
A rearrangement of saccharin-derived cyclic ketimines with 3-chlorooxindoles leading to spiro-1,3-benzothiazine oxindoles†
Rui-Li
Li
,
Qing-Yun
Fang
,
Mei-Yuan
Li
,
Xiang-Shan
Wang
and
Li-Ming
Zhao
 *
*
School of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou 221116, Jiangsu, China. E-mail: lmzhao@jsnu.edu.cn
First published on 5th October 2021
Abstract
An unusual rearrangement of saccharin-derived cyclic ketimines (SDCIs) and 3-chlorooxindoles has been developed to provide a series of spiro-1,3-benzothiazine oxindoles. The reaction features simple manipulations, short reaction times, mild reaction conditions and inexpensive reagents. It is the first example where SDCIs serve as a ring-opening reagent in organic synthesis.
Saccharin-derived cyclic ketimines (SDCIs) are a family of valuable and versatile building blocks in organic synthesis, and have been widely applied in various types of reactions for the synthesis of structurally diverse benzosultam derivatives.1 Currently, SDCI-involved reactions can be mainly divided into four categories (Scheme 1): (a) C–H functionalizations, in which SDCIs act as a directing group to assist C–H cleavage of phenyl moiety at the 3-position;2 (b) nucleophilic addition reactions, in which SDCIs participate as electrophiles reacting with various nucleophiles for the production of benzosultam-based adducts or cyclized products;3 (c) electrophilic addition reactions. In 3-alkyl substituted SDCIs, the α-proton is somewhat acidic due to the electron-withdrawing effect of the imine group. Thus, they can also be used as nucleophiles in organic synthesis;4 and (4) cycloaddition reactions of 3-vinyl substituted SDCIs serving as a 4- or 2-atom synthon for the construction of six- or five-membered heterocycles.5
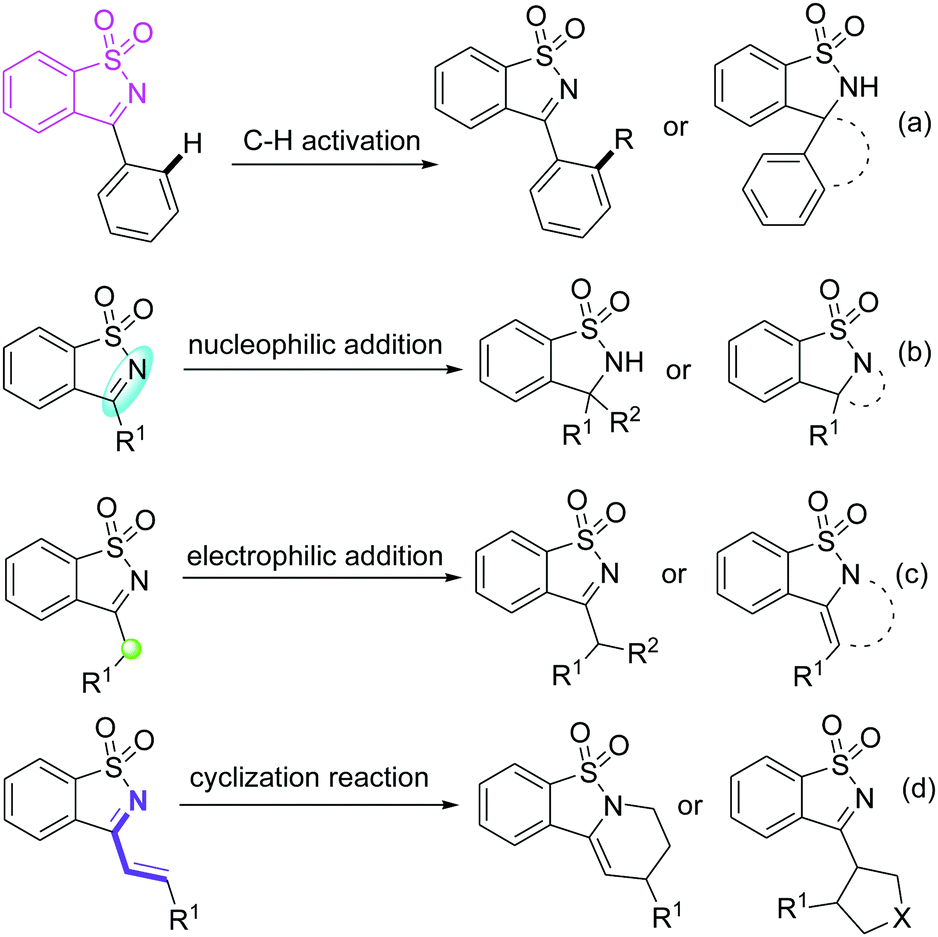 | ||
| Scheme 1 Reaction profiles of SDCIs. | ||
In view of the importance of this building block in organic synthesis and based on our interest in the construction of spirocyclic compounds,6 we hope to open further its application to the construction of a spiro-aziridine oxindole compound with potential biological activities and thus devise a formal [2+1] cycloaddition reaction of SDCI with 3-chlorooxindole (Scheme 