
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular recognition of sialoglycans by streptococcal Siglec-like adhesins: toward the shape of specific inhibitors†
Cristina
Di Carluccio
a,
Rosa Ester
Forgione
a,
Andrea
Bosso
b,
Shinji
Yokoyama
c,
Yoshiyuki
Manabe
 c,
Elio
Pizzo
b,
Antonio
Molinaro
ac,
Koichi
Fukase
c,
Marco
Fragai
de,
Barbara A.
Bensing
fg,
Roberta
Marchetti
*a and
Alba
Silipo
*a
c,
Elio
Pizzo
b,
Antonio
Molinaro
ac,
Koichi
Fukase
c,
Marco
Fragai
de,
Barbara A.
Bensing
fg,
Roberta
Marchetti
*a and
Alba
Silipo
*a
aDepartment of Chemical Sciences, University of Naples Federico II, Via Cinthia 4, 80126, Naples, Italy. E-mail: marchetti@unina.it; silipo@unina.it
bDepartment of Biology, University of Naples Federico II, Via Cinthia 4, 80126, Naples, Italy
cDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan
dMagnetic Resonance Center (CERM), University of Florence, and Consorzio Interuniversitario Risonanze Magnetiche di Metallo Proteine (CIRMMP), Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy
eDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3-13, 50019 Sesto Fiorentino, Italy
fDivision of Infectious Diseases, Veterans Affairs Medical Center, Department of Medicine, University of California, San Francisco, California 94121, USA
gNorthern California Institute for Research and Education, San Francisco, California 94121, USA
First published on 18th October 2021
Abstract
Streptococcus gordonii and Streptococcus sanguinis, commensal bacteria present in the oral cavity of healthy individuals, upon entry into the bloodstream can become pathogenic, causing infective endocarditis (IE). Sialic acid-binding serine-rich repeat adhesins on the microbial surface represent an important factor of successful infection to cause IE. They contain Siglec-like binding regions (SLBRs) that variously recognize different repertoires of O-glycans, with some strains displaying high selectivity and others broader specificity. We here dissect at an atomic level the mechanism of interaction of SLBR-B and SLBR-H from S. gordonii with a multivarious approach that combines NMR spectroscopy and computational and biophysical studies. The binding pockets of both SLBRs are broad enough to accommodate extensive interactions with sialoglycans although with key differences related to strain specificity. Furthermore, and significantly, the pattern of interactions established by the SLBRs are mechanistically very different from those reported for mammalian Siglecs despite them having a similar fold. Thus, our detailed description of the binding modes of streptococcal Siglec-like adhesins sparks the development of tailored synthetic inhibitors and therapeutics specific for Streptococcal adhesins to counteract IE, without impairing the interplay between Siglecs and glycans.
Introduction
In infective endocarditis (IE) disease, microbes colonize and proliferate in the endocardium, resulting in the formation of emboli and strokes if untreated. Despite the development of therapies based on long-term antibiotic treatment, due to the high risk of incidence and mortality and the rise in antimicrobial resistance, improving treatments to counteract IE is urgently required.1 The pathogenesis and etiology of IE have been partially defined and typically originate when commensal bacteria transit into the bloodstream.2 Whereas some species such as Staphylococcus aureus may infect native or prosthetic valves and cause acute disease, the Mitis group of oral streptococci tend to infect damaged valves and cause more chronic, sub-acute disease.3,4 Pieces of evidence suggest that the adherence of oral streptococci to platelets represents a crucial step in the pathogenesis of infective endocarditis and this process is mediated in part by the presence of serine-rich repeat (SRR) proteins anchored to the bacterial cell wall.5–7SRR adhesins are organized with an N-terminal ∼90 amino acid signal peptide (SP), followed by a short serine-rich region (SRR1), a ligand binding region (BR), a long serine-rich repeat region (SRR2), and a C-terminal cell wall anchor (CWA) (Fig. 1a). Depending on the organism to which the SRR adhesins belong, the BRs can vary in amino acid length and sequence, and in secondary structure and folding, and these characteristics define the ligand specificity for different bacterial strains. The SRR adhesins expressed by the oral commensal species Streptococcus gordonii and Streptococcus sanguinis typically have “Siglec-like” BRs (SLBRs). SLBR-B and SLBR-H are two of the most extensively characterized SLBRs, derived from the SRR adhesins expressed by S. gordonii strains M99 and DL1, respectively, and are involved in the recognition of terminal Neu5Ac-α-(2,3)–Gal glycans exposed on MG2/MUC7 salivary mucins and/or the human platelet glycoprotein GPIbα.8
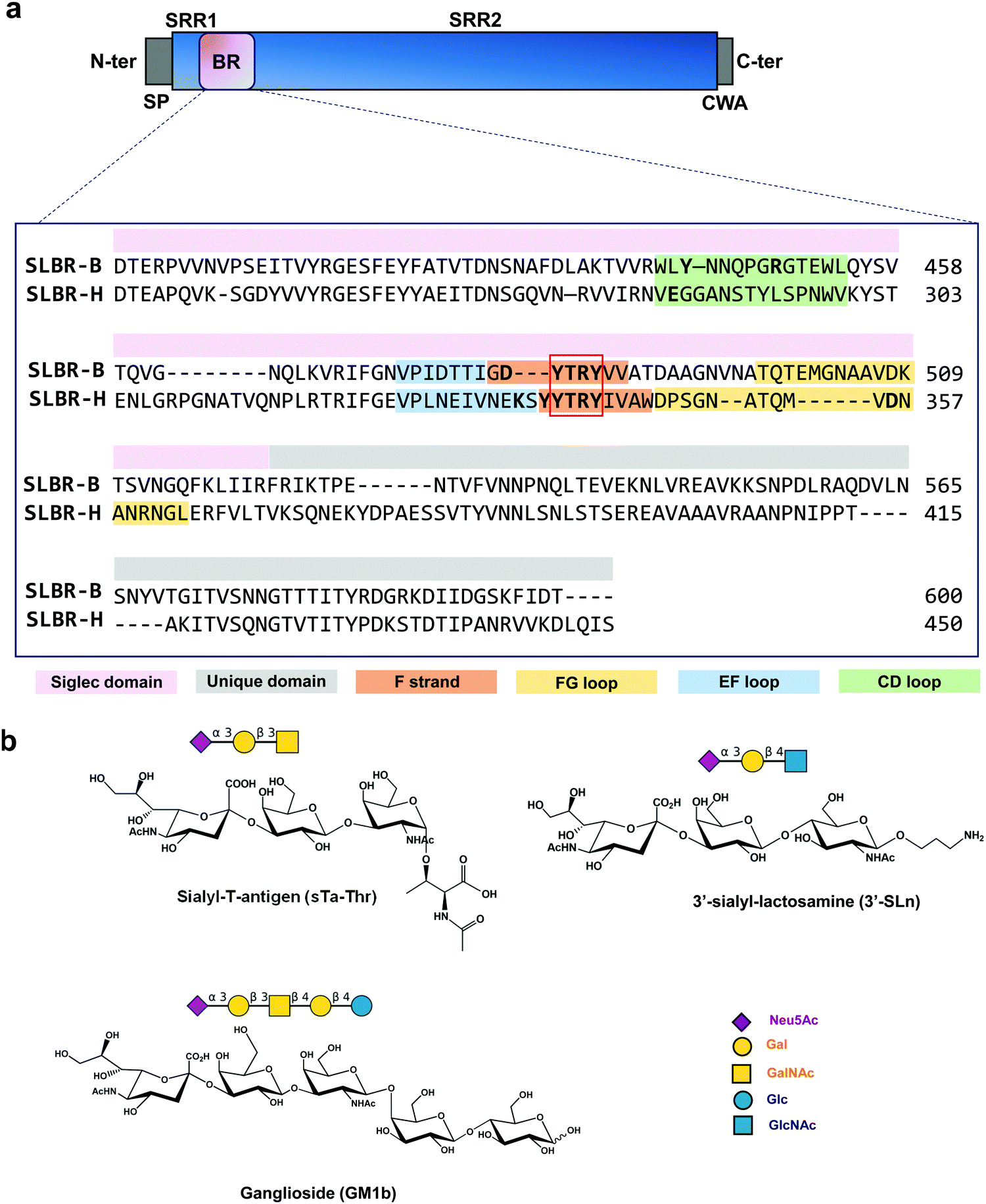 | ||
| Fig. 1 (a) SLBR-B and SLBR-H alignment using Python in PyMol. The upper panel shows the general domain organization of SRR adhesins. SP, signal peptide; SRR1, serine-rich region 1; BR, ligand binding region; SRR2, serine-rich region 2; CWA, cell wall anchor domain. SLBR-B includes only the Siglec and Unique domains (lacking the CnaA domain, present instead in GspB). The sequence corresponding to the Siglec domain of SLBR-B and -H is indicated in pink, and the sequence of the Unique domain in grey. The amino acids in the FG, EF and CD loops are colored in yellow, blue and green, respectively. The YTRY consensus sequence is indicated by the red square and the F-strand that hosts the YTRY motif is shown in orange. Key amino acids are highlighted in bold. (b) Sialoglycans studied in the molecular interactions with SLBR-H and SLBR-B. From top to bottom: sialyl-T-antigen linked to threonine (sTa-Thr), 3′-sialylactosamine (3′-SLn) and ganglioside GM1b, also displayed with SNFG representation. | ||
SLBR-B and SLBR-H are composed of two conserved domains important for sialoglycan binding: a V-set Ig fold Siglec subdomain, highly similar to that found in mammalian Siglecs (sialic acid binding immunoglobulin-like lectins) in terms of topology and strand inserts (hence the name “Siglec-like” adhesins), and the Unique domain, not directly involved in the interaction with carbohydrates, though possibly modulating the conformation of the nearby Siglec domain. A third domain, called CnaA, is also present in the serine-reach repeat adhesin GspB but does not contribute to glycan binding.9 The Unique and Siglec subdomains of SRR adhesins play key roles in mediating bacterial recognition of host sialoglycans.10,11 In particular, a YTRY consensus sequence, further refined to a ΦTRX motif in the broader family of SLBRs, is present in the F-strand of the Siglec domain (Fig. 1a), establishing crucial contacts with Neu5Ac-α-(2,3)-Gal containing ligands.12Although SLBR-B and SLBR-H show moderate sequence identity, the selectivity of their BRs toward sialoglycan structures is different;13 indeed, SLBR-B strictly recognizes sialyl-T-antigen (sTa), while SLBR-H binds a repertoire of glycans with different shapes and topologies, including sTa, 3′-sialylactosamine (3′-SLn) and related structures.14 Also, the impact of the Siglec-like adhesins on the virulence of these streptococcal pathogens differs with respect to the bound sialoglycan: for example, strains that bind sialyl-T-antigen are more virulent compared with a strain that binds core 2 O-glycans.9 This emphasizes the need for selective inhibition of binding to the former O-glycan structure.
