
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular nanoparticles self-assembled from reduction-responsive cabazitaxel prodrugs for effective cancer therapy†
Jiahui
Jin‡
ab,
Jianqin
Wan‡
a,
Xiaoxiao
Hu
a,
Tao
Fang
c,
Zhijian
Ye
c and
Hangxiang
Wang
 *a
*a
aThe First Affiliated Hospital, Zhejiang University School of Medicine, NHC Key Laboratory of Combined Multi-Organ Transplantation, Key Laboratory of Organ Transplantation, Research Center for Diagnosis and Treatment of Hepatobiliary Diseases, Zhejiang Province, Hangzhou, 310003, P. R. China. E-mail: wanghx@zju.edu.cn
bXingzhi College, Zhejiang Normal University, Zhejiang Province, Jinhua, 321100, P. R. China
cJinhua People's Hospital, Zhejiang Province, Jinhua, 321000, P. R. China
First published on 15th January 2021
Abstract
Using hydrophobic cabazitaxel as a target anticancer drug, we show that the conjugation of oligo(ethylene glycol)–oligolactide (OEG–OLA) via a self-immolative linkage induces the self-assembly of the resulting prodrug into injectable nanoparticles. The nanoparticles release chemically unmodified cabazitaxel after endocytosis in cancer cells. With the optimal conjugate, the nanotherapy not only potently induces tumor regression but also has a higher safety margin in animals than the free drug administered in its clinical formulation. Our studies highlight the design rationale that attaching a short amphiphilic oligomer to a toxic drug can convert it to a self-deliverable and safe nanotherapy.
Supramolecular chemistry involves molecular assemblies formed by self-assembling a number of individual molecular components via noncovalent interactions.1 Harnessing the reversible and dynamic feature of nanoassemblies, Hamachi and colleagues proposed novel sensing systems in which the detection signal is turned on due to the dissociation of self-assembled nanoaggregation of various probes in response to protein recognition.2 In addition to the use of molecular sensing and imaging, this bottom-up approach also opens up possibilities for the creation of fascinating nanoassemblies to deliver a wide range of pharmacologically active compounds.3 Extensive studies on the development of nanoparticle drug delivery approaches have been encouraged by the clinical success of some nanotherapeutics, including Abraxane, a human serum albumin-based paclitaxel formulation for cancer therapy.4 In particular, “smart” nanotherapies that are responsive to physiological or biochemical stimuli have the potential to treat the evolving cancer.
Intrigued by the advantages of molecular self-assembly, we conceived reduction-responsive prodrugs that are capable of recapitulating self-assembly to construct injectable nanosystems for the in vivo delivery of cytotoxic agents. As a proof-of-principle demonstration, we selected a toxic anticancer agent, cabazitaxel (CTX), for testing the validity of our “self-assembling reduction-triggered prodrug” strategy. CTX is capable of binding with β-tubulin, stabilizing microtubules, and arresting cells in the G2/M phase, which eventually induces cell apoptosis.5 This new taxane agent has the potential to overcome multidrug resistance because of its lower affinity for p-glycoprotein (P-gp) relative to paclitaxel and docetaxel. Despite this great potential, CTX exhibited substantially higher toxicity than other taxanes in clinical trials, limiting its clinical use.6 To date, CTX has only been approved as a second-choice regimen for patients with metastatic castration-resistant prostate cancer (mCRPC) who are refractory to docetaxel therapy. For clinical use, CTX is formulated in polysorbate 80 and ethanol, under the tradename of Jevtana, for intravenous injection due to its poor water solubility. This may cause severe sensitivity reactions in patients.7 Hence, there is an urgent need for the development of novel surfactant-free delivery approaches to improve its therapeutic index and to alleviate its toxicity.
