
Copper-catalyzed enantioselective synthesis of bridged bicyclic ketals from 1,1-disubstituted-4-methylene-1,6-hexanediols and related alkenols†
Ameya S.
Burde
 ,
Shuklendu D.
Karyakarte‡
,
Eric D.
Sylvester
and
Sherry R.
Chemler
*
,
Shuklendu D.
Karyakarte‡
,
Eric D.
Sylvester
and
Sherry R.
Chemler
*
Chemistry Department, Natural Science Complex, State University of New York at Buffalo, Buffalo, New York 14260, USA. E-mail: schemler@buffalo.edu
First published on 8th December 2020
Abstract
Bridged bicyclic ketals display a range of bioactivities. Their catalytic enantioselective synthesis from acyclic 1,1-disubstituted alkene diols is disclosed. This reaction combines asymmetric catalysis with a distal radical migration. Alkynes and arenes undergo the group transfer.
Bridged bicyclic ketals (e.g. those in Fig. 1) are rigid organic structures composed of saturated cyclic ethers that can place substituents in precise relative locations in three dimensions.1 Bridged bicyclic ketal natural products include the zaragozic acids,2 and the saliniketals.3 Designed bioactive bridged bicyclic ketals include Steglatro4 and a cytotoxic alkyne.5
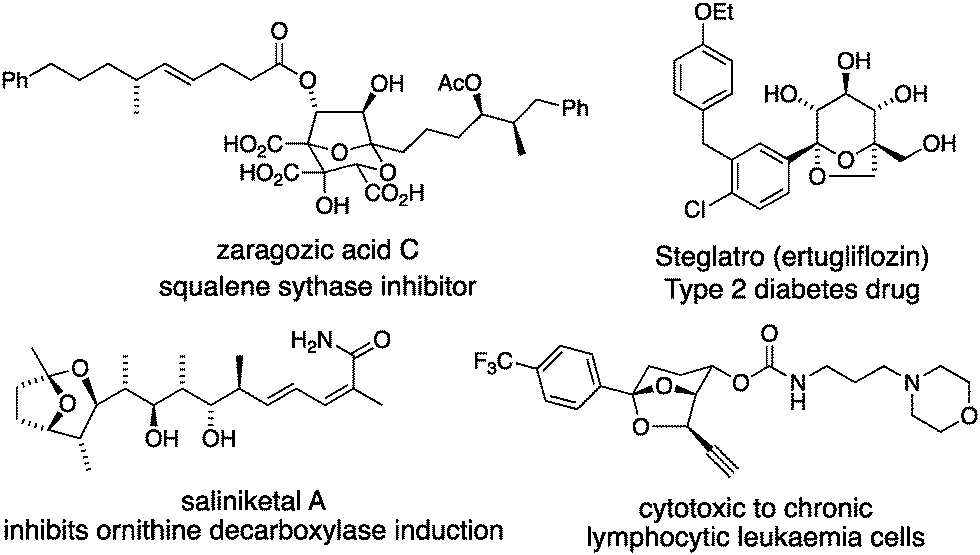 | ||
| Fig. 1 Bioactive bridged bicyclic ketals. | ||
Microbial production is a common route to complex bridged bicyclic ketals.6,7 Considerable effort has also been put toward their de novo chemical synthesis.1 A ketal stereocenter is frequently created via cyclization of two pendant alcohols onto a ketone, where the lowest energy diastereomer is typically favored.1 The acid-catalyzed addition of an alcohol to an enol ether is a related approach.8 Transition metal-catalyzed [including (Pt),9 (Ag)10 and (Au)11] additions of alcohols, carbonyls or carbenes [catalyzed by (Rh)12] to alkynes have also been applied.1
In recent years, excellent progress has also been made toward the catalytic enantioselective synthesis of spirocyclic ketals.13–16 All such methods reported to date involve enantioselective conjugate addition of various nucleophiles to enones, followed by diastereoselective addition of pendant alcohols to the resident carbonyl (Scheme 1a). Along these lines, Shi and co-workers have reported a [Pd]-catalyzed method.17 Liu and co-workers have reported a Brønsted acid catalyzed method.18 Related organocatalytic methods have been reported independently by Pan,19 Jorgensen,20 and Franzen.21 A copper-catalyzed cyclization of a ketone onto an alkene in the presence of DMSO for the synthesis of (±)-benzobicyclo[3.2.1]octanes has also been disclosed.22
