
A trinuclear salen–Al complex for copolymerization of epoxides and anhydride: mechanistic insight into a cocatalyst-free system†
Ranlong
Duan
ab,
Yanchuan
Zhou
a,
Yuezhou
Huang
ab,
Zhiqiang
Sun
a,
Han
Zhang
a,
Xuan
Pang
 *ab and
Xuesi
Chen
ab
*ab and
Xuesi
Chen
ab
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: xpang@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, China
First published on 20th November 2020
Abstract
A rare example of a trinuclear salen–Al complex is reported. Due to the intramolecular interaction, the trinuclear salen–Al complex can catalyze copolymerization with high activity in the absence of any cocatalyst. On the basis of a covalent coordination insertion mechanism, ABC(1)C(2) type tetrablock copolymers were produced using an hydroxyl initiator.
The global consumption of traditional plastics has reached enormous proportions in recent years. Although traditional plastics have promoted social development and brought convenience, waste plastics have caused an environmental crisis because they are difficult to degrade.1 Polyester is a type of degradable material, and it can be used as a substitute for traditional plastics in many fields.2 Among the synthetic methods used for polyesters, ring-opening copolymerization of epoxides and cyclic anhydrides has advantages, such as a wide range of monomer resources, and the polymers obtained have low-dispersity (Đ) and controlled molecular weights.3 Since the first successful copolymerization of epoxides and cyclic anhydrides was reported, many metal complexes have been used as catalysts in the copolymerization.4 Among these, salen–Al complexes show excellent performance. Williams and coworkers explored salen–Al complexes as switch catalysts to prepare multi-block polymers from a mixture of propylene oxide (PO), lactide (LA) and phthalic anhydride (PA), which is difficult to obtain using conventional procedures.5 Lu and coworkers reported bimetallic salen–Al complexes containing a binaphthol-linked ligand, which could catalyze the copolymerization of racemic terminal epoxides or meso-epoxides with cyclic anhydrides. The catalyst system exhibited high regioselectivity and enantioselectivity and isotactic semicrystalline polymers were produced.6 To maintain activity, salen–Al catalysts usually work in the form of bicomponent systems with the aid of a nucleophilic