Given its specific metabolism, the tumor microenvironment (TME) is characterized by a high intracellular concentration of reductive glutathione (GSH, typically ∼10 mM).8 Based on this unique feature, reduction-responsive drug delivery systems have been widely successful for potentiating their therapeutic efficacy.9 In these platforms, chemical linkages (e.g., disulfide bond) for drug ligation are stable in the bloodstream after intravenous injection. Once accumulated in tumor tissues and internalized by tumor cells, therapeutically active drugs can be rapidly released from nanocarriers due to the high concentration of intracellular GSH.10 With this design rationale, we conjugated the CTX molecule to an oligo(ethylene glycol) through a hydrophobic segment (i.e., oligo- or polylactide) and a self-immolative linker (i.e., a disulfide bond), which induces self-assembly of the resulting amphiphiles into supramolecular nanoparticles (NPs).
The CTX molecule possesses two hydroxyl groups. The C2′-hydroxyl is exposed outside and is more reactive for chemical derivatization via the formation of the ester bond.3c The synthesis of two prodrug conjugates with varying molecular weights of oligo or polylactide chains follows the scheme summarized in Scheme S1 (ESI†). First, a hydrophilic OEG fragment was selected as an initiator for polymerization to furnish OEG–OLA15 and OEG–PLA50 with a terminal hydroxyl. Next, the hydroxyl groups of bis(2-hydroxyethyl) disulfide were activated by p-nitrophenyl chloroformate to yield carbonate 5 and then further reacted with CTX to produce intermediate 6. Finally, OEG–OLA15 and OEG–PLA50 were selectively conjugated with 6 to produce the final conjugates, OEG–OLA15–CTX and OEG–PLA50–CTX, respectively. Both prodrugs were purified by silica gel chromatography and were characterized by 1H and 13C NMR spectra (Fig. S1–S10, ESI†). High-performance liquid chromatography (HPLC) analysis demonstrated the absence of unreacted CTX in the purified adducts. The characteristic peaks of the protons of OLA15 (5.17–5.26 ppm for –OCH, 1.52–1.60 ppm for –CH3, and 3.38 ppm for –OCH3 at the end of the OLA15 and CTX aromatic protons) are clearly observed in Fig. S7 (ESI†). The peak integration via1H NMR indicated that nearly one CTX molecule was successfully conjugated to each polymer chain. Measurements via gel permeation chromatography (GPC) further verified the prodrug conjugates with monomodal mass-distribution and low polydispersities, and the molecular weights were in accordance with the results from 1H NMR spectra (Fig. S11, ESI†).
In our prodrug design, hydrophobic fragments such as the drug and OLA or PLA provide the driving force for self-assembly, whereas the OEG chain constitutes the hydrophilic shell for stabilizing the overall nanoassemblies. We therefore investigated the self-assembling behaviors of the prodrug constructs in aqueous solutions by exploiting a reprecipitation protocol. Interestingly, both conjugates showed the capacity to form nanostructures with spherical morphologies, as evidenced by transmission and scanning electron microscopy (TEM and SEM, respectively). Compared with the relatively small size and narrow distributions for the OEG–OLA15–CTX conjugate-assembled supramolecular NPs (OEG–OLA15–CTX SNPs, Fig. 1b and c), broader size distributions were found for OEG–PLA50–CTX conjugates that assembled to form OEG–PLA50–CTX SNPs (Fig. S12, ESI†).
