
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic investigation of benzene esterification by K2CO3/TiO2: the catalytic role of the multifunctional interface†
Jittima
Meeprasert
a,
Guanna
Li
 *bc and
Evgeny A.
Pidko
*a
*bc and
Evgeny A.
Pidko
*a
aInorganic Systems Engineering, Department of Chemical Engineering, Delft University of Technology, The Netherlands. E-mail: e.a.pidko@tudelft.nl
bBiobased Chemistry and Technology, Wageningen University & Research, The Netherlands. E-mail: guanna.li@wur.nl
cLaboratory of Organic Chemistry, Wageningen University & Research, The Netherlands
First published on 19th July 2021
Abstract
Potassium carbonate dispersed over a defective TiO2 support (K2CO3/TiO2) is an efficient catalyst for benzene esterification with CO2 and CH3OH. Density functional theory calculations reveal that this unique catalytic reactivity originates from the cooperation of the Ti3+/K+ surface sites. The K2CO3 promotor steers the stabilization of surface intermediates thus preventing catalyst deactivation.
CO2 conversion into valuable chemicals has received much attention due to the environmental concerns associated with the growing atmospheric concentrations of greenhouse gases. Many efforts have been invested to develop the economic carbon-neutral system by recycling the carbon resource of CO2 from industrial emission to the production of chemicals.1–3 The carboxylation of aromatics by CO2 is one of the attractive routes for the valorization of CO2, because the produced aromatic carboxylic acids and their derivatives can serve as important chemical feedstocks.4,5 Conventionally, such a carboxylation coupling reaction is carried out in the presence of a strong base or Lewis acid such as NaH,6 AlCl3,4 and AlBr3.7 The strong base is needed to cleave the C–H bond of arene to form a nucleophilic carbon atom which can further interact with the weak CO2 electrophile. The role of the Lewis acid is to activate the CO2 molecule before the arene C–H bond carboxylation.8 However, both these strategies usually provide rather low yields of the target products due to the low electrophilicity of CO2 and side reactions caused by excessive reactivity of the mediators.7,8 Therefore, the development of alternative catalytic procedures avoiding the usage of strong base or acid is highly desired but also represents one of the great challenges for this reaction.
Recently, Kanan et al. reported that alkali carbonates (K2CO3 and Cs2CO3) finely dispersed over a TiO2 support can promote the two-step cycle of benzene esterification with CO2 and CH3OH to produce methyl benzoate with both high yield (80%) and high selectivity (100%) in the absence of stoichiometric additives.9 It is important to note that bare TiO2 can also promote the first step of benzene C–H bond carboxylation, however, the catalyst became deactivated after just one catalyst recycling. In contrast, no carboxylation products were observed when K2CO3 or Cs2CO3 powders as the only catalyst component were used. Thus, it was hypothesized that dispersing alkali carbonate over TiO2 would engender catalytic carboxylation activity towards hydrocarbon substrates because of the disruption of the bulk alkali carbonate structure. However, the mechanistic aspects of this system, such as the nature of the active site, initiation of the reaction and the exact role of the different catalyst components, remained moot.8 This inspired us to carry out a comprehensive mechanistic study of benzene carboxylation with CO2 and subsequent reaction with CH3OH over the K2CO3/TiO2 catalyst by periodic density functional theory (DFT) calculations. Our main objective was to identify the role and function of each catalyst component and to propose the origin of the deactivation of bare TiO2 catalyst.
In this work, all calculations were performed using the Vienna Ab Initio Simulation Package (VASP) 5.3.5.10,11 DFT calculations were carried out using PBE functional based on the generalized gradient approximation (GGA).12 Grimme's DFT-D3 method with Becke-Jonson damping was used to account for the dispersion interactions.13 The DFT+U method was applied to the 3d orbitals of Ti to correct the on-site Coulomb interactions. The U value used in this work is 4.2 eV.14 The energy cutoff and convergence criteria for the electronic and ionic loops were 400 eV, 10−5 eV, and 0.05 eV Å−1, respectively. Transition states were determined by the nudged-elastic band method with the improved tangent estimate (CI-NEB) and subsequent frequency analysis. The model of K2CO3/TiO2 catalyst was built following the experimental evidence of the very fine dispersion of K2CO3 on the surface of TiO2.9 Fig. S1 in the ESI† shows the catalyst model featuring the K2CO3 species deposited on the defective anatase TiO2(101) surface. We hypothesized that the interface of coordination-unsaturated surface Ti site (Ti3+) together with the adjacent K2CO3 cluster form the reactive ensemble because neither the bulk crystalline K2CO3 nor the pristine TiO2 surface are active.9,15
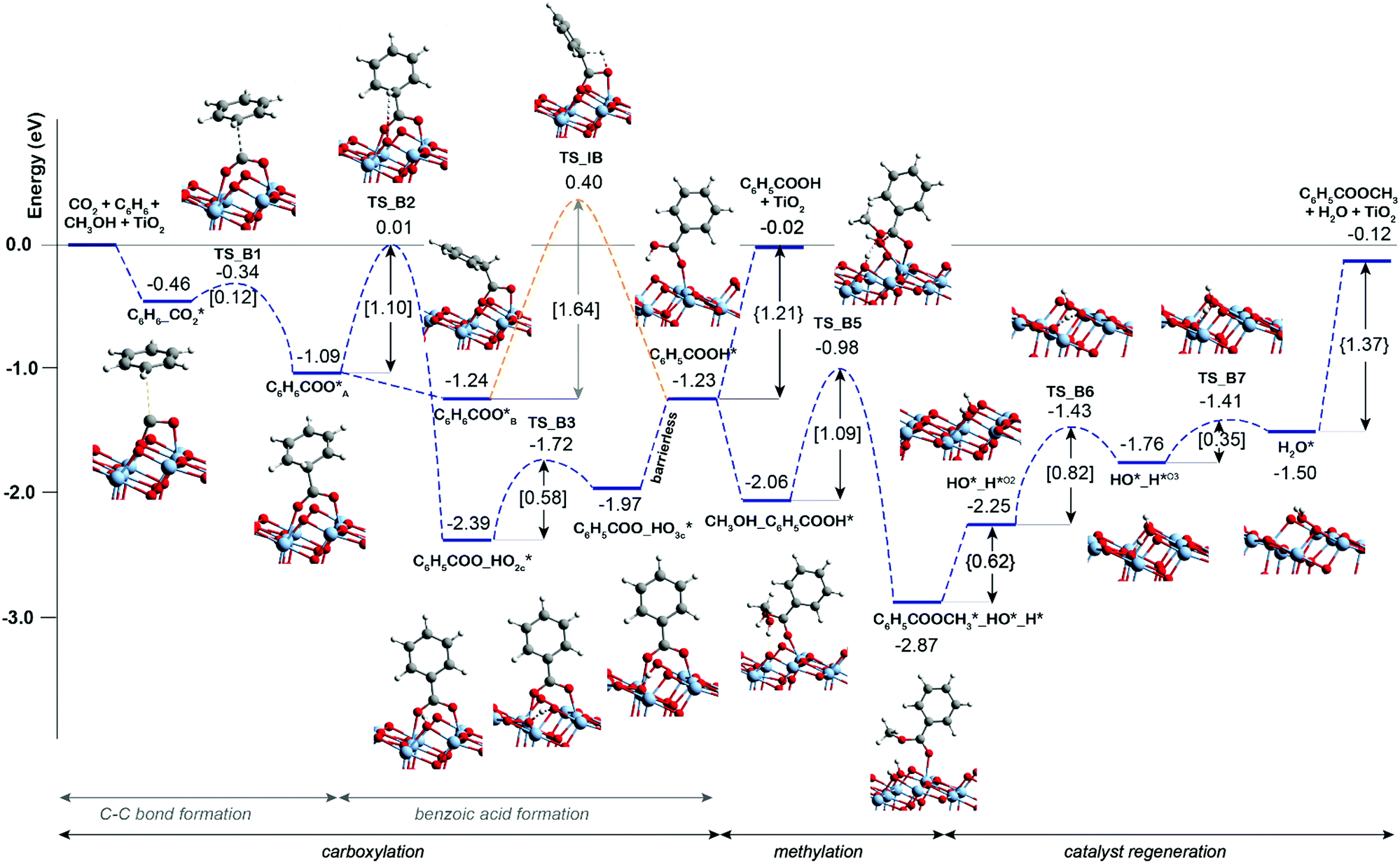
Scheme 1 presents a proposed reaction mechanism for the esterification of benzene with CO2 and CH3OH by the K2CO3/TiO2 catalyst. The computed reaction energy profile is shown in Fig. 1. Firstly, the two possible mechanisms of C6H6 C–H bond deprotonation were evaluated and it was found that the direct C–H carboxylation of benzene with activated CO2 is preferred over the C6H6 deprotonation (Fig. S2 and discussion, ESI†). Therefore, we proposed that the reaction starts with the adsorption of CO2 through bidentate coordination with the interface Ti3+ and K+ sites. The presence of K2CO3 prevents a bidentate adsorption mode of CO2 with two Ti3+ surface atoms which occur on the bare defective TiO2 surface. In the next step, the C–C bond formation between the co-adsorbed benzene and CO2 occurs to form the C6H6COO* intermediate (C6H6_CO2* → C6H6COO*). This step is endothermic with ΔE = 0.46 eV and it proceeds with an activation energy (E‡) of 0.72 eV. Next, the C–H bond of the activated C6H6 fragment is cleaved to form benzoic acid (C6H5COOH*) or benzoate (C6H5COO*) surface intermediate. The former is formed via a direct intramolecular H-transfer from the C6H6 moiety of C6H6COO* to terminal O of the carboxylate moiety. The computed activation barrier for this step is 1.31 eV. An alternative path involves a two-step surface-assisted H*-transfer, upon which the C6H6COO* intermediate is first deprotonated by the vicinal basic surface O sites (E‡ = 1.19 eV) followed by the C6H5COO* and H* recombination (E‡ = 1.19 eV). The highest activation energy of the indirect route is only ca. 0.10 eV lower than that of the direct pathway, indicating that both reaction routes can contribute to the catalytic reaction. The hydrogen transfer from the C6H6COO* to form C6H5COO* is predicted to be more difficult than the initial coupling of CO2 and benzene, which is consistent with the experimentally observed kinetic isotopic effect results.9 Electronic analysis further indicated that effective charge transfer between intermediates and the defective catalyst surface facilitates the C–C coupling and deprotonation reaction processes (Fig. S3 and S4, ESI†). The desorption of benzoic acid to regenerate the catalytic interface is endothermic by 0.95 eV.
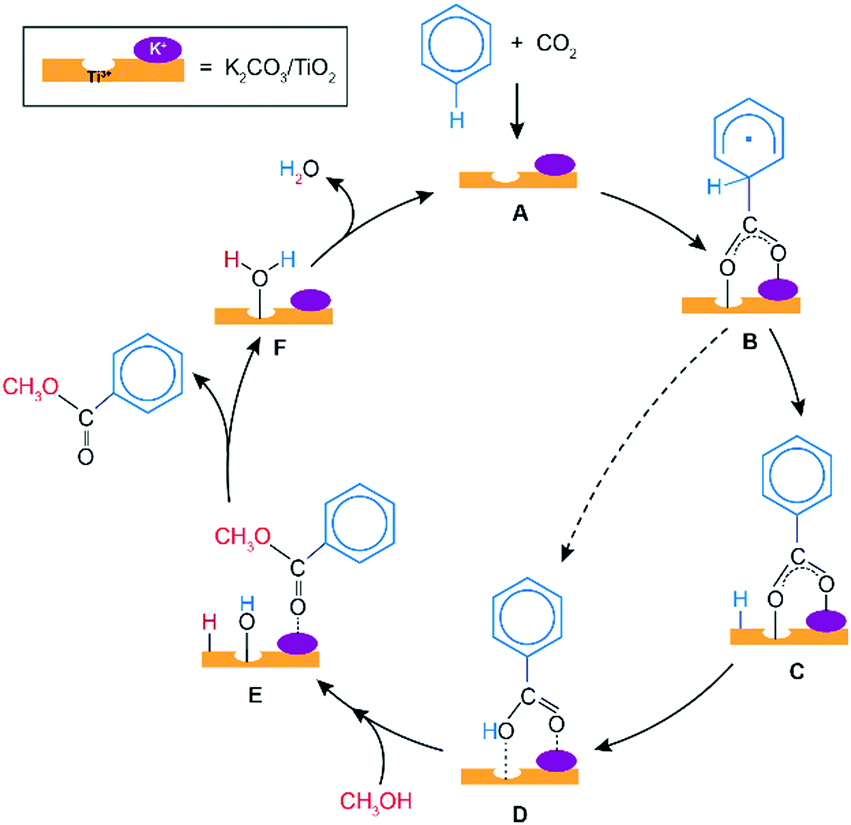 | ||
| Scheme 1 Proposed mechanism of benzene esterification with CO2 and CH3OH on K2CO3/TiO2 catalyst. The A + CO2 + C6H6 → D conversion involves the carboxylation of benzene with CO2via the one-step direct (dashed) or stepwise indirect (solid path) mechanisms to yield the adsorbed benzoic acid. The latter is methylated with CH3OH (D + CH3OH → F + C6H5COOCH3) to yield methyl benzoate product and the adsorbed water. Subsequent desorption of H2O by-product (F → A + H2O) regenerates the catalytic surface ensemble. | ||
 | ||
| Fig. 1 DFT-computed reaction energy diagram for the benzene esterification with CO2 and CH3OH on K2CO3/TiO2. | ||
The closure of the catalytic cycle can be facilitated in the presence of methanol, which reacts with the surface benzoate intermediate (CH3OH_C6H5COOH*) to produce methyl benzoate product (C6H5COOCH3*_HO*_H*). During the methylation process, a CH3OH molecule is added to the system and co-adsorbed at the neighboring surface oxygen atom of benzoic acid. Then, the methyl benzoate is generated by the formation of the C–O bond between CH3OH and benzoic acid. The simultaneous deprotonation of CH3OH* and cleavage of the C–OH bond of C6H5COOH* result in the generation of two hydroxyl groups on the surface. This concerted step is slightly exothermic and proceeds with an activation energy of 0.73 eV. In the next step, the methyl benzoate product is desorbed from the surface with ΔE of 0.30 eV. The last step is the recombination of surface OH groups to form H2O and regenerate the catalytic interface sites (HO*_H*O2 → H2O*). This dehydration step is barrierless but proceeds with a barrier of 1.00 eV associated with the surface migration of H*. The hydrogen transfer step following the C–C bond formation is identified as the most difficult step of the carboxylation reaction having the highest activation barrier of 1.31 eV along the reaction path.
Previous experimental studies9 showed that the carbonate-free defective TiO2 can also promote benzene carboxylation but it loses the activity already after the first reaction cycle. We therefore hypothesized that the reaction intermediates or the reaction products (e.g. methyl benzoate or water) might block the active site of the bare TiO2 catalyst, while the presence of K2CO3 species protects catalyst from such poisoning. To check this hypothesis, the DFT analysis was extended to the mechanism of benzene carboxylation followed by methylation on the bare and defective anatase TiO2 (101) surface. Fig. 2 presents the respective DFT-computed reaction energy diagram. In this case, the C–C bond formation between CO2 and benzene is thermodynamically and kinetically more favorable than on the interface site (ΔE = −0.63 eV, E‡ = 0.12 eV). However, the subsequent H* transfer to directly form adsorbed benzoic acid is in this case 0.3 eV higher than over the K2CO3/TiO2. A much more favorable reaction is the benzoate formation via the hydroxylation of the TiO2 surface. This step has a barrier of 1.1 eV and stabilizes the system by ΔE of −1.3 eV. Almost identical energetics was observed for the K2CO3/TiO2 catalyst. For the subsequent concerted benzoic acid methylation reaction, the activation energy over defective TiO2 is 0.36 eV higher than that of K2CO3/TiO2 catalyst reaction (TS_B5 = 1.09 eV vs. TS_K5 = 0.73 eV).
 | ||
| Fig. 2 Reaction energy diagram for the benzene esterification with CO2 and CH3OH on defective TiO2 surface. | ||
The comparison of the reaction profiles in Fig. 1 and 2 reveals that the activation energies of all elementary steps (except the initial CO2 coupling with benzene) over the defective TiO2 surface and K2CO3-promoted TiO2 are quite close and comparable suggesting that indeed both catalysts can enable the esterification reaction of benzene with CO2 and CH3OH to form methyl benzoate. However, we find that most of the reaction intermediates on the defective TiO2 surface are significantly more stable than those on K2CO3/TiO2. The energies of all reaction intermediates on the defective TiO2 surface are in the range of 0.00 to −3.00 eV, while those on K2CO3/TiO2 catalyst fall in the range of +1.00 to −2.00 eV relative to the reactant state. The stronger binding interaction on the bare TiO2 surface impedes the desorption of the products and regeneration of the active site for the next catalytic cycle (Fig. S5, ESI†). Specifically, the desorption energies of methyl benzoate and H2O on the defective TiO2 surface are 0.39 and 0.32 eV higher than that on K2CO3/TiO2, respectively (ΔEC6H5COOCH3: 1.37 eV vs. 0.98 eV; ΔEH2O: 0.62 eV vs. 0.30 eV).
Based on these results, we conclude that both bare TiO2 with oxygen vacancy and K2CO3/TiO2 catalysts are able to activate CO2 and benzene to form benzoate products. However, the excessive reactivity of the surface sites in the former results in surface poisoning by the reaction products/byproducts. The presence of potassium carbonate species partially deactivates the reactive sites on the titanium surface to facilitate the product desorption and the regeneration of the catalytic interface sites. Although the K2CO3 species cannot act as an active site alone for this reaction, it steers the local structures of the transition states and reaction intermediates, and thus facilitates the products desorption and catalyst regeneration. These insights shed light onto the role of multicomponent reaction environments on the catalytic surface for the efficient CO2 valorization and they can form a base for further development of efficient and stable catalysts for the direct carboxylation with CO2 of other more challenging hydrocarbon substrates such as ethane, methane and ethylene.
Authors acknowledge financial support from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 725686) J. M. gratefully acknowledges the Royal Thai Government Scholarships for the financial support. The use of supercomputer facilities was sponsored by NWO Domain Science.
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- A. D. N. Kamkeng, M. Wang, J. Hu, W. Du and F. Qian, Chem. Eng. J., 2021, 409, 128138 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- G. A. Olah, B. Török, J. P. Joschek, I. Bucsi, P. M. Esteves, G. Rasul and G. K. Surya Prakash, J. Am. Chem. Soc., 2002, 124, 11379–11391 CrossRef CAS PubMed.
- A. S. Lindsey and H. Jeskey, Chem. Rev., 1957, 57, 583–620 CrossRef CAS.
- J. Luo, S. Preciado, P. Xie and I. Larrosa, Chem. – Eur. J., 2016, 22, 6798–6802 CrossRef CAS PubMed.
- K. Nemoto, H. Yoshida, N. Egusa, N. Morohashi and T. Hattori, J. Org. Chem., 2010, 75, 7855–7862 CrossRef CAS PubMed.
- J. Luo and I. Larrosa, ChemSusChem, 2017, 10, 3317–3332 CrossRef CAS PubMed.
- D. J. Xiao, E. D. Chant, A. D. Frankhouser, Y. Chen, A. Yau, N. M. Washton and M. W. Kanan, Nat. Chem., 2019, 11, 940–947 CrossRef CAS PubMed.
- G. F. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. F. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. B. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 3865–3868 CrossRef CAS PubMed.
- G. S. Stefan, E. Stephan and G. Lars, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- W. Song, S. Ma, L. Wang, J. Liu and Z. Zhao, ChemCatChem, 2017, 9, 4340–4344 CrossRef CAS.
- D. C. Sorescu, W. A. Al-Saidi and K. D. Jordan, J. Chem. Phys., 2011, 135, 124701 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02513a |
| This journal is © The Royal Society of Chemistry 2021 |