cocatalyst. Coates and coworkers researched the mechanism of the copolymerization using the bicomponent salen–Al catalyst system. The simplified mechanism is shown in Scheme 1. Salen–Al and the cocatalyst bis(triphenylphosphine)iminium chloride (PPNCl) formed a bis(alkoxide) intermediate, which worked as the initiator at the beginning of copolymerization. Polymer chain propagation was achieved through the interaction of several five- or six-coordinated alkoxide and carboxylated intermediates. In the propagation process, the insertion step of the cyclic anhydride was fast, and the activation and insertion steps of the epoxide were the rate limiting steps.7
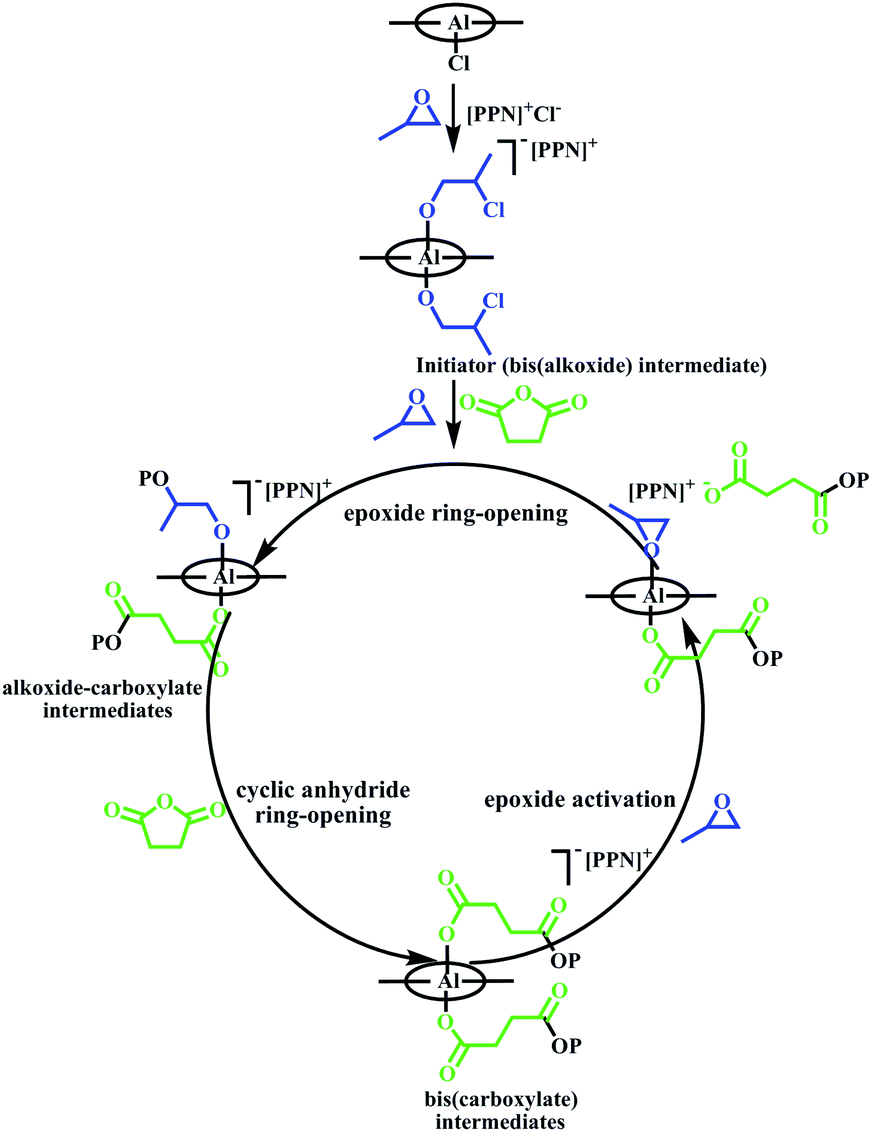 | ||
| Scheme 1 Simplified mechanism of the copolymerization of epoxides and cyclic anhydride using the bicomponent system.7b | ||
Although the propagation of polymer chains using the bicomponent catalytic system was demonstrated nicely by this mechanism, the disadvantage of this approach was apparent in that the addition of cocatalyst increased the complexity of the reaction system. As described in Scheme 1, the epoxide was activated and ring-opened under the combined action of PPNCl and the salen–Al complex to obtain the bis(alkoxide) intermediate. This intermediate, as the precatalyst, can initiate ring-opening and insertion of cyclic anhydride to form the first repeat unit. Therefore, whether the axial substituent of salen–Al was Cl- or alkoxide group, the salen–Al complexes would produce copolymer with the end group of the Cl ring-opened epoxides.
This restricted the direct participation of hydroxy compounds as initiators in the copolymerization. For example, polyolefin and polyester multi-block copolymers could be easily obtained by using a functional hydroxyl initiator, while they were difficult to obtain via the above-mentioned methods.8 Although the synthesis of multi-block copolymers of epoxides, anhydrides and other monomers can be achieved through specially substituted PPNX or chain transfer agents,5,9 the direct use of a functional hydroxyl initiator and salen–Al complexes to catalyze the copolymerization could provide a simple way to produce multi-block copolymers of epoxides, anhydrides and other monomers. In order to achieve this goal, it is necessary to eliminate the influence of the cocatalyst in the initiation step.