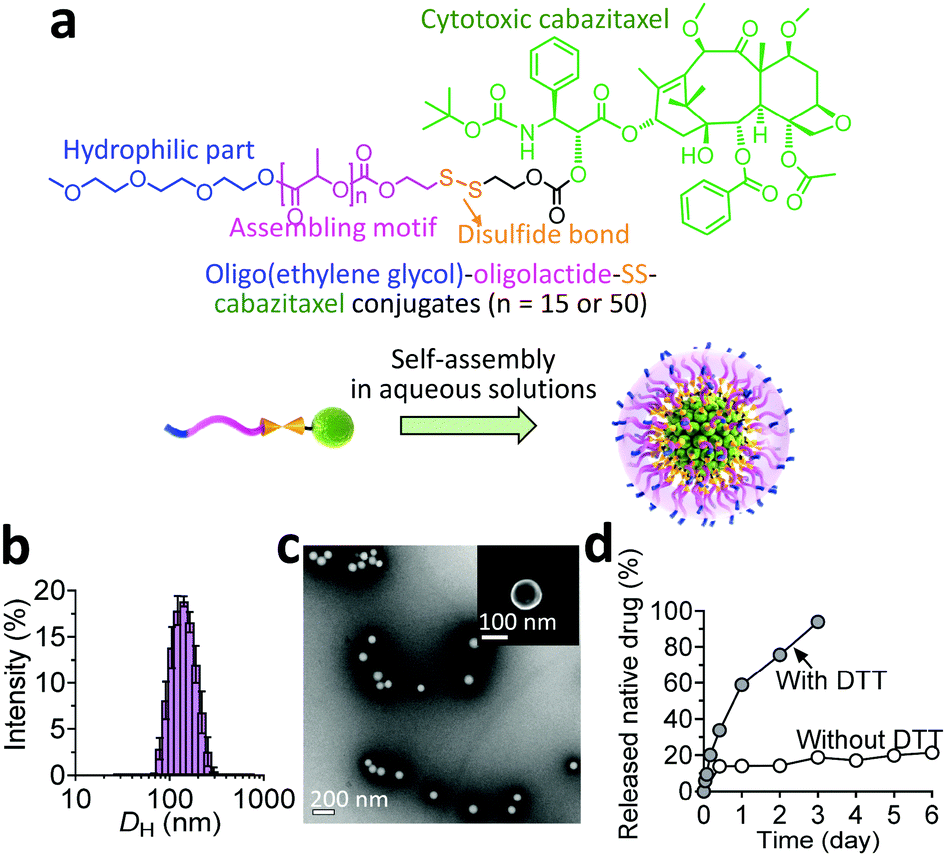 | ||
| Fig. 1 Characterization of the self-assembled prodrug NPs. (a) Chemical structure of CTX prodrugs used in this study and self-assembly of amphiphilic prodrugs in water. (b) Hydrodynamic diameter (DH) of OEG–OLA15–CTX NPs measured by DLS. (c) TEM image of OEG–OLA15–CTX NPs. Inset: SEM image. (d) Release profiles of active CTX from NPs in the presence or absence of DTT using a dialysis method at 37 °C. | ||
Their differences in hydrodynamic diameters (DH) were further examined by dynamic light scattering (DLS) analysis. The results showed that the DH values for OEG–OLA15–CTX SNPs and OEG–PLA50–CTX SNPs were 145.8 ± 41.4 nm and 177.9 ± 73.4 nm, respectively, which is consistent with the TEM results. The hydrophobic component (i.e., the polylactide chain) of the entire prodrug could prominently dictate the particle size; that is, assembling the prodrug with higher hydrophobicity resulted in the formation of larger particles. Owing to its small polydispersity index (PDI) and excellent dispersity in water, we thus selected the OEG–OLA15–CTX SNPs for the next experiments. Notably, stability studies showed that OEG–OLA15–CTX SNPs were stable in deionized (DI) water or DI water containing 10% or 50% FBS, and no variation in their diameters or PDI was observed for several days (Fig. S13, ESI†).
