
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Voltage-induced fluorescence lifetime imaging of a BODIPY derivative in giant unilamellar vesicles as potential neuron membrane mimics†
Dumitru
Sirbu
 *a,
Lingli
Zeng
b,
Paul G.
Waddell
c,
Nikolai V.
Tkachenko
d,
Stanley W.
Botchway
e and
Andrew C.
Benniston
*b
*a,
Lingli
Zeng
b,
Paul G.
Waddell
c,
Nikolai V.
Tkachenko
d,
Stanley W.
Botchway
e and
Andrew C.
Benniston
*b
aSchool of Mathematics, Statistics & Physics, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: dumitru.sirbu@ncl.ac.uk
bMolecular Photonics Laboratory, Chemistry-School of Natural & Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: andrew.benniston@ncl.ac.uk
cCrystallography Laboratory, Chemistry-School of Natural & Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
dFaculty of Engineering and Natural Sciences, Tampere University, Tampere, Finland
eCentral Laser Facility, Rutherford Appleton Laboratory, Harwell, Oxford, OX11 0QX, UK
First published on 3rd November 2021
Abstract
Fluorescence lifetime imaging (FLIM) was used to study the behaviour of a BODIPY dye in a giant unilamellar vesicle (GUV) in the presence of an electric field. The modulation of the electric field resulted in distinctive fluorescence lifetime changes in line with environment alterations within the membrane mimic.
Voltage sensitive dyes (VSDs) have been the workhorse in the neuroscience field for monitoring electrical processes in cells and tissues by their fluorescence response.1 They operate by a change to their absorption or emission spectra based on a voltage change across a thin membrane.2 Generally speaking there are two types of VSDs according to their response speed; one type is termed a fast-response probe and the other one is a slow-response probe.3 The disparate response mechanisms by which the two types of VSDs operate offer different levels of information, with fast responsive affording a low signal-to-noise (S/N) ratio but more critical detail of frequent spikes.4 Slow responsive probes offer a superior S/N but miss subtle details. The ultimate objective is to have a VSD that is fast and has an excellent S/N ratio.5 Many fast responsive VSDs rely on small changes in fluorescence levels induced by the modulation in the electric field across a membrane.6 Although modest progress has been made in improving fast VSDs there appears to be an impasse in making major future advancements, requiring a radical approach to create a next generation neuroscience technology. The “holy grail” in the end is to be able to see in real time the workings of the nervous system, with neurons receiving excitatory and inhibitory postsynaptic potentials.7
Fluorescence lifetime imaging microscopy (FLIM) is viewed as one of the most powerful techniques to determine the spatial distribution of excited state lifetimes in a sample together with picosecond time response. FLIM can be used in a number of places, as it is extremely sensitive, non-invasive, non-destructive and able to provide useful biochemical interactions information within the molecular scale that is not assessable to steady-state fluorescence technology.8 Moreover, fluorescence steady-state measurements and imaging is prone to errors such as probe concentration, detector non-linearity and photobleaching. These are rarely corrected for during confocal imaging alone. FLIM takes advantage of the fact that the lifetime of a fluorophore has a concentration-independent property, which means it depends on its environment rather than the concentration.9 Hence, the effects of molecules on fluorescence lifetime can be investigated independently without knowing the concentration of the fluorophore. We envisaged that the marriage of FLIM to VSDs could open up the prospect of voltage-induced lifetime imaging (VILI) as a new technology.
As the first stage development of the new concept giant unilamellar vesicles (GUVs) are introduced as the mimic of the plasma membrane. They represent free-standing bilayer vesicles with diameters ranging between 10–100 μm and are capable of supporting fluorescent dyes without any major perturbation of the GUV structure.10 In this paper, a new low molecular weight BODIPY VSD derivative aJBD was tested. The dye is neutral and lipophilic and the julolidine acts as electron donor while the BODIPY core is the electron acceptor to create intramolecular CT character. The interaction of the external electric field and the CT state was envisaged to perturb the emitting state of the dye by altering their mutual electronic coupling. Rather than measuring fluorescence intensity changes the lifetime alterations were expected to correlate to voltage modulations. Such a new method would hopefully have application in such an area as mammalian brain slice imaging which is still a challenging task.11
The preparation of the target dye is shown in Scheme 1 starting from BD which was synthesized according to an adapted method reported elsewhere.12 The compound aJBD was synthesised through the Knoevenagel condensation between BD and JUL-CHO as a black solid with a green metallic colour.
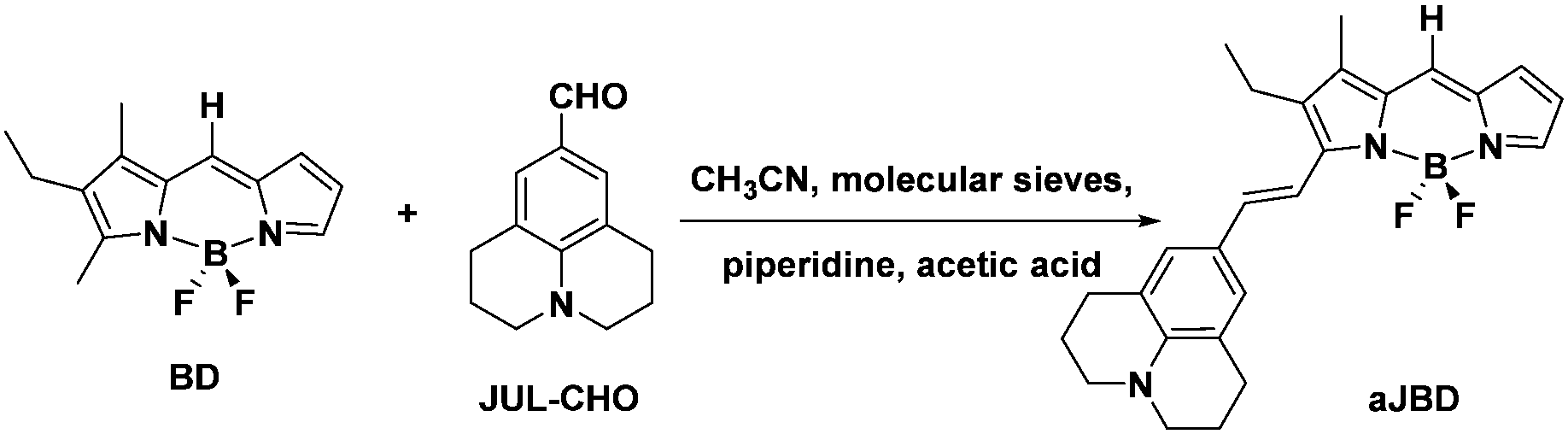 | ||
| Scheme 1 Preparation of the FLIM responsive dye aJBD containing the donor julolidine group and BODIPY acceptor unit. | ||
1H NMR, 13C NMR, COSY, HSQC and HMBC NMR spectra for aJBD were consistent with the proposed structure (see ESI†). Interestingly, the resonances for the two alkene protons appeared as a pseudo-triplet with a coupling constant in the order of 17Hz rather than two doublets. A triplet peak found for the 11B NMR spectrum at 1.03 ppm and the quartet (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1) for the 19F NMR spectrum at −143.06 ppm, with a coupling constant of around 32–34 Hz are typical for BODIPYs and further corroborate the structure. High-resolution mass spectrometry and HPLC measurements confirmed the high purity of the material (see ESI†). An X-ray structure determination also confirmed the trans carbon–carbon double bond (see ESI†).
:1:1:1) for the 19F NMR spectrum at −143.06 ppm, with a coupling constant of around 32–34 Hz are typical for BODIPYs and further corroborate the structure. High-resolution mass spectrometry and HPLC measurements confirmed the high purity of the material (see ESI†). An X-ray structure determination also confirmed the trans carbon–carbon double bond (see ESI†).
The electronic absorption spectrum for aJBD (Fig. 1) in hexane shows a typical strong absorption band at λABS = 623 nm, which is associated with the S0–S1 BODIPY-centred electronic transition. A further vibrational shoulder on the higher-energy side at around 580 nm is also observed. Both bands are strongly red-shifted when compared to the parent unsubstituted BODIPY.13 The relative narrow fluorescence profile is located at λEM = 644 nm and a small vibrational shoulder on the lower-energy side. The Stokes shift (SS) is small (20 nm, 511 cm−1) which is typical for a BODIPY compound. The fluorescence quantum yield (ϕFLU) reaches near unity.
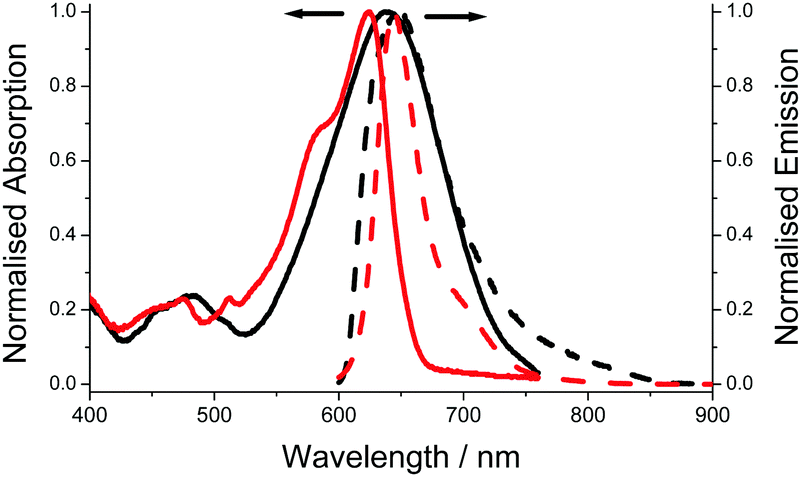 | ||
| Fig. 1 Absorption (solid) and fluorescence (dash) spectra recorded for aJBD in dilute hexane (red) and DMF (black). | ||
Compared to the UV-visible absorption spectrum in the non-polar solvent hexane, the spectrum in a polar solvent such as DMF shows very different behaviour. The main electronic transition is significantly broadened and shifted to 648 nm. Unlike similar julolidine BODIPY compounds, where the two units are connected at the meso-BODIPY position and display an additional broad charge transfer CT band in the red/near-IR region,14 no obvious CT band is observed for aJBD in DMF. The intense fluorescence observed in the very low polarity solvent hexane is almost extinguished in DMF, supporting that an efficient quenching process is introduced.
Fluorescence quantum yields for aJBD were recorded in a series of alkanol and aprotic solutions of varying dielectric constants and viscosities (see ESI†). Generally speaking, the ϕFLU values cover a wide range from 0.01 to almost 1. It is evident that the fluorescence quantum yields changed significantly with the dielectric constant. The increase of dielectric constant from hexane (ε = 1.9) to DMSO (ε = 46.7) is accompanied by a dramatic decrease in the fluorescence quantum yield. The emission is essentially extinguished at ε = 9 or above. Given the presence of the nitrogen atom on the julolidine unit, which is likely to hydrogen bond with alkanols, we specifically investigated a small range of alkanols to see if the fluorescence quantum yields varied systematically with viscosity. Indeed, in addition to the dependence on the dielectric constant the quantum yields showed a clear dependence on the viscosity of the solvent as highlighted by a good fit to the Förster–Hoffmann equation (see ESI†).15 The presence of a single bond at the julolidine para position should allow rotation of the two subunits as illustrated in Fig. S15 (see ESI†). Hence, formation of a twisted intramolecular charge transfer (TICT) state is feasible, caused by this rotation resulting in efficient non-radiative decay. From these two studies it is clear that strong fluorescence should be observed from aJBD when aligned in a membrane of low polarity and high viscosity.
To further understand the excited state dynamics of aJBD, fluorescence lifetimes were measured in a number of non-protiated solvents using the time-correlated single photon counting technique. The fluorescence decay profile was satisfactory fitted to a double exponential decay. In hexane, τ = 3.1 ns (contribution of ca. 87%), corresponding to a radiative rate constant kRAD = 3.2 × 108 s−1. This is assigned to the lifetime of the radiative relaxation of the S1 state. A second slightly longer-lived component (τ = 7.2 ns) but with a smaller contribution of ca. 13% was also detected. The second contribution is attributed to the repopulation of the S1 state from a CT state (i.e., delayed fluorescence). For this mechanism to operate in very non-polar solvents the CT state must be sufficiently decoupled from the ground state to avoid rapid deactivation, but this is enhanced as the polarity increases thus dramatically decreasing both the quantum yield and lifetime. Indeed as the polarity of the solvents increased the lifetime decreased to below the instrument response limit (<0.1 ns).
To gain a more comprehensive picture of the excited-state behaviour of aJBD in hexane and DMSO, femtosecond pump–probe transient absorption spectroscopy was employed (Fig. 2). In hexane there is a clear ground-state bleach centred around 650 nm at around 0.2 ps which fits with the rapid formation of the S1 excited state. A further short-lived species (1 ps) is also evident from the spectral records, which presumably arises from a conformational change in the molecule as it vibrationally cools. A further transient signal (119 ps) is also observed which displays two prominent features at around 640 nm and 720 nm; the latter band is in the range where there is limited ground-state absorption and is assigned to emission from the CT state. The 640 nm feature is consistent with ground-state bleach and in agreement with an equilibrium between the singlet and CT states (vide supra). The global fit provides a long lifetime of the latter feature (>1.6 ns), limited by the instrument time-window, which is in line with the fluorescence lifetime value (see ESI†).
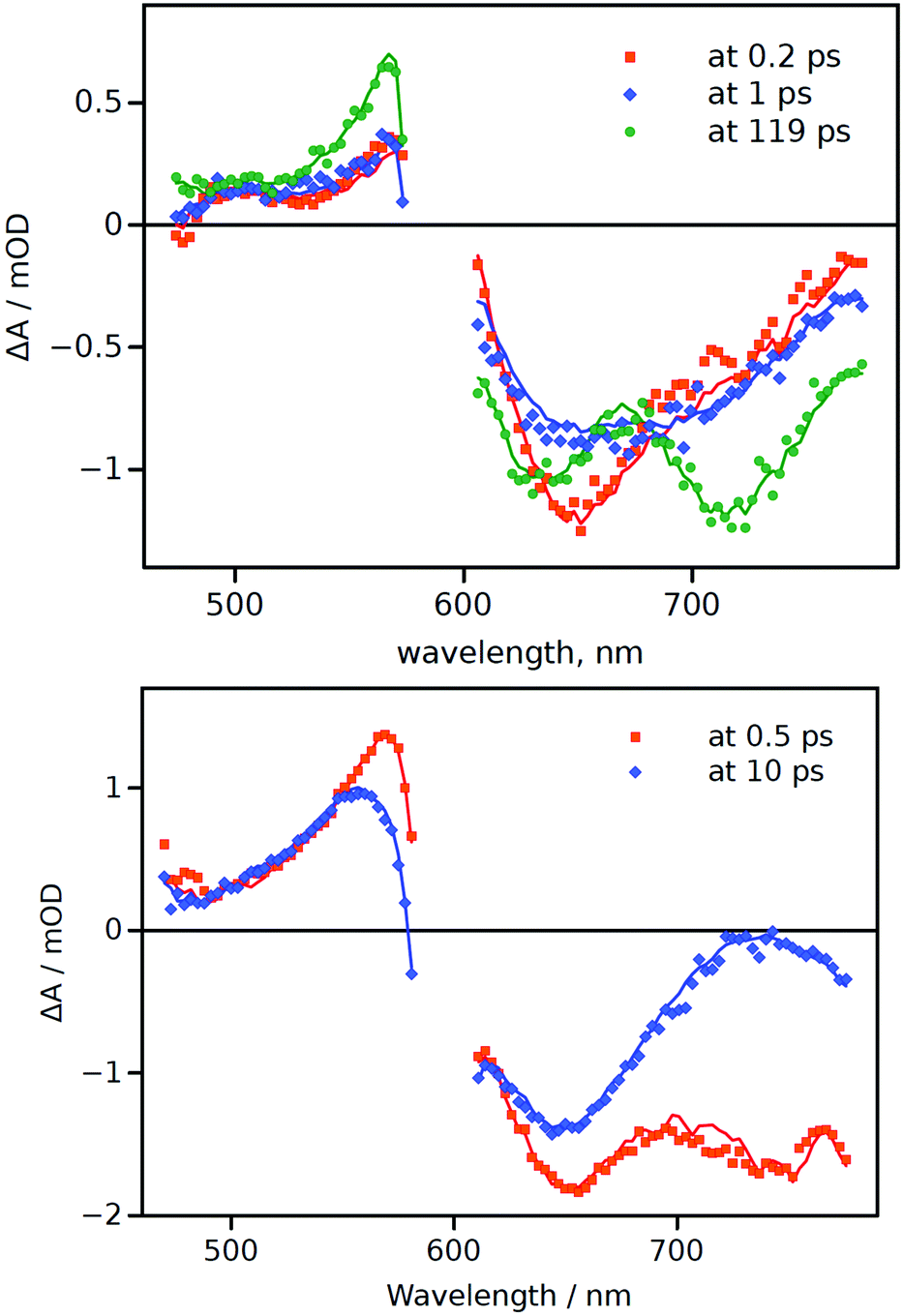 | ||
| Fig. 2 Differential transient-absorption spectra at a few selected delay times (indicated in the plots) for aJBD in hexane (top) and DMSO (bottom). | ||
Compared to aJBD in hexane, the global fit in DMSO only required the use of two lifetimes and there was no sign of any long-lived component. At short delay time (0.5 ps) the transient spectrum contains features similar to those seen in hexane, but at longer delay (10 ps) there is a well-defined alteration to the profile. This change implies a structural perturbation of the structure, again in agreement with the proposed TICT model as discussed from the fluorescence viscosity measurements.
The overall picture is that in very non-polar solvents the excited state dynamics is governed by the interplay of the S1 and CT states. With an increase in the solvent polarity the CT state drops well below the emissive S1 state and becomes an efficient deactivation pathway. Importantly the results illustrate that the excited state lifetime for aJLB is very responsive to its local surrounding through the modulation of the CT state energy and its tight connection with the S1 state. This positive feature boded well for a voltage-induced lifetime effect.
To test the VILI approach GUV models of a cellular membrane were grown by applying alternating current (AC) frequency of 10 Hz with an output voltage of 3.3 Vpp to a platinum wire covered with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) in a sucrose solution. Staining with aJBD showed the formation of well-defined GUVs on the surface of the electrode (see ESI†). An isolated GUV is illustrated in Fig. 3a as its intensity map, and the corresponding FLIM measurements performed at different locations of the spherical surface. Fitting the decay trace required at least two exponentials and performing this for each pixel produced the lifetime map images as shown in Fig. 3a (middle and right). The short-lived component (τ1 ∼1 ns) always showed a higher contribution (∼80%), whilst the long-lived component (τ2 >3 ns) had a smaller contribution to the fluorescence lifetime distribution (Fig. 3b). It is noticeable that τ2 is on par and in agreement with the singlet lifetime measured in the non-polar environment of hexane, whereas τ1 would suggest that a proportion of aJBD molecules are located in an environment of a different polarity. Importantly, whilst τ2 was relatively constant, τ1 varied significantly in the range of 0.9 to 1.8 ns. Furthermore, the intensity-weighed lifetime distribution (Fig. 3c) across the GUV membrane showed two defined maxima τ′ and τ′′ at around 1 ns and 2 ns. In order to check the electric-field nature of this effect, the voltage applied at the platinum wire was ramped up, resulting in a linear increase of the longer component contribution and thus the higher ratio τ′′/τ′ (Fig. 3d and e). This process was reversible and could be restored to the original state upon switching off the external bias, as shown in the step 5 from Fig. 3d. The cycle could be repeated over 50 ON–OFF cycles. As a control the well-known VSD Di-4-ANEPPS was also tested in a GUV under identical conditions (see ESI†). The dye readily distributes within the membrane but the distinct two lifetime distribution was not observed; the lifetime spread is much less pronounced.
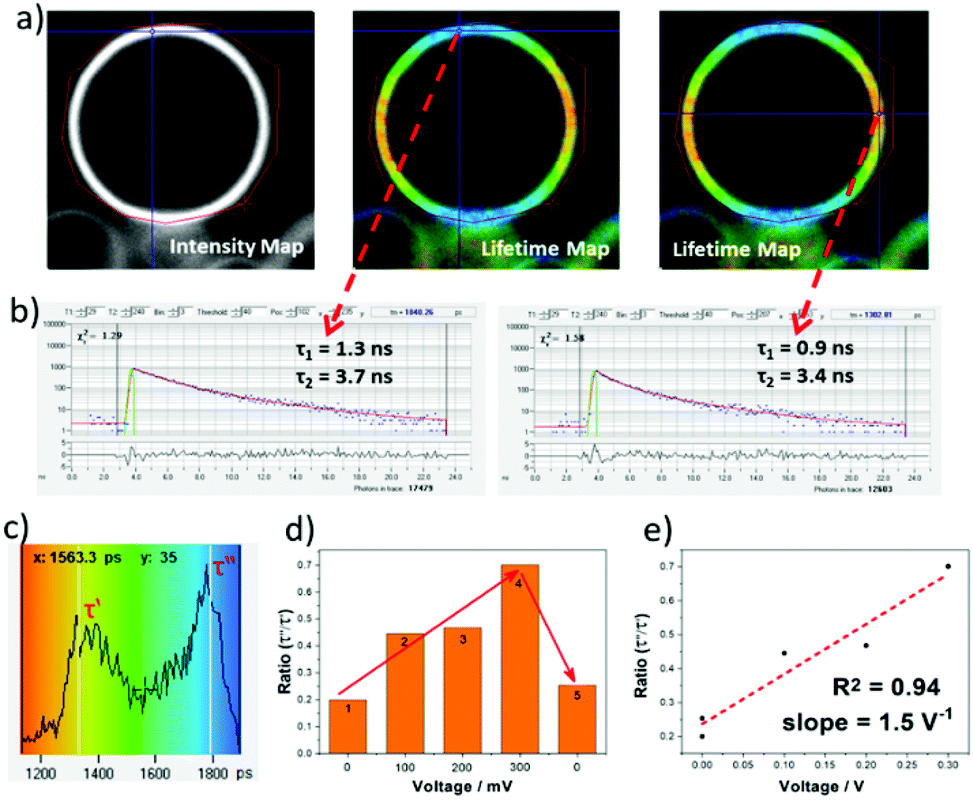 | ||
| Fig. 3 (a) Steady-state intensity and lifetime map of a GUV labelled with aJBD under 633 nm one-photon excitation. The lifetime distributions are colour-coded where red represents short lifetimes while blue represents long lifetimes. (b) Kinetic traces from two different pixels on the GUV membrane and (c) the intensity-weighted lifetime distribution map of the shorter fluorescence lifetime component τ1. (d) Changes induced to τ1 by varying the voltage at the platinum electrode with the recovery of the initial state at the end. (e) Plot of the lifetime distribution ratio vs the applied voltage. | ||
It is noted that previous work has focussed on using FLIM to measure membrane potential16 and membrane tension17 in mitochondria. Interestingly recent studies by Miller et al.18 observed that FLIM could be used to predict the performance of VSDs in cardiomyocytes and neurons. A distinct fluorescence lifetime change per mV was witnessed for patch clamped HEK cells. However, the modulated ramping effect with voltage change for aJBD would appear to be unique, and supports that the dye's design offers an alternative method to monitor voltage alterations in membranes.
The main focus of the work was to exam the voltage sensitive property of aJBD in GUVs. The results displayed a good lifetime performance as a function of the potential change (ON–OFF cycle, ramping voltage) when in the GUV membrane. The action potential of an axon usually ranges from 90 to 130 mV and the duration is about 0.5 ∼ 2.0 ms. The measured lifetimes and collection times are certainly within this time window which is a positive feature, but at present a more precise control of the size and step-time for the voltage change is required. It is worth noting that the unoptimised fluorescence ratio change per V is ca. 1.5 V−1 meaning that an axon potential change of ca. 10 mV could produce an output signal change of around 1.5%.
To conclude we have demonstrated that the fluorescence lifetime of a carefully designed BODIPY dye will respond to a voltage change across the membrane of a GUV. The monitoring based around using a lifetime distribution method, which has not been employed in any previously published studies, is the basis for VILI applications.
We thank the CLF Octopus/Ultra Facilities for the financial support and access to the Octopus FLIM facilities. The National Mass Spectrometry Facility at Swansea University is thanked for collecting mass spectra.
Conflicts of interest
There are no conflicts to declare.References
- (a) P. Yan, C. D. Acker and L. M. Loew, ACS Sens., 2018, 3, 2621–2628 CrossRef CAS PubMed; (b) N. A. Sayresmith, A. Saminathan, J. K. Sailer, S. M. Patberg, K. Sandor, Y. Krishnan and M. G. Walter, J. Am. Chem. Soc., 2019, 141, 18780–18790 CrossRef CAS PubMed.