2a). To our surprise, this reaction failed to afford the anticipated spiro-aziridine oxindole product. Instead, we observed the formation of an unknown product in this reaction. Finally, the structure of the product was determined using single-crystal X-ray diffraction analysis and was identified as spiro-1,3-benzothiazine oxindole 3aa (CCDC†2098947; see the ESI†), which apparently arose from an unusual rearrangement. This rearrangement broke an ArS(O2)–N bond under very mild conditions in a short time, leading to an arylsulfonyl group migration from N to C involving a sulfonynamide to sulfone rearrangement (Scheme 2b). This finding is very interesting because cleavage of the S(O2)–N bond generally requires relatively drastic conditions such as harsh reagents, long reaction times, and high reaction temperatures, as well as strongly acidic or basic conditions.7 Furthermore, exploration of the new role of SDCIs as a ring-opening reagent in a rearrangement reaction has not been previously reported.
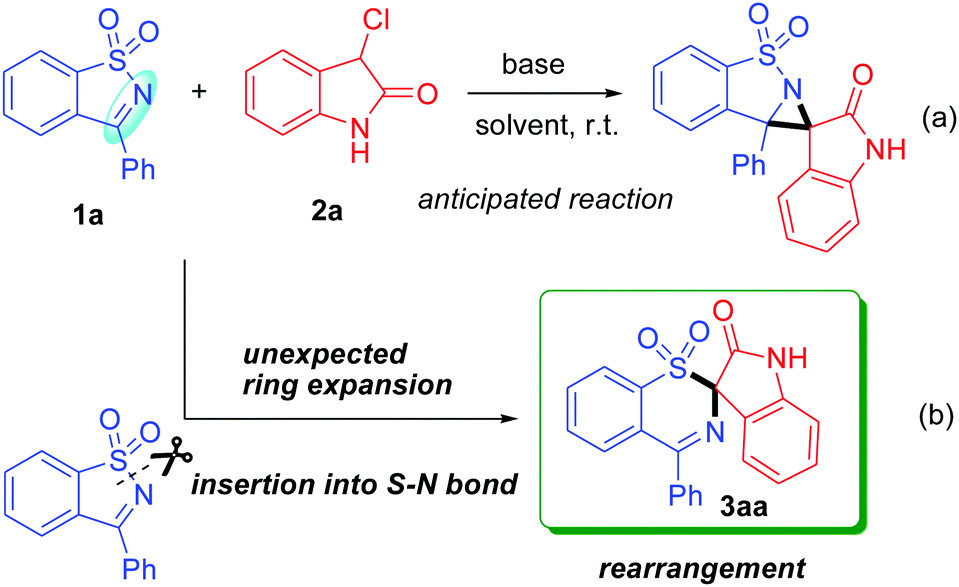 | ||
| Scheme 2 The present work. | ||
1,3-Benzothiazines are the core structural motifs of many biologically and pharmaceutically active molecules.8 Although there are several ways to prepare 1,3-benzothiazines,9–12 they are limited in structural diversification. For example, the chemistry on the construction of a spiro 1,3-benzothiazine framework is still underdeveloped.13 Spirocyclic scaffolds, especially spirooxindoles, are of great interest because they are not only useful intermediates in organic synthesis but offer great potential in drug discovery.14
Considering the above facts, we decided to develop this unexpected S–N bond insertion reaction as a general way to prepare spiro-1,3-benzothiazine oxindoles. Note that such a ring expansion/rearrangement not only enriches the chemistry of SDCIs but is also an extremely rare example of the construction of structurally interesting yet synthetically challenging spirocyclic systems that have potential synthetic usefulness and biological activities. In addition, although 3-chlorooxindoles have been widely used as one-atom synthons in organic synthesis, employing them in a rearrangement reaction is still unappreciated.