When the prodrugs encounter reductive substances, the thiol resulting from GSH cleavage is expected to cyclize into the proximate carbonyl group of the linker, resulting in the release of CTX in its native form (Scheme S2, ESI†).11 We thus assessed drug release profiles when the conjugates were assembled into NPs. Upon dialyzing against solutions with or without DTT, a series of oligolactide-tethered CTX derivatives will be released from OEG–OLA15–CTX SNPs except for free CTX, complicating the analysis using HPLC. To simplify the quantification of the released drugs, the released samples were subjected to treatment with sodium hydroxide to hydrolyze the CTX derivatives into benzoic acid according to our previously established protocol.12 The results indicated that OEG–OLA15–CTX SNPs exhibited excellent stability in the absence of DTT, with only 20% of CTX released from the NPs after incubation for 6 days (Fig. 1d). As expected, the OEG–OLA15–CTX SNPs responded to the DTT treatment, resulting in 90% of active CTX released in 3 days. Moreover, when the OEG–OLA15–CTX conjugate was incubated in the solution of DMSO/PBS (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) containing 10 mM GSH, rapid activation of free CTX was observed (Fig. S14, ESI†). Hence, OEG–OLA15–CTX SNPs are expected to be stable during systemic circulation but respond to reducing environmental changes in cells to liberate active CTX, leading to efficient cell apoptosis. Such a reduction-triggered drug release behavior could benefit sufficient delivery of NPs and subsequent drug activation at tumor sites.
:1, v/v) containing 10 mM GSH, rapid activation of free CTX was observed (Fig. S14, ESI†). Hence, OEG–OLA15–CTX SNPs are expected to be stable during systemic circulation but respond to reducing environmental changes in cells to liberate active CTX, leading to efficient cell apoptosis. Such a reduction-triggered drug release behavior could benefit sufficient delivery of NPs and subsequent drug activation at tumor sites.
We next evaluated the cytotoxicity of the nanotherapies in two melanoma cell lines B16F10 and A375, and in one mouse macrophage cell line RAW 264.7. Free CTX dissolved in DMSO was included as a comparison. Following a 72 h exposure to the drugs, the cell viability was determined by the CCK-8 assay. The dose–response curves and IC50 values are indicated in Fig. 2a. Cells treated with drug-free OEG–OLA15 fragments did not show cell death. OEG–OLA15–CTX SNPs exhibited slightly reduced cytotoxicity in cells compared with free CTX, which could be attributable to the covalent conjugation of the assembling motifs to CTX at the C2′-OH position critical for drug activity. In contrast, in slowly dividing normal RAW 264.7 macrophages, free CTX exhibited reduced cytotoxic effect, and the IC50 value reflecting the in vitro potency of the NPs also increased accordingly.