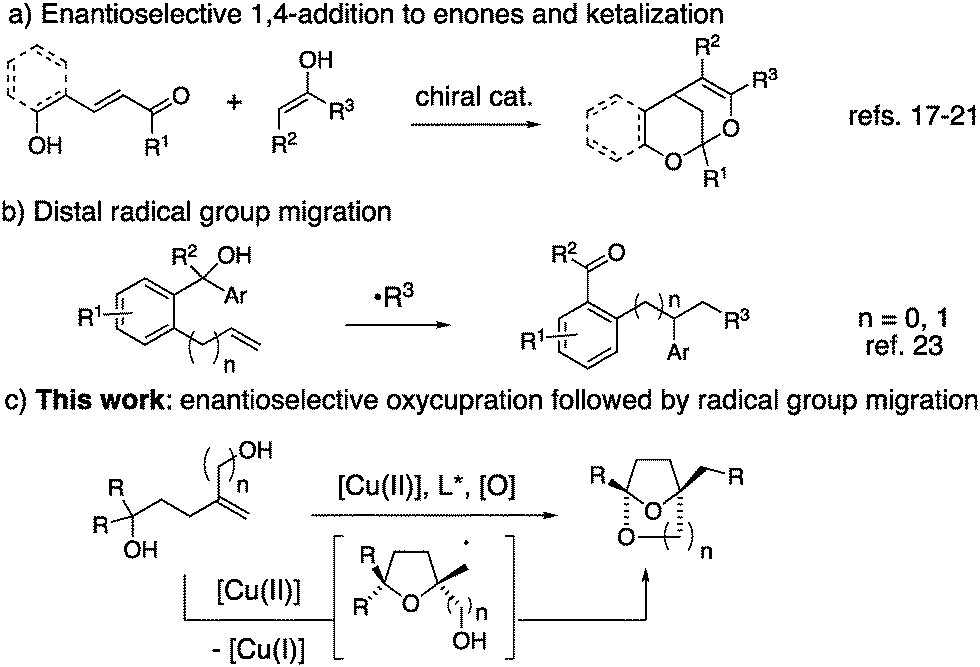 | ||
| Scheme 1 Bridged bicyclic ketals and distal group migration. | ||
We report herein a strategy for the enantioselective synthesis of chiral bridged bicyclic spiroketals that involves a radical group transfer step (Scheme 1c). Radical migrations enable the scission and transfer of unsaturated groups within a molecule. A number of recent reports have highlighted the power of the distal radical migration strategy in organic synthesis (e.g.Scheme 1b).23–26 Herein we report the first distal radical migration reaction to occur in the context of asymmetric catalysis.
In recent years, our group has established that enantioenriched tetrahydrofurans can be synthesized from copper-catalyzed carboetherifications of 4-pentenols.27–30 Enantioselective cis-oxycupration across the alkene,26 followed by in situ homolysis of the resulting C–Cu(II) bond is proposed to result in carbon radical formation (Schemes 1c and 4, vide infra). Subsequent C–C bond formation can occur via addition of the carbon radical to styrenes28,29 or pendant arenes,27,30 which, in prior reports, underwent subsequent oxidation under the conditions to form higher substituted alkenes or arenes (net C–H functionalization). We have also shown that copper(II)-promoted oxyamination and dioxygenation can occur in the absence of π-bond radical group acceptors,31 where the carbon–heteroatom bond formation is thought to occur via a Cu(III) intermediate and a Kharash-Sosnovsky type mechanism.32,33 Although we anticipated 1,1-diphenyl-4-methylene-1,6-hexanediol (1a) would undergo the oxycupration step, it was unclear if the resulting radical intermediate would favor 1,4-phenyl transfer, followed by cyclization to form ketal 2a, or a Cu(III)-facilitated dioxygenation to form bis(ether) 3a (Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Change from standard conditions | 2a + 3a (%) |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3ab :3ab |
ee (%) 2ac |
|
a Reactions run on 0.1 mmol of 1a in 1 mL of PhCF3 with a [Cu]:L ratio of 1:1.25 in a sealed tube (heated in 120 °C oil bath). Isolated yield following chromatography on silica gel, unless otherwise noted.
b Ratio obtained from analysis of the crude 1H NMR.
c Enantiomeric excess measured by chiral HPLC.
d Estimated crude 1H NMR yield. Nd = not determined.