It is worth noting that salen–Al complexes exhibit excellent performance in the ring-opening polymerization of lactide, with the absence of cocatalyst, and the polymerization mechanism as shown in Scheme S1 (ESI†). The covalent intermediate initiated ring-opening and insertion of lactide to achieve the polymer chain.10 Inspired by this cocatalyst-free system, we explored the feasibility of cocatalyst-free salen–Al catalysts in the copolymerization of epoxides and cyclic anhydrides. Our group reported a series of trinuclear salen–Al complexes.11 In the study of mononuclear and trinuclear salen–Al complexes (Scheme 2), the electronic spectra of the trinuclear complex 1 indicate that complex 1 is not a simple superposition of three salen–Al units with similar structures to the mononuclear complex 2. It was believed that an electronic interaction existed between the three salen–Al subunits via the phloroglucinol moieties. This electronic interaction changed the electrophilicity of the Al atom and hence significantly improved the activity of complex 1 in the ring-opening polymerization of cyclic esters.11 Herein, complex 1 was investigated as a catalyst for the copolymerization of epoxides and cyclic anhydrides in the presence of 2-propanol. For comparison, the mononuclear salen–Al complex 2 was also prepared.
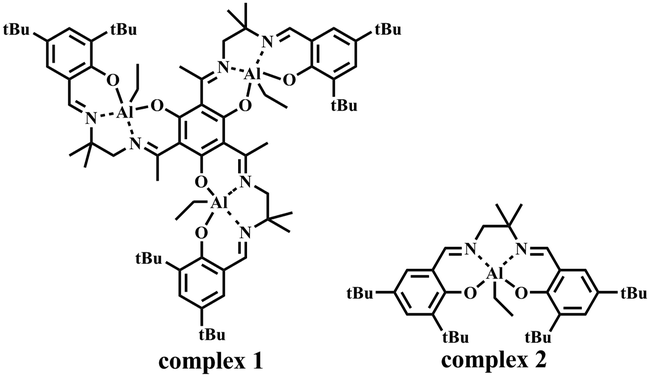 | ||
| Scheme 2 The complexes used in this study. | ||
As shown in Table 1, in the presence of PPNCl as a cocatalyst, complex 2 can catalyze the copolymerization of epoxides and cyclic anhydrides (Table 1, entry 4). Complex 2 lost its activity in the absence of the cocatalyst (Table 1, entry 2). Compared with complex 2, the trinuclear complex 1 retained its activity regardless of the presence of the cocatalyst (Table 1, entries 1 and 3). The copolymer obtained by using the bicomponent catalytic system of complex 2 and PPNCl had a wide molecular weight distribution (Table 1, entry 4). However, the copolymer produced using complex 1 alone as the catalyst had a narrow molecular weight distribution. This result indicated that, compared with the bicomponent catalytic system of complex 2 and cocatalyst, complex 1 had better controllability for the initiation and growth of polymer chains with the absence of cocatalyst. Under the same conditions, using complex 1 in the presence of PPNCl, the molecular weight of the obtained copolymer decreases and the molecular weight distribution becomes wider (Table 1, entries 1 and 3).
| Entry | Catalyst | [PPNCl]b | [PA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Cat]:[i-PrOH]b :[Cat]:[i-PrOH]b |
Temp./°C | Time/h | Conv./%c | M n/(kg mol−1)d | Đ |
|---|---|---|---|---|---|---|---|---|
|
a
The designed amounts of catalyst and [PPNCl] were dissolved in 2 mL PO and carefully added into a 25 mL autoclave, [Cat]:[PO] = 1:3000 using complex 1 and [Cat]:[PO] = 1:1000 using complex 2. The autoclave was heated up to a suitable temperature.
b Molar ratio.
c The results determined by 1H NMR analysis of the conversion of PA in the crude reaction mixture.
d Determined by gas-phase chromotography using CH2Cl2 as solution, calibrated with polystyrene standard.
|
||||||||
| 1 | 1 | 0 | 300:1:3 |
80 | 14 | 99 | 15.6 | 1.16 |
| 2 | 2 | 0 | 100:1:1 |
80 | 14 | 0 | — | — |
| 3 | 1 | 1 | 300:1:3 |
80 | 14 | 99 | 9.5 | 1.36 |
| 4 | 2 | 1 | 100:1:1 |
80 | 14 | 99 | 14.2 | 1.51 |
| 5 | 1 | 0 | 400:1:3 |
80 | 12 | 99 | 17.3 | 1.18 |
| 6 | 1 | 0 | 500:1:3 |
80 | 13 | 59 | 10.9 | 1.16 |
| 7 | 1 | 0 | 300:1:3 |
60 | 24 | 78 | 12.5 | 1.15 |
| 8 | 1 | 0 | 600:1:3 |
100 | 18 | 90 | 28.1 | 1.17 |
This result may be attributed to the increase in initiator groups caused by the addition of PPNCl. In the absence of cocatalyst, the activity of complex 1 increased with increase in temperature (Table 1 entries 1, 7 and 8).
To better understand the influence of intramolecular interactions on the activity of complex 1, the kinetic behavior of complex 1 with or without cocatalyst was studied (for details see ESI†). The kinetic studies indicated that complex 1 maintained high activity in the copolymerization without cocatalyst (Fig. S1, ESI†). In the absence of PPNCl, first-order kinetics in PA and the catalyst were observed (Fig. S3 and S4, ESI†). A linear relationship between PA conversion and Mn was observed (Fig. S5, ESI†). The influence of the concentration of PO and PA was also investigated. As described in Fig. 1a, with decrease in PA concentration the activity of copolymerization clearly decreased, and the conversion of PA increased with increase in PO concentration (Fig. 1b). These results indicate that the copolymerization was not zero-order-dependent on PA, and that the polymerization activity was influenced by the concentration of PA.
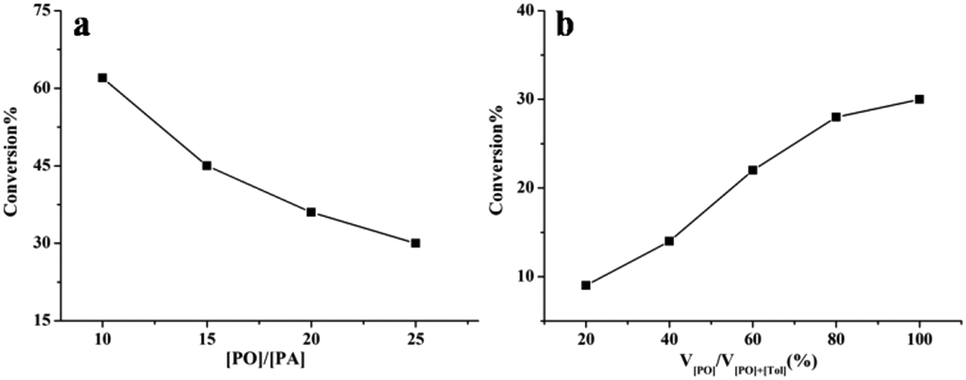 | ||
| Fig. 1 (a) The conversion of PA vs. concentration using complex 1 without the presence of a cocatalyst. [1]:[i-PrOH]:[PA]:[PO] = 1:3:600:6000 to 1:3:600:15000, 80 °C, 14 h. (b) Conversion of PA vs. concentration of PO using complex 1 without the presence of cocatalyst. [1]:[i-PrOH]:[PA] = 1:3:600, [PA] = 0.54 mol L−1, 80 °C, 14 h. | ||
As reported by Coates and coworkers, for copolymerization using the bicomponent catalytic system (PPNCl and salen–Al), after complete conversion of the anhydride, the six-coordinate bis(alkoxide) and alkoxide–carboxylate salen–Al complexes were produced, and the color of the reaction system changed from yellow to red due to the presence of these two complexes.7b
In order to illustrate the coordination of the active species in the copolymerization using complex 1, the color change of the reaction system was studied. Due to the low boiling point of PO, high boiling point POCl was used. As shown in Fig. 2a, in the presence of PPNCl, the copolymerization proceeded very rapidly. The conversion of PA was higher than 99% within 1 hour and the color of the reaction system quickly turned red and then remained stable. In the absence of cocatalyst, the activity of copolymerization was lower; with conversion of PA higher than 99% occurring after 8 h.
 | ||
| Fig. 2 Photographs of copolymerization solutions at the specified time points. [1]:[i-PrOH]:[PA]:[POCl] = 1:3:300:3000, 100 °C. (a) [1]:[PPNCl] = 1:1; (b) PPNCl free. | ||
As shown in Fig. 2b, the color of the reaction system remained yellow all the time. After 24 h, upon addition of PPNCl, the color quickly turned red (Fig. S7, ESI†). Compared with previous research, here, in the absence of cocatalyst, the bis(alkoxide) and alkoxide-carboxylate intermediates (as shown in Scheme 1) might not be produced in the copolymerization, and the propagation of the copolymer chain might be initiated by the covalent-active species (as shown in Fig. 3). In order to further determine the mechanism of initiation, the MALDI-TOF mass spectrum of the copolymer was investigated. The results indicated that the copolymer was end-capped with an isopropyl group, showing that the initiator was the aluminum alkoxide intermediate (Fig. S8, ESI†).
 | ||
| Fig. 3 Proposed mechanism for copolymerization of PA and PO using complex 1 with the absence of cocatalyst. | ||
On the basis of the above research, it was hypothesized that complex 1 might select a coordination insertion mechanism in the copolymerization in the absence of the nucleophilic cocatalyst. As shown in Fig. 3, the five-coordinate alkoxide initiator was obtained by rapid reaction of complex 1 with 2-propanol. Then, anhydrides were activated by the alkoxide intermediate, and ring-opening and insertion of anhydride between Al and alkoxide groups formed the carboxylate intermediate. This carboxylate intermediate would further activate and ring-open PO to achieve propagation of the copolymer chain.
It has been reported that the catalytic system of salen–Al and PPNCl can catalyze the copolymerization of epoxides, cyclic anhydrides and lactide (LA),5 and that PPNCl influences the initiation of propagation. In these copolymerizations, the hydroxyl compounds usually act as a chain transfer agent rather than as an initiator.5 The mechanism described in Fig. 3 provides a new possibility for copolymerization initiated directly by hydroxyl compounds. Therefore, ternary copolymerization using complex 1 was investigated in the absence of the cocatalyst. As shown in Table S1 (ESI†), the copolymers obtained by means of ternary copolymerization have a narrow molecular weight distribution. The narrow molecular weight distribution can still be maintained even at high molecular weights (Fig. S9, ESI†).
Similar to previous studies,12 copolymerization of PA and PO was superior to the ring-opening polymerization of LA (Fig. S10, ESI†). Based on the interesting finding that complex 1 and 2-propanol can catalyze copolymerization, a simple method for the preparation of multi-block copolymer containing epoxides and anhydrides was provided by using the hydroxyl initiator. This method makes it possible to use the macromolecular hydroxyl initiator methoxy polyethylene glycol to prepare ABC-type triblock copolymers by terpolymerization of LA, PA and PO. The triblock copolymer had a narrow molecular weight distribution (Fig. S11 and S12, ESI†). It is worth noting that when ε-caprolactone (CL) was added at the end of the ternary copolymerization, the copolymerization continued, as shown in Fig. S13, S14 and Table S1 (ESI†) (entry 9). With participation of CL, the ABC(1)C(2)-type tetrablock copolymer can be obtained.
In summary, a trinuclear salen–Al complex was prepared for copolymerization of epoxide and cyclic anhydride. The three salen–Al subunits on the phloroglucinol moieties permitted intramolecular electronic communication to optimize the electrophilicity of the Al atom. This change made the trinuclear salen–Al complex show high activity in the copolymerization of epoxide and anhydride without the presence of a cocatalyst. Copolymerization kinetics indicated the influence of monomer concentration on activity and provided new mechanistic insight into the copolymerization. The mechanism indicates that the copolymerization can be carried out in situ by an hydroxyl initiator and the trinuclear salen–Al complex. Furthermore, multi-block copolymers were synthesized using a macromolecular initiator and this mechanism. This method may provide a simple way to synthesize multi-block copolymers containing epoxide and anhydride blocks.
This work was funded by the National Key Research and Development Program of China (2016YFC1100701) and the National Natural Science Foundation of China (No. 51773200 and 52073272).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 5 CrossRef; (b) J. Ma, G. D. Sheng and P. O'Connor, Environ. Pollut., 2020, 264, 10 Search PubMed; (c) C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC; (d) R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS.
- (a) J. B. Zhang, L. B. Wang, S. F. Liu and Z. B. Li, J. Polym. Sci., 2020, 58, 803–810 CrossRef CAS; (b) Y. J. Shin and M. L. Becker, Polym. Chem., 2020, 11, 3313–3321 RSC.
- (a) Z. Q. Wan, W. M. Ren, S. Yang, M. R. Li, G. G. Gu and X. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 17636–17640 CrossRef CAS; (b) H. Y. Ji, D. P. Song, B. Wang, L. Pan and Y. S. Li, Green Chem., 2019, 21, 6123–6132 RSC; (c) Z. Q. Wan, J. M. Longo, L. X. Liang, H. Y. Chen, G. J. Hou, S. Yang, W. P. Zhang, G. W. Coates and X. B. Lu, J. Am. Chem. Soc., 2019, 141, 14780–14787 CrossRef CAS.
- (a) F. Santulli, I. D'Auria, L. Boggioni, S. Losio, M. Proverbio, C. Costabile and M. Mazzeo, Organometallics, 2020, 39, 1213–1220 CrossRef CAS; (b) H. K. Ryu, D. Y. Bae, H. Lim, E. Lee and K. S. Son, Polym. Chem., 2020, 11, 3756–3761 RSC; (c) F. Isnard, F. Santulli, M. Cozzolino, M. Lamberti, C. Pellecchia and M. Mazzeo, Catal. Sci. Technol., 2019, 9, 3090–3098 RSC; (d) H. Li, J. P. Zhao and G. Z. Zhang, ACS Macro Lett., 2017, 6, 1094–1098 CrossRef CAS; (e) A. M. DiCiccio, J. M. Longo, G. G. Rodriguez-Calero and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 7107–7113 CrossRef CAS; (f) D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS; (g) L. Y. Wu, D. D. Fan, X. Q. Lu and R. Lu, Chin. J. Polym. Sci., 2014, 32, 768–777 CrossRef CAS.
- T. Stoesser, D. Mulryan and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 16893–16897 CrossRef CAS.
- (a) J. Li, B. H. Ren, Z. Q. Wan, S. Y. Chen, Y. Liu, W. M. Ren and X. B. Lu, J. Am. Chem. Soc., 2019, 141, 8937–8942 CrossRef CAS; (b) J. Li, B. H. Ren, S. Y. Chen, G. H. He, Y. Liu, W. M. Ren, H. Zhou and X. B. Lu, ACS Catal., 2019, 9, 1915–1922 CrossRef CAS.
- (a) B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS; (b) M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS.
- (a) Y. Wang, Y. Zhao, Y. Ye, H. Peng, X. Zhou, X. Xie, X. Wang and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 3593–3597 CrossRef CAS; (b) H. Dong, Y. J. Zhu, Z. J. Li, J. X. Xu, J. J. Liu, S. Q. Xu, H. X. Wang, Y. Gao and K. Guo, Macromolecules, 2017, 50, 9295–9306 CrossRef CAS; (c) Y. Y. Zhang, G. W. Yang and G. P. Wu, Macromolecules, 2018, 51, 3640–3646 CrossRef CAS.
- T. Stosser, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 9 CrossRef.
- (a) Z. H. Tang, X. S. Chen, X. Pang, Y. K. Yang, X. F. Zhang and X. B. Jing, Biomacromolecules, 2004, 5, 965–970 CrossRef CAS; (b) N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef CAS.
- X. Pang, R. Duan, X. Li, C. Hu, X. Wang and X. Chen, Macromolecules, 2018, 51, 906–913 CrossRef CAS.
- Y. C. Zhou, C. Y. Hu, T. H. Zhang, X. W. Xu, R. L. Duan, Y. Luo, Z. Q. Sun, X. Pang and X. S. Chen, Macromolecules, 2019, 52, 3462–3470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06874h |
| This journal is © The Royal Society of Chemistry 2021 |