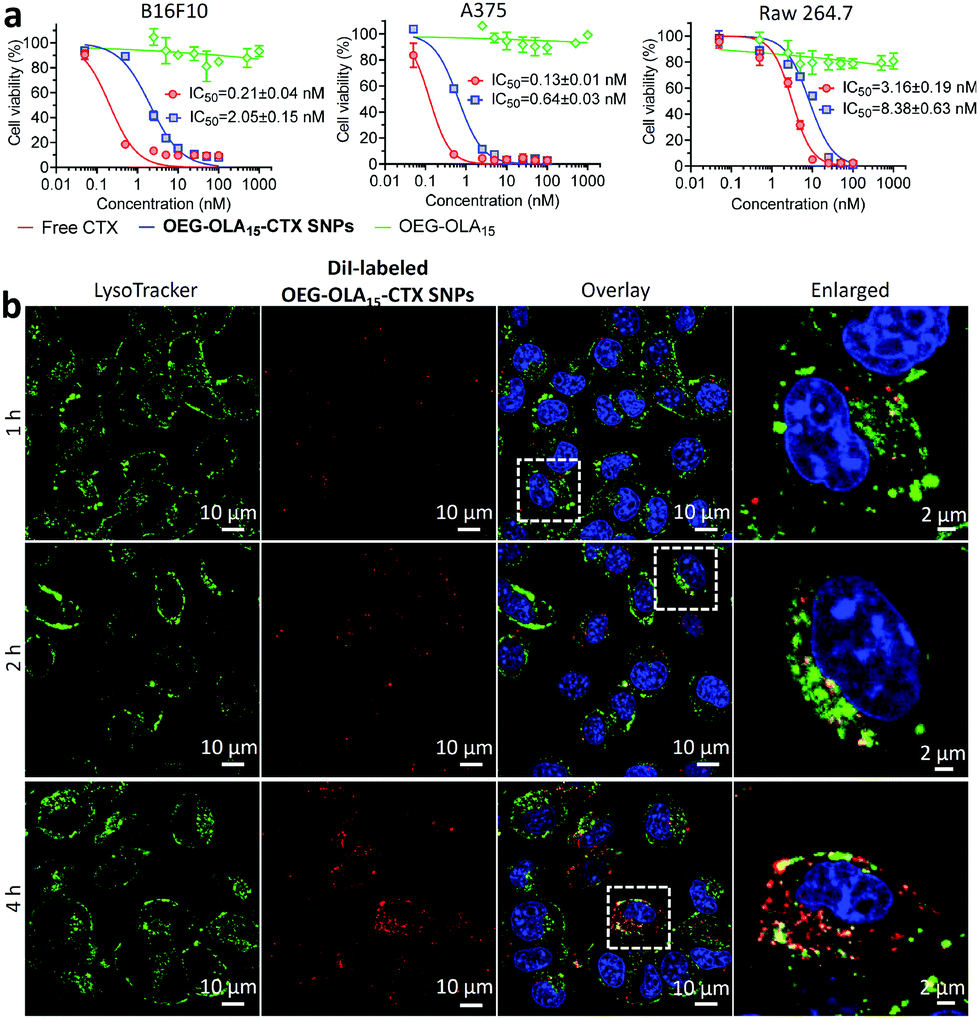 | ||
| Fig. 2 (a) In vitro cytotoxicity of prodrug-assembled NPs. The dose–response curves of free CTX or OEG–OLA15–CTX SNPs for 72 h treatment. Cell viability was determined using the CCK-8 assay in melanoma B16F10 and A375, and in mouse macrophage RAW 264.7 cells. (b) Time-lapse fluorescence images of B16F10 cells costained with LysoTracker Green (0.1 μM) and DiI-labeled OEG–OLA15–CTX SNPs (0.2 μM DiI). | ||
To confirm that the cell growth inhibition was a consequence of CTX-induced apoptosis, an acridine orange/ethidium bromide (AO/EB) staining assay was performed. AO enables the staining of all cells, whereas EB is only able to label necrotic or apoptotic cells. Quantitative analysis of the apoptotic index (i.e., ratios of orange- or red-stained cells/green-stained cells) revealed that OEG–OLA15–CTX SNPs induced notably high levels of apoptosis in the tested cell lines (50.8% ± 2.4% for B16F10 and 49.5% ± 8.2% for A375) compared with the untreated group (4.1% ± 2.0% for B16F10 and 10.0% ± 2.3% for A375) (Fig. S15, ESI†).
We next explored the ability of OEG–OLA15–CTX SNPs to inhibit cell proliferation. After exposure to drugs, click-iT 5-ethynyl-2′-deoxyuridine (EdU) was used to label the proliferating cells because of its capacity to be incorporated into synthesizing DNA. High inhibitory rates on proliferation were manifested in OEG–OLA15–CTX SNP-treated cells, which were 1.8-fold for B16F10 cells and 1.2-fold for A375 cells relative to untreated cells (Fig. S16, ESI†). In addition, due to the stabilization of microtubules resulting from CTX, cell nuclei treated with the drugs exhibited obvious malformation. In general, the prodrug strategy and nanoformulation of the prodrugs significantly reduce the in vitro potency by requiring the release of the prodrugs from the carriers and subsequent cleavage of the prodrugs into their active form. Fortunately, the cytotoxicity of the nanoparticle scaffold was maintained within a comparable range relative to free CTX. These data highlight the success of engineering hydrophobic drugs into surfactant-free, self-assembling NPs that specifically and spontaneously release active drugs inside tumor cells.