The role of the Siglec-like adhesins in the pathogenesis of infective endocarditis has been widely demonstrated in vitro and in vivo.10 The presence of GspB or Hsa on the Streptococcus gordonii surface mediates the bacterial binding to human platelets and contributes to the ability of the organism to colonize damaged heart valves. Indeed, the deletion or the introduction of single point mutations within gspB or hsa resulted in a significant reduction of the virulence of the organism, as determined using animal models of infective endocarditis.6,7 Although the features of SLBR-H and SLBR-B binding pockets have been investigated,15 the mechanisms of sialoglycan recognition and binding by Siglec-like adhesins have not been determined, including the different affinity and binding specificity together with the dynamic range of conformations adopted by the SLBR–sialoglycan complexes. Therefore, we here dissect, at an atomic level, the recognition profiles and binding modes of different sialoglycans by SLBR-B and SLBR-H adhesins (Fig. 1b)16, describe the 3D complexes, map the binding epitopes and the bioactive conformations, and discuss the structural and conformational features driving the different affinity and selectivity of SLBR-B and SLBR-H toward sialoglycans17 (3′-sialylactosamine (3′-SLn), sialyl-T-antigen (sTa-Thr), GM1b ganglioside).
Results
The Siglec and Unique domains of SLBR-B and SLBR-H were expressed as GST fusion proteins to promote a greater expression, purification and solubility, without affecting the interactions with the glycans under study.8 The binding profiles of different sialoglycans recognized by SLBR-B and SLBR-H were investigated by using complementary approaches. Steady-state fluorescence analysis (Fig. S1, ESI†) was undertaken to provide the binding affinities of SLBR-B and SLBR-H for α-2,3-sialoglycans. A concentration dependent reduction in fluorescence intensity upon sialoglycan binding was used to monitor the interaction and the dissociation constant (Kd) was determined by non-linear regression analysis (Fig. S1, ESI†). The Kd values of SLBR-B and SLBR-H in the interaction with sTa were almost comparable, indicating similar recognition of the substrate, while a slightly stronger affinity was found for the interaction of SLBR-H with 3′-SLn.Three ligands were investigated when bound to SLBR-B and SLBR-H, after first being characterized through canonical NMR experiments: sialyl-T-antigen [Neu5Ac-α-(2,3)–Gal-β-(1,3)–GalNAc-α-Thr, sTa-Thr], 3′-sialylactosamine [Neu5Ac-α-(2,3)–Gal-β-(1,4)–GlcNAc-β-O-(CH2)3NH2, 3′-SLn] and GM1b ganglioside [Neu5Ac-α-(2,3)–Gal-β-(1,3)–GalNAc-β-(1,4)–Gal-β-(1,4)–Glc–OH]. Saturation transfer difference NMR18,19 (STD NMR) analysis was employed to map the interacting epitopes of the ligands upon binding, transferred-NOESY20,21 (tr-NOESY) experiments to describe the bioactive conformation of the ligands, and computational studies to depict the 3D complex.
SLBR-H interaction with sTa-Thr
As indicated by the numerous and highly intense signals in the STD NMR spectroscopy performed on a mixture of SLBR-H with sTa-Thr (Fig. 2a, upper panel), the SLBR-H binding pocket was large enough to accommodate the entire trisaccharide. In the resulting epitope mapping (Fig. 2b, upper panel), indeed, all three glycan residues showed STD enhancement and thus contributed to the binding process, although the GalNAc unit interacted with SLBR-H to a lesser extent, as testified by the lower STD signals. The sialic acid (Neu5Ac) H7 proton, as well as H4 and H6 of the Gal unit, showed high relative STD percentages in the binding epitope (above 80%). Strong STD effects, above 60%, were also observed for H4, H6 and H9 of Neu5Ac and for H5 of Gal. The other protons of Neu5Ac and H3 of Gal displayed discrete STD signals, as did the GalNAc H5 and H6 protons. Lower STD intensities were detected for the H3ax of Neu5Ac, the H2 of Gal, the acetyl group of GalNAc, and the threonine moiety, suggesting that these protrude from the binding pocket of the protein.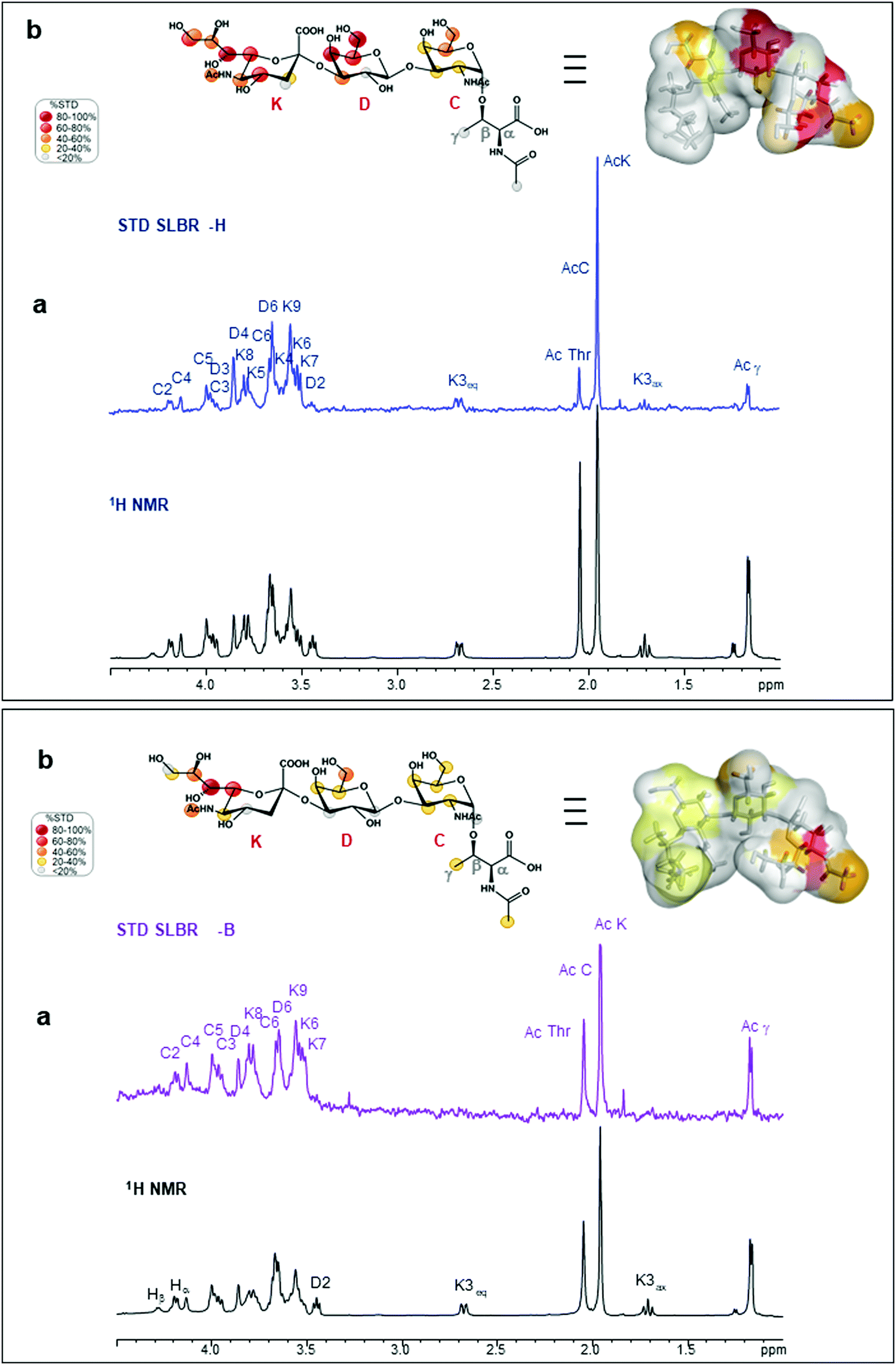 | ||
| Fig. 2 NMR analysis of sTa-Thr bound to SLBR-H and SLBR-B. Upper panel: SLBR-H–sTa-Thr binding analysis. (a) STD-NMR spectrum (blue) of SLBR-H–sTa-Thr and the unsaturated reference spectrum (black). (b) Epitope map of the ligand recognized by SLBR-H; the STD-derived epitope mapping of sTa-Thr in its bioactive conformation with the surface coloured according to the STD effects is also shown. Lower panel: SLBR-B–sTa-Thr binding analysis. (a) STD-NMR spectrum (purple) of SLBR-B–sTa-Thr and the unsaturated reference spectrum (black). (b) Epitope map of the ligand recognized by SLBR-B and its bioactive conformation with the surface coloured according to the STD effects. | ||
The STD NMR results were then complemented with a conformational analysis based on tr-NOESY and molecular modelling. The sTa-Thr structure was manually docked in the SLBR-H binding site, a 100 ns MD simulation was performed, and the molecular interactions were monitored and then compared with the experimental data. The results indicated that the sTa-Thr shape and conformation are guided by the glycosidic torsion angles, namely φ(C1–C2–O–C3′)/ψ(C2–O–C3′–H3′) around Neu5Ac-α-(2,3)-Gal and φ(H1–C1–O–C3′)/ψ(C1–O–C3′–H3′) around the Gal-β-(1,3)–GalNAc bonds. The glycosidic torsion trajectories were sampled during the MD simulations in the free and bound states (Fig. S2, ESI†) to evaluate the conformational behavior of sTa-Thr when complexed with SLBR-H. No significant differences were found upon binding, except for the φ torsion angle along the Neu5Ac-α-(2,3)–Gal unit, which in the bound state populated exclusively the family around −60° and in the free state also adopted a value of 180°. Additionally, the NOE contacts between ROESY (Fig. S3a, ESI†) and tr-NOESY spectra (Fig. S3b, ESI†) confirmed a comparable conformational behavior. The contacts that sTa-Thr established with SLBR-H, analyzed during the MD simulation, mainly involved the amino acids of the ΦTRY motif of the F-strand, which plays an essential role in the sialoglycan binding. An extended network of polar interactions stabilized the complex, and numerous significant H-bonds, remained during the simulation, were established at the protein–glycan interface (Fig. 3a and c). Among the most stable there were those involving the Thr339, Arg340 and Tyr341 of the ΦTRY motif with the Neu5Ac and Gal residues (Fig. S2f, ESI†). Stable polar interactions, above 90% of the simulation time, were established between the carboxylate group of Neu5Ac and both the backbone and the hydroxyl group of the lateral chain of Thr339 (H and HG1 in Fig. S2f, ESI†) as well as those between the guanidinium group of Arg340 and O8 and O9 of Neu5Ac.
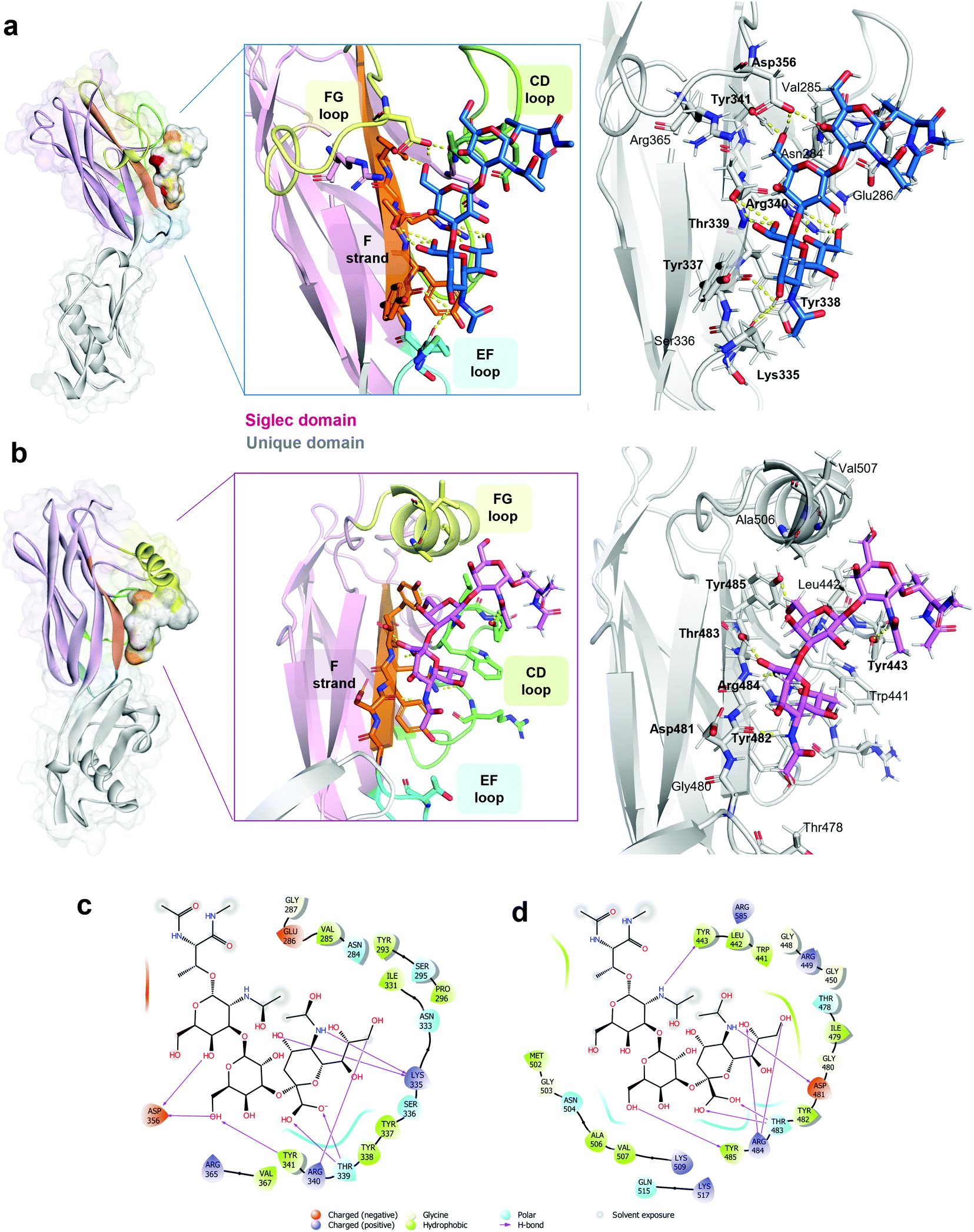 | ||
| Fig. 3 3D view of SLBR-H–sTa-Thr and SLBR-B–sTa-Thr complexes. (a) 3D view of the SLBR-H–sTa-Thr complex according to STD, tr-NOESY and MD data. The binding site is coloured according to the sequence in Fig. 1a. The starting pose of the SLBR-H–sTa-Thr complex was obtained by manual docking using the published structure of the protein in complex with sialyl-T-antigen as the reference (6EFD). (b) 3D view of the SLBR-B–sTa-Thr complex in accordance with STD, tr-NOESY and MD data, with the binding site coloured according to the sequence in Fig. 1a. (c) Two-dimensional plot of the SLBR-H–sTa-Thr complex highlighting the interactions established between SLBR-H and sTa-Thr: solid arrows represent hydrogen bonds with the functional groups of the backbone; the other residues in the binding pocket participate in polar and hydrophobic interactions. (d) Two-dimensional plot of the interactions established at the SLBR-B–sTa-Thr interface. | ||
Furthermore, the Tyr341 hydroxyl group also formed a significant H-bond with the O6 of Gal (87%), and the interactions of the Asp356 carboxylate group with the hydrogen of the hydroxyl group at position 6 of Gal explained the high contribution to the binding of this region of the Gal residue (Fig. 2b, upper panel). Consistent with the STD NMR results, OH at position 4 of Neu5Ac interacted with the backbone carboxyl group of Lys335, and the amide group at the C5 of Neu5Ac also served as a H-bond donor to the oxygen of the carboxyl group of Tyr337. Furthermore, polar interactions of the Asp356 chain were found with OH at positions 4 and 6 of GalNAc, in line with the STD NMR results which showed a partial involvement of the N-acetyl-galactosamine in the interaction with SLBR-H (Fig. 2, upper panel). In addition, the presence of aromatic amino acids in the binding pocket of SLBR-H contributed to the network of interactions with sTa-Thr, including aromatic hydrogen bonds and hydrophobic interactions (Fig. 3c). Among these, pi–alkyl interactions involving the aromatic rings of Tyr338 and Tyr341, both belonging to the ΦTRY consensus sequence, with positions C8 of Neu5Ac and C6 of Gal, respectively, were found. Finally, the hydrophobic interaction established between the H7 proton of Neu5Ac and the proximal CH of Tyr338 created a contact that likely determined the strongest STD response.
SLBR-B interaction with sTa-Thr
Interactions between the more selective SLBR-B and sTa-Thr, as well as the ligand conformation upon binding, were also assessed (Fig. 2, lower panel). The STD NMR spectrum clearly showed that the sialic acid displayed the highest STD contribution (Fig. 2a and b, lower panel), indicative of its proximity to the SLBR-B binding pocket. Among Neu5Ac protons recognized by the adhesin, those belonging to the glycerol chain showed a strong contribution to the binding, together with the acetyl group at position 5. The H6 of Gal also received a good magnetization transfer from SLBR-B, with a STD percentage around 40%. Although giving STD signals, the rest of the sTa-Thr protons were less involved in the interaction with the protein, showing %STD values below 40% (Fig. 2b).The comparison between ROESY (Fig. S3a, ESI†) and tr-NOESY (Fig. S3c, ESI†) experiments indicated that the ligand conformation was unchanged in complex with SLBR-B. Once the bioactive conformation and ligand interacting epitopes were clarified, the binding profile of the complex of SLBR-B with sTa-Thr was further investigated via computational approaches. A 100 ns MD simulation was performed on the complex, built by docking the ligand in the published crystal structure of the protein (pdb: 5IUC). Regarding the Neu5Ac-α-(2,3)–Gal glycosidic linkage (Fig. S5a, ESI†), the energetic minimum populated in the bound state corresponded to the most populated in the free state (Fig. S2a, ESI†). Even for the Gal-β-(1,3)–GalNAc glycosidic bond, the trajectories monitored in the bound state were mostly maintained with respect to the free state, with φ/ψ torsion angles assuming the most stable minimum around 40°/−20° (Fig. S5b, ESI†). The stability of the complex was also supported by RMSD (root-mean-square deviation) variation during the MD simulation (Fig. S5c, ESI†), confirming that the ligand atomic positions did not change during the simulation and indicating the stability of the ligand in the SLBR-B binding pocket.
A map of the molecular interactions occurring within the complex of SLBR-B and sTa-Thr (Fig. 3b and d) helped to dissect the sTa-Thr arrangement in the protein binding pocket. A few polar interactions remained in the complex and only three H-bonds were stably present for more than 50% of the MD simulation time. The major contribution came from the Neu5Ac unit, while Gal and GalNAc residues were less recognized by SLBR-B, supporting the STD NMR data (Fig. 2, lower panel). The most stable contact occurred between the amide group of Neu5Ac and the Asp481 backbone, present for around 70% of the MD simulation time. The Neu5Ac carboxyl group also established H-bonds with both the backbone and side chain of Thr483 (H and HG1 in Fig. S5d, ESI†), belonging to the ΦTRY consensus sequence (Fig. 3b). The O9 and O8 of the glycerol chain of Neu5Ac interacted with the guanidinium group of Arg484, although these contacts were stable for only 30% of the simulation time (see the table in Fig. S5d, ESI†). Regarding the galactose, only the OH at position 6 established a H-bond interaction with the OH of Tyr485; no other significant contacts with SLBR-B were found. Moreover, the GalNAc similarly to the Gal unit did not exhibit important interactions, although a weak H-bond between the oxygen of the acetyl group and the Tyr443 hydroxyl group was present (Fig. S5d, ESI†).
The above interactions were in complete agreement with the STD NMR results, showing the main involvement of Neu5Ac and the recognition of Gal and GalNAc to a lesser extent (Fig. 2). Thus, combining the analysis of the contacts monitored by MD simulations, the epitope mapping and bioactive conformation obtained using NMR data, a 3D view of the SLBR-B–sTa-Thr complex was achieved (Fig. 3b).
SLBR-H interaction with 3′-SLn
Given the broader capability of SLBR-H to bind to different α-2,3 sialylated substrates, its molecular interaction with 3′-sialylactosamine (3′-SLn) was investigated. The conformational behavior of the ligand in the SLBR-H binding pocket was assessed via NMR and computational studies. In the free state (Fig. S4, ESI†), an equilibrium of three different conformations around the Neu5Ac-α-(2,3)–Gal glycosidic linkage was detected (corresponding to ϕ −60°/60°/180°).22,23 Conversely, NOE and binding data revealed that, upon accommodation of 3′-SLn into the SLBR-H binding site, a bioactive conformation, stable during the MD simulation of the SLBR-H–3′-SLn complex, was preferentially selected (Fig. S6, ESI†). As indicated by key inter-proton distances in the tr-NOESY (Fig. 4a), the NOE occurring between H3 Gal and H8 Neu5Ac (B3-K8) and the absence of the NOEs between H3 Gal and the diastereotopic H3 (axial and equatorial) protons of Neu5Ac were crucial for the discrimination of the ligand conformation and supported the selection of specific conformation (Fig. S6d, ESI†), confirming the experimental tr-NOESY data (Fig. 4a). The epitope map of the ligand obtained by STD NMR (Fig. 4b) showed that all three monosaccharides of the 3′-SLn were recognized by SLBR-H, with the main contribution coming from the sialic acid. Indeed, the highest STD effect was attributed to the acetyl group of sialic acid; strong STD responses also corresponded to the glycerol chain and to the H4, H5 and H6 of Neu5Ac, to the H4 and H6 of Gal as well as to the H2 and the acetyl group of GlcNAc. The other protons displayed STD effects lower than 40% and the diastereotopic protons of Neu5Ac (H3ax and H3eq) were consequently oriented away from the SLBR-H binding pocket. Complementing STD and tr-NOESY NMR experiments with computational studies provided a global view of the 3D SLBR-H–3′-SLn complex (Fig. 4c).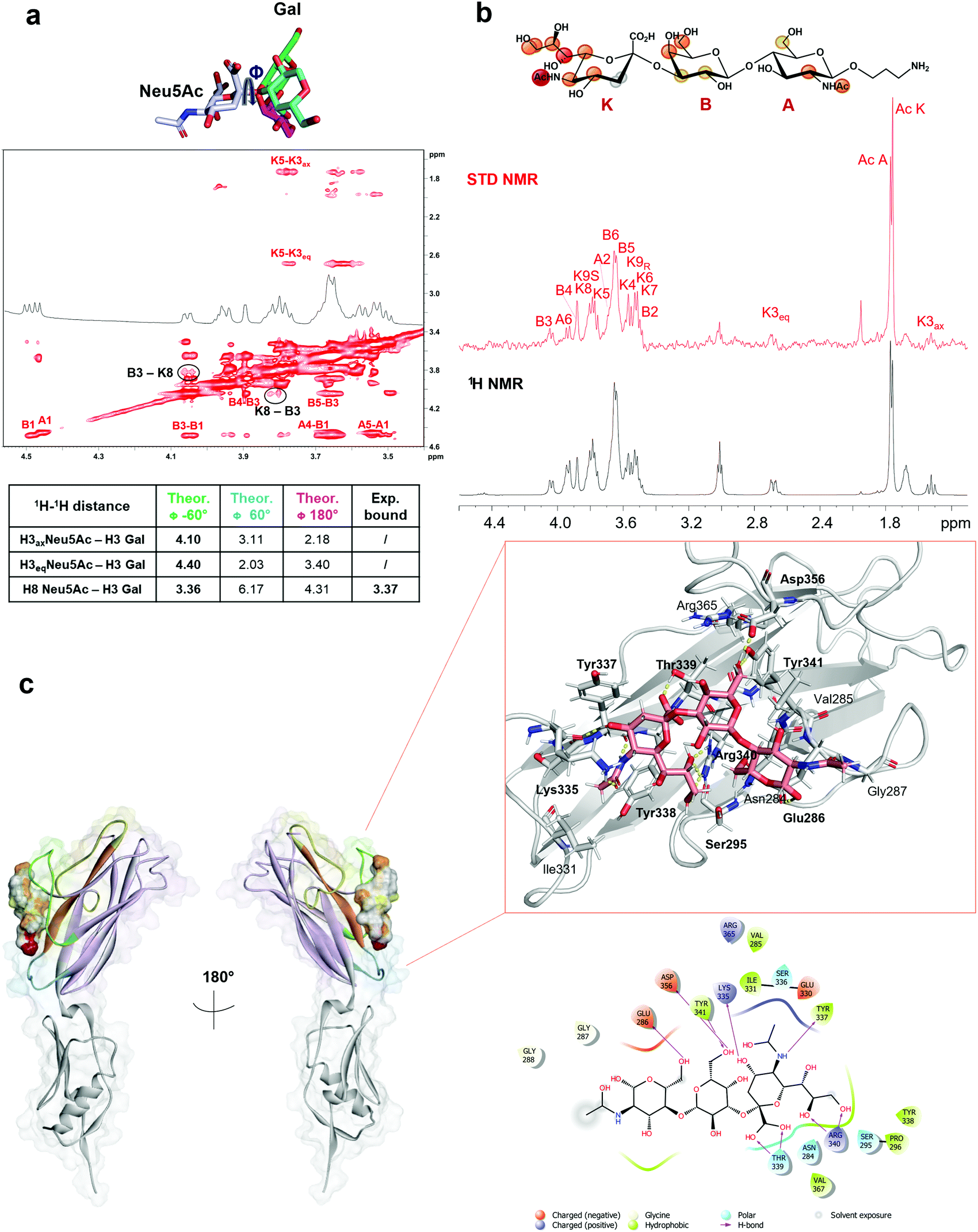 | ||
| Fig. 4 SLBR-H–3′-SLn binding analysis. (a) Conformational analysis of 3′-SLn bound to SLBR-H. Three different φ values (−60°/60°/180°) can be assumed around the Neu5Ac–Gal glycosidic bond of the ligand in the free state. As indicated by the inter-proton distances reported in the table, calculated by tr-NOESY, the presence of key NOE B3-K8 and the absence of NOEs B3-K3ax/K3eq allowed affirmation of a conformer selection with φ = −60° in the bound state. (b) Epitope map of the ligand recognized by SLBR-H; STD-NMR spectrum (red) and unsaturated reference spectrum (black). (c) 3D view of the complex according to STD, tr-NOESY and MD data with the two-dimensional plot of the complex highlighting the interactions established between SLBR-H and 3′-SLn. | ||
As shown in Fig. 4, the sialic acid was primarily recognized by the recurring amino acids of the ΦTRY sequence, as Thr339 and Arg340. The hydrogen bond established between the carboxyl group of Neu5Ac and the hydroxyl group of the Thr339 side chain was the strongest contact observed during the MD simulation, present for above 90% of the MD simulation (Fig. S6e, ESI†); the backbone of Tyr339 also interacted with the carboxyl group of Neu5Ac. Further, the guanidinium group of Arg340 formed a H-bond with the O8 of Neu5Ac, thus reinforcing binding and orientation of 3′-SLn within the SLBR-H binding pocket, and with Neu5Ac tuning the accommodation of the whole sialoglycan in the binding site. Then, the strong STD NMR signal of the acetyl group of Neu5Ac was supported by the polar interaction between the carbonyl oxygen of Tyr337 and the amide nitrogen of Neu5Ac. Another significant contact was observed for OH at C4 of Neu5Ac, forming a hydrogen bond with the backbone of Lys335, stable during the MD simulation, also supported by the high STD contribution of H4 (around 60%). In addition, the guanidinium group of Arg340 established H-bonds with hydroxyl protons 8 and 9 of the glycerol chain, consistent with the high contribution of the glycerol chain in the STD spectra. Interestingly, Tyr338 played a key role in tuning the 3′-SLn accommodation, as evidenced by its proximity to the sialic acid residue and established a crucial hydrophobic interaction with the H7 of Neu5Ac, providing a strong contribution to the binding. As for the other residues, the main interactions were polar and included H-bonds established by the Gal and GlcNAc residues. In particular, the O6 of Gal interacted with the OH of Tyr341, found in the ΦTRY motif of SLBR-H, while the proton of the same Gal hydroxyl group established a H-bond with the carboxylate group of Asp356. Furthermore, the OH-6 of GlcNAc acted as a hydrogen donor to the carboxylate of Glu286. Despite the hydrogen bonds mostly participating in the stabilization of the complex, weaker interactions could also be detected at the protein–ligand interface (Fig. 4c). As shown by STD NMR and MD simulations, all the three residues composing 3′-SLn were recognized by SLBR-H (Fig. 4).
Since water molecules are often important mediators of protein–glycan interactions, WaterLOGSY (Water–Ligand Observation with Gradient SpectroscopY)24 experiments were employed to detect this type of molecular interaction between SLBR-H and 3′-SLn.17,25 Generally, a small ligand in the free state or not interacting with the protein shows negative peaks, while the signals corresponding to exchangeable protons or ligand resonances experiencing magnetization transfer originating from water molecules exhibit a positive phase in WaterLOGSY spectra.26 However, depending on the protein–ligand exchange regime, signals from a bound molecule can remain negative, with their intensity reduced with respect to the reference spectrum.
This is the case of the SLBR-H–3′-SLn system, where a decrease of all the negative signals was observed, meaning a recognition of 3′-SLn from the protein, according to the STD NMR data, with no strong influence from water molecules. Interestingly, by comparing the WaterLOGSY spectra of 3′-SLn in the absence and in the presence of SLBR-H, we observed a change in the sign for only one peak ascribable to position 9 of sialic acid (Fig. 5a and Fig. S7a, ESI†). It is worth noting that H9 gave an STD signal and the hydroxyl group established a H-bond with the guanidinium group of Arg340 (Fig. 4). Nevertheless, the positive signal in the WaterLOGSY spectrum was indicative of the existence of specific water molecules at the interface of the terminal glycerol chain of Neu5Ac and SLBR-H.
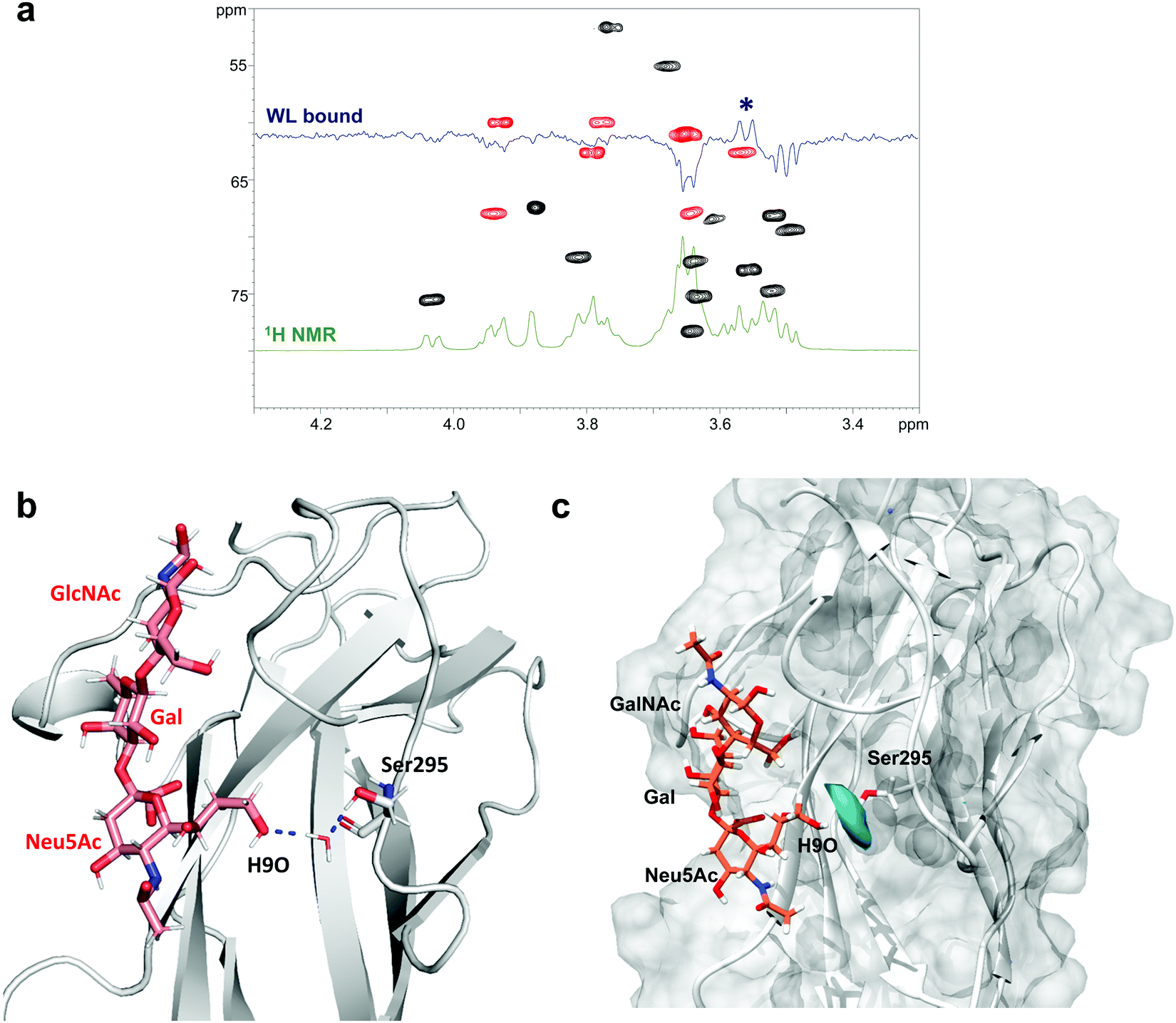 | ||
| Fig. 5 WaterLOGSY analysis of the SLBR-H–3′-SLn complex. (a) WaterLOGSY NMR experiment of the free (green) and bound (blue) states superimposed on the HSQC of the ligand; the only positive signal refers to position 9 of sialic acid (indicated by an asterisk). (b) 3D view of a complex showing the polar interactions established between the H9O of 3′-SLn and the hydroxyl group of Ser295 of SLBR-H mediated by a water molecule. (c) Calculation of water occupancy (in cyan) around 4 Å from Ser295 obtained using VMD software highlighting the proximity of the water molecules to position 9 of Neu5Ac. | ||
Thus, using the CPPTRAJ program implemented in Amber, we obtained details on water molecules simultaneously interacting with both ligand and protein residues during MD simulation, thus acting as a bridge and mediating protein–ligand interactions. We found that the hydroxyl proton of Ser295 interacted with OH at C9 of Neu5Ac by means of a resident water molecule in the protein binding site of SLBR-H (Fig. 5b), as shown by the water density calculated around Ser295 derived from MD simulations (Fig. 5c). The stability of the occurring H-bonds, measured as distances between the atoms involved in the bridging interactions, was then monitored and confirmed during the entire MD simulation (Fig. S7b, ESI†). Conversely, no noteworthy results were obtained in the WaterLOGSY spectra acquired for the other SLBR–glycan pairs under study (data not shown).
GM1b interaction with SLBR-H and SLBR-B
In order to get additional insights into the different binding properties of SLBR-H and SLBR-B, we also used GM1b as a substrate, which is a natural ligand containing the Neu5Ac-α-(2,3)–Gal-β-(1,3)–GalNAc epitope as a terminal extension of a glycolipid. STD NMR analysis mapped the binding epitopes of GM1b in interaction with SLBR-H and SLBR-B (Fig. S8, ESI†). The presence of STD signals in both spectra demonstrated the accommodation of GM1b in the binding sites of both proteins, with the Neu5Ac-α-(2,3)–Gal-β-(1,3)–GalNAc portion (sialyl-T-antigen-like) being principally involved. Indeed, Gal and Glc at the reducing end did not give significant STD signals.In detail, regarding SLBR-H (Fig. S8a, ESI†), a strong saturation transfer was detected for the sialic acid (K) of GM1b. All the protons of the residue, indeed, exhibited STD enhancement, showing %STD values higher than 40%; moreover, the highest STD signal belonged to the acetyl group of K. The Gal (D) and GalNAc (C) residues displayed similar STD percentages, with the respective H3 and H4 protons showing the STD range between 40 and 60%, and the H2 and H6 protons of GalNAc showing STD percentages between 20 and 40%. The acetyl group of GalNAc (C) had a very low %STD, indicating that it was solvent-exposed. The Gal (B) and the reducing Glc (A) units pointed far from the binding pocket of HSA, as confirmed by the absence of the isolated STD signals of that residue (black labels on the off-resonance spectrum, Fig. S8a, ESI†). Thus, the sialic acid was mostly recognized by the protein, giving significant %STD values, while the subterminal Gal and GalNAc residues showed %STD lower than 60%, with the acetyl group of GalNAc being solvent-exposed.
Regarding SLBR-B (Fig. S8b, ESI†), the region including Neu5Ac-α-(2,3)–Gal-β-(1,3)–GalNAc was extensively recognized. The acetyl group of Neu5Ac gave the strongest STD signal (100%), followed by the H8 of Neu5Ac and the H4 of GalNAc which also showed high STD signals. Then, the rest of the glycerol chain protons and the H5 of Neu5Ac as well as the H1 and H3 of Gal (D) and the H2 and H3 of GalNAc (C) exhibited %STD higher than 60%. The other protons of the sialyl-T-antigen-like portion were also saturated by SLBR-B, with %STD between 40% and 60%, except for the acetyl group of GalNAc (around 30%) and the diastereotopic H3 protons of Neu5Ac (H3eq less than 20%, H3ax completely excluded from the recognition). Finally, low STD signals and relative percentages could be observed for the Gal (B) residue, while the isolated signals belonging to the A residue were not involved in the binding with SLBR-B.
The MD analysis of the sugar portion of the ganglioside GM1b bound to SLBR-H (Fig. S9a, ESI†) and SLBR-B (Fig. S9b, ESI†) provided the 3D binding profiles of the complexes and helped to understand how the ligand is accommodated into the binding pocket of the protein. Fig. S9 (ESI†) shows the superimposition of representative poses of the ligand bound to SLBR-H and SLBR-B. According to the STD NMR data, the Neu5Ac-α-(2,3)–Gal-β-(1,3)–GalNAc portion of the ganglioside was enveloped by each SLBR, with the reducing end Gal and Glc residues being exposed to the solvent. Moreover, when comparing the two complexes, the proximity of the GalNAc (residue C) to the helix in the FG loop of SLBR-B (Fig. S9b, ESI†) may explain the higher %STD values displayed in the ligand epitope mapping (Fig. S8b, ESI†). Thus, the Neu5Ac-α-(2,3)–Gal-β-(1,3)–GalNAc portion of the ganglioside was recognized by both Siglec-like adhesins, with nearly all the protons showing STD higher than 60% when complexed with SLBR-B, due in part to the presence of the helix in the FG loop (Fig. S9b and S10, ESI†) that allows interactions with the GalNAc residue of GM1b.
Discussion and conclusions
The role of Siglec-like adhesins in the mediation of streptococcal attachment to host glycans and glycoproteins on mammalian cells has been extensively documented.5,9,14,27 However, a detailed description of sialoglycan recognition and binding by Siglec-like adhesins is far from complete. We here propose a global view of the recognition modes of bacterial adhesins, SLBR-H and SLBR-B, normally expressed on the S. gordonii surface, interacting with different sialoglycans. We analyzed the glycan behavior upon binding, mapped the key ligand epitopes recognized by the protein, defined the ligand conformational shape adopted into the binding pocket of the receptors (bioactive conformation), and described the 3D complex and the structural and topological features driving the different recognition and binding.In detail, the analysis and comparison of SLBR-H and SLBR-B binding to sTa covalently linked to a threonine (sTa-Thr) was performed to evaluate the recognition of a natural O-glycan typically exposed on platelet or salivary glycoproteins. Apart from the known repertoire of synthetic and natural ligands,14 the recognition of other glycan substrates like gangliosides, ubiquitous in many tissues, including the brain, gut and vessels, cannot be excluded (yet).28–30 Therefore, we also show how GM1b is a potential ligand for both SLBR-B and SLBR-H, through the sTa epitope present on the terminal portion of the glycan chain, and mainly accommodated in the protein binding pocket. Given the broader specificity of SLBR-H, we also investigated its binding with 3′-sialylactosamine (3′-SLn), identifying the ligand region directly involved in the interaction and highlighting the presence of resident water molecules in the protein–ligand complex.
As described by Stubbs et al.,12 the YTRY motif of the F-strand was refined to ΦTRX, and the threonine and arginine of the sequence were previously determined to be important residues for binding. Previous site-directed mutagenesis studies highlighted the importance of specific consensus motif amino acids of both adhesins in the binding to sialylated glycans, including R484E or Y485F in GspB, and T339V or R340E in Hsa.10,14 These substitutions were indeed shown to abolish binding to sialylated targets.10,14
Our study further elaborates the essential role of the YTRY amino acids in the α-2,3 sialoglycan binding with both adhesins. In fact, the central TR of the ΦTRY sequence established the most stable H-bonds with the sialic acid of α-2,3 sialoglycans. Furthermore, the first tyrosine of the ΦTRY sequence was particularly relevant in establishing a hydrophobic interaction with the H7 of sialic acid in both Siglec-like adhesins, while the last tyrosine made a recurrent H-bond with Gal at position 6 of Neu5Ac-α-(2,3)–Gal containing sialoglycans. This may also explain the absence of binding by 6 sulfo-Gal modified 3′-SLn.13 Moreover, the accommodation of the sialoglycans in SLBR-B and SLBR-H binding pockets was governed by the critical involvement of the flexible CD, EF and FG loops in the control of ligand orientation.12
In SLBR-H the wide binding pocket allows either 3′-SLn or sTa to be fully accommodated (Fig. 2 and 4). The interactions that SLBR-H established with 3′-SLn and sTa-Thr were comparable, mainly involving the common Neu5Ac-α-(2,3)–Gal epitope, which is therefore fundamental for the recognition process. Notably, the third sugar, β-1,3-linked in sTa and β-1,4-linked in 3′-SLn, is differently recognized from SLBR-H, being involved in the interaction of the FG loop with Asp356 and the CD loop with Glu286, respectively. Urano-Tashiro et al.31 observed the critical involvement of two arginine residues in SLBR-H, Arg340 and Arg365 in the recognition of sialoglycans since their mutation to asparagine (R340N and R365N) demonstrated a reduction of the SLBR-H interaction to human erythrocytes and platelets. In our results, Arg340 and Arg365 were consistently found in the binding site of SLBR-H; however, we found that only Arg340 of the ΦTRY motif established crucial contacts with sialoglycans. Regarding SLBR-B, the presence of the FG loop helix close to the binding pocket of the protein shapes the binding site, driving a highly specific arrangement of sialyl-T-antigen. Compared with SLBR-H, we showed a similar epitope of sTa-Thr in the interaction with SLBR-B, with lower contributions of Gal and GalNAc to the recognition and binding process. This is likely due to the interactions between the threonine moiety of sTa-Thr and the helix of the SLBR-B FG loop, which may contribute to the movement of the Gal and GalNAc units further from the protein surface (Fig. 2). MD analysis supported the NMR data, with a reduced stability of the network of interactions established during the simulation of the SLBR-B–sTa-Thr complex (Fig. S5d, ESI†). The YTRY sequence in the F strand of SLBR-B and SLBR-H displayed the same interactions with Neu5Ac (Fig. 2d and 3d), consistent with its importance in mediating sialoglycan recognition. However, the amide of Neu5Ac interacted with different residues of the Siglec-like adhesins, namely with Asp481 in SLBR-B and Tyr337 in SLBR-H, although both amino acids belonged to the F-strand. Another difference could be detected in the recognition of the O6 of GalNAc, that in SLBR-B interacts with Tyr443 of the CD loop, while that in SLBR-H interacts with Asp356 of the FG loop. Furthermore, the orientation of the Thr of sTa-Thr is comparable in both SLBR-H and SLBR-B complexes. The threonine portion is mostly solvent-exposed, which is consistent with streptococcal adherence to host O-glycoproteins, with the sialoglycan linked to a protein chain (Fig. S10, ESI†). In this context, it has been suggested that mucin conformations are influenced by the peptide backbone to which the glycan portion is linked.32 Indeed, the presence of proline residues to Ser or Thr, often associated with O-glycosylation sites, characterizes the glycopeptide shape that assumes an extended rodlike structure form.33 The different flexibility of serine-linked and threonine-linked O-glycans and/or the higher rigidity of threonine linked-O-glycans toward the peptide backbone may favor the interaction with adhesins like SLBR-H and SLBR-B.34
Our overall findings indicate that, despite a shared general architecture of the V-set Ig domains in terms of loops and β-sheets, the nature of interactions in the binding site of mammalian Siglecs and bacterial Siglec-like adhesins is mechanistically very different. The diverse conformations of the loops in the sialic acid-binding V-set domain tune the capability and the selectivity of SLBR-H and SLBR-B toward sialoglycan recognition, which is mechanistically distinct from binding via the CC′ loop of the analogous sialic acid-binding domain in mammalian Siglecs.35 Indeed, we demonstrated that the entire glycan moiety is accommodated into the Siglec-like BRs, establishing a wide range of interactions with several amino acids of the protein binding pocket (Fig. 6 and Fig. S11, ESI†). On the contrary, previously reported Siglec–sialoglycan 3D complexes clearly show predominant interactions of the protein with the sialic acid, minimal interactions with galactose, and none with the third sugar residue (Fig. 6 and Fig. S11, ESI†). Indeed the sialoglycan is anchored to the Siglec through its extreme terminal portion, while the reducing end of the glycan protrudes away from the binding site.
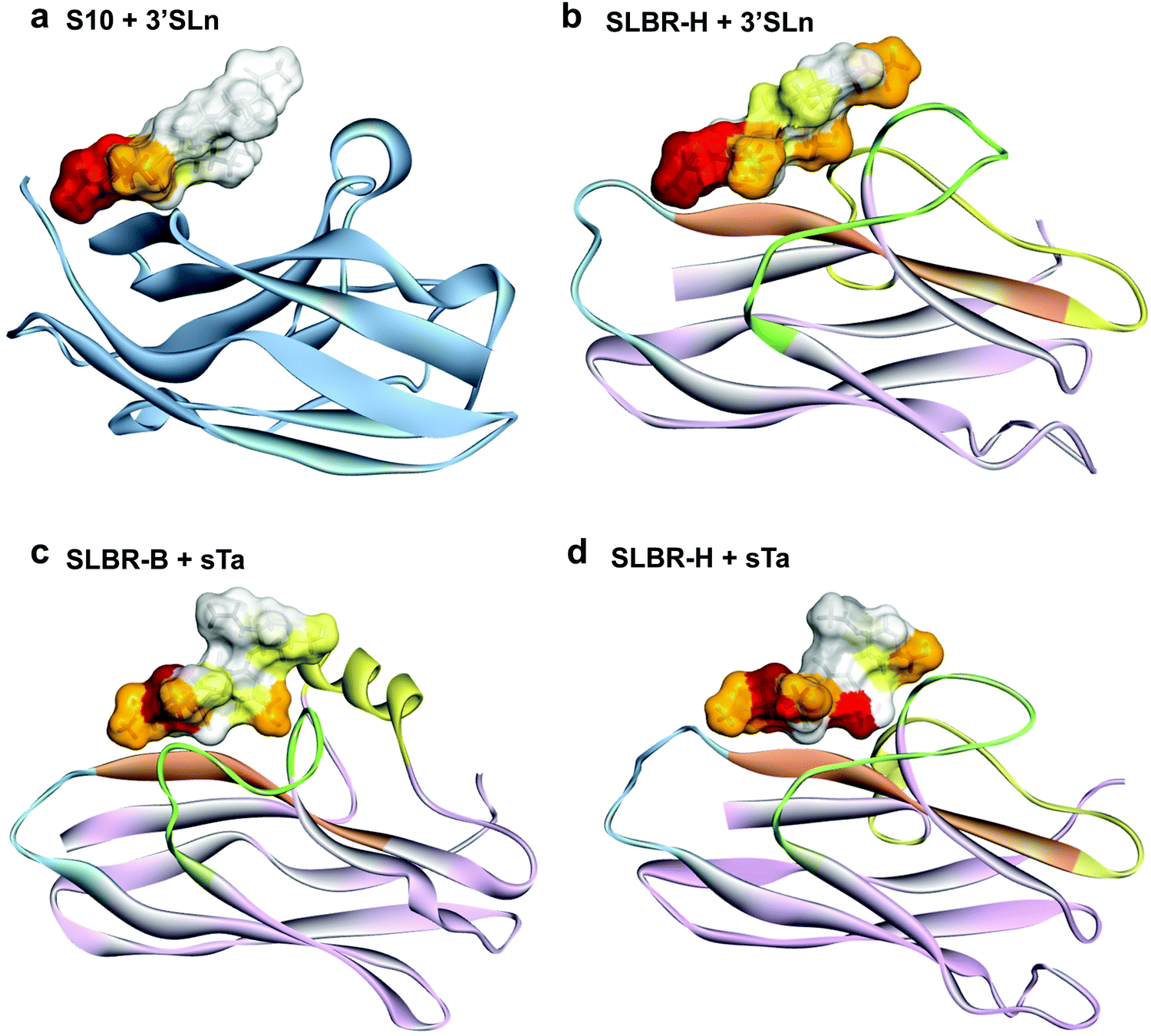 | ||
| Fig. 6 Comparison of sialoglycan recognition by Siglecs and Siglec-like adhesins. (a) The epitope mapping of the 3′-sialylactosamine bound to Siglec-10, obtained from previously published22 STD NMR results is reported as an example of the binding mode of Siglecs. The 3D complexes of (b) SLBR-H–3′-sialylactosamine, (c) SLBR-B–sialyl-T-antigen and (d) SLBR-H–sialyl-T-antigen show the accommodation of the entire ligand in the binding pocket. | ||
The N-terminal V-set Ig domain in the Siglec family contains common structural determinants, such as a conserved arginine in the F strand, a key residue for the formation of a salt bridge with the carboxyl group of sialic acid.22,35,36 As for the Siglec-like adhesins SLBR-H and SLBR-B, despite the proximity in the binding site of arginine residues (Arg340SLBR-H and Arg484SLBR-B) to the sialic acid unit of the ligands, no similar ionic interactions with the carboxyl group of Neu5Ac were detected, but important hydrogen bonds were found with the glycerol moiety, in particular with the O8 and O9 of Neu5Ac. Instead, the carboxylate group of Neu5Ac established interactions with the threonine residue of the ΦTRY motif (Thr339SLBR-H and Thr483SLBR-B). Another key difference is that an intra-sheet disulfide bond between specific β-strands of the mammalian Siglec binding site allows the exposure of hydrophobic residues, in particular a conserved aromatic amino acid (usually a Trp) which interacts with the glycerol side chain of the sialic acid.37 In the SLBR-H and SLBR-B binding sites, we found aromatic residues which established hydrophobic interactions with the sialoglycan, e.g. with the H7 of Neu5Ac, but the binding pocket of the two Siglec-like adhesins provided mainly polar interactions, with a high level of H-bond interactions.
In conclusion, the BRs of SLBR-B and SLBR-H are considered attractive molecular targets for drug development due to their role in infective endocarditis (IE). It is noteworthy that there is no vaccine or anti-adhesive drug approved against IE. Thus, unveiling the molecular mechanisms of glycan recognition by Siglec-like adhesins can explain the different selectivity and flexibility of the streptococcal adhesins towards sialoglycans and can give a boost to the design of novel therapeutics to prevent or treat IE. In particular, the detailed understanding of SLBR-Siglec differences may enable the design of tailored inhibitors that do not interfere with normal, important Siglec interactions.
Author contributions
A. S. and R. M. conceived the study. A. S., R. M., C. D. C., and R. E. F. designed the research. All the authors conducted the research and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by PRIN 2017 (2017XZ2ZBK, 2019-2022) to AS, by H2020-MSCA-ITN-2020 – grant agreement 956758 (GLYTUNES) to AS and MF, and by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme – grant agreement No. 851356 to RM. FSE, PON Ricerca e Innovazione 2014-2020, Azione I.1 “Dottorati Innovativi con caratterizzazione Industriale” is acknowledged for funding the PhD grant to REF.References
- T. L. Holland, L. M. Baddour, A. S. Bayer, B. Hoen, J. M. Miro and V. G. Fowler Jr, Infective Endocarditis, Nat. Rev. Dis. Primers, 2017, 2, 16059 CrossRef PubMed.
- K. Werdan, S. Dietz, B. Löffler, S. Niemann, H. Bushnaq, R. E. Silber, G. Peters and U. Müller-Werdan, Mechanisms of infective endocarditis: pathogen-host interaction and risk states, Nat. Rev. Cardiol., 2014, 11(1), 35–50 CrossRef CAS PubMed.
- S. Chamat-Hedemand, A. Dahl, L. Østergaard, M. Arpi, E. Fosbøl, J. Boel, L. B. Oestergaard, T. K. Lauridsen, G. Gislason, C. Torp-Pedersen and N. E. Bruun, Prevalence of Infective Endocarditis in Streptococcal Bloodstream Infections Is Dependent on Streptococcal Species, Circulation, 2020, 142(8), 720–730 CrossRef CAS PubMed.
- C. Del Giudice, E. Vaia, D. Liccardo, F. Marzano, A. Valletta, G. Spagnuolo, N. Ferrara, C. Rengo, A. Cannavo and G. Rengo, Infective Endocarditis: A Focus on Oral Microbiota, Microorganisms, 2021, 9, 1218 CrossRef PubMed.
- B. A. Bensing, Q. Li, D. Park, C. B. Lebrilla and P. M. Sullam, Streptococcal Siglec-like adhesins recognize different subsets of human plasma glycoproteins: implications for infective endocarditis, Glycobiology, 2018, 28, 601–611 CrossRef CAS PubMed.
- Y. Q. Xionga, B. A. Bensing, A. S. Bayer, H. F. Chambers and P. M. Sullam, Microb. Pathog., 2008, 45(4), 297–301 CrossRef PubMed.
- Y. Takahashi, E. Takashima, K. Shimazu, H. Yagishita, T. Aoba and K. Konishi, Contribution of sialic acid-binding adhesin to pathogenesis of experimental endocarditis caused by treptococcus gordonii DL1, Infect. Immun., 2006, 74(1), 740–743 CrossRef CAS PubMed.
- D. Takamatsu, B. A. Bensing, H. Cheng, G. A. Jarvis, I. R. Siboo, J. A. López, J. McLeod Griffiss and P. M. Sullam, Binding of the Streptococcus gordonii surface glycoproteins SLBR-B and SLBR-H to specific carbohydrate structures on platelet membrane glycoprotein Ibα, Mol. Microbiol., 2005, 58(2), 380–392 CrossRef CAS PubMed.
- B. A. Bensing, L. Li, O. Yakovenko, M. Wong, K. N. Barnard, T. M. Iverson, C. B. Lebrilla, C. R. Parrish, W. E. Thomas, Y. Xiong and P. M. Sullam, Recognition of specific sialoglycan structures by oral streptococci impacts the severity of endocardial infection, PLoS Pathog., 2019, 15, e1007896 CrossRef CAS PubMed.
- T. M. Pyburn, B. A. Bensing, Y. Q. Xiong, B. J. Melancon, T. M. Tomasiak, N. J. Ward, V. Yankovskaya, K. M. Oliver, G. Cecchini, G. A. Sulikowski, M. J. Tyska, P. M. Sullam and T. M. Iverson, A Structural Model for Binding of the Serine-Rich Repeat Adhesin SLBR-B to Host Carbohydrate Receptors, PLoS Pathog., 2011, 7, e1002112 CrossRef CAS PubMed.
- R. Agarwal, B. A. Bensing, D. Mi, P. N. Vinson, J. Baudry, T. M. Iverson and J. C. Smith, Structure based virtual screening identifies small molecule effectors for the sialoglycan binding protein SLBR-H, Biochem. J., 2020, 477(19), 3695–3707 CrossRef CAS PubMed.
- H. E. Stubbs, B. A. Bensing, I. Yamakawa, P. Sharma, H. Yu, X. Chen, P. M. Sullam and T. M. Iverson, Tandem sialoglycan-binding modules in a Streptococcus sanguinis serine-rich repeat adhesin create target dependent avidity effects, J. Biol. Chem., 2020, 295(43), 14737–14749 CrossRef CAS PubMed.
- L. Deng, B. A. Bensing, S. Thamadilok, H. Yu, K. Lau, X. Chen, S. Ruhl, P. M. Sullam and A. Varki, Oral Streptococci Utilize a Siglec-Like Domain of Serine-Rich Repeat Adhesins to Preferentially Target Platelet Sialoglycans in Human Blood, PLoS Pathog., 2014, 10(12), e1004540 CrossRef PubMed.
- B. A. Bensing, Z. Khedri, L. Deng, H. Yu, A. Prakobphol, S. J. Fisher, X. Chen, T. M. Iverson, A. Varki and P. M. Sullam, Novel aspects of sialoglycan recognition by the Siglec-like domains of streptococcal SRR glycoproteins, Glycobiology, 2016, 26(11), 1221–1233 CAS.
- B. A. Bensing, L. V. Loukachevitc, R. Agarwal, I. Yamakawa, K. Luong, A. Hadadianpour, H. Yuf, K. P. Fialkowski, M. A. Castrog, Z. Wawrzak, X. Chen, J. Baudry, J. C. Smith, P. M. Sullam and T. M. Iverson, Selectivity and engineering of the sialoglycan-binding spectrum in Siglec-like adhesins, bioRxiv, 2019, 796912 Search PubMed.
- K. Cheng, Y. Zhou and S. Neelamegham, DrawGlycan-SNFG: a robust tool to render glycans and glycopeptides with fragmentation information, Glycobiology, 2017, 27(3), 200–205 CAS.
- C. Di Carluccio, M. C. Forgione, S. Martini, F. Berti, A. Molinaro, R. Marchetti and A. Silipo, Investigation of protein-ligand complexes by ligand-based NMR methods, Carbohydr. Res., 2021, 503, 108313 CrossRef CAS PubMed.
- M. Mayer and B. Meyer, Characterization of ligand binding by saturation transfer difference NMR spectroscopy, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS PubMed.
- C. D. Owen, L. E. Tailford, S. Monaco, T. Šuligoj, L. Vaux, R. Lallement, Z. Khedri, H. Yu, K. Lecointe, J. Walshaw, S. Tribolo, M. Horrex, A. Bell, X. Chen, G. L. Taylor, A. Varki, J. Angulo and N. Juge, Unravelling the specificity and mechanism of sialic acid recognition by the gut symbiont Ruminococcus gnavus, Nat. Commun., 2017, 19, 8, 1(.), 2196 CrossRef PubMed.
- B. Meyer and T. Peters, NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors, Angew. Chem., Int. Ed., 2003, 42, 864–890 CrossRef CAS PubMed.
- R. Marchetti, S. Perez, A. Arda, A. Imberty, J. Jimenez-Barbero, A. Silipo and A. Molinaro, “Rules of Engagement” of proteins-glycoconjugates interactions: a molecular view achievable by NMR spectroscopy and molecular modeling, ChemistryOpen, 2016, 5, 274–296 CrossRef CAS PubMed.
- R. E. Forgione, C. Di Carluccio, J. Guzman-Caldentey, R. Gaglione, F. Battista, F. Chiodo, Y. Manabe, A. Arciello, P. Del Vecchio, K. Fukase, A. Molinaro, S. Martín-Santamaría, P. R. Crocker, R. Marchetti and A. Silipo, Unveiling molecular recognition of sialoglycans by human siglec-10, iScience, 2020, 23, 6, 10, 1231 Search PubMed.
- R. E. Forgione, C. Di Carluccio, M. Kubota, Y. Manabe, K. Fukase, A. Molinaro, T. Hashigushi, R. Marchetti and A. Silipo, Structural basis for Glycan-receptor binding by mumps virus hemagglutinin-neuraminidase, Sci. Rep., 2020, 10, 1589 CrossRef CAS PubMed.
- C. Dalvit, P. Pevarello, M. Tatò, M. Veronesi, A. Vulpetti and M. Sundström, Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water, J. Biomol. NMR, 2000, 18, 65–68 CrossRef CAS PubMed.
- C. Raingeval, O. Cala, B. Brion, M. Le Borgne, R. Eliot Hubbard and I. Krimm, 1D NMR WaterLOGSY as an efficient method for fragment-based lead discovery, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 1218–1225 CrossRef CAS PubMed.
- R. Huang and I. K. H. Leung, Methods Enzymol., 2019, 477–500 CAS.
- Y. Narimatsu, et al., An Atlas of Human Glycosylation Pathways Enables Display of the Human Glycome by Gene Engineered Cells, Mol. Cell, 2019, 75, 394–407 CrossRef CAS PubMed.
- R. K. Yu, Y. T. Tsai, T. Ariga and M. Yanagisawa, Structures, biosynthesis, and functions of gangliosides—An overview, J. Oleo Sci., 2011, 60(10), 537–544 CrossRef CAS PubMed.
- F.-Q. Wen, A. A. Jabbar, D. A. Patel, T. Kazarian and L. A. Valentino, Atherosclerotic Aortic Gangliosides Enhance Integrin-Mediated Platelet Adhesion to Collagen, Arterioscler., Thromb., Vasc. Biol., 1999, 19, 519–524 CrossRef CAS PubMed.
- N. Sasaki and M. Toyoda, Vascular Diseases and Gangliosides, Int. J. Mol. Sci., 2019, 20, 6362 CrossRef CAS PubMed.
- Y. Urano-Tashiro, Y. Takahashi, R. Oguchi and K. Konishi, Two Arginine Residues of Streptococcus gordonii Sialic Acid-Binding Adhesin Hsa Are Essential for Interaction to Host Cell Receptors, PLoS One, 2016, 11(4), e0154098 CrossRef PubMed.
- T. L. Gururaja, N. Ramasubbu, P. Venugopalan, M. S. Reddy, K. Ramalingam and M. J. Levine, Structural features of the human salivary mucin, MUC7, Glycoconjugate J., 1998, 15, 457–467 CrossRef CAS PubMed.
- G. C. Hansson, Mucins and the Microbiome, Annu. Rev. Biochem., 2020, 89, 769–793 CrossRef CAS PubMed.
- G. A. Naganagowda, T. L. Gururaja, J. Satyanarayana and M. J. Levine, NMR analysis of human salivary mucin (MUC7) derived O-linked model glycopeptides: comparison of structural features and carbohydrate-peptide interactions, J. Pept. Res., 1999, 54, 290–310 CrossRef CAS PubMed.
- C. Di Carluccio, R. E. Forgione, A. Molinaro, P. R. Crocker, R. Marchetti and A. Silipo, Exploring the fascinating world of sialoglycans in the interplay with Siglecs, Carbohydr. Chem., 2021, 44, 31–55 CAS.
- C. Di Carluccio, E. Crisman, Y. Manabe, R. E. Forgione, A. Lacetera, J. Amato, B. Pagano, B. A. Randazzo, A. Zampella and R. Lanzetta, et al., Characterization of the dynamic interactions between complex N-glycans and human CD22, ChemBioChem, 2020, 21, 129–140 CrossRef CAS PubMed.
- A. Varki, R. D. Cummings and J. D. Esko, et al., Essentials of Glycobiology, Cold Spring Harbor, NY, 3rd edn, 2015–2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; Fig. S1–S11. See DOI: 10.1039/d1cb00173f |
| This journal is © The Royal Society of Chemistry 2021 |