|
||||
| 1 | None | 64 | >10:1 |
92 |
| 2 | L1, 48 h | 61 | 5:1 |
-- |
| 3 | L3 | 68 | 4:1 |
57 |
| 4 | 24 h | 55 | >10:1 |
92 |
| 5 | 15 mol% [Cu], 48 h | 46 | >20:1 |
nd |
| 6 | 25 mol% [Cu], 72 h | 72 | 5:1 |
92 (3a:86) |
| 7 | 30 mol% [Cu], 48 h | 62 | 3:1 |
nd |
| 8 | 60 mol% [Cu], 48 h | 70 | 1.7:1 |
nd |
| 9d | 50 mol% [Cu], L1, 48 h | ca. 90 | 2:1 |
nd |
| 10 | Ag2CO3 (1.5 equiv.) instead of MnO2 | 76 | 5:1 |
nd |
| 11 | 25 mol% [Cu], 10% KMnO4 combined with the MnO2 | 68 | 1.4:1 |
nd |
| 12 | 25 mol% [Cu], PhCH3 instead of PhCF3, 48 h | 51 | 4:1 |
78 |
| 13 | DCE instead of PhCF3, 48 h | 38 | >10:1 |
50 |
| 14d | Cu(NTf2)2 instead of Cu(OTf)2, 48 h | 53 | 1.5:1 |
nd |
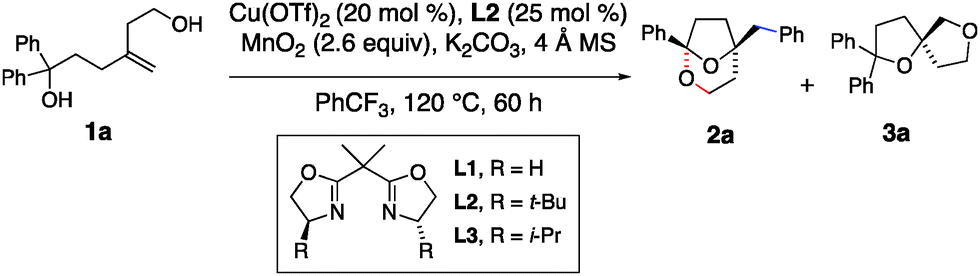
Various reaction conditions for the oxidative transformation of 1a are presented in Table 1. Bridged bicyclic ketal 2a is formed in 64% yield, >10:1 selectivity of 2a over 3a, in 92% ee, using 20 mol% Cu(OTf)2 and 25 mol% of (S,S)-t-BuBox (L2) as catalyst, MnO2 (2.6 equiv.) as oxidant, with K2CO3 and 4 Å mol. sieves as additives, in PhCF3 at 120 °C for 60 h (Table 1, entry 1). The first indication of the major product's identity as a ketal was its distinctive 13C NMR signal at 106 ppm.
A ligand effect on product ratio was observed: when the reaction was run using the achiral bis(oxazoline) ligand (L1), a 5:1 ratio of racemic ketal 2a and bis(ether) 3a was obtained in a combined 61% yield (entry 2). When (S,S)-i-Pr-Box (L3) was applied, a 4:1 mixture of 2a and 3a was observed where 2a was formed in 57% ee (entry 3). Catalyst loading had a more significant impact on the ratio of products. Increased catalyst loading decreased the ratio of 2a:3a, tending to form more 3a as the loading increased (entries 6–8). At 60 mol% [Cu] loading, the ratio of products was close to 1:1. At 25 mol% [Cu] loading, using (S,S)-t-Bu-Box (L2), the ratio of 2a to 3a was 5:1; bis(ether) 3a was isolated and its enantiomeric excess was measured at 86% (entry 6). Changing the oxidant to Ag2CO3 did not strongly impact the reaction while addition of KMnO4 (10 mol%) increased the amount of bis(ether) 3a produced (entries 10 and 11). While the reaction worked in both toluene and DCE (1,2-dichloroethane, entries 12 and 13), neither reaction was superior to the reaction in α,α,α-trifluorotoluene. Cu(NTf2)2 was tried as an alternative source but was found to give lower 2a:3a selectivity for substrate 1a (entry 14). The ratio of 2a and 3a is kinetically controlled; both products were re-subjected to the reaction conditions and were not observed to interconvert.
1,1-Diaryl-4-methylene-1,6-hexanediols bearing various aryl substituents were next examined in the enantioselective [3.2.1]-bridged bicyclic ketal synthesis (Table 2). While the parent 1,1-diphenyl substrate gave ketal 2a in 64% yield and 92% ee, 4- and 3-toluyl analogs gave ketals 2b and 2c in moderate yields (40–42%) but good enantioselectivities (87% and 92%, respectively, Table 2, entries 2 and 3). The remainder of the mass was attributed to oxepanes 4, formed via a competing SN1 process enabled by presumably more stabilized benzylic carbocations. Under the standard conditions, oxepane 4e was the predominant product (61% yield) with bis(4-methoxyphenyl)hexanediol 1e as substrate (Table 2, entry 6). By changing the catalyst to Cu(NTf)2, we were able to obtain ketal 2e in a modest 40% isolated yield and in 70% ee (Table 2, entry 7). Substrates with electron-withdrawing substituents fared better in the ketal synthesis reaction. 4-Trifluoromethylphenyl, 4-chlorophenyl, 3-chlorophenyl and 4-sulfonamidophenyl substituted hexane diols underwent the cyclization in 52–73% yield and with 88–97% ee (entries 4, 8, 9 and 10). Reaction of the 4-trifluoromethylphenyl substituted hexane diol was performed on 1 mmol scale (entry 5). Surprisingly, a 4-phenylbenzene substrate gave ketal 2i in 59% yield but with only 20% ee under standard conditions (entry 11). The enantioselectivity was improved to 56% ee by changing to the less sterically demanding (S,S)-i-Pr-Box ligand (L3) (entry 12). Ketal 2i was crystalline and its X-ray structure enabled its rigorous structural assignment (see ESI,† for details). Reaction of 4-methylene-1,1-diphenylheptane-1,7-diol, whose more substituted alcohol chain is more conformationally pre-disposed toward cyclization, led to the [4.2.1]-bridged bicyclic ketal 2j, (entry 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | n | Product | Yieldb (%) | eec (%) |
| a Table 1, entry 1 conditions were applied with 0.1 mmol of substrate 1. b Isolated yield of major product following chromatography on silica gel. c Enantiomeric excess measured by chiral HPLC. d 30–40% of an SN1 product 4 was also formed. e 25 mol% Cu(OTf)2 and 31 mol% (S,S)-t-Bu-Box was used. f 1 mmol of 1d was used. 34% of starting 1d was recovered. g Cu(NTf2)2 was used. h (S,S)-i-Pr-Box was used, reaction temperature was 105 °C (at 120 °C, 58% of 2i was obtained in 50% ee). | |||||
| 1 | Ph | 1 | 2a | 64 | 92 |
| 2d | 4-MeC6H4 | 1 | 2b | 40 | 87 |
| 3d | 3-MeC6H4 | 1 | 2c | 42 | 92 |
| 4e | 4-CF3C6H4 | 1 | 2d | 71 | >95 |
| 5f | 4-CF3C6H4 | 1 | 2d | 51 | >95 |
| 6 | 4-MeOC6H4 | 1 | 4e | 61 | — |
| 7d,g | 4-MeOC6H4 | 1 | 2e | 40 | 70 |
| 8 | 4-ClC6H4 | 1 | 2f | 53 | 88 |
| 9g | 3-ClC6H4 | 1 | 2g | 58 | 94 |
| 10 | 4-TsMeNC6H4 | 1 | 2h | 73 | 90 |
| 11 | 4-PhC6H4 | 1 | 2i | 59 | 20 |
| 12h | 4-PhC6H4 | 1 | 2i | 64 | 56 |
| 13 | Ph | 2 | 2j | 68 | 93 |
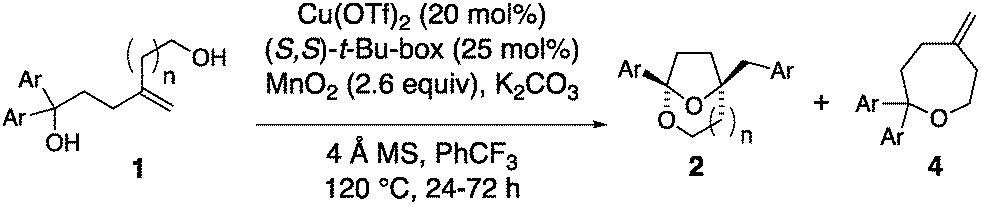
1,1-Diynyl-4-methylene-1,6-hexane diols readily underwent the cyclization and group transfer process to provide bridged bicyclic ketals 2k and 2l in moderate yield and very good enantioselectivity (Scheme 2). The absolute configuration of 2l was assigned by X-ray crystallography.