Cellular uptake of the prodrug NPs was investigated using confocal fluorescence microscopy in B16F10 cells. OEG–OLA15–CTX SNPs were fluorescently labeled with a lipophilic dye DiI via a coassembly protocol. The localization of the NPs in the cells was observed by costaining of live cells with LysoTracker Green (a lysosome probe). Time-lapse fluorescence images showed that the fluorescence of DiI almost overlaid with that of LysoTracker Green at early time-points (Fig. 2b). After 4 h of incubation, most of the NPs were not colocalized with lysosomes, which indicates efficient endosomal escape. We also examined the endocytosis pathways for the NPs by using various inhibitors. Flow cytometry analysis uncovered that the clathrin-coated pit endocytosis inhibitor chlorpromazine reduced intracellular uptake, suggesting that this pathway should be involved in the internalization of the NPs (Fig. S17, ESI†).
Clinical trials showed that clinical CTX formulations (Jevtana) caused significant systemic toxicity, such as hepatorenal toxicities, in patients. The maximum tolerated dose (MTD) was only 25 mg m−2 in Phase I studies, which was lower than that of other taxanes, with MTD values for paclitaxel and docetaxel of 175 mg m−2 and 60–100 mg m−2, respectively.13 The surfactant (i.e., polysorbate 80) used in the Jevtana formula may induce hemolytic damage to red blood cells (RBCs). Thus, a hemolysis assay was conducted to evaluate the hemocompatibility of OEG–OLA15–CTX SNPs, and free CTX formulated in the Jevtana-mimicking formulation was included as a reference. As depicted in Fig. S18 (ESI†), compared with the severe hemolysis caused by polysorbate 80, the NPs showed negligible hemolysis (<1%) even at a concentration of 3 mg mL−1, indicating that the OEG–OLA15–CTX SNPs were applicable for intravenous injection.
Intrigued by this result, we further hypothesized that the self-assembling prodrug nanoparticle approach was able to alleviate the toxicity induced by CTX. To validate this assumption, healthy ICR mice were intravenously injected with free CTX (in the Jevtana-mimicking formulation) or NPs via the tail vein. Following three injections, variations in body weight (Fig. S19, ESI†) and mouse survival (Table S1, ESI†) for each group were recorded to evaluate the toxicity of the drug formulations. Encouragingly, mice treated with OEG–OLA15–CTX SNPs showed no drop in body weight even at the highest dose of 20 mg kg−1 (CTX equivalent), whereas free CTX resulted in a significant loss of body weight. In particular, a dose of 10 mg kg−1 of free CTX induced a 15% loss of body weight and led to only one survival out of three mice. Neutropenia, typically characterized by reduced white blood cell (WBC) counts, is a dose-limiting toxicity observed in most of patients administered with taxane drugs.13 The mice that received intravenous injections of free CTX showed a substantial reduction of the WBC counts (Fig. S20, ESI†). Injection of OEG–OLA15–CTX SNPs at the same dosage did not exhibit myelosuppression during the observation period. Moreover, other blood parameters remained at normal ranges.
To further assess the damage of the drugs to major organs (e.g., heart, liver, spleen, lungs and kidneys), we performed histological analysis (Fig. S21 and S22, ESI†). Obviously, in free CTX-treated mice, the liver was significantly damaged, and hepatocyte edema was observed around the central veins. In contrast, the liver sections of the nanotherapy-treated mice showed normal histology. These results indicated that OEG–OLA15–CTX SNPs had the potential to mitigate the toxicity resulting from CTX and substantially improved drug safety profiles.