We chose to optimize the synthesis of 3aavia the ring-expansion rearrangement using SDCI 1a and 3-chlorooxindole 2a as the model reaction, primarily examining the behavior of different bases and solvents on the reaction (Table 1). When the reaction was performed in THF at room temperature in the presence of DBU, the ring-expanded product 3aa was formed in 73% yield within 30 min (entry 1). DBN gave a similar reaction yield with DBU (entry 2), whereas the use of other organic bases such as DABCO and Et3N failed to improve the reaction yield even after 12 h (entries 3 and 4), presumably because of their lower basicity. Subsequent optimization with other bases revealed that Cs2CO3 was the most effective for the current reaction, affording the product 3aa in 79% yield (entries 5–7). Further screenings showed that the reaction was not very sensitive to the solvent used and occurred smoothly in various solvents including ether solvents (e.g., DME and Et2O), halogen solvents (e.g., CHCl3), aprotic solvents (e.g., MeCN, DMF and DMSO), protic solvents (e.g., MeOH), and aromatic solvents (e.g., toluene) (entries 8–15). DME gave a better result and was chosen as the optimal solvent for the reaction (entry 8). Finally, the best result was achieved by conducting the reaction with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of 1a and 2a in the presence of Cs2CO3 in DME at room temperature (entry 16).
:2 ratio of 1a and 2a in the presence of Cs2CO3 in DME at room temperature (entry 16).
| Entry | Base | Solvent | Time | 3aa |
|---|---|---|---|---|
|
a Unless otherwise noted, reactions were carried out with 1a (0.1 mmol), 2a (0.15 mmol), and a base in a solvent (1 mL) at room temperature.
b Isolated yields.
c
1a:2a = 1:2. DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; DBN, 1,5-diazabicyclo[4.3.0]non-5-ene; DABCO, 1,4-diazabicyclo[2.2.2]octane; DME, 1,2-dimethoxyethane; DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; THF, tetrahydrofuran.
|
||||
| 1 | DBU | THF | 30 min | 73 |
| 2 | DBN | THF | 30 min | 72 |
| 3 | DABCO | THF | 12 h | 37 |
| 4 | Et3N | THF | 12 h | 36 |
| 5 | Na2CO3 | THF | 30 min | 74 |
| 6 | Cs2CO3 | THF | 30 min | 79 |
| 7 | t BuOK | THF | 30 min | 74 |
| 8 | Cs2CO3 | DME | 30 min | 87 |
| 9 | Cs2CO3 | Et2O | 30 min | 84 |
| 10 | Cs2CO3 | CHCl3 | 30 min | 81 |
| 11 | Cs2CO3 | MeCN | 30 min | 79 |
| 12 | Cs2CO3 | DMF | 30 min | 61 |
| 13 | Cs2CO3 | DMSO | 30 min | 65 |
| 14 | Cs2CO3 | MeOH | 30 min | 83 |
| 15 | Cs2CO3 | Toluene | 30 min | 73 |
| 16c | Cs2CO3 | DME | 30 min | 90 |
Various substituted SDCIs 1 and 3-chlorooxindoles 2 were subjected to the optimized reaction conditions. Compared with 1a and 2a, the substituted substrates required slightly longer times (1 h) for the rearrangement process. Table 2 shows that our ring-expansion rearrangement is compatible with functionalized and hindered R1, R2 and R3 groups. Generally, with respect to the 3-aryl substituted SDCIs 1, both electron-donating and -withdrawing groups, regardless of the substitution patterns, on the arene moiety were well tolerated, giving the rearranged products in good to excellent yield (entries 1–7). When electron-rich substituents such as a methoxy group are present in the ortho, meta or para positions, the reactions proceeded smoothly, leading to the formation of products 3ba–3da in 83–90% yield (entries 1–3). The bulky t-butyl substituent (1e) was well tolerated without any deleterious effect on the reaction efficacy being observed compared with a smaller methoxy group (1d) (entries 4 vs. 3). Substrates 1f and 1g bearing halogen substituents worked equally efficiently, affording 3fa and 3ga smoothly in 83% and 82% yields, respectively (entries 5 and 6). In addition to substrates with monosubstituted phenyl groups, those bearing disubstituted phenyl groups such as 1h were also well tolerated, producing the corresponding product 3ha in 86% yield (entry 7). The naphthalyl derived substrate 1i also efficiently rearranged into the six-membered product 3ia in 84% yield (entry 8). Next, the tolerance of 3-chlorooxindoles 2 was examined. 5-Methyl- and 6-methyl-3-chlorooxindoles (2b and 2c) reacted very well in the reaction, affording the corresponding products 3ab and 3ac in 84% and 86% yields, respectively (entries 9 and 10). However, when the same substituent was moved to the 4-position, the desired reaction did not occur anymore (data not shown), and was probably due to steric hindrance. Substrates with a strong electron-donating methoxy group at the C5-position (2d) of the phenyl ring gave lower yields than their counterparts substituted with a methyl group (entries 11 vs. 9). For 3-chlorooxindoles 2 with halo-substituents at the C5-position, such as 2e–2g, the reaction efficiency was decreased to afford the products 3ae–3ag in 56–63% yields, which was due to the low conversion of 1a and the concomitant formation of unidentified byproducts (entries 12–14). For the C6 halogenated substrates (2h and 2i), good reaction yields and clean reactions were retained (entries 15 and 16). When a methyl group was introduced to the nitrogen atom of 3-chlorooxindoles, the yield of the rearranged product 3aj decreased significantly (entry 17). No reaction occurred when using a Boc group in place of the methyl group on the nitrogen atom (entry 18). Finally, various substituted SDCIs 1 and 3-chlorooxindoles 2 were subjected to the above optimal reaction conditions. As expected, when there were substituents on both phenyl rings, the reactions could also afford the sulfonative products 3cd, 3ec, 3bh, 3fb, and 3fh in good yields, irrespective of the electronic property and substitution pattern of the substituent (entries 19–23). These results highlighted the broad substrate scope of the Cs2CO3-mediated new rearrangement reactions of SDCIs and 3-chlorooxindoles.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | 2 | R1/R2/R3 | 3 | Yieldb (%) |
| a Reactions were carried out with 1 (0.1 mmol), 2 (0.2 mmol), and Cs2CO3 (0.2 mmol) in DME (1 mL) at room temperature for 1 h. b Isolated yields. | |||||
| 1 | 1b | 2a | 2-OMeC6H4/H/H | 3ba | 89 |
| 2 | 1c | 2a | 3-OMeC6H4/H/H | 3ca | 90 |
| 3 | 1d | 2a | 4-OMeC6H4/H/H | 3da | 83 |
| 4 | 1e | 2a | 4-tBuC6H4/H/H | 3ea | 86 |
| 5 | 1f | 2a | 3-FC6H4/H/H | 3fa | 83 |
| 6 | 1g | 2a | 4-ClC6H4/H/H | 3ga | 82 |
| 7 | 1h | 2a | 3,5-Me2C6H3/H/H | 3ha | 86 |
| 8 | 1i | 2a | 2-Naphth/H/H | 3ia | 84 |
| 9 | 1a | 2b | Ph/5-Me/H | 3ab | 84 |
| 10 | 1a | 2c | Ph/6-Me/H | 3ac | 86 |
| 11 | 1a | 2d | Ph/5-OMe/H | 3ad | 76 |
| 12 | 1a | 2e | Ph/5-F/H | 3ae | 57 |
| 13 | 1a | 2f | Ph/5-Cl/H | 3af | 56 |
| 14 | 1a | 2g | Ph/5-Br/H | 3ag | 63 |
| 15 | 1a | 2h | Ph/6-Cl/H | 3ah | 75 |
| 16 | 1a | 2i | Ph/6-Br/H | 3ai | 70 |
| 17 | 1a | 2j | Ph/H/Me | 3aj | 43 |
| 18 | 1a | 2k | Ph/H/Boc | 3ak | 0 |
| 19 | 1c | 2d | 3-OMeC6H4/5-OMe/H | 3cd | 73 |
| 20 | 1e | 2c | 4-tBuC6H4/6-Me/H | 3ec | 87 |
| 21 | 1b | 2h | 2-OMeC6H4/6-Cl/H | 3bh | 86 |
| 22 | 1f | 2b | 3-FC6H4/5-Me/H | 3fb | 87 |
| 23 | 1f | 2h | 3-FC6H4/6-Cl/H | 3fh | 74 |
For the formation of the rearranged product 3, we put forward two plausible pathways as shown in Scheme 3. Both pathway I and pathway II are initiated by the nucleophilic attack of the initially formed enolate A15 on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of 1 to generate a nitrogen anion B, which is further transformed into the aziridine Cvia an intramolecular SN2-type nucleophilic substitution.16 Pathway I involves the 1,2-shift of an arylsulfonyl group from a N atom to a C atom with a concomitant S–N cleavage followed by rapid cleavage of the C–C bond to yield the rearranged sulfone 3. Another possibility is that intermediate C undergoes the homolysis of the S–N bond to afford biradical D, which would undergo aziridine ring opening to form a more stable carbon-center radical E.17 Finally, product 3 is produced by bond formation between the S and C radical species (pathway II).
N bond of 1 to generate a nitrogen anion B, which is further transformed into the aziridine Cvia an intramolecular SN2-type nucleophilic substitution.16 Pathway I involves the 1,2-shift of an arylsulfonyl group from a N atom to a C atom with a concomitant S–N cleavage followed by rapid cleavage of the C–C bond to yield the rearranged sulfone 3. Another possibility is that intermediate C undergoes the homolysis of the S–N bond to afford biradical D, which would undergo aziridine ring opening to form a more stable carbon-center radical E.17 Finally, product 3 is produced by bond formation between the S and C radical species (pathway II).
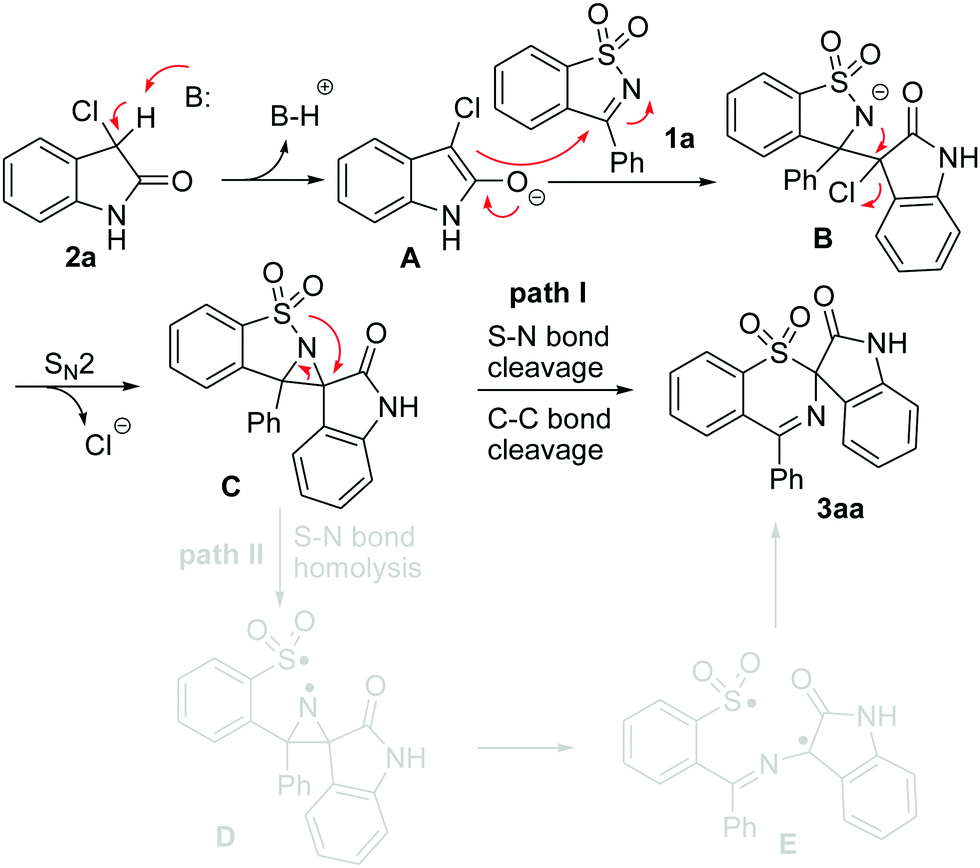 | ||
| Scheme 3 Two potential mechanistic pathways. | ||
To understand the above proposed mechanisms, control experiments of 1a with 2a were conducted in the presence of a radical-trapping reagent, such as hydroquinone, 2,6-di-tert-butyl-4-methylphenol (BHT), and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) (Scheme 4). It was observed that the addition of hydroquinone or BHT did not inhibit the reaction and 3aa was obtained in an 80% or 85% yield, respectively. When TEMPO was added to the reaction, 3aa was obtained in a ca. 30% yield. These results indicate that the radical process might not be involved in the present reaction. On the basis of these observations, we consider that it is possible that the reaction proceeded via the former mechanism.
 | ||
| Scheme 4 Control experiments. | ||
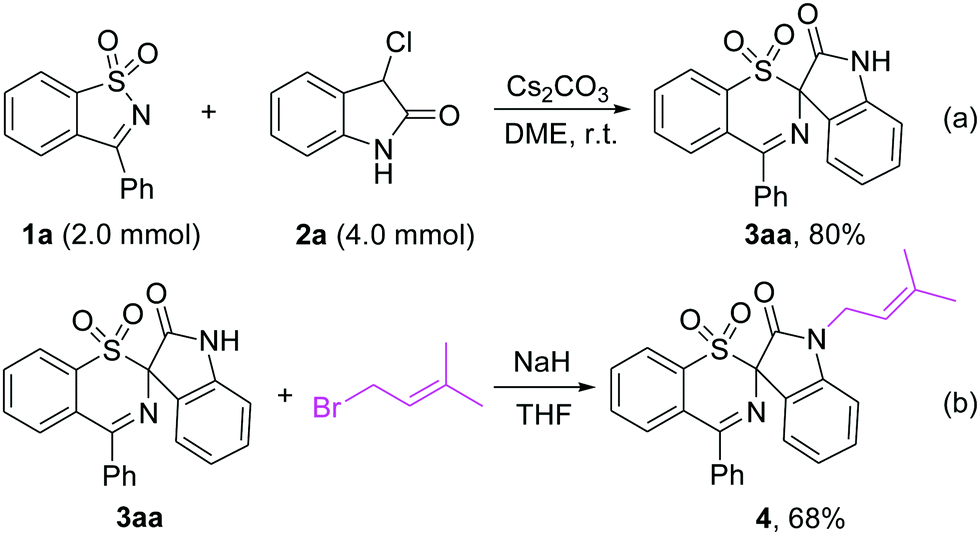
Performing the rearrangement reaction on a larger scale using 2.0 mmol of 1a and 4.0 mmol of 2a as substrates gave 3aa in 80% yield (Scheme 5a). When 3aa was treated with prenyl bromide in the presence of NaH in THF, the prenylated indole 4 was obtained in 68% yield, which is a privileged structural unit present in many N-prenylindole alkaloids such as the fumitremorgin B and flustramine families of alkaloids (Scheme 5b).18
 | ||
| Scheme 5 Scale-up synthesis and further transformation of 3aa. | ||
In conclusion, an unusual rearrangement reaction between SDCIs and 3-chlorooxindoles under mild conditions has been developed. This reaction occurred via a S–N bond insertion process followed by an arylsulfonyl group transfer, thus leading to a novel reaction mode involving SDCIs and the construction of novel and structurally interesting spirocyclic compounds. Moreover, this work uncovered the hidden potential of SDCIs in organic synthesis, which would be helpful for the further development of new reactions with SDCIs.
This work was supported by the Science and Technology Support Program of Jiangsu Province (BE2017643).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D.-J. Cheng and Y.-D. Shao, Adv. Synth. Catal., 2018, 360, 3614 CrossRef CAS.
- (a) A. Mishra, U. Mukherjee, T. K. Vats and I. Deb, J. Org. Chem., 2018, 83, 3756 CrossRef CAS PubMed; (b) M. Maraswami, G. Chen and T.-P. Loh, Adv. Synth. Catal., 2018, 360, 416 CrossRef CAS; (c) X. Zhang, Y. Gao, J. Chen, R. Fan, G. Shi, Z. He and B. Fan, Adv. Synth. Catal., 2019, 361, 4495 CrossRef CAS; (d) W. Sarkar, A. Bhowmik, S. Das, A. B. Sulekha, A. Mishra and I. Deb, Org. Biomol. Chem., 2020, 18, 7074 RSC; (e) R. Yabe, Y. Ebe and T. Nishimura, Synthesis, 2021, 3051 CAS; (f) T. Nishimura, Y. Ebe and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 2092 CrossRef CAS PubMed; (g) M. V. Pham and N. Cramer, Chem. – Eur. J., 2016, 22, 2270 CrossRef CAS PubMed; (h) B. Liu, P. Hu, Y. Zhang, Y. Li, D. Bai and X. Li, Org. Lett., 2017, 19, 5402 CrossRef CAS PubMed; (i) A. Mishra, U. Mukherjee, W. Sarkar, S. L. Meduri, A. Bhowmik and I. Deb, Org. Lett., 2019, 21, 2056 CrossRef CAS PubMed.
- (a) G. Yang and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7540 CrossRef CAS PubMed; (b) X. Zhang, J. Chen, Y. Gao, K. Li, Y. Zhou, W. Sun and B. Fan, Org. Chem. Front., 2019, 6, 2410 RSC; (c) Y. Gao, X. Zhang, R. D. Laishram, J. Chen, K. Li, K. Zhang, G. Zeng and B. Fan, Adv. Synth. Catal., 2019, 361, 3991 CrossRef CAS; (d) M.-F. Li, A.-Q. Miao, H.-Y. Zhu, R. Wang, W.-J. Hao, S.-J. Tu and B. Jiang, J. Org. Chem., 2020, 85, 13602 CrossRef CAS PubMed; (e) M. Rommel, T. Fukuzumi and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 17266 CrossRef CAS PubMed; (f) S. Zhang, L. Li, L. Xin, W. Liu and K. Xu, J. Org. Chem., 2017, 82, 2399 CrossRef CAS PubMed; (g) R.-R. Liu, J.-P. Hu, J.-J. Hong, C.-J. Lu, J.-R. Gao and Y.-X. Jia, Chem. Sci., 2017, 8, 2811 RSC.
- (a) Q. An, J. Li, J. Shen, N. Butt, D. Liu, Y. Liu and W. Zhang, Chem. Commun., 2015, 51, 885 RSC; (b) K. Yuan, F. Dong, X. Yin, S.-S. Li, L. Wang and L. Xu, Org. Chem. Front., 2020, 7, 3868 RSC; (c) M. Prakash, R. Lodhi and S. Samanta, J. Org. Chem., 2021, 86, 6721 CrossRef CAS PubMed; (d) A. S. Kumar, S. Chauhan and K. C. K. Swamy, Org. Lett., 2021, 23, 1123 CrossRef CAS PubMed.
- (a) X. Feng, Z. Zhou, C. Ma, X. Yin, R. Li, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 14173 CrossRef CAS PubMed; (b) X.-R. Ren, J.-B. Lin, X.-Q. Hu and P.-F. Xu, Org. Chem. Front., 2019, 6, 2280 RSC; (c) S. Zhang, L. Bacheley, C. M. Young, D. G. Stark, T. O'Riordan, A. M. Z. Slawin and A. D. Smith, Asian J. Org. Chem., 2020, 9, 1562 CrossRef CAS; (d) H. Lin, H. Yang, Q. Gong, S. Luo, J. Gu, X. Cao, B. Mao, Y. Ge and C. Yuan, RSC Adv., 2021, 11, 20118 RSC; (e) H.-P. Lv, X.-P. Yang, B.-L. Wang, H.-D. Yang, X.-W. Wang and Z. Wang, Org. Lett., 2021, 23, 4715 CrossRef CAS PubMed.
- (a) D.-F. Li, Y. Gu, J.-R. Zhang, K. Liu and L.-M. Zhao, J. Org. Chem., 2019, 84, 879 CrossRef CAS PubMed; (b) Q.-Y. Fang, H.-S. Jin, R.-B. Wang and L.-M. Zhao, Org. Lett., 2020, 22, 7358 CrossRef CAS PubMed; (c) J.-R. Zhang, H.-S. Jin, J. Sun, J. Wang and L.-M. Zhao, Eur. J. Org. Chem., 2020, 4988 CrossRef CAS; (d) Q.-Y. Fang, M.-H. Yi, X.-X. Wu and L.-M. Zhao, Org. Lett., 2020, 22, 5266 CrossRef CAS PubMed.
- (a) C. Goulaouic-Dubois, A. Guggisberg and M. Hesse, J. Org. Chem., 1995, 60, 5969 CrossRef CAS; (b) H. Knowles, A. F. Parsons and R. M. Pettifer, Synlett, 1997, 271 CrossRef CAS; (c) A. F. Parsons and R. M. Pettifer, Tetrahedron Lett., 1996, 37, 1667 CrossRef CAS; (d) B. Nyasse, L. Grehn and U. Ragnarsson, Chem. Commun., 1997, 1017 RSC; (e) D. A. Alonso and P. G. Andersson, J. Org. Chem., 1998, 63, 9455 CrossRef CAS; (f) E. Alonso, D. J. Ramón and M. Yus, Tetrahedron, 1997, 53, 14355 CrossRef CAS; (g) A. Yasuhara and T. Sakamoto, Tetrahedron Lett., 1998, 39, 595 CrossRef CAS; (h) S. Chandrasekhar and S. Mohapatra, Tetrahedron Lett., 1998, 39, 695 CrossRef CAS; (i) G. Sabitha, B. V. S. Reddy, S. Abraham and J. S. Yadav, Tetrahedron Lett., 1999, 40, 1569 CrossRef CAS.
- (a) H. Kimura, Y. Sato, Y. Tajima, H. Suzuki, H. Yukitake, T. Imaeda, M. Kajino, H. Oki, M. Takizawa and S. Tanida, Chem. Biol., 2010, 17, 1282 CrossRef CAS PubMed; (b) T. Mizuhara, S. Oishi, H. Ohno, K. Shimura, M. Matsuoka and N. Fujii, Bioorg. Med. Chem., 2012, 20, 6434 CrossRef CAS PubMed; (c) B. Wolff, N. Jaensch, W. O. Sugiarto, S. Fruehschulz, M. Lang, R. Altintas, I. Oehme and E.-J. Meyer-Almes, Eur. J. Med. Chem., 2019, 184, 111756 CrossRef CAS PubMed; (d) J. Li, X. Fan, J. Deng, Y. Liang, S. Ma, Y. Lu, J. Zhang, T. Shi, W. Tan and Z. Wang, Bioorg. Med. Chem., 2020, 28, 115526 CrossRef CAS PubMed.
- (a) A. S. Shestakov, N. V. Gusakova, K. S. Shikhaliev and A. G. Timoshkina, Russ. J. Org. Chem., 2007, 43, 1825 CrossRef CAS; (b) H.-H. Wang, T. Shi, W.-W. Gao, H.-H. Zhang, Y.-Q. Wang, J.-F. Li, Y.-S. Hou, J.-H. Chen, X. Peng and Z. Wang, Org. Biomol. Chem., 2017, 15, 8013 RSC.
- D. K. Thakur and Y. D. Vankar, Synthesis, 1983, 223 CrossRef CAS.
- L.-R. Wen, C.-C. Zhou, M.-Z. Zhu, S.-G. Xie, W.-S. Guo and M. Li, Org. Biomol. Chem., 2019, 17, 3356 RSC.
- T. Kentaro, Chem. Lett., 1990, 2205 Search PubMed.
- E. F. Ewies and F. A. A. El-Hag, J. Heterocycl. Chem., 2020, 57, 163 CrossRef CAS.
- (a) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673 CrossRef CAS PubMed; (b) M. Benabdallah, O. Talhi, F. Nouali, N. Choukchou-Braham, K. Bachari and A. M. S. Silva, Curr. Med. Chem., 2018, 25, 3748 CrossRef CAS PubMed; (c) L. Wu, G. Yang and W. Zhang, CCS Chem., 2019, 1, 623 Search PubMed; (d) L. Wu, Q. Shao, L. Kong, J. Chen, Q. Wei and W. Zhang, Org. Chem. Front., 2020, 7, 862 RSC.
- (a) A. Noole, M. Ošeka, T. Pehk, M. Öeren, I. Järving, M. R. J. Elsegood, A. V. Malkov, M. Lopp and T. Kanger, Adv. Synth. Catal., 2013, 355, 829 CrossRef CAS; (b) J.-H. Li, T.-F. Feng and D.-M. Du, J. Org. Chem., 2015, 80, 11369 CrossRef CAS PubMed.
- (a) A. P. Sakla, P. Kansal and N. Shankaraiah, Org. Biomol. Chem., 2020, 18, 8572 RSC; (b) J. Li, T. Du, G. Zhang and Y. Peng, Chem. Commun., 2013, 49, 1330 RSC.
- (a) I. Elghamry, D. Döpp and G. Henkel, Synthesis, 2001, 1223 CAS; (b) I. Elghamry, D. Döpp and G. Henkel, J. Heterocycl. Chem., 2007, 44, 849 CrossRef CAS.
- (a) M. Yamazaki, K. Sasago and K. Miyaki, Chem. Commun., 1974, 408 RSC; (b) P. B. Holst, U. Anthoni, C. Christophersen and P. H. Nielsen, J. Nat. Prod., 1994, 57, 997 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2098947. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04179g |
| This journal is © The Royal Society of Chemistry 2021 |