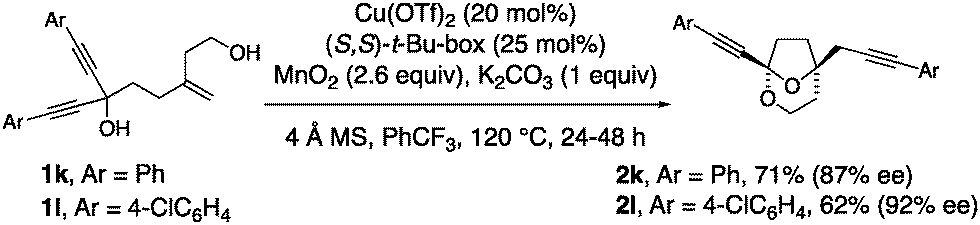 | ||
| Scheme 2 Alkyne-substituted examples. | ||
1,1-Diaryl-4-methylene-1,5-pentane diols 1m and 1n were investigated for their ability to form [2.2.1]-bridged bicyclic ketals (Scheme 3). Surprisingly, the 1,1-diphenyl substrate 1n exclusively formed spirooxetane 3m. Conversely, bis(biaryl) allylic alcohol 1n provided the [2.2.1]-bicyclic ketal exclusively.
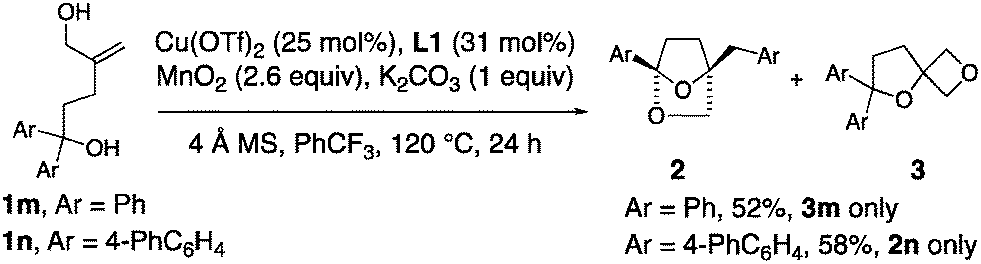 | ||
| Scheme 3 Reactions of allylic alcohols. | ||
A mechanistic analysis is illustrated in Scheme 4. Complexation of the [Cu(II)] catalyst to the tertiary alcohol leads to intermediate I, which can enter the oxycupration TS-A, generating tetrahydrofuran II enantioselectively. In this transition state, the alkene terminal carbon approaches the copper anti to the t-Bu group on the nearest oxazoline ring.28 Organocopper intermediate II undergoes C–[Cu(II)] homolysis to give the carbon radical III. Carbon radical III can undergo ipso aryl addition, to give IV, which undergoes subsequent C–C bond cleavage/group transfer to give the alkoxy-stabilized carbon radical V. Oxidation of V then gives oxonium ion VI and subsequent ketalization provides bicyclic ketal 2 (Scheme 4A). A path for diminished enantioselectivity is ring opening of carbon radical III to give alkoxy radical VII, which could cyclize to give a racemic tetrahydrofuranyl radical (±)-III (Scheme 4B).27 Alternatively, homolysis of the RO-[Cu(II)] bond of intermediate I could also give radical VII. Substrates with resonance-donating aryl substituents (e.g.1d, R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ph and 