Given the promising cytotoxicity and tolerance observed with OEG–OLA15–CTX SNPs, we thus explored the therapeutic efficacy in a preclinical mouse model of human melanoma patient-derived xenografts (PDXs). This model was established and characterized in our recently published studies.14 The fifth passage of xenografts was used for efficacy testing. Following three intravenous injections, OEG–OLA15–CTX SNPs effectively inhibited the tumor burden, whereas rapid tumor growth was observed in saline-treated mice (Fig. S23a, ESI†). At the endpoint of the study, the tumor volume of the mice receiving nanotherapy was significantly smaller than that in the saline-treated mice (p < 0.01 versus saline) (Fig. S23c, ESI†). Of note, administration of OEG–OLA15–CTX SNPs at 12 mg kg−1 elicited durable tumor shrinkage, and this dose was well tolerated by the animals, as evidenced by quickly rebounding body weights after cessation of treatment. In contrast, dosing of free CTX was effective in inhibiting tumor growth but showed significant toxicity, resulting in substantial weight loss even at the endpoint of the study (Fig. S23b,ESI†).
The tumor sections excised from the PDXs were subjected to histological analysis to examine the antitumor activity (Fig. S23d, ESI†). TUNEL staining revealed that OEG–OLA15–CTX SNPs induced extensive intratumoral apoptosis, correlating with the results of H&E staining (Fig. S24, ESI†). Ki67 is regarded as a specific marker of cell proliferation.15 Compared to the saline group, a decreased level of Ki67 was observed in drug-treated mouse tumors, revealing that NP treatment efficiently inhibited the proliferation of tumor cells. Collectively, OEG–OLA15–CTX SNPs were favorable formulations for cancer therapy. The detailed characterization of the toxicities and efficacies in animals highlights the successful vectorization of this agent, which deserves further investigation.
Finally, the efficacy of the NPs was tested in another separate B16F10 xenograft-bearing mouse model (Fig. S25, ESI†). The OEG–OLA15 pro-moiety used for drug derivatization had no therapeutic effect and showed comparable tumor growth with the saline treatment (p > 0.05, versus saline). Intriguingly, intravenous injection of OEG–OLA15–CTX SNPs substantially delayed tumor growth in this highly aggressive and fast-growing model.
Through structure-based rational engineering of a taxane drug, we successfully synthesized two novel amphiphilic OEG–OLAn–CTX prodrugs that form self-assembling NPs in an aqueous medium. Notably, we showed for the first time that chemical derivatization with short OEG–OLA fragments induced the self-assembly of the prodrug entities. The nanoparticles degraded specifically and released active compounds in response to the intracellular reducing environment, thereby showing high in vitro cytotoxic potency as well as alleviating in vivo drug toxicity by reducing systemic drug exposure. In the preclinical melanoma PDX model, administration of OEG–OLA15–CTX SNPs provoked robust and lasting tumor inhibitory activity at a tolerable dose. Given the high drug loading and biocompatibility for each component, we also expect the approach to be a generalizable approach for the reformulation of other therapeutic agents with high toxicity and low efficiency.
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (No. LR19H160002), the National Natural Science Foundation of China (No. 82073296, 81773193, and 81571799), the Science and Technology Research Program of Jinhua City (2017-3-020), and the Social Development Project of Public Welfare Technology Research in Zhejiang Province (2017c37139). We thank Chenyu Yang in the Center of Cryo-Electron Microscopy (CCEM), Zhejiang University for her technical assistance on TEM analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. K. Smith, Chem. Commun., 2018, 54, 4743–4760 RSC; (b) W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS; (c) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- (a) Y. Takaoka, T. Sakamoto, S. Tsukiji, M. Narazaki, T. Matsuda, H. Tochio, M. Shirakawa and I. Hamachi, Nat. Chem., 2009, 1, 557–561 CrossRef CAS; (b) K. Mizusawa, Y. Ishida, Y. Takaoka, M. Miyagawa, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 7291–7293 CrossRef CAS; (c) Y. Takaoka, Y. Sun, S. Tsukiji and I. Hamachi, Chem. Sci., 2011, 2, 511–520 RSC.