- (a) E. Fluhler, V. G. Burnham and L. M. Loew, Biochemistry, 1985, 24, 5749–5755 CrossRef CAS PubMed; (b) V. Grenier, B. R. Daws, P. Liu and E. W. Miller, J. Am. Chem. Soc., 2019, 141, 1349–1358 CrossRef CAS PubMed; (c) P. Liu, V. Grenier, W. Hong, V. R. Muller and E. W. Miller, J. Am. Chem. Soc., 2017, 139, 17334–17340 CrossRef CAS PubMed.
- P. E. Z. Klier, J. G. Martin and E. W. Miller, J. Am. Chem. Soc., 2021, 143, 4095–4099 CrossRef CAS PubMed.
- B. Kuhn and C. J. Roome, Front. Cell. Neurosci., 2019, 13, 321 CrossRef CAS PubMed.
- (a) E. W. Miller, Curr. Opin. Chem. Biol., 2016, 33, 74–80 CrossRef CAS PubMed; (b) J. E. Gonzalez and R. Y. Tsien, Chem. Biol., 1997, 4, 269–277 CrossRef CAS PubMed.
- (a) R. U. Kulkarni, D. J. Kramer, N. Pourmandi, K. Karbasi, H. S. Bateup and E. W. Miller, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2813–2818 CrossRef CAS PubMed; (b) S. C. Boggess, J. R. Lazzari-Dean, B. K. Raliski, D. M. Mun, A. Y. Li, J. L. Turnbull and E. W. Miller, RSC Chem. Biol., 2021, 2, 248–258 RSC.
- Y. Bando, C. Grimm, V. H. Cornejo and R. Yuste, BMC Biol., 2019, 17, 71 CrossRef PubMed.
- (a) E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879–886 RSC; (b) M. Li, H. Ge, V. Mirabello, R. L. Arrowsmith, G. Kociok-Köhn, S. W. Botchway, W. Zhu, S. I. Pascu and T. D. James, Chem. Commun., 2017, 53, 11161–11164 RSC.
- W. Becker, J. Microsc., 2012, 247, 119–136 CrossRef CAS PubMed.
- (a) J. Steinkühler, P. De Tillieux, R. L. Knorr, R. Lipowsky and R. Dimova, Sci. Rep., 2018, 8, 1–9 Search PubMed; (b) D. O’Connor, A. Byrne and T. E. Keyes, RSC Adv., 2019, 9, 22805–22816 RSC; (c) R. B. Lira, L. Benk, E. Ewins, J. P. Spatz, R. Lipowsky, I. Platzman and R. Dimova, Biophys. J., 2018, 114, 94a–95a CrossRef.
- K. Dorgans, B. Kuhn and M. Y. Uusisaari, Front. Cell. Neurosci., 2020, 14, 607483 Search PubMed.
- X. Zhou, C. Yu, Z. Feng, Y. Yu, J. Wang, E. Hao, Y. Wei, X. Mu and L. Jiao, Org. Lett., 2015, 17, 4632–4635 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- D. Sirbu, J. B. Butcher, P. G. Waddell, P. Andras and A. C. Benniston, Chem. – Eur. J., 2017, 23, 14639–14649 CrossRef CAS PubMed.
- T. Förster and G. Hoffmann, Z. Phys. Chem., 1971, 75, 63–76 CrossRef.
- I. A. Okkelman, D. B. Papkovsky and R. I. Dmitriev, Cytometry, 2020, 97A, 471–482 CrossRef PubMed.
- A. Goujon, A. Colom, K. Straková, V. Mercier, D. Mahecic, S. Manley, N. Sakai, A. Roux and S. Matile, J. Am. Chem. Soc., 2019, 141, 3380–3384 CrossRef CAS PubMed.
- S. C. Boggess, J. R. Lazzari-Dean, B. K. Raliski, D. M. Mun, A. Y. Li, J. L. Turnbull and E. W. Miller, RSC Chem. Biol., 2021, 2, 248–258 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and spectra, X-ray data and additional spectra. CCDC 2091629. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc03756k |
| This journal is © The Royal Society of Chemistry 2021 |