1e, ROMe, Scheme 4) give 2d and 2e in lower enantioselectivity (Table 2) compared to substrates with neutral or electron-withdrawing substituents (e.g.1a, RH, 1d, RCF3, Scheme 4 and Table 2). Electron-donating arene substituents could lower the oxidation potential of the alcohol.34
Ph and 1e, ROMe, Scheme 4) give 2d and 2e in lower enantioselectivity (Table 2) compared to substrates with neutral or electron-withdrawing substituents (e.g.1a, RH, 1d, RCF3, Scheme 4 and Table 2). Electron-donating arene substituents could lower the oxidation potential of the alcohol.34
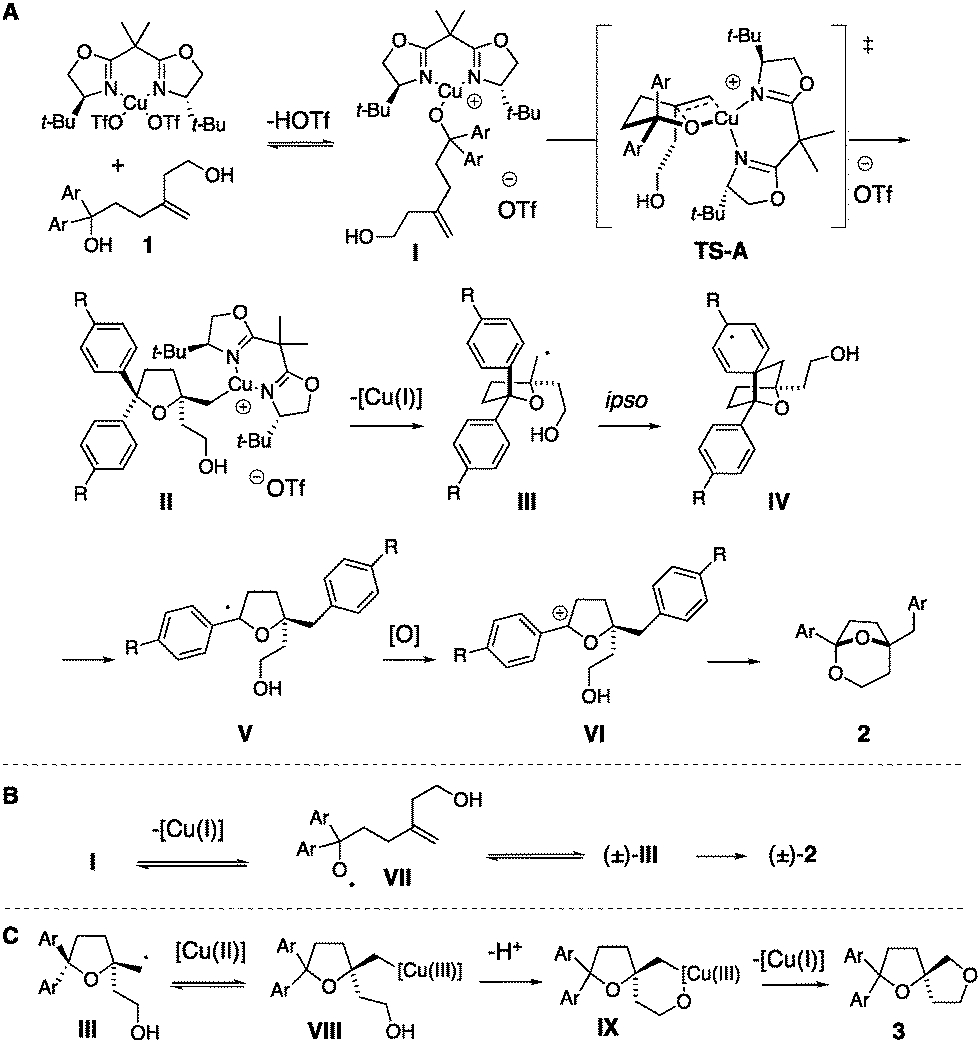 | ||
| Scheme 4 Proposed mechanism to 2 and 3. | ||
A route to bis(ether) 3a involves radical III combining with [Cu(II)] to give [Cu(III)] intermediate VIII. Alcohol coordination then gives IX, and reductive elimination provides 3a.32,33
That the bis(biaryl) allylic alcohol 1n forms ketal 2n, while the diphenyl allylic alcohol 1m forms oxetane 3m may be due to a faster rate of ipso addition to the phenyl-substituted arene.25
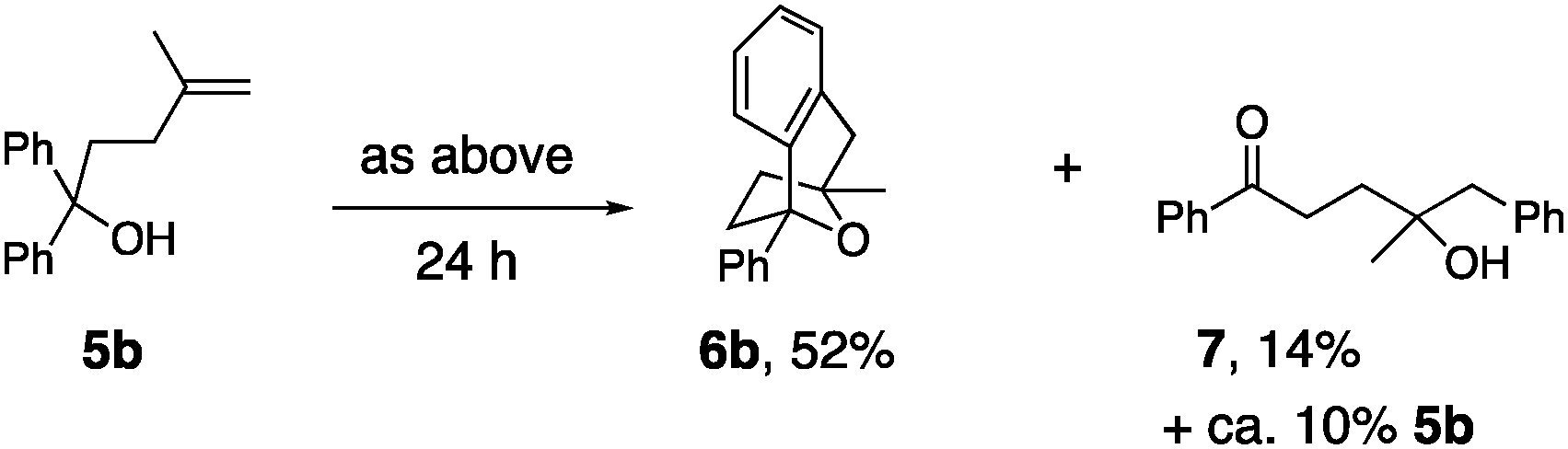
Substrate 5a, that lacks the second alcohol, forms bridged bicyclic [3.2.1] heterocycle 6a (eqn (1)).27 The 1,1-disubstituted alkene 5b, differing from 5a only in alkene substitution, provided adduct 6b and 14% of ketone 7 (eqn (2)). Ketone 7 is likely formed via group transfer and subsequent hydrolysis of the resulting intermediate. The higher substitution of 5b results in greater bond angle compression of its carbon radical intermediate, placing the radial closer to the arene ipso carbon, leading to 7.
In conclusion, a new route to enantioenriched bridged bicyclic ketals from acyclic alkenols has been developed. The polar/radical cross-over mechanistic abilities of copper(II) catalysis enable both enantioselective alkene addition and group transfer reactivity. Some reactivity trends associated with radical stability have been identified; these observations can be used to guide new reaction design.
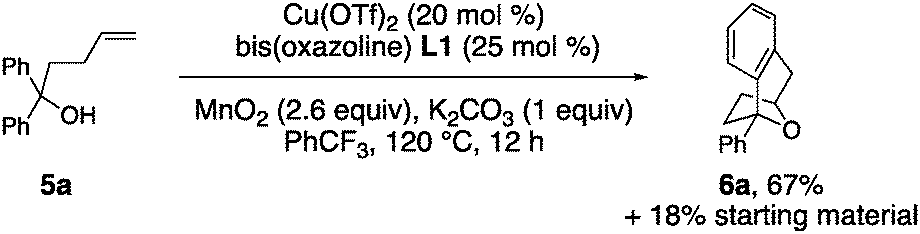 | (1) |
 | (2) |
We thank the National Institutes of Health (GM078383) for support of this work and Prof. Jason Benedict for assisting E. D. S. with the X-ray structures of 2i and 2l.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Bera, B. Chatterjee and D. Mondal, Eur. J. Org. Chem., 2018, 5337 CrossRef CAS.
- J. D. Bergstrom, M. M. Kurtz, D. J. Rew, A. M. Amend, J. D. Karkas, R. G. Bosterdor, V. S. Bansal, C. Dufresne, F. L. VanMiddlesworth and O. D. Hensens, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 80 CrossRef CAS PubMed.
- P. G. Williams, R. N. Asolkar, T. Kondratyuk, J. M. Pezzuto, P. R. Jensen and W. Fenical, J. Nat. Prod., 2007, 70, 83 CrossRef CAS PubMed.
- V. Mascitti, et al. , J. Med. Chem., 2011, 54, 2952 CrossRef CAS PubMed.
- L. Milroy, G. Zinzalla, F. Loiseau, Z. Qian, G. Prencipe, C. Pepper, C. Fegan and S. V. Ley, ChemMedChem, 2008, 3, 1922 CrossRef CAS PubMed.
- M. J. Dawson, A. Baxter, R. M. Tait, N. S. Watson, D. Noble, A. Shuttleworth, H. G. Wildman and M. V. Hayes, WO9212156 A1, 1992.
- M. C. Wilson, T. A. M. Gulder, T. Mahmud and B. S. Moore, J. Am. Chem. Soc., 2010, 132, 12757 CrossRef CAS PubMed.
- I. Paterson, M. Razzak and E. A. Anderson, Org. Lett., 2008, 10, 3295 CrossRef CAS PubMed.
- J. Liu and J. K. De Brabander, J. Am. Chem. Soc., 2009, 131, 12562 CrossRef CAS PubMed.
- C. H. Oh, H. J. Yi and J. H. Lee, New J. Chem., 2007, 31, 835 RSC.
- S. Das, B. Induvadana and C. V. Ramana, Tetrahedron, 2013, 53, 45 Search PubMed.
- Y. Hirata, S. Nakamura, N. Watanabe, O. Kataoka, T. Kurosaki, M. Anada, S. Kitagaki, M. Shiro and S. Hashimoto, Chem. – Eur. J., 2006, 12, 8898 CrossRef CAS PubMed.
- L. Cala, F. J. Fananas and F. Rodriguez, Org. Biomol. Chem., 2014, 12, 5324 RSC.
- J. Y. Hamilton, S. L. Rossler and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 8082 CrossRef CAS PubMed.
- N. Yoneda, Y. Fukata, K. Asano and S. Matsubara, Angew. Chem., Int. Ed., 2015, 54, 15497 CrossRef CAS PubMed.
- A. Midya, S. Maity and P. Ghorai, Chem. – Eur. J., 2017, 23, 11216 CrossRef CAS PubMed.
- F. Wang, F. Chen, M. Qu, T. Li, Y. Liu and M. Shi, Chem. Commun., 2013, 49, 3360 RSC.
- X. Zhang, X. Lv, J. Pei, R. Tan and Y. Liu, Org. Chem. Front., 2020, 7, 292 RSC.
- M. Balha and S. C. Pan, J. Org. Chem., 2018, 83, 14703 CrossRef CAS PubMed.
- B. M. Paz, L. Klier, L. Naesborg, V. H. Lauridsen, F. Jensen and K. A. Jorgensen, Chem. – Eur. J., 2016, 22, 16810 CrossRef CAS PubMed.
- M. F. Polat, L. Hettmanczyk, W. Zhang, Z. Szabo and J. Franzen, ChemCatChem, 2013, 5, 1334 CrossRef CAS.
- C. K. Chan, Y. L. Tsai and M. Y. Chang, Org. Lett., 2017, 19, 1870 CrossRef CAS PubMed.
- L. Li, Q. Gu, N. Wang, P. Song, Z. Li, X. Li, F. Wangand and X. Liu, Chem. Commun., 2017, 53, 4038 RSC.
- W. Li, W. Xu, J. Xie, S. Yu and C. Zhu, Chem. Soc. Rev., 2018, 47, 654 RSC.
- J. Fang, W. L. Dong, G. Q. Xu and P. F. Xu, Org. Lett., 2019, 21, 4480 CrossRef CAS PubMed.
- L. Li, Z. L. Li, F. L. Wang, Z. Guo, Y. F. Cheng, N. Wang, X. W. Dong, C. Fang, J. Liu, C. Hou, B. Tan and X. Y. Liu, Nat. Commun., 2016, 7, 13852, DOI:10.1038/ncomms13852.
- Y. Miller, L. Miao, A. S. Hosseini and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 12149 CrossRef CAS PubMed.
- M. T. Bovino, T. W. Liwosz, N. E. Kendel, Y. Miller, N. Tyminska, E. Zurek and S. R. Chemler, Angew. Chem., Int. Ed., 2014, 53, 6383 CrossRef CAS PubMed.
- D. Chen and S. R. Chemler, Org. Lett., 2018, 20, 6453 CrossRef CAS PubMed.
- S. D. Karyakarte, C. Um, I. A. Berhane and S. R. Chemler, Angew. Chem., Int. Ed., 2018, 57, 12921 CrossRef CAS PubMed.
- F. C. Sequeira and S. R. Chemler, Org. Lett., 2012, 14, 4482 CrossRef CAS PubMed.
- J. A. Mayoral, S. Rodriguez-Rodriguez and L. Salvatella, Chem. – Eur. J., 2008, 14, 9274 CrossRef CAS PubMed.
- L. M. Huffman, A. Casitas, M. Font, M. Canta, M. Costas, X. Ribas and S. S. Stahl, Chem. – Eur. J., 2011, 17, 10643 CrossRef CAS PubMed.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2020673 and 1978089. Crystal structures of 2i and 2l have been deposited in the Cambridge Database. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06404a |
| ‡ Current address: Southern Research Institute, Birmingham, Alabama, 35255, USA. |
| This journal is © The Royal Society of Chemistry 2021 |