- (a) Q. Pei, X. Hu, X. Zheng, S. Liu, Y. Li, X. Jing and Z. Xie, ACS Nano, 2018, 12, 1630–1641 CrossRef CAS; (b) Q. Pei, X. Hu, S. Liu, Y. Li, Z. Xie and X. Jing, J. Controlled Release, 2017, 254, 23–33 CrossRef CAS; (c) H. Wang, Z. Lu, L. Wang, T. Guo, J. Wu, Z. Li, D. Jiang, P. Song, H. Xie, L. Zhou, X. Xu and S. Zheng, Cancer Res., 2017, 77, 6963–6974 CrossRef CAS; (d) T. Fang, Z. Ye, J. Wu and H. Wang, Chem. Commun., 2018, 54, 9167–9170 RSC.
- V. Roy, B. R. Laplant, G. G. Gross, C. L. Bane and F. M. Palmieri, Ann. Oncol., 2008, 20, 449–453 CrossRef.
- M. D. Galsky, A. Dritselis, P. Kirkpatrick and W. K. Oh, Nat. Rev. Drug Discovery, 2010, 9, 677–678 CrossRef CAS.
- C. S. M. Albuquerque, J. A. Rinck, T. V. Costa, C. N. M. Ferreira, A. A. Dettino, I. H. F. Cunha and V. F. Calsavara, J. Clin. Oncol., 2017, 35, e566 Search PubMed.
- W. Shelley, N. Talanin and E. D. Shelley, The Lancet, 1995, 345, 1312–1313 CrossRef CAS.
- (a) C. A. Lyssiotis and A. C. Kimmelman, Trends Cell Biol., 2017, 27, 863–875 CrossRef CAS; (b) A. Bansal and M. C. Simon, J. Cell Biol., 2018, 217, 2291–2298 CrossRef CAS.
- (a) C. Chu, H. Lin, H. Liu, X. Wang, J. Wang, P. Zhang, H. Gao, C. Huang, Y. Zeng, Y. Tan, G. Liu and X. Chen, Adv. Mater., 2017, 29, 1605928 CrossRef; (b) C. Wang, F. Cao, Y. Ruan, X. Jia, W. Zhen and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 9846–9850 CrossRef CAS.
- S. Raina and D. Missiakas, Ann. Rev. Microbiol., 1997, 51, 179–202 CrossRef CAS.
- L. Zhou, H. Xie, X. Chen, J. Wan, S. Xu, Y. Han, D. Chen, Y. Qiao, L. Zhou, S. Zheng and H. Wang, Acta Biomater., 2020, 113, 464–477 CrossRef CAS.
- J. Wan, Y. Qiao, X. Chen, J. Wu, L. Zhou, J. Zhang, S. Fang and H. Wang, Adv. Funct. Mater., 2018, 28, 1804229 CrossRef.
- (a) A. C. Mita, L. J. Denis, E. K. Rowinsky, J. S. DeBono, A. D. Goetz, L. Ochoa, B. Forouzesh, M. Beeram, A. Patnaik, K. Molpus, D. E. Semiond, M. L. Besenval and A. W. Tolcher, Clin. Cancer Res., 2009, 15, 723 CrossRef CAS; (b) S. E. Jones, J. Erban, B. Overmoyer, G. T. Budd, L. Hutchins, E. Lower, L. Laufman, S. Sundaram, W. J. Urba, K. I. Pritchard, R. Mennel, D. Richards, S. Olsen, M. L. Meyers and P. M. Ravdin, J. Clin. Oncol., 2005, 23, 5542–5551 CrossRef CAS.
- (a) L. Shi, Y. Wang, Q. Wang, Z. Jiang, L. Ren, Y. Yan, Z. Liu, J. Wan, L. Huang, B. Cen, W. Han and H. Wang, J. Controlled Release, 2020, 324, 289–302 CrossRef CAS; (b) L. Huang, X. Chen, Q. Bian, F. Zhang, H. Wu, H. Wang and J. Gao, J. Controlled Release, 2020, 328, 325–338 CrossRef CAS.
- A. Urruticoechea, I. E. Smith and M. Dowsett, J. Clin. Oncol., 2005, 23, 7212–7220 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary figures. See DOI: 10.1039/d0cc06854c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |