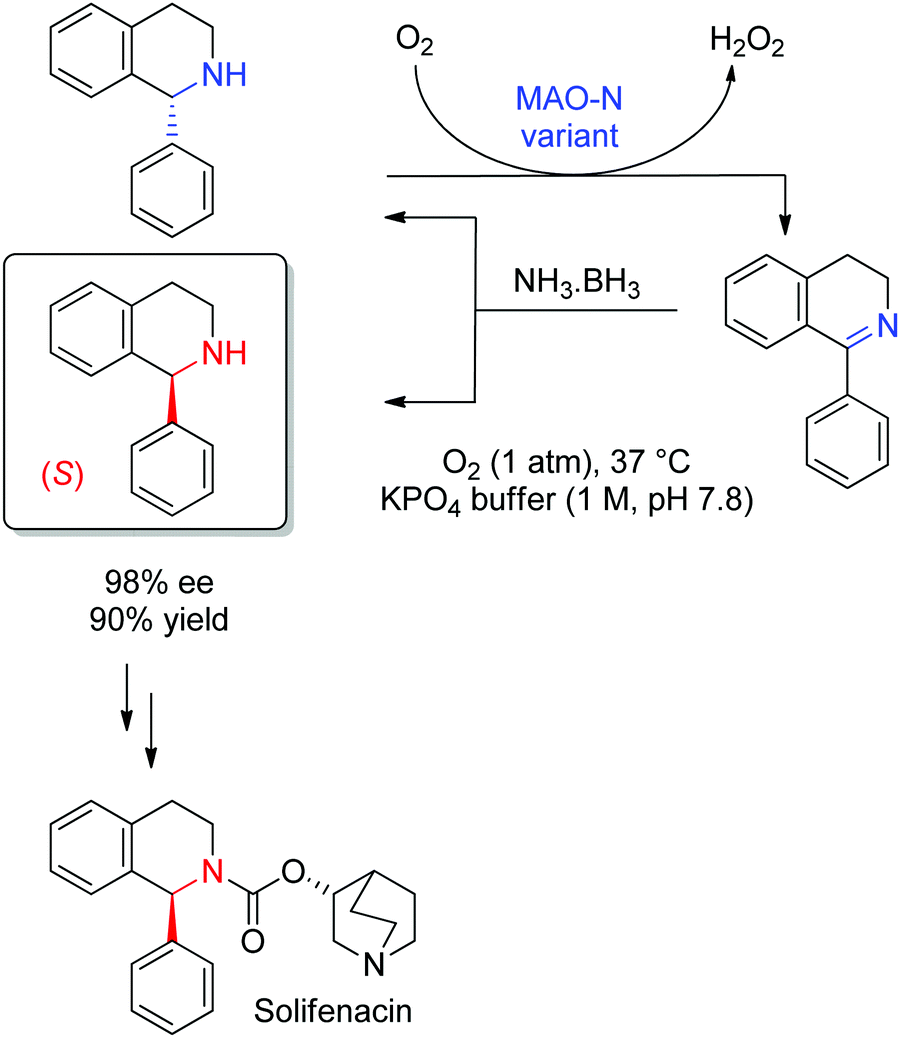 | ||
| Scheme 34 Cyclic deracemization of rac-1-phenyl-1,2,3,4-tetrahydroisoquinoline through enantioselective oxidation by a variant of monoamine oxidase from Aspergillus niger (MAO-N) and nonselective reduction with ammonia–borane complex. The (S)-amine was then employed to access solifenacin in two steps.295 | ||
3. Atroposelective reactions
Enzymatic protocols used in asymmetric synthesis are predominantly applied to the generation of central chirality, mostly tetrasubstituted carbon atoms, occasionally chiral sulfur atoms (see Section 2.1.2). Nevertheless, stereoselective enzymatic strategies also exist to generate products with axial chirality in enantiopure form. Cases in which planar chirality is involved will not be discussed here. For larger sampling of existing examples dealing with noncentral chirality with enzymes, the reader is referred to excellent reviews and articles broadly covering the topic.301–3053.1. (Dynamic) kinetic resolution of axially chiral molecules
Initial studies dedicated to asymmetric biotransformations involving noncentral chirality were focusing on reactions catalyzed by hydrolytic enzymes. Those were employed in the kinetic resolution of racemic axially chiral molecules in both reaction directions (hydrolysis of esters and transesterification reaction). For instance, pig liver esterase was found to hydrolyze racemic allenic esters with some enantioselectivity (E value up to 42), leading to formation of the corresponding acids in 83% ee,306,307 while lipases were used for the acylation of primary allenic alcohols by vinyl butyrate and vinyl acetate. Excellent enantioselectivities were obtained with a few substrates under perfect kinetic resolution (E value >200).308,309 Later, that system could be upgraded to a dynamic kinetic resolution with the introduction of a palladium-catalyzed isomerization of the substrate that was compatible with the biocatalyzed acylation and led to the formation of optically active allenes in up to 83% yield and 89% ee.310Such biocatalytic hydrolytic kinetic resolutions have been successfully applied to atropisomeric biaryl compounds. Several enzyme preparations (porcine pancreatic lipase and cholesterol esterase for instance) could efficiently catalyze the atroposelective hydrolysis of racemic 1,1′-binaphthyl-2,2′-diol based diesters, leading to the formation of the diol with excellent ee value and E value up to 105 (Scheme 35).311,312 Lipase-catalyzed monoacylation reactions were also developed for binaphthol and proceeded with high stereoselectivity.313
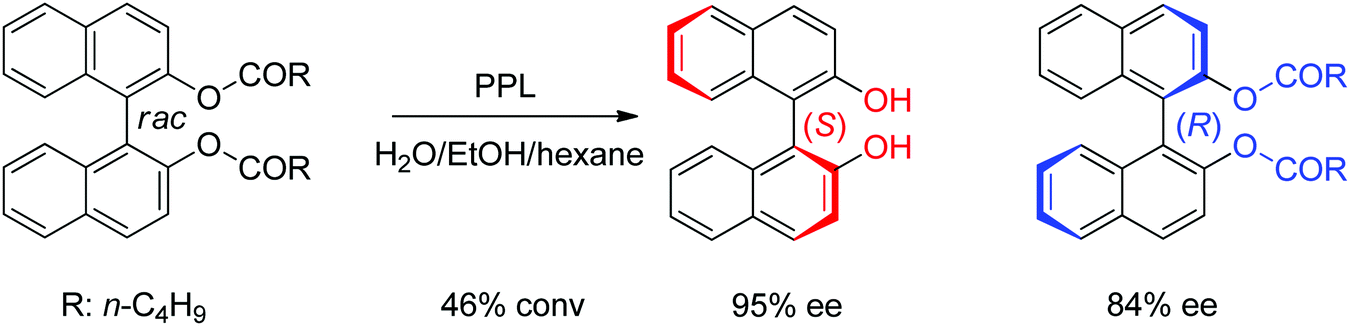 | ||
| Scheme 35 Kinetic resolution of racemic 1,1′-binaphthyl-2,2′-diol based diesters through atroposelective hydrolysis to corresponding diols catalyzed by porcine pancreatic lipase (PPL).311 | ||
Recently, a dynamic kinetic resolution of racemic axially chiral 2,2′-dihydroxy-1,1′-biaryls was achieved by including to the biocatalytic monoacylation step an in situ racemization by a Ru(II)-based catalyst. Several biaryl diols could be obtained in high yields and with up to 98% ee after final base-mediated deacylation.314
Importantly, although still underexplored, atroposelective kinetic resolutions have also been established via enzymatic redox chemistry. Early studies reported the microbial full oxidation of α-allenic alcohols to their corresponding acids, however obtained ee values remained modest.315,316 In one of the first reported examples with biaryl compounds, baker's yeast was applied to the reduction of non C2-symmetric substrates such as 2-formyl-1,1′-binaphthyls, here again the ee values of the product did not exceed 70%.317 More recently, perfect enantioselectivity could be obtained by employing commercial ADHs in the reduction of biaryl monoaldehydes displaying additional N-oxide functionalization. When applied to configurationally unstable substrates, an elegant dynamic kinetic resolution could be obtained. This was possible due to the more stable chiral axis in the resulting product, which was finally obtained in quantitative yield and >95% ee in a stereocomplementary manner with enzymes of opposite selectivity (Scheme 36).318 The products were employed as Lewis base organocatalysts in asymmetric allylation of aldehydes.
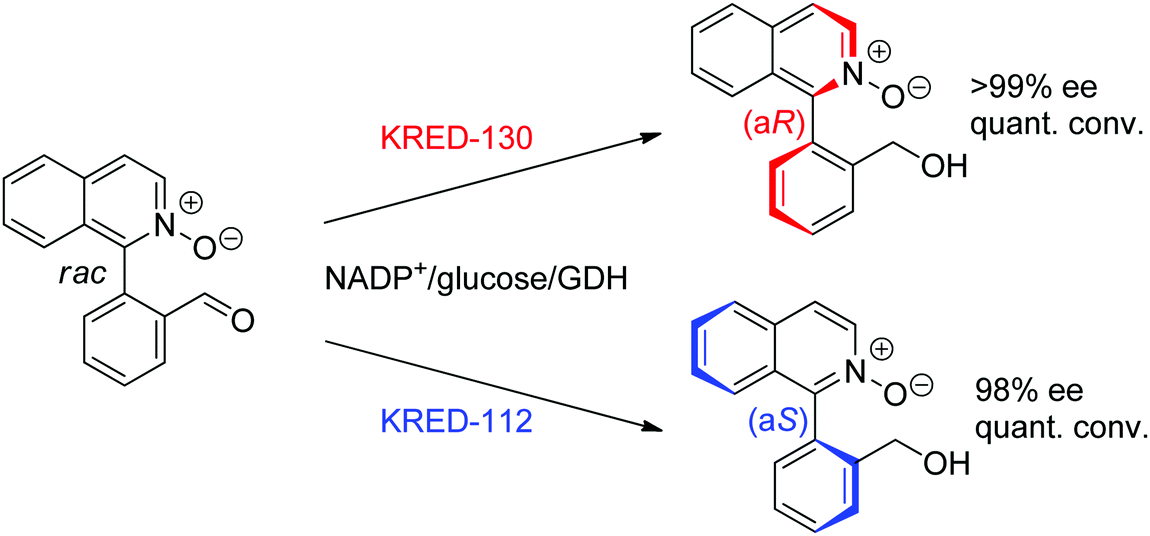 | ||
| Scheme 36 Dynamic kinetic resolution of configurationally unstable substrates via atroposelective reduction by commercial alcohol dehydrogenases (KRED-130 and KRED-112) toward formation of atropisomeric biaryl N-oxides (GDH: glucose dehydrogenase).318 | ||
3.2. Conversion of prochiral compounds into axially chiral compounds
Axially chiral compounds exist in enantiopure form in nature (e.g., gossypol, vancomycin). Their synthesis often involves oxidative coupling reactions catalyzed by cytochrome P450 enzymes or laccases.319 A few studies have shown the successful coupling of two units via enzymatic phenol coupling reactions using laccases.320,321 Atroposelective behavior could be confirmed with several fungal laccases in the regioselective dimerization of γ-naphthopyrone to both atropisomers of ustilaginoidin A.322 Other stereoselective dimerization systems based on laccases and dirigent proteins323–326 and P450s327–329 are emerging. Though promising, such approaches are currently limited to the synthesis of natural product derivatives and will necessitate further improvements to be employed with nonnatural molecules in broadly applicable synthetic routes.Outlook
Biocatalysis could be established over the past two decades as a powerful methodology in asymmetric synthesis mostly owing to its interdisciplinary character. Many of the examples treated in this review highlight the advantages of merging biocatalysis with methodologies of biotechnology, molecular biology, protein engineering, bioinformatics, or synthetic biology, to name a few complementary areas. Several waves of technological development have been recognized by experts over the last decade.330,331 Writing 2021, a new revolution – more than a wave – triggered by artificial intelligence (AI) is already on the rise. The recent spectacular example of AlphaFold from DeepMind in accurately predicting the structure of a protein from its amino acid sequence will certainly become disruptive to the whole field of protein science.332,333 Computational tools have long contributed to revolutionize the field of biocatalysis, including protein engineering campaign, modelling and docking, and mechanistic investigations. In particular in asymmetric synthesis, the accurate prediction of protein 3D structures, combined with other methodologies such as molecular docking and protein engineering, will become big assets in speeding up the development of robust and selective biocatalysts to generate a broad range of enantiopure molecules useful in the fine chemical and pharmaceutical sector.334In addition to the increasing availability and diversity of commercial enzymes in form of off-the-shelf or customized biocatalysts,38 other aspects are contributing to rendering enzymes accessible to chemists willing to incorporate these catalysts in their synthesis planning. Retrobiosynthesis for instance is an emerging area that provides keys to apply biosynthetic logic to the design of artificial synthesis routes.335,336 While mimicking the retrosynthetic approach developed by Corey,337,338 the focus remains on key enzymatic steps to inspire retrobiocatalytic strategies.339,340 Biocatalysis offers many ways to access key functional group interconversions and computational tools are now available341–343 to help identify disconnections – a key strategy in retrosynthesis – and suitable starting materials with corresponding enzymes toward a target molecule.
The field of catalysis in general has revolutionized how (chiral) molecules were made during the 20th century. Being able to combine the advantages of the many disciplines in catalysis (metal, organo-, bio, photo-catalysis) in cascade processes344 will define how chemicals of the 21st century are made.345–348 Several examples are paving the way toward the merging of enzymes with chemical methods in elaborated (cascade) synthesis.349–353
Finally, the growing importance of (meta)genomics and ultrahigh-throughput screening, such as seen with microfluidic approaches,354 will raise new challenges. Generating large data set from vast and diverse protein libraries is not technically limited anymore, instead the interpretation of the rapidly growing amount of information remains currently an obstacle to advancing further and faster. Beyond helping in solving the ‘protein folding problem’, AI is mature to tackle the big data problem emerging in biocatalysis. Game changer to the field of asymmetric biocatalysis will be the successful prediction of enzyme structure–activity–stereoselectivity pattern. While this is already possible for some classes of enzymes, and has led to the establishment of popular rules (the ‘Prelog’ rule with alcohol dehydrogenases86 or the ‘Kazlauskas’ rule with esterases and lipases355), many more enzyme classes still rely on large screening efforts to identify the ideal substrate-enzyme pair for the generation of the targeted product, despite mechanistic understanding at the molecular level.
As witnessed in the currently challenging pandemic situation worldwide, accessing biologically active molecules quickly has become vital and the merging of all the above-mentioned disciplines will be essential to making an impact in this area. The recent example of the multi-step fully biocatalytic synthesis of islatravir, a potential drug for treatment of HIV developed by Merck & Co, Inc.,256 indicates that the future of biocatalysis in the field of asymmetric synthesis is bright,255,257 in particular in the context of a multi-disciplinary science.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Austrian Science Fund (FWF, P32815-N) and by the University of Graz is gratefully acknowledged.Notes and references
- Modern Biocatalysis: Advances towards synthetic biological systems, ed. G. Williams and M. Hall, Royal Society of Chemistry, Croydon, 2018 Search PubMed.
- Biocatalysis for Practitioners: Techniques, Reactions and Applications, ed. G. de Gonzalo and I. Lavandera, Wiley-VCH, Weinheim, 2021 Search PubMed.
- U. T. Bornscheuer and K. Buchholz, Eng. Life Sci., 2005, 5, 309–323 Search PubMed.
- C. M. Heckmann and F. Paradisi, ChemCatChem, 2020, 12, 6082–6102 Search PubMed.
- M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 Search PubMed.
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G. D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 Search PubMed.
- Y. F. Miao, R. Metzner and Y. Asano, ChemBioChem, 2017, 18, 451–454 Search PubMed.
- D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190–195 Search PubMed.
- E. Malito, A. Alfieri, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13157–13162 Search PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 Search PubMed.
- K. Chen and F. H. Arnold, Nat. Cat., 2020, 3, 203–213 Search PubMed.
- N. P. Dunham and F. H. Arnold, ACS Catal., 2020, 10, 12239–12255 Search PubMed.
- S. C. Hammer, A. M. Knight and F. H. Arnold, Curr. Opin. Green Sustain. Chem., 2017, 7, 23–30 Search PubMed.
- R. J. K. Zhang, X. Y. Huang and F. H. Arnold, Curr. Opin. Chem. Biol., 2019, 49, 67–75 Search PubMed.
- S. K. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 Search PubMed.
- B. Martín-Matute and J.-E. Bäckvall, in Asymmetric Organic Synthesis with Enzymes, ed. V. Gotor, I. Alfonso and E. García-Urdiales, Wiley-VCH, 2008, pp. 87–113 Search PubMed.
- M. T. Reetz, Curr. Opin. Chem. Biol., 2002, 6, 145–150 Search PubMed.
- K. D. Daiha, R. Angeli, S. D. de Oliveira and R. V. Almeida, PLoS One, 2015, 10, e0131624 Search PubMed.
- N. J. Turner, in Advanced Asymmetric Synthesis, ed. G. R. Stephenson, Springer, Dordrecht, 1996, pp. 260–274 Search PubMed.
- N. J. Turner, in Asymmetric Organic Synthesis with Enzymes, ed. V. Gotor, I. Alfonso and E. García-Urdiales, Wiley-VCH, 2008, pp. 115–131 Search PubMed.
- Y. Simeó, W. Kroutil and K. Faber, in Biocatalysis in the Pharmaceutical and Biotechnology Industries, ed. R. N. Patel, CRC Press, 1st edn, 2006, ch. 2 DOI:10.1201/9781420019377.
- A. Diaz-Rodriguez, I. Lavandera and V. Gotor, Curr. Green Chem., 2015, 2, 192–211 Search PubMed.
- U. T. Strauss, U. Felfer and K. Faber, Tetrahedron: Asymmetry, 1999, 10, 107–117 Search PubMed.
- N. J. Turner, Curr. Opin. Biotechnol., 2003, 14, 401–406 Search PubMed.
- J. M. Palomo and Z. Cabrera, Curr. Org. Synth., 2012, 9, 791–805 Search PubMed.
- P. Berglund, Biomol. Eng., 2001, 18, 13–22 Search PubMed.
- F. Hollmann, D. J. Opperman and C. E. Paul, Angew. Chem., Int. Ed., 2021, 60, 5644–5665 Search PubMed.
- H. S. Toogood and N. S. Scrutton, ACS Catal., 2018, 8, 3532–3549 Search PubMed.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 Search PubMed.
- K. Faber and M. Hall, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 213–260 Search PubMed.
- K. Durchschein, S. Wallner, P. Macheroux, W. Schwab, T. Winkler, W. Kreis and K. Faber, Eur. J. Org. Chem., 2012, 4963–4968 Search PubMed.
- D. J. Mansell, H. S. Toogood, J. Waller, J. M. X. Hughes, C. W. Levy, J. M. Gardiner and N. S. Scrutton, ACS Catal., 2013, 3, 370–379 Search PubMed.
- H. S. Toogood, A. N. Cheallaigh, S. Tait, D. J. Mansell, A. Jervis, A. Lygidakis, L. Humphreys, E. Takano, J. M. Gardiner and N. S. Scrutton, ACS Synth. Biol., 2015, 4, 1112–1123 Search PubMed.
- K. Saito, D. J. Thiele, M. Davio, O. Lockridge and V. Massey, J. Biol. Chem., 1991, 266, 20720–20724 Search PubMed.
- S. Velikogne, W. B. Breukelaar, F. Hamm, R. A. Glabonjat and W. Kroutil, ACS Catal., 2020, 10, 13377–13382 Search PubMed.
- B. J. Brown, Z. Deng, P. A. Karplus and V. Massey, J. Biol. Chem., 1998, 273, 32753–32762 Search PubMed.
- R. M. Kohli and V. Massey, J. Biol. Chem., 1998, 273, 32763–32770 Search PubMed.
- M. Hall, in Biocatalysis for Practitioners: Techniques, Reactions and Applications, ed. G. de Gonzalo and I. Lavandera, Wiley-VCH, Weinheim, 2021, pp. 181–224 Search PubMed.
- H. S. Toogood, J. M. Gardiner and N. S. Scrutton, ChemCatChem, 2010, 2, 892–914 Search PubMed.
- E. Brenna, G. Fronza, C. Fuganti and F. Parmeggiani, Isot. Environ. Health Stud., 2015, 51, 24–32 Search PubMed.
- N. G. Turrini, R. C. Cioc, D. J. H. van der Niet, E. Ruijter, R. V. A. Orru, M. Hall and K. Faber, Green Chem., 2017, 19, 511–518 Search PubMed.
- S. K. Padhi, D. J. Bougioukou and J. D. Stewart, J. Am. Chem. Soc., 2009, 131, 3271–3280 Search PubMed.
- E. D. Amato and J. D. Stewart, Biotechnol. Adv., 2015, 33, 624–631 Search PubMed.
- M. Kataoka, A. Kotaka, A. Hasegawa, M. Wada, A. Yoshizumi, S. Nakamori and S. Shimizu, Biosci., Biotechnol., Biochem., 2002, 66, 2651–2657 Search PubMed.
- M. Hall, C. Stueckler, W. Kroutil, P. Macheroux and K. Faber, Angew. Chem., Int. Ed., 2007, 46, 3934–3937 Search PubMed.
- A. Müller, B. Hauer and B. Rosche, Biotechnol. Bioeng., 2007, 98, 22–29 Search PubMed.
- C. Stueckler, N. J. Mueller, C. K. Winkler, S. M. Glueck, K. Gruber, G. Steinkellner and K. Faber, Dalton Trans., 2010, 39, 8472–8476 Search PubMed.
- C. Stueckler, C. K. Winkler, M. Bonnekessel and K. Faber, Adv. Synth. Catal., 2010, 352, 2663–2666 Search PubMed.
- M. A. Swiderska and J. D. Stewart, Org. Lett., 2006, 8, 6131–6133 Search PubMed.
- D. Mangan, I. Miskelly and T. S. Moody, Adv. Synth. Catal., 2012, 354, 2185–2190 Search PubMed.
- C. Stueckler, M. Hall, H. Ehammer, E. Pointner, W. Kroutil, P. Macheroux and K. Faber, Org. Lett., 2007, 9, 5409–5411 Search PubMed.
- G. Oberdorfer, K. Gruber, K. Faber and M. Hall, Synlett, 2012, 1857–1864 Search PubMed.
- E. Brenna, G. Fronza, C. Fuganti and F. G. Gatti, Eur. J. Org. Chem., 2010, 5077–5084 Search PubMed.
- E. Brenna, F. G. Gatti, A. Manfredi, D. Monti and F. Parmeggiani, Adv. Synth. Catal., 2012, 354, 2859–2864 Search PubMed.
- E. Brenna, M. Crotti, F. G. Gatti, D. Monti, F. Parmeggiani, R. W. Powell, S. Santangelo and J. D. Stewart, Adv. Synth. Catal., 2015, 357, 1849–1860 Search PubMed.
- M. Crotti, F. Parmeggiani, E. E. Ferrandi, F. G. Gatti, A. Sacchetti, S. Riva, E. Brenna and D. Monti, Front. Bioeng. Biotechnol., 2019, 7, 89 Search PubMed.
- G. Tasnádi, C. K. Winkler, D. Clay, N. Sultana, W. M. F. Fabian, M. Hall, K. Ditrich and K. Faber, Chem. – Eur. J., 2012, 18, 10362–10367 Search PubMed.
- C. Stueckler, C. K. Winkler, M. Hall, B. Hauer, M. Bonnekessel, K. Zangger and K. Faber, Adv. Synth. Catal., 2011, 353, 1169–1173 Search PubMed.
- C. K. Winkler, C. Stueckler, N. J. Mueller, D. Pressnitz and K. Faber, Eur. J. Org. Chem., 2010, 6354–6358 Search PubMed.
- T. Classen, M. Korpak, M. Schölzel and J. Pietruszka, ACS Catal., 2014, 4, 1321–1331 Search PubMed.
- S. Venturi, E. Brenna, D. Colombo, M. W. Fraaije, F. G. Gatti, P. Macchi, D. Monti, M. Trajkovic and E. Zamboni, ACS Catal., 2020, 10, 13050–13057 Search PubMed.
- C. K. Winkler, K. Faber and M. Hall, Curr. Opin. Chem. Biol., 2018, 43, 97–105 Search PubMed.
- N. G. Turrini, M. Hall and K. Faber, Adv. Synth. Catal., 2015, 357, 1861–1871 Search PubMed.
- I. Janicki, P. Kielbasinski, N. G. Turrini, K. Faber and M. Hall, Adv. Synth. Catal., 2017, 359, 4190–4196 Search PubMed.
- F. Nagy, I. Gyujto, G. Tasnadi, B. Barna, D. Balogh-Weiser, K. Faber, L. Poppe and M. Hall, J. Biotechnol., 2020, 323, 246–253 Search PubMed.
- H. Li, W. H. Xiao, P. P. Xie and L. Y. Zheng, Enzyme Microb. Technol., 2018, 109, 66–73 Search PubMed.
- H. Li, X. M. Cui and L. Y. Zheng, J. Microbiol. Biotechnol., 2019, 29, 607–616 Search PubMed.
- F. Tentori, T. Bavaro, E. Brenna, D. Colombo, D. Monti, R. Semproli and D. Ubiali, Catalysts, 2020, 10, 260 Search PubMed.
- T. W. Thorpe, S. P. France, S. Hussain, J. R. Marshall, W. Zawodny, J. Mangas-Sanchez, S. L. Montgomery, R. M. Howard, D. S. B. Daniels, R. Kumar, F. Parmeggiani and N. J. Turner, J. Am. Chem. Soc., 2019, 141, 19208–19213 Search PubMed.
- K. Heckenbichler, A. Schweiger, L. A. Brandner, A. Binter, M. Toplak, P. Macheroux, K. Gruber and R. Breinbauer, Angew. Chem., Int. Ed., 2018, 57, 7240–7244 Search PubMed.
- M. V. Shah, J. Antoney, S. W. Kang, A. C. Warden, C. J. Hartley, H. Nazem-Bokaee, C. J. Jackson and C. Scott, Catalysts, 2019, 9, 868 Search PubMed.
- C. Greening, T. Jirapanjawat, S. Afroze, B. Ney, C. Scott, G. Pandey, B. M. Lee, R. J. Russell, C. J. Jackson, J. G. Oakeshott, M. C. Taylor and A. C. Warden, Front. Microbiol., 2017, 8, 1000 Search PubMed.
- S. Mathew, M. Trajkovic, H. Kumar, Q. T. Nguyen and M. W. Fraaije, Chem. Commun., 2018, 54, 11208–11211 Search PubMed.
- M. Hall, C. Stueckler, B. Hauer, R. Stuermer, T. Friedrich, M. Breuer, W. Kroutil and K. Faber, Eur. J. Org. Chem., 2008, 1511–1516 Search PubMed.
- M. Klimacek, K. L. Kavanagh, D. K. Wilson and B. Nidetzky, Chem. – Biol. Interact., 2003, 143, 559–582 Search PubMed.
- K. Niefind, J. Müller, B. Riebel, W. Hummel and D. Schomburg, J. Mol. Biol., 2003, 327, 317–328 Search PubMed.
- T. M. Penning, Chem. – Biol. Interact., 2015, 234, 236–246 Search PubMed.
- A. Weckbecker and W. Hummel, Biocatal. Biotransform., 2006, 24, 380–389 Search PubMed.
- J. H. An, Y. Nie and Y. Xu, Crit. Rev. Biotechnol., 2019, 39, 366–379 Search PubMed.
- H. Gröger, W. Hummel, S. Borchert and M. Kraußer, in Enzyme catalysis in organic synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2012, vol. 2, pp. 1037–1110 Search PubMed.
- T. Matsuda, R. Yamanaka and K. Nakamura, Tetrahedron: Asymmetry, 2009, 20, 513–557 Search PubMed.
- T. Moody, S. Mix, G. Brown and D. Beecher, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 421–458 Search PubMed.
- C. V. Voss, C. C. Gruber and W. Kroutil, Synlett, 2010, 991–998 Search PubMed.
- C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789–1805 Search PubMed.
- J. C. Moore, D. J. Pollard, B. Kosjek and P. N. Devine, Acc. Chem. Res., 2007, 40, 1412–1419 Search PubMed.
- V. Prelog, Pure Appl. Chem., 1964, 9, 119–130 Search PubMed.
- N. Itoh, Appl. Microbiol. Biotechnol., 2014, 98, 3889–3904 Search PubMed.
- I. Lavandera, A. Kern, B. Ferreira-Silva, A. Glieder, S. de Wildeman and W. Kroutil, J. Org. Chem., 2008, 73, 6003–6005 Search PubMed.
- I. Lavandera, A. Kern, V. Resch, B. Ferreira-Silva, A. Glieder, W. M. F. Fabian, S. de Wildeman and W. Kroutil, Org. Lett., 2008, 10, 2155–2158 Search PubMed.
- H. M. Li, D. M. Zhu, L. Hua and E. R. Biehl, Adv. Synth. Catal., 2009, 351, 583–588 Search PubMed.
- H. Man, K. Kedziora, J. Kulig, A. Frank, I. Lavandera, V. Gotor-Fernandez, D. Rother, S. Hart, J. P. Turkenburg and G. Grogan, Top. Catal., 2014, 57, 356–365 Search PubMed.
- M. D. Truppo, D. Pollard and P. Devine, Org. Lett., 2007, 9, 335–338 Search PubMed.
- X. Chen, H. L. Zhang, M. A. Maria-Solano, W. D. Liu, J. Li, J. H. Feng, X. T. Liu, S. Osuna, R. T. Guo, Q. Q. Wu, D. M. Zhu and Y. H. Ma, Nat. Cat., 2019, 2, 931–941 Search PubMed.
- M. A. Maria-Solano, A. Romero-Rivera and S. Osuna, Org. Biomol. Chem., 2017, 15, 4122–4129 Search PubMed.
- C. M. Nealon, M. M. Musa, J. M. Patel and R. S. Phillips, ACS Catal., 2015, 5, 2100–2114 Search PubMed.
- E. L. Noey, N. Tibrewal, G. Jimenez-Oses, S. Osuna, J. Park, C. M. Bond, D. Cascio, J. Liang, X. Y. Zhang, G. W. Huisman, Y. Tang and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E7065–E7072 Search PubMed.
- A. A. Koesoema, D. M. Standley, T. Senda and T. Matsuda, Appl. Microbiol. Biotechnol., 2020, 104, 2897–2909 Search PubMed.
- J. Lalonde, Curr. Opin. Biotechnol., 2016, 42, 152–158 Search PubMed.
- G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 Search PubMed.
- R. J. Fox, S. C. Davis, E. C. Mundorff, L. M. Newman, V. Gavrilovic, S. K. Ma, L. M. Chung, C. Ching, S. Tam, S. Muley, J. Grate, J. Gruber, J. C. Whitman, R. A. Sheldon and G. W. Huisman, Nat. Biotechnol., 2007, 25, 338–344 Search PubMed.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 Search PubMed.
- J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. N. Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193–198 Search PubMed.
- J. Xu, Y. Z. Peng, Z. G. Wang, Y. J. Hu, J. J. Fan, H. Zheng, X. F. Lin and Q. Wu, Angew. Chem., Int. Ed., 2019, 58, 14499–14503 Search PubMed.
- J. H. Schrittwieser, S. Velikogne and W. Kroutil, Adv. Synth. Catal., 2015, 357, 1655–1685 Search PubMed.
- G. Grogan and N. J. Turner, Chem. – Eur. J., 2016, 22, 1900–1907 Search PubMed.
- M. Höhne, Nat. Cat., 2019, 2, 841–842 Search PubMed.
- L. Ducrot, M. Bennett, G. Grogan and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2021, 363, 328–351 Search PubMed.
- S. Fademrecht, P. N. Scheller, B. M. Nestl, B. Hauer and J. Pleiss, Proteins, 2016, 84, 600–610 Search PubMed.
- P. N. Scheller, S. Fademrecht, S. Hofelzer, J. Pleiss, F. Leipold, N. J. Turner, B. M. Nestl and B. Hauer, ChemBioChem, 2014, 15, 2201–2204 Search PubMed.
- M. Lenz, S. Fademrecht, M. Sharma, J. Pleiss, G. Grogan and B. M. Nestl, Protein Eng., Des. Sel., 2018, 31, 109–120 Search PubMed.
- D. Wetzl, M. Berrera, N. Sandon, D. Fishlock, M. Ebeling, M. Müller, S. Hanlon, B. Wirz and H. Iding, ChemBioChem, 2015, 16, 1749–1756 Search PubMed.
- G. A. Aleku, H. Man, S. P. France, F. Leipold, S. Hussain, L. Toca-Gonzalez, R. Marchington, S. Hart, J. P. Turkenburg, G. Grogan and N. J. Turner, ACS Catal., 2016, 6, 3880–3889 Search PubMed.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985–11015 Search PubMed.
- P. Matzel, M. Gand and M. Hohne, Green Chem., 2017, 19, 385–389 Search PubMed.
- S. Hussain, F. Leipold, H. Man, E. Wells, S. P. France, K. R. Mulholland, G. Grogan and N. J. Turner, ChemCatChem, 2015, 7, 579–583 Search PubMed.
- H. Li, Z. J. Luan, G. W. Zheng and J. H. Xu, Adv. Synth. Catal., 2015, 357, 1692–1696 Search PubMed.
- P. N. Scheller, M. Lenz, S. C. Hammer, B. Hauer and B. M. Nestl, ChemCatChem, 2015, 7, 3239–3242 Search PubMed.
- T. Huber, L. Schneider, A. Prag, S. Gerhardt, O. Einsle and M. Müller, ChemCatChem, 2014, 6, 2248–2252 Search PubMed.
- T. Knaus, W. Bohmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 Search PubMed.
- F. F. Chen, S. C. Cosgrove, W. R. Birmingham, J. Mangas-Sanchez, J. Citoler, M. P. Thompson, G. W. Zheng, J. H. Xu and N. J. Turner, ACS Catal., 2019, 9, 11813–11818 Search PubMed.
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg, J. L. Petit, A. Mariage, A. Debard, V. Pellouin, A. Perret, V. de Berardinis, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, Nat. Cat., 2019, 2, 324–333 Search PubMed.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 Search PubMed.
- J. Mangas-Sanchez, M. Sharma, S. C. Cosgrove, J. I. Ramsden, J. R. Marshall, T. W. Thorpe, R. B. Palmer, G. Grogan and N. J. Turner, Chem. Sci., 2020, 11, 5052–5057 Search PubMed.
- S. P. France, R. M. Howard, J. Steflik, N. J. Weise, J. Mangas-Sanchez, S. L. Montgomery, R. Crook, R. Kumar and N. J. Turner, ChemCatChem, 2018, 10, 510–514 Search PubMed.
- G. D. Roiban, M. Kern, Z. Liu, J. Hyslop, P. L. Tey, M. S. Levine, L. S. Jordan, K. K. Brown, T. Hadi, L. A. F. Ihnken and M. J. B. Brown, ChemCatChem, 2017, 9, 4475–4479 Search PubMed.
- S. L. Montgomery, A. Pushpanath, R. S. Heath, J. R. Marshall, U. Klemstein, J. L. Galman, D. Woodlock, S. Bisagni, C. J. Taylor, J. Mangas-Sanchez, J. I. Ramsden, B. Dominguez and N. J. Turner, Sci. Adv., 2020, 6, eaay9320 Search PubMed.
- W. Zawodny, S. L. Montgomery, J. R. Marshall, J. D. Finnigan, N. J. Turner and J. Clayden, J. Am. Chem. Soc., 2018, 140, 17872–17877 Search PubMed.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G. D. Roiban, Nat. Cat., 2019, 2, 909–915 Search PubMed.
- E. Tassano and M. Hall, Chem. Soc. Rev., 2019, 48, 5596–5615 Search PubMed.
- M. M. Musa, F. Hollmann and F. G. Mutti, Catal. Sci. Technol., 2019, 9, 5487–5503 Search PubMed.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 Search PubMed.
- Y. P. Xue, C. H. Cao and Y. G. Zheng, Chem. Soc. Rev., 2018, 47, 1516–1561 Search PubMed.
- M. J. Abrahamson, E. Vazquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969–3972 Search PubMed.
- M. J. Abrahamson, J. W. Wong and A. S. Bommarius, Adv. Synth. Catal., 2013, 355, 1780–1786 Search PubMed.
- H. Y. Wang, G. Qu, J. K. Li, J. A. Ma, J. G. Guo, Y. C. Miao and Z. T. Sun, Catal. Sci. Technol., 2020, 10, 5945–5952 Search PubMed.
- V. Tseliou, T. Knaus, M. F. Masman, M. L. Corrado and F. G. Mutti, Nat. Commun., 2019, 10, 3717 Search PubMed.
- A. A. Caparco, E. Pelletier, J. L. Petit, A. Jouenne, B. R. Bommarius, V. de Berardinis, A. Zaparucha, J. A. Champion, A. S. Bommarius and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2020, 362, 2427–2436 Search PubMed.
- Y. Asano, K. Yamaguchi and K. Kondo, J. Bacteriol., 1989, 171, 4466–4471 Search PubMed.
- H. Chen, S. J. Collier, J. Nazor, J. Sukumaran, D. Smith, J. C. Moore, G. Hughes, J. Janey, G. Huisman, S. Novick, N. Agard, O. Alvizo, G. Cope, W. L. Yeo and S. N. Minor, US Pat., US9193957 B2, 2015 Search PubMed.
- J. F. Kirsch, G. Eichele, G. C. Ford, M. G. Vincent, J. N. Jansonius, H. Gehring and P. Christen, J. Mol. Biol., 1984, 174, 497–525 Search PubMed.
- B. Y. Hwang, B. K. Cho, H. Yun, K. Koteshwar and B. G. Kim, J. Mol. Catal. B: Enzym., 2005, 37, 47–55 Search PubMed.
- D. Koszeleivski, I. Lavandera, D. Clay, G. M. Guebitz, D. Rozzell and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 9337–9340 Search PubMed.
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 Search PubMed.
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 Search PubMed.
- H. Hayashi, J. Biochem., 1995, 118, 463–473 Search PubMed.
- Y. Katsura, M. Shirouzu, H. Yamaguchi, R. Ishitani, O. Nureki, S. Kuramitsu, H. Hayashi and S. Yokoyama, Proteins: Struct., Funct., Bioinf., 2004, 55, 487–492 Search PubMed.
- S. Hutson, Prog. Nucleic Acid Res. Mol. Biol., 2001, 70, 175–206 Search PubMed.
- T. Yoshimura, K. Nishimura, J. Ito, N. Esaki, H. Kagamiyama, J. M. Manning and K. Soda, J. Am. Chem. Soc., 1993, 115, 3897–3900 Search PubMed.
- S. A. Kelly, S. Mix, T. S. Moody and B. F. Gilmore, Appl. Microbiol. Biotechnol., 2020, 104, 4781–4794 Search PubMed.
- S. E. Payer, J. H. Schrittwieser and W. Kroutil, Eur. J. Org. Chem., 2017, 2553–2559 Search PubMed.
- R. C. Simon, B. Grischek, F. Zepeck, A. Steinreiber, F. Belaj and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 6713–6716 Search PubMed.
- S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 Search PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 Search PubMed.
- M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965–6982 Search PubMed.
- F. Parmeggiani, N. J. Weise, S. T. Ahmed and N. J. Turner, Chem. Rev., 2018, 118, 73–118 Search PubMed.
- B. Wu, W. Szymanski, M. M. Heberling, B. L. Feringa and D. B. Janssen, Trends Biotechnol., 2011, 29, 352–362 Search PubMed.
- N. J. Turner, Curr. Opin. Chem. Biol., 2011, 15, 234–240 Search PubMed.
- S. Bartsch and A. Vogel, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 291–312 Search PubMed.
- N. J. Weise, S. T. Ahmed, F. Parmeggiani, E. Siirola, A. Pushpanath, U. Schell and N. J. Turner, Catal. Sci. Technol., 2016, 6, 4086–4089 Search PubMed.
- F. Parmeggiani, S. L. Lovelock, N. J. Weise, S. T. Ahmed and N. J. Turner, Angew. Chem., Int. Ed., 2015, 54, 4608–4611 Search PubMed.
- H. Raj, W. Szymanski, J. de Villiers, H. J. Rozeboom, V. P. Veetil, C. R. Reis, M. de Villiers, F. J. Dekker, S. de Wildeman, W. J. Quax, A. M. W. H. Thunnissen, B. L. Feringa, D. B. Janssen and G. J. Poelarends, Nat. Chem., 2012, 4, 478–484 Search PubMed.
- J. L. Zhang, H. G. Fu, P. G. Tepper and G. J. Poelarends, Adv. Synth. Catal., 2019, 361, 2433–2437 Search PubMed.
- H. G. Fu, J. L. Zhang, M. Saifuddin, G. Cruiming, P. G. Tepper and G. J. Poelarends, Nat. Cat., 2018, 1, 186–191 Search PubMed.
- H. J. Wijma, R. J. Floor, S. Bjelic, S. J. Marrink, D. Baker and D. B. Janssen, Angew. Chem., Int. Ed., 2015, 54, 3726–3730 Search PubMed.
- R. F. Li, H. J. Wijma, L. Song, Y. L. Cui, M. Otzen, Y. E. Tian, J. W. Du, T. Li, D. D. Niu, Y. C. Chen, J. Feng, J. Han, H. Chen, Y. Tao, D. B. Janssen and B. Wu, Nat. Chem. Biol., 2018, 14, 664–670 Search PubMed.
- S. K. Wu, J. Liu and Z. Li, ACS Catal., 2017, 7, 5225–5233 Search PubMed.
- A. T. Li and Z. Li, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 479–505 Search PubMed.
- M. Hobisch, D. Holtmann, P. Gomez de Santos, M. Alcalde, F. Hollmann and S. Kara, Biotechnol. Adv., 2020, 107615 Search PubMed.
- A. Kantz and G. T. Gassner, Biochemistry, 2011, 50, 523–532 Search PubMed.
- H. Lin, Y. Liu and Z. L. Wu, Tetrahedron: Asymmetry, 2011, 22, 134–137 Search PubMed.
- A. Schmid, K. Hofstetter, H. J. Feiten, F. Hollmann and B. Witholt, Adv. Synth. Catal., 2001, 343, 732–737 Search PubMed.
- H. Toda, R. Imae and N. Itoh, Tetrahedron: Asymmetry, 2012, 23, 1542–1549 Search PubMed.
- H. Lin, Y. Liu and Z. L. Wu, Chem. Commun., 2011, 47, 2610–2612 Search PubMed.
- S. K. Wu, A. T. Li, Y. S. Chin and Z. Li, ACS Catal., 2013, 3, 752–759 Search PubMed.
- S. Wu, Y. Zhou, T. Wang, H. P. Too, D. I. C. Wang and Z. Li, Nat. Commun., 2016, 7, 11917 Search PubMed.
- C. Cui, C. Guo, H. Lin, Z. Y. Ding, Y. Liu and Z. L. Wu, Enzyme Microb. Technol., 2020, 132, 109391 Search PubMed.
- H. Xiao, S. Dong, Y. Liu, X. Q. Pei, H. Lin and Z. L. Wu, Catal. Sci. Technol., 2021, 11, 2195–2201 Search PubMed.
- S. Panke, A. Meyer, C. M. Huber, B. Witholt and M. G. Wubbolts, Appl. Environ. Microbiol., 1999, 65, 2324–2332 Search PubMed.
- M. G. Wubbolts, J. Hoven, B. Melgert and B. Witholt, Enzyme Microb. Technol., 1994, 16, 887–894 Search PubMed.
- M. J. Desmet, B. Witholt and H. Wynberg, J. Org. Chem., 1981, 46, 3128–3131 Search PubMed.
- M. J. Desmet, H. Wynberg and B. Witholt, Appl. Environ. Microbiol., 1981, 42, 811–816 Search PubMed.
- H. Fu, M. Newcomb and C. H. Wong, J. Am. Chem. Soc., 1991, 113, 5878–5880 Search PubMed.
- A. Q. H. Habetscrutzen, S. J. N. Carlier, J. A. M. Debont, D. Wistuba, V. Schurig, S. Hartmans and J. Tramper, Enzyme Microb. Technol., 1985, 7, 17–21 Search PubMed.
- C. A. G. M. Weijers, C. G. Vanginkel and J. A. M. Debont, Enzyme Microb. Technol., 1988, 10, 214–218 Search PubMed.
- A. T. Li, S. K. Wu, J. P. Adams, R. Snajdrova and Z. Li, Chem. Commun., 2014, 50, 8771–8774 Search PubMed.
- A. T. Li, J. Liu, S. Q. Pham and Z. Li, Chem. Commun., 2013, 49, 11572–11574 Search PubMed.
- N. N. Ma, Z. F. Chen, J. Chen, J. F. Chen, C. Wang, H. F. Zhou, L. S. Yao, O. Shoji, Y. Watanabe and Z. Q. Cong, Angew. Chem., Int. Ed., 2018, 57, 7628–7633 Search PubMed.
- S. Bormann, A. G. Baraibar, Y. Ni, D. Holtmann and F. Hollmann, Catal. Sci. Technol., 2015, 5, 2038–2052 Search PubMed.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 Search PubMed.
- R. Ulrich, J. Nuske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 Search PubMed.
- D. H. Anh, R. Ullrich, D. Benndorf, A. Svatos, A. Muck and M. Hofrichter, Appl. Environ. Microbiol., 2007, 73, 5477–5485 Search PubMed.
- G. Gröbe, R. Ullrich, M. J. Pecyna, D. Kapturska, S. Friedrich, M. Hofrichter and K. Scheibner, AMB Express, 2011, 1, 31 Search PubMed.
- M. C. Sigmund and G. J. Poelarends, Nat. Catal., 2020, 3, 690–702 Search PubMed.
- M. M. C. H. van Schie, A. T. Kaczmarek, F. Tieves, P. G. de Santos, C. E. Paul, I. W. C. E. Arends, M. Alcalde, G. Schwarz and F. Hollmann, ChemCatChem, 2020, 12, 3186–3189 Search PubMed.
- C. Guo, M. Saifuddin, T. Saravanan, M. Sharifi and G. J. Poelarends, ACS Catal., 2019, 9, 4369–4373 Search PubMed.
- G. C. Xu, M. Crotti, T. Saravanan, K. M. Kataja and G. J. Poelarends, Angew. Chem., Int. Ed., 2020, 59, 10374–10378 Search PubMed.
- V. Resch and U. Hanefeld, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 261–290 Search PubMed.
- R. M. Demming, S. C. Hammer, B. M. Nestl, S. Gergel, S. Fademrecht, J. Pleiss and B. Hauer, Angew. Chem., Int. Ed., 2019, 58, 173–177 Search PubMed.
- S. E. Payer, K. Faber and S. M. Glueck, Adv. Synth. Catal., 2019, 361, 2402–2420 Search PubMed.
- S. E. Payer, H. Pollak, S. M. Glueck and K. Faber, ACS Catal., 2018, 8, 2438–2442 Search PubMed.
- C. Wuensch, J. Gross, G. Steinkellner, K. Gruber, S. M. Glueck and K. Faber, Angew. Chem., Int. Ed., 2013, 52, 2293–2297 Search PubMed.
- M. Brovetto, D. Gamenara, P. S. Mendez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 Search PubMed.
- Y. F. Miao, M. Rahimi, E. M. Geertsema and G. J. Poelarends, Curr. Opin. Chem. Biol., 2015, 25, 115–123 Search PubMed.
- V. Resch, J. H. Schrittwieser, E. Siirola and W. Kroutil, Curr. Opin. Biotechnol., 2011, 22, 793–799 Search PubMed.
- L. E. Zetzsche and A. R. H. Narayan, Nat. Rev. Chem., 2020, 4, 334–346 Search PubMed.
- P. Clapes, in Science of Synthesis - Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 31–92 Search PubMed.
- M. Pohl, C. Wechsler and M. Müller, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 93–132 Search PubMed.
- K. Faber, Biotransformations in Organic Chemistry, Springer, Heidelberg, 7th edn, 2018 Search PubMed.
- M. Pickl, Curr. Opin. Green Sustain. Chem., 2021, 30, 100476 Search PubMed.
- P. Lehwald, M. Richter, C. Rohr, H. W. Liu and M. Muller, Angew. Chem., Int. Ed., 2010, 49, 2389–2392 Search PubMed.
- V. Laurent, E. Darii, A. Aujon, M. Debacker, J. L. Petit, V. Helaine, T. Liptaj, M. Breza, A. Mariage, L. Nauton, M. Traikia, M. Salanoubat, M. Lemaire, C. Guerard-Helaine and V. de Berardinis, Angew. Chem., Int. Ed., 2018, 57, 5467–5471 Search PubMed.
- K. Maruyama, J. Biochem., 1990, 108, 327–333 Search PubMed.
- P. Clapes and X. Garrabou, Adv. Synth. Catal., 2011, 353, 2263–2283 Search PubMed.
- V. Laurent, L. Gourbeyre, A. Uzel, V. Helaine, L. Nauton, M. Traikia, V. de Berardinis, M. Salanoubat, T. Gefflaut, M. Lemaire and C. Guerard-Helaine, ACS Catal., 2020, 10, 2538–2543 Search PubMed.
- R. Marin-Valls, K. Hernandez, M. Bolte, T. Parella, J. Joglar, J. Bujons and P. Clapes, J. Am. Chem. Soc., 2020, 142, 19754–19762 Search PubMed.
- I. Dreveny, C. Kratky and K. Gruber, Protein Sci., 2002, 11, 292–300 Search PubMed.
- S. Nanda, Y. Kato and Y. Asano, Tetrahedron, 2005, 61, 10908–10916 Search PubMed.
- J. N. Andexer, N. Staunig, T. Eggert, C. Kratky, M. Pohl and K. Gruber, ChemBioChem, 2012, 13, 1932–1939 Search PubMed.
- J. Zuegg, K. Gruber, M. Gugganig, U. G. Wagner and C. Kratky, Protein Sci., 1999, 8, 1990–2000 Search PubMed.
- K. Steiner, A. Glieder and M. Gruber-Khadjawi, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 2, pp. 1–30 Search PubMed.
- T. Purkarthofer, K. Gruber, M. Gruber-Khadjawi, K. Waich, W. Skranc, D. Mink and H. Griengl, Angew. Chem., Int. Ed., 2006, 45, 3454–3456 Search PubMed.
- M. Gruber-Khadjawi, T. Purkarthofer, W. Skranc and H. Griengl, Adv. Synth. Catal., 2007, 349, 1445–1450 Search PubMed.
- K. Fuhshuku and Y. Asano, J. Biotechnol., 2011, 153, 153–159 Search PubMed.
- C. Dresen, M. Richter, M. Pohl, S. Ludeke and M. Müller, Angew. Chem., Int. Ed., 2010, 49, 6600–6603 Search PubMed.
- E. Kasparyan, M. Richter, C. Dresen, L. S. Walter, G. Fuchs, F. J. Leeper, T. Wacker, S. L. A. Andrade, G. Kolter, M. Pohl and M. Müller, Appl. Microbiol. Biotechnol., 2014, 98, 9681–9690 Search PubMed.
- X. Y. Chen, Z. G. Wang, Y. J. Lou, Y. Z. Peng, Q. Y. Zhu, J. Xu and Q. Wu, Angew. Chem., Int. Ed., 2021, 60, 9326–9329 Search PubMed.
- E. Zandvoort, E. M. Geertsema, B. J. Baas, W. J. Quax and G. J. Poelarends, Angew. Chem., Int. Ed., 2012, 51, 1240–1243 Search PubMed.
- Y. F. Miao, P. G. Tepper, E. M. Geertsema and G. J. Poelarends, Eur. J. Org. Chem., 2016, 5350–5354 Search PubMed.
- J. Y. van der Meer, H. Poddar, B. J. Baas, Y. F. Miao, M. Rahimi, A. Kunzendorf, R. van Merkerk, P. G. Tepper, E. M. Geertsema, A. M. W. H. Thunnissen, W. J. Quax and G. J. Poelarends, Nat. Commun., 2016, 7, 10911 Search PubMed.
- N. Fujieda, H. Ichihashi, M. Yuasa, Y. Nishikawa, G. Kurisu and S. Itoh, Angew. Chem., Int. Ed., 2020, 59, 7717–7720 Search PubMed.
- S. E. Payer, X. Sheng, H. Pollak, C. Wuensch, G. Steinkellner, F. Himo, S. M. Glueck and K. Faber, Adv. Synth. Catal., 2017, 359, 2066–2075 Search PubMed.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. B. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 Search PubMed.
- X. Q. Huang, B. J. Wang, Y. J. Wang, G. D. Jiang, J. Q. Feng and H. M. Zhao, Nature, 2020, 584, 69–74 Search PubMed.
- C. G. Page, S. J. Cooper, J. S. DeHovitz, D. G. Oblinsky, K. F. Biegasiewicz, A. H. Antropow, K. W. Armbrust, J. M. Ellis, L. G. Hamann, E. J. Horn, K. M. Oberg, G. D. Scholes and T. K. Hyster, J. Am. Chem. Soc., 2021, 143, 97–102 Search PubMed.
- N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 Search PubMed.
- M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712–10723 Search PubMed.
- F. Tieves, S. J. P. Willot, M. M. C. H. van Schie, M. C. R. Rauch, S. H. H. Younes, W. Y. Zhang, J. J. Dong, P. G. de Santos, J. M. Robbins, B. Bommarius, M. Alcalde, A. S. Bommarius and F. Hollmann, Angew. Chem., Int. Ed., 2019, 58, 7873–7877 Search PubMed.
- W. Y. Zhang, B. O. Burek, E. Fernandez-Fueyo, M. Alcalde, J. Z. Bloh and F. Hollmann, Angew. Chem., Int. Ed., 2017, 56, 15451–15455 Search PubMed.
- D. Baud, P. L. Saaidi, A. Monfleur, M. Harari, J. Cuccaro, A. Fossey, M. Besnard, A. Debard, A. Mariage, V. Pellouin, J. L. Petit, M. Salanoubat, J. Weissenbach, V. de Berardinis and A. Zaparucha, ChemCatChem, 2014, 6, 3012–3017 Search PubMed.
- D. Baud, O. Peruch, P. L. Saaidi, A. Fossey, A. Mariage, J. L. Petit, M. Salanoubat, C. Vergne-Vaxelaire, V. de Berardinis and A. Zaparucha, Adv. Synth. Catal., 2017, 359, 1563–1569 Search PubMed.
- A. Amatuni, A. Shuster, A. Adibekian and H. Renata, Cell Chem. Biol., 2020, 27, 1318–1326 Search PubMed.
- A. Ilie, K. Harms and M. T. Reetz, J. Org. Chem., 2018, 83, 7504–7508 Search PubMed.
- G. D. Roiban, R. Agudo, A. Ilie, R. Lonsdale and M. T. Reetz, Chem. Commun., 2014, 50, 14310–14313 Search PubMed.
- R. Agudo, G. D. Roiban and M. T. Reetz, ChemBioChem, 2012, 13, 1465–1473 Search PubMed.
- K. Neufeld, B. Henssen and J. Pietruszka, Angew. Chem., Int. Ed., 2014, 53, 13253–13257 Search PubMed.
- I. Kaluzna, T. Schmitges, H. Straatman, D. van Tegelen, M. Müller, M. Schürmann and D. Mink, Org. Process Res. Dev., 2016, 20, 814–819 Search PubMed.
- A. Bokel, A. Ruhlmann, M. C. Hutter and V. B. Urlacher, ACS Catal., 2020, 10, 4151–4159 Search PubMed.
- S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 Search PubMed.
- J. Manning, M. Tavanti, J. L. Porter, N. Kress, S. P. De Visser, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2019, 58, 5668–5671 Search PubMed.
- W. Y. Zhang, E. Fernandez-Fueyo, Y. Ni, M. van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55–62 Search PubMed.
- S. Grobe, C. P. S. Badenhorst, T. Bayer, E. Hamnevik, S. K. Wu, C. W. Grathwol, A. Link, S. Koban, H. Brundiek, B. Grossjohann and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 753–757 Search PubMed.
- M. Hall, in The Enzymes, ed. P. Chaiyen and F. Tamanoi, Academic Press, 2020, vol. 47, pp. 37–62 Search PubMed.
- G. Grogan, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 3, pp. 285–312 Search PubMed.
- P. B. Brondani, M. W. Fraaije and G. de Gonzalo, in Green biocatalysis, ed. R. N. Patel, John Wiley & Sons, Hoboken, New Jersey, 2016, ch. 6, pp. 149–164 Search PubMed.
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 1063–1080 Search PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. J. Xu and H. Yang, Science, 2019, 366, 1255–1259 Search PubMed.
- P. D. de Maria, G. de Gonzalo and A. R. Alcantara, Catalysts, 2019, 9, 802 Search PubMed.
- S. Colonna, N. Gaggero, G. Carrea and P. Pasta, Chem. Commun., 1997, 439–440 Search PubMed.
- G. de Gonzalo, D. E. T. Pazmino, G. Ottolina, M. W. Fraaije and G. Carrea, Tetrahedron: Asymmetry, 2005, 16, 3077–3083 Search PubMed.
- G. de Gonzalo, D. E. T. Pazmino, G. Ottolina, M. W. Fraaije and G. Carrea, Tetrahedron: Asymmetry, 2006, 17, 130–135 Search PubMed.
- A. Rioz-Martinez, G. de Gonzalo, D. E. T. Pazmino, M. W. Fraaije and V. Gotor, Eur. J. Org. Chem., 2010, 6409–6416 Search PubMed.
- G. Carrea, B. Redigolo, S. Riva, S. Colonna, N. Gaggero, E. Battistel and D. Bianchi, Tetrahedron: Asymmetry, 1992, 3, 1063–1068 Search PubMed.
- N. M. Kamerbeek, D. B. Janssen, W. J. H. van Berkel and M. W. Fraaije, Adv. Synth. Catal., 2003, 345, 667–678 Search PubMed.
- F. Secundo, G. Carrea, S. Dallavalle and G. Franzosi, Tetrahedron: Asymmetry, 1993, 4, 1981–1982 Search PubMed.
- Y. K. Bong, S. W. Song, J. Nazor, M. Vogel, M. Widegren, D. Smith, S. J. Collier, R. Wilson, S. M. Palanivel, K. Narayauaswamy, B. Mijts, M. D. Clay, R. Fong, J. Colbeck, A. Appaswami, S. Muley, J. Zhu, X. Y. Zhang, J. Liang and D. Entwistle, J. Org. Chem., 2018, 83, 7453–7458 Search PubMed.
- B. A. Sandoval, A. J. Meichan and T. K. Hyster, J. Am. Chem. Soc., 2017, 139, 11313–11316 Search PubMed.
- J. Latham, E. Brandenburger, S. A. Shepherd, B. R. K. Menon and J. Micklefield, Chem. Rev., 2018, 118, 232–269 Search PubMed.
- A. V. Fejzagic, J. Gebauer, N. Huwa and T. Classen, Molecules, 2019, 24, 4008 Search PubMed.
- S. Grüschow, D. R. M. Smith, D. S. Gkotsi and R. J. M. Goss, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 3, pp. 313–359 Search PubMed.
- C. H. Zhao, S. Yan, Q. Li, H. C. Zhu, Z. Y. Zhong, Y. Ye, Z. X. Deng and Y. H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 9478–9484 Search PubMed.
- M. L. Hillwig and X. Y. Liu, Nat. Chem. Biol., 2014, 10, 921–923 Search PubMed.
- A. J. Mitchell, Q. Zhu, A. O. Maggiolo, N. R. Ananth, M. L. Hillwig, X. Y. Liu and A. K. Boal, Nat. Chem. Biol., 2016, 12, 636–640 Search PubMed.
- T. Hayashi, M. Ligibel, E. Sager, M. Voss, J. Hunziker, K. Schroer, R. Snajdrova and R. Buller, Angew. Chem., Int. Ed., 2019, 58, 18535–18539 Search PubMed.
- S. Duewel, L. Schmermund, T. Faber, K. Harms, V. Srinivasan, E. Meggers and S. Hoebenreich, ACS Catal., 2020, 10, 1272–1277 Search PubMed.
- M. Voss, S. H. Malca and R. Buller, Chem. – Eur. J., 2020, 26, 7336–7345 Search PubMed.
- E. Romero, B. S. Jones, B. N. Hogg, A. R. Casamajo, M. A. Hayes, S. L. Flitsch, N. J. Turner and C. Schnepel, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202014931.
- L. A. Harwood, L. L. Wong and J. Robertson, Angew. Chem., Int. Ed., 2021, 60, 4434–4447 Search PubMed.
- A. Liese, K. Seelbach, A. Buchholz and J. Haberland, in Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, Wiley-VCH, 2nd edn, 2006, pp. 273–349 Search PubMed.
- A. Liese, K. Seelbach, A. Buchholz and J. Haberland, in Industrial Biotransformations, ed. A. Liese, K. Seelbach and C. Wandrey, Wiley-VCH, 2nd edn, 2006, pp. 350–446 Search PubMed.
- M. Pickl, A. Swoboda, E. Romero, C. K. Winkler, C. Binda, A. Mattevi, K. Faber and M. W. Fraaije, Angew. Chem., Int. Ed., 2018, 57, 2864–2868 Search PubMed.
- J. Xu, Y. J. Hu, J. J. Fan, M. Arkin, D. Y. Li, Y. Z. Peng, W. H. Xu, X. F. Lin and Q. Wu, Angew. Chem., Int. Ed., 2019, 58, 8474–8478 Search PubMed.
- G. de Gonzalo, W. J. H. van Berkel and M. W. Fraaije, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgart, 2015, vol. 3, pp. 187–233 Search PubMed.
- K. Konigsberger and H. Griengl, Biorg. Med. Chem., 1994, 2, 595–604 Search PubMed.
- N. Berezina, E. Kozma, R. Furstoss and V. Alphand, Adv. Synth. Catal., 2007, 349, 2049–2053 Search PubMed.
- A. Rioz-Martinez, G. de Gonzalo, D. E. T. Pazmino, M. W. Fraaije and V. Gotor, J. Org. Chem., 2010, 75, 2073–2076 Search PubMed.
- C. Rodriguez, G. de Gonzalo, A. Rioz-Martinez, D. E. T. Pazmino, M. W. Fraaije and V. Gotor, Org. Biomol. Chem., 2010, 8, 1121–1125 Search PubMed.
- N. Berezina, V. Alphand and R. Furstoss, Tetrahedron: Asymmetry, 2002, 13, 1953–1955 Search PubMed.
- A. Cuetos, A. Rioz-Martinez, F. R. Bisogno, B. Grischek, I. Lavandera, G. de Gonzalo, W. Kroutil and V. Gotor, Adv. Synth. Catal., 2012, 354, 1743–1749 Search PubMed.
- D. Kalaitzakis and I. Smonou, in Synthetic methods for biologically active molecules, ed. E. Brenna, Wiley-VCH, Weinheim, 2014, pp. 307–327 Search PubMed.
- A. Cuetos, A. Díaz-Rodríguez and Í. Lavandera, in Synthetic methods for biologically active molecules, ed. E. Brenna, Wiley-VCH, Weinheim, 2014, pp. 85–111 Search PubMed.
- J. A. Friest, Y. Maezato, S. Broussy, P. Blum and D. B. Berkowitz, J. Am. Chem. Soc., 2010, 132, 5930–5931 Search PubMed.
- E. Tassano, K. Faber and M. Hall, Adv. Synth. Catal., 2018, 360, 2742–2751 Search PubMed.
- L. Pollegioni and G. Molla, in Science of Synthesis – Biocatalysis in organic synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Thieme, Stuttgard, 2015, vol. 3, pp. 235–284 Search PubMed.
- N. J. Turner, Chem. Rev., 2011, 111, 4073–4087 Search PubMed.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 Search PubMed.
- C. J. Dunsmore, R. Carr, T. Fleming and N. J. Turner, J. Am. Chem. Soc., 2006, 128, 2224–2225 Search PubMed.
- M. Alexeeva, A. Enright, M. J. Dawson, M. Mahmoudian and N. J. Turner, Angew. Chem., Int. Ed., 2002, 41, 3177–3180 Search PubMed.
- V. F. Batista, J. L. Galman, D. C. G. A. Pinto, A. M. S. Silva and N. J. Turner, ACS Catal., 2018, 8, 11889–11907 Search PubMed.
- E. O'Reilly, C. Iglesias, D. Ghislieri, J. Hopwood, J. L. Galman, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 2447–2450 Search PubMed.
- T. Li, J. Liang, A. Ambrogelly, T. Brennan, G. Gloor, G. Huisman, J. Lalonde, A. Lekhal, B. Mijts, S. Muley, L. Newman, M. Tobin, G. Wong, A. Zaks and X. Y. Zhang, J. Am. Chem. Soc., 2012, 134, 6467–6472 Search PubMed.
- M. Ogasawara, Tetrahedron: Asymmetry, 2009, 20, 259–271 Search PubMed.
- S. C. Yu and S. M. Ma, Chem. Commun., 2011, 47, 5384–5418 Search PubMed.
- G. Y. Ma and M. P. Sibi, Chem. – Eur. J., 2015, 21, 11644–11657 Search PubMed.
- B. Skrobo, J. D. Rolfes and J. Deska, Tetrahedron, 2016, 72, 1257–1275 Search PubMed.
- C. Gagnon, E. Godin, C. Minozzi, J. Sosoe, C. Pochet and S. K. Collins, Science, 2020, 367, 917–921 Search PubMed.
- S. Ramaswamy, R. A. H. F. Hui and J. B. Jones, J. Chem. Soc., Chem. Commun., 1986, 1545–1546 Search PubMed.
- M. Pietzsch, O. Vielhauer, D. Pamperin, B. Ohse and H. Hopf, J. Mol. Catal. B: Enzym., 1999, 6, 51–57 Search PubMed.
- B. C. N. M. Jones, J. V. Silverton, C. Simons, S. Megati, H. Nishimura, Y. Maeda, H. Mitsuya and J. Zemlicka, J. Med. Chem., 1995, 38, 1397–1405 Search PubMed.
- J. Deska and J. E. Backvall, Org. Biomol. Chem., 2009, 7, 3379–3381 Search PubMed.
- J. Deska, C. D. Ochoa and J. E. Backvall, Chem. – Eur. J., 2010, 16, 4447–4451 Search PubMed.
- S. Miyano, K. Kawahara, Y. Inoue and H. Hashimoto, Chem. Lett., 1987, 355–356 Search PubMed.
- R. J. Kazlauskas, J. Am. Chem. Soc., 1989, 111, 4953–4959 Search PubMed.
- M. Inagaki, J. Hiratake, T. Nishioka and J. Oda, Agric. Biol. Chem., 1989, 53, 1879–1884 Search PubMed.
- G. A. I. Moustafa, Y. Oki and S. Akai, Angew. Chem., Int. Ed., 2018, 57, 10278–10282 Search PubMed.
- M. Bertrand, E. Ferre, G. Gil, J. Lepetit and L. Deveze, Tetrahedron Lett., 1980, 21, 1711–1714 Search PubMed.
- E. Ferre, G. Gil, M. Bertrand and J. Lepetit, Appl. Microbiol. Biotechnol., 1985, 21, 258–266 Search PubMed.
- K. Kawahara, M. Matsumoto, H. Hashimoto and S. Miyano, Chem. Lett., 1988, 1163–1164 Search PubMed.
- S. Staniland, R. W. Adams, J. J. W. McDouall, I. Maffucci, A. Contini, D. M. Grainger, N. J. Turner and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 10755–10759 Search PubMed.
- H. Aldemir, R. Richarz and T. A. M. Gulder, Angew. Chem., Int. Ed., 2014, 53, 8286–8293 Search PubMed.
- K. G. Hugentobler and M. Müller, Biorg. Med. Chem., 2018, 26, 1374–1377 Search PubMed.
- L. Frtges, S. Obermaier, W. Thiele, S. Foegen and M. Mller, ChemBioChem, 2019, 20, 1928–1932 Search PubMed.
- S. Obermaier, W. Thiele, L. Furtges and M. Müller, Angew. Chem., Int. Ed., 2019, 58, 9125–9128 Search PubMed.
- B. Pickel, M. A. Constantin, J. Pfannstiel, J. Conrad, U. Beifuss and A. Schaller, Angew. Chem., Int. Ed., 2010, 49, 202–204 Search PubMed.
- J. Y. Hu, H. Li and Y. H. Choo, J. Am. Chem. Soc., 2019, 141, 8068–8072 Search PubMed.
- W. Thiele, S. Obermaier and M. Müller, ACS Chem. Biol., 2020, 15, 844–848 Search PubMed.
- S. S. Kim and E. S. Sattely, J. Am. Chem. Soc., 2021, 143, 5011–5021 Search PubMed.
- L. S. Mazzaferro, W. Huttel, A. Fries and M. Müller, J. Am. Chem. Soc., 2015, 137, 12289–12295 Search PubMed.
- S. Obermaier and M. Müller, Biochemistry, 2019, 58, 2589–2593 Search PubMed.
- C. C. Forneris and M. R. Seyedsayamdost, Angew. Chem., Int. Ed., 2018, 57, 8048–8052 Search PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 Search PubMed.
- U. T. Bornscheuer, Philos. Trans. R. Soc., A, 2018, 376, 20170063 Search PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. L. Qin, A. Zidek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 Search PubMed.
- E. Callaway, Nature, 2020, 588, 203–204 Search PubMed.
- B. Hauer, ACS Catal., 2020, 10, 8418–8427 Search PubMed.
- N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 Search PubMed.
- R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem. – Eur. J., 2017, 23, 12040–12063 Search PubMed.
- E. J. Corey, Chem. Soc. Rev., 1988, 17, 111–133 Search PubMed.
- E. J. Corey, Angew. Chem., Int. Ed. Engl., 1991, 30, 455–465 Search PubMed.
- F. Parmeggiani, A. R. Casamajo, D. Colombo, M. C. Ghezzi, J. L. Galman, R. A. Chica, E. Brenna and N. J. Turner, Green Chem., 2019, 21, 4368–4379 Search PubMed.
- A. Stamm, A. Biundo, B. Schmidt, J. Brucher, S. Lundmark, P. Olsen, L. Fogelstrom, E. Malmstrom, U. T. Bornscheuer and P. O. Syren, ChemBioChem, 2019, 20, 1664–1671 Search PubMed.
- N. Hadadi and V. Hatzimanikatis, Curr. Opin. Chem. Biol., 2015, 28, 99–104 Search PubMed.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, Nat. Catal., 2021, 4, 98–104 Search PubMed.
- Y. Kim, J. Y. Ryu, H. U. Kim, W. D. Jang and S. Y. Lee, Biotechnol. J., 2021, 16, 2000605 Search PubMed.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 Search PubMed.
- M. Honig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942–8973 Search PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Groger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 Search PubMed.
- H. Groger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 Search PubMed.
- S. Martinez, L. Veth, B. Lainer and P. Dydio, ACS Catal., 2021, 11, 3891–3915 Search PubMed.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–103 Search PubMed.
- E. J. Craven, J. Latham, S. A. Shepherd, I. Khan, A. Diaz-Rodriguez, M. F. Greaney and J. Micklefield, Nat. Catal., 2021, 4, 385–394 Search PubMed.
- Z. C. Litman, Y. J. Wang, H. M. Zhao and J. F. Hartwig, Nature, 2018, 560, 355–359 Search PubMed.
- H. Sato, W. Hummel and H. Groger, Angew. Chem., Int. Ed., 2015, 54, 4488–4492 Search PubMed.
- S. Gonzalez-Granda, I. Lavandera and V. Gotor-Fernandez, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202015215.
- P. Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 Search PubMed.
- R. J. Kazlauskas, A. N. E. Weissfloch, A. T. Rappaport and L. A. Cuccia, J. Org. Chem., 1991, 56, 2656–2665 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |