
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ball milling – a new concept for predicting degradation profiles in active pharmaceutical ingredients†
Reinhard P.
Kaiser
a,
Everaldo F.
Krake
 b,
Laura
Backer
c,
Jonas
Urlaub
c,
Wolfgang
Baumann
b,
Norbert
Handler
d,
Helmut
Buschmann
d,
Torsten
Beweries
*b,
Ulrike
Holzgrabe
*c and
Carsten
Bolm
*a
b,
Laura
Backer
c,
Jonas
Urlaub
c,
Wolfgang
Baumann
b,
Norbert
Handler
d,
Helmut
Buschmann
d,
Torsten
Beweries
*b,
Ulrike
Holzgrabe
*c and
Carsten
Bolm
*a
aRWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, 52074 Aachen, Germany. E-mail: carsten.Bolm@oc.rwth-aachen.de
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: torsten.beweries@catalysis.de
cInstitut für Pharmazie und Lebensmittelchemie, Am Hubland, 97074 Würzburg, Germany. E-mail: ulrike.holzgrabe@uni-wuerzburg.de
dRD&C Research, Development & Consulting GmbH, Neuwaldegger Strasse 35/2/3, 1170 Vienna, Austria
First published on 27th October 2021
Abstract
A method for forced oxidative mechanochemical degradation of active pharmaceutical ingredients (APIs) using clopidogrel hydrogensulfate as a model compound is presented. Considerable and selective formation of degradants occurs already after very short reaction times of less than 15 minutes and the nature of the products is strongly dependent on the used oxidant.
Virtually all pharmaceutical formulations are multicomponent and multiphase systems in (un)stable matrices. Upon approval application at regulatory authorities (such as the FDA and EMA) stability data must be submitted. However, there is a significant lack of predictive tools for solid-state characteristics, especially with respect to solid-state stability and degradation.1 Also, kinetics and decomposition products of solid-state degradation processes are unique for each compound, making the development of stability models very time-consuming and costly. Available prediction methods in aqueous environments result in high failure rates as non-relevant degradants may be formed resulting in high development risk for the manufacturer of new drugs and for the patient.
Clopidogrel hydrogensulfate (Clp) is a well-established drug substance showing direct inhibition of adenosine diphosphate (ADP) binding to its receptor and thus the ADP-mediated activation of the glycoprotein GPIIb/IIIa complex.2 It acts as platelet aggregation inhibitor and is indicated for the treatment and management of heart attack and stroke, coronary and artery occlusion. The active pharmaceutical ingredient (API) is marketed in various oral drug formulations mainly as tablets with immediate release but also modified release formulations. The characteristics of Clp are summarised in Fig. 1a.
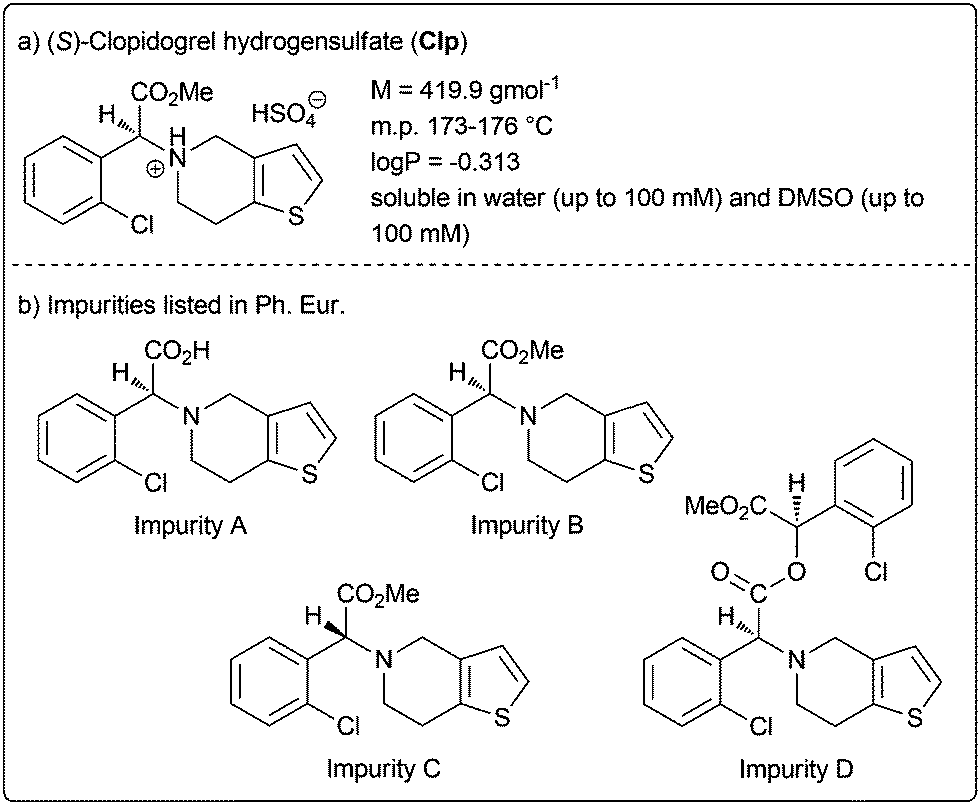 | ||
| Fig. 1 Clopidogrel hydrogensulfate (Clp) and impurities listed in the European Pharmacopoeia (Ph. Eur.). | ||
Currently, the European Pharmacopoeia (Ph. Eur.) reveals the four impurities for Clp (impurity A–D, Fig. 1b), however, no oxidative impurity is specified.3,4 To evaluate potential oxidative degradation products, usually forced degradation conditions in solution (i.e. with aqueous H2O2) are applied. Although stability indicating analytical methods were described, none included complete forced degradation studies elucidating all chemical structures of (potential) degradation products.5,6 For example, Singh and co-workers characterised several degradation products under solid-state stress conditions applying accelerated ICH7 conditions (40 °C/75% relative humidity) and solid stressors such as oxalic acid and Na2CO3 over a period of 1 or 3 months.8 These studies identified several structures including oxidative and hydrolytic degradants, although no explicit oxidative conditions were applied.
With the goal to overcome apparent deficits of current forced degradation studies (long incubation time, not all stressors available as solids, inefficient reactions, complex and misleading degradation profiles) we started looking for an innovative, fast, and reproducible method to mimic solid-state degradation of APIs like Clp. In particular we became interested in producing oxidative degradants avoiding commonly used solutions like H2O2, which often result in complex degradation profiles or even irrelevant products due to harsh conditions, undesired reaction pathways and strong solvent effects. After in-depth screening of suitable methods, we finally identified mechanochemistry as a highly versatile, efficient, and reproducible preparative application for our purposes.
Mechanochemistry has proven relevant in various field of research,9 including organic synthesis10 and pharmaceutical,11 medicinal,12 and agrochemical sciences.13 By applying mechanochemical techniques, existing reaction pathways could be affected leading to higher efficiencies compared to established protocols. For example, catalyst loadings could be reduced14 and product distributions be altered.10,15 Surprisingly, despite the success of mechanochemistry, its applications in pharmaceutical sciences have been limited mostly to the study of polymorphism of solid drugs and to the formation of co-crystals containing APIs.16 However, the existing knowledge on mechanosyntheses of organic molecules17 was foreseen as an ideal platform for inducing solid-state oxidative transformations of functional groups present in APIs. To best of our knowledge, although ball milling has been proven to be predictive for degradation processes of a drug,18 a systematic mechanochemical approach has not been applied to date to overcome the above-mentioned shortcomings of forced degradation studies.
The quality of drugs is controlled by the Ph. Eur. by means of a test for related substances, which can limit synthesis by- and starting products as well as degradation products. The Ph. Eur. methods in the Clp monograph3 make use of an ion-pair RP-HPLC method including Na-pentanesulfonate and phosphoric acid as mobile phase additives, which is not MS compatible for structure elucidation of unknown mechanochemically-generated degradation products. In contrast, both Singh and co-workers8 and Mashelkar and Renapurkar6 reported similar stability-indicating RP-HPLC methods for the elucidation of degradation products occurring on various stressing conditions. Those methods were taken as a starting point for development, optimisation and validation of a method, which is able to separate the Clp degradation impurities produced by ball-milling. The separation was carried out using a C8 double-end capped column with a carbon load of 7% and a gradient elution with water and MeCN/HCOOH (0.1%). The method is sufficiently sensitive, accurate and precise (see ESI† for details) for degradation product profiling. Since especially the oxidant KNO3 (but also with KMnO4) produced a tailing of the main degradation product (Fig. S2, ESI†) maybe by continuously forming and destroying a complex with this product, the mobile phase was slightly modified and HCOOH was replaced by 0.1% trifluoroacetic acid, which increased the efficiency likely by forming a more stable ion pair. Even though the retention time of the peaks was increased, the elution order and separation were similar (Fig. S2–S4, ESI†), but no additional peaks were observed by this orthogonal approach even though the non-tailing of the degradant opens “space” for peaks of related substances.
Clp shows polymorphism. Six different polymorphic forms and an amorphous form of the drug have been identified.19,20 Depending on the modification, different degradation products can be formed.8 In order to clarify in which form the API is present, a PXRD pattern was recorded (Fig. S1, ESI†). The comparison with literature patterns,19 showed that Clp used is present in polymorphic form I.
For the evaluation of the mechanochemical degradation under oxidative conditions, we have used three oxidants, KMnO4, KNO3, as well as KHSO5·0.5KHSO4·0.5K2SO4 (oxone®). Mixtures of these compounds and Clp were mixed with a defined amount of inert SiO2 and stressed at a frequency of 30 Hz in ZrO2 jars and balls using a mixer ball mill for t = 1–15 min. Milling times of 15 minutes were sufficient for conversion of 30–40% of Clp (determined by 1H NMR), depending on the oxidant used. The physical changes observed during solid-state oxidative degradation are shown in Fig. 2.
 | ||
| Fig. 2 Photographs of ball-milled Clp samples using KMnO4 (left), oxone® (centre), and KNO3 (right) as the oxidant after t = 15 min. | ||
Reducing the milling frequency to values below 20 Hz gave no conversion of Clp. Longer reaction times resulted in higher degradation of the API but were not necessary for the identification of characteristic degradation profiles. As an example, the LC-MS analysis of a reaction of Clp with KNO3 for 120 min is shown in Fig. S9 (ESI†).
All samples remained solid, albeit with pronounced colour differences: whereas samples treated with KMnO4 showed an intense brown colour (due to formation of MnO2) after ball milling, mixtures obtained after milling with KNO3 and oxone were pale yellow to yellow, indicating the formation of degradants that possess chromophoric groups. Workup of these samples was done by washing with MeCN, followed by filtration and concentration to dryness in vacuum. Thus obtained oily residues were free of residual oxidant and subsequently analysed by HPLC-MS21 using the described method (Fig. S8–S12, ESI†), as well as NMR and ATR-IR spectroscopy. Especially 1H NMR analysis is well suited as structural changes affecting the heterocycle moieties of Clp should be readily detectable at lower field (Fig. 3).
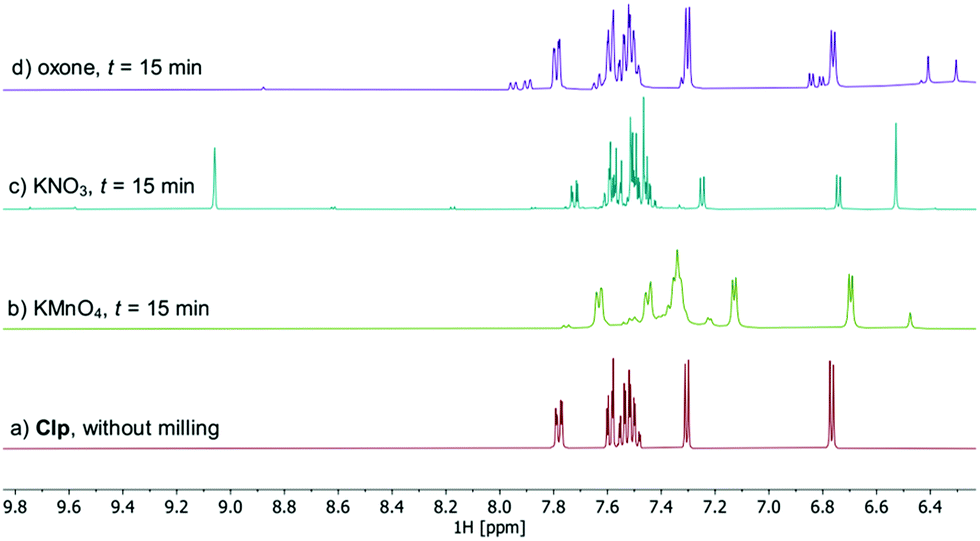 | ||
| Fig. 3 Comparison of the low-field region of 1H NMR spectra recorded after ball milling of Clp with SiO2 and one equivalent of oxidant for t = 15 min (CD3CN, 25 °C, 400 MHz). Full spectra and peak assignment are shown in Fig. S14–S20 (ESI†). | ||
HPLC analysis of reaction mixtures obtained after using KMnO4 showed the presence of one main degradant with a retention time of approximately 10 min (Fig. S3, ESI†). LC-MS analysis identified this species as the endo-iminium impurity (3,22m/z 319.9, Scheme 1). Furthermore, formation of traces of the free Clp acid (1, m/z 308.0), its iminium form (m/z 306.0), and degradants containing pyridine fragments (m/z 318.0, 2; 334.0, 4) became evident. Notably, no decarboxylation of Clp to produce ticlopidine-type structures (Table S2, ESI†)8 was observed, most likely due to the absence of an aqueous medium. Furthermore, C–N cleavage to produce thienopyridines, as it was described before using KMnO4 in solution, was not found.23 Formation of compound 3 as the main product could also be confirmed by 1H NMR spectroscopic analysis (Fig. 3b and Fig. S17, ESI†). ATR-IR spectra (Fig. 4) show bands at ν 1650 and 1580 cm−1 which can be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds present in 3 and to pyridine containing fragments such as in 4, respectively.
N bonds present in 3 and to pyridine containing fragments such as in 4, respectively.
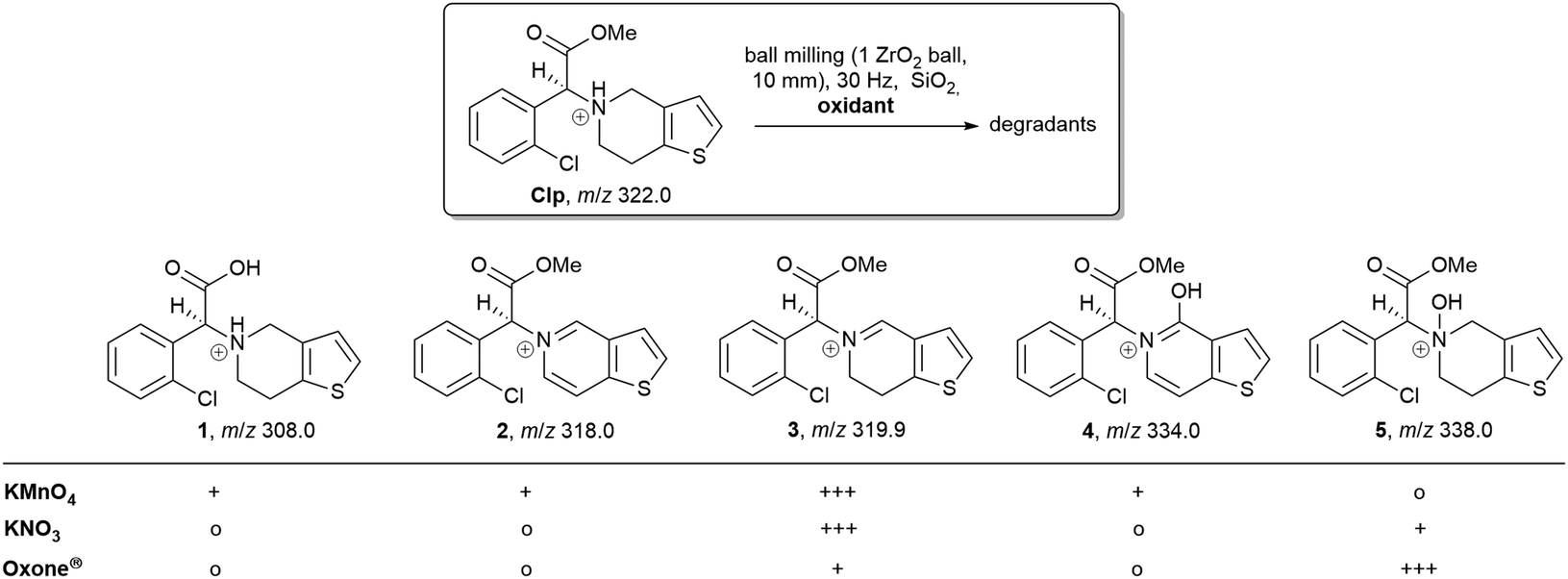 | ||
| Scheme 1 Overview of main reaction products detected by LC-MS (+++ main product, + product was detected, o no formation of this product). For compound 4 only the literature-reported tautomer8 is shown. | ||
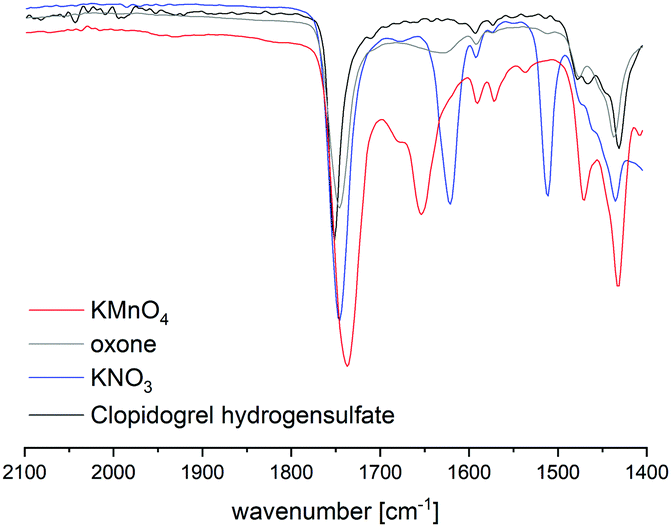 | ||
| Fig. 4 Comparison of ATR IR spectra of Clp and reaction mixtures after ball milling in the presence of different oxidants after t = 15 min. Shown is the spectral range that is characteristic for CN bonds. Full spectra are depicted in the ESI.† | ||
With KNO3 the same main degradant was observed by HPLC analysis (Fig. S2, ESI†) and identified as the endo-iminium compound 3, which explains the intense yellow colour of the reaction mixture (Fig. 2, right). UV analysis of the main HPLC signal at 11.4 min reveals absorption maxima at λmax = 216, 306 nm (Fig. S5 and S6, ESI†), values that are similar to those reported already for compound 3.22a Formation of this species occurs with greater selectivity compared to reactions that used KMnO4 as the oxidant. The formation of compound 3 was further corroborated using 1H and 13C NMR spectroscopic analysis of the reaction mixture in CD3CN, which showed characteristic singlet resonances at δ 6.48/9.06 ppm (CHCOOMe/CHN), and 163.1 ppm (CHN),22 respectively (Fig. 3c and Fig. S14–S16, ESI†). In addition, several minor products could be detected, out of which an N-oxide species 5 could be identified by LC-MS (m/z 338.0, Scheme 1). ATR-IR spectroscopy shows similar pattern as for reactions with KMnO4, albeit with the above-mentioned bands being more resolved (Fig. S12, ESI† and Fig. 4).
A different degradation profile was found when Oxone® was used as the oxidant (Scheme 1). According to HPLC (Fig. S4, ESI†), formation of compound 3 occurs in comparably small amounts, resulting in off-white reaction mixtures (Fig. 2, right). Instead, apart from several minor degradants, a main species with a retention time of approximately 17 minutes could be identified as the N-oxide 5 by LC-MS (m/z 338.0). Selective transformation of Clp into this compound was achieved recently by application of oxone® in aqueous/organic solution.241H NMR spectroscopic data of our mixtures are well in line with those reported before, showing characteristic signals for both diastereomers of 5 (e.g. δ 6.84, 6.80 ppm, d, 3J = 5.2 Hz; Fig. 3d and Fig. S18–S20, ESI†). In agreement with HPLC data, ATR-IR spectra show the above-mentioned bands at 1680 cm−1 that correspond to the CN bond in compound 3 (Fig. 4). Selective formation of clopidogrel N-oxide 5 by solid-state degradation has not been observed so far. Formation of N-oxidised degradants of Clp was described before,6 however, in this case also hydrolysis of the ester group was evident. Mechanochemical oxidation of thiophene moieties, also using oxone® was observed before.25 Analogous reaction products could not be found in any of our reactions.
Several publications deal with oxidative degradation products of Clp where several relevant structures were identified (Table S2, ESI†). These studies were mainly performed in aqueous environment and required harsh conditions with high concentration of H2O2, high temperature and/or long reaction times. Interestingly, Singh and co-workers reported oxidation even with ambient oxygen, although very long reaction times of up to 3 months under elevated temperature were needed.8 However, also concurrent hydrolytic reactions were observed as the reactions took place in open vials at 40 °C/75% relative humidity yielding corresponding acid analogues of oxidised Clp. Such degradants will not likely occur in solid-state APIs and formulations due to the low availability and mobility of water and slow reaction kinetics of hydrolytic cleavage, whereas the hydrolytic pathway seems to be preferred in water-based solutions as described in literature.8 Overall, the published artificial degradation profiles tend to be rather complex.
In contrast, our study shows high specificity and efficiency of mechanochemical formation of oxidative products of Clp, the obtained degradation product is clearly depending on the oxidant applied. The reaction was highly efficient and produced meaningful degradation profiles that may be used for the prediction of API stability under realistic conditions already after 15 minutes. Additionally, side reactions such as hydrolysis can be neglected as all oxidants delivered mainly oxidised ester products.
We thank Benjamin Andres, Andreas Koch, Susanne Schareina, Dr Henrik Lund and Dr Marcus Klahn for assistance. Financial support by the Leibniz-Gemeinschaft through the project PHARMSAF (K136/2018) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. C. Waterman and R. C. Adami, Int. J. Pharm., 2005, 293, 101–125 CrossRef CAS PubMed; (b) M. Blessy, R. D. Patel, P. N. Prajapati and Y. K. Agrawal, J. Pharm. Anal., 2014, 4, 159–165 CrossRef CAS PubMed.
- P. J. Sharis, C. P. Cannon and J. Loscalzo, Ann. Intern. Med., 1998, 129, 394–405 CrossRef CAS PubMed.
- Clopidogrel hydrogensulfate, Monograph 2531 (01/2017), European Pharmacopoeia 10.0, EDQM Strasbourg, 2020.
- Further salt forms of clopidogrel described in Ph. Eur. are clopidogrel besilate and clopidogrel hydrochloride.
- P. R. Deshmukh, V. L. Gaikwad, P. K. Tamane, K. R. Mahadik and R. N. Purohit, J. Pharm. Biomed. Anal., 2019, 165, 346–356 CrossRef CAS PubMed.
- U. C. Mashelkar and S. D. Renapurkar, Int. J. ChemTech Res., 2010, 2, 822–829 CAS.
- ICH: International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.
- K. Raijada, B. Prasad, A. Paudel, R. P. Shah and S. Singh, J. Pharm. Biomed. Anal., 2010, 52, 332–344 CrossRef PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- (a) J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed; (b) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (c) T. Frisčǐc, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed; (d) I. N. Egorov, S. Santra, D. S. Kopchuk, I. S. Kovalev, G. V. Zyryanov, A. Majee, B. C. Ranu, V. L. Rusinov and O. N. Chupakhin, Green Chem., 2020, 22, 302–315 RSC; (e) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- (a) S. Hasa and W. Jones, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef PubMed; (b) M. Pérez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef.
- (a) D. Tan, L. Loots and T. Frisčǐc, Chem. Commun., 2016, 52, 7760–7781 RSC; (b) E. Colacino, A. Porcheddu, C. Charnay and F. Delogu, React. Chem. Eng., 2019, 4, 1179–1188 RSC; (c) P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- L. Casali, L. Mazzei, O. Shemchuk, L. Sharma, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2019, 7, 2852–2859 CrossRef CAS.
- (a) G. N. Herrmann, M. T. Unruh, S.-H. Jung, M. Krings and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 10723–10727 CrossRef PubMed; (b) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS.
- S. Mateti, M. Mathesh, Z. Liu, T. Tao, T. Ramireddy, A. M. Glushenkov, W. Yang and Y. I. Chen, Chem. Commun., 2021, 57, 1080–1092 RSC.
- (a) T. Stolar, S. Lukin, M. Tireli, I. Sović, B. Karadeniz, I. Kereković, G. Matijašić, M. Gretić, Z. Katančić, I. Dejanović, M. d. Michiel, I. Halasz and K. Užarević, ACS Sustainable Chem. Eng., 2019, 7, 7102–7110 CrossRef CAS; (b) C. Medina, D. Daurio, K. Nagapudi and F. Alvarez-Nunez, J. Pharm. Sci., 2010, 99, 1693–1696 CrossRef CAS PubMed.
- (a) F. Krauskopf, K.-N. Truong, K. Rissanen and C. Bolm, Org. Lett., 2021, 23, 2699–2703 CrossRef PubMed; (b) J.-H. Schöbel, P. Elbers, K.-N. Truong, K. Rissanen and C. Bolm, Adv. Synth. Catal., 2021, 363, 1322–1329 CrossRef; (c) K. J. Ardila-Fierro, S. Lukin, M. Etter, K. Užarević, I. Halasz, C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2020, 59, 13458–13462 CrossRef CAS PubMed.
- (a) H. H. Buschmann and N. Handler, WIPO (PCT) world patent, WO2018/096066 A1, 2018 Search PubMed; (b) J. Urlaub, R. P. Kaiser, O. Scherf-Clavel, C. Bolm and U. Holzgrabe, Electrophoresis, 2021, 42, 1790–1799 CrossRef CAS PubMed.
- V. Koradia, G. Chawla and A. K. Bansal, Acta Pharm., 2004, 54, 193–204 CAS.
- It is not possible to make a general statement about the polymorphic form in market products because there are many generic products, and some conversion can occur during the manufacture and formulation of the products.
- For details of the MS analysis see the ESI† (Fig. S7–S12).
- (a) A. Mohan, M. Hariharan, E. Vikraman, G. Subbaiah, B. R. Venkataraman and D. Saravanan, J. Pharm. Biomed. Anal., 2008, 47, 183–189 CrossRef CAS PubMed; (b) Y. Zhu and J. Zhou, ACS Med. Chem. Lett., 2012, 3, 844–849 CrossRef CAS PubMed.
- K. S. Byadagi, R. V. Hosahalli, S. T. Nandibewoor and S. A. Chimatadar, Ind. Eng. Chem. Res., 2011, 50, 10962–10971 CrossRef CAS.
- E. F. Krake, H. Jiao and W. Baumann, J. Mol. Struct., 2022, 1247, 131309 CrossRef CAS.
- (a) G. Cravotto, D. Garella, D. Carnaroglio, E. C. Gaudino and O. Rosati, Chem. Commun., 2012, 48, 11632–11634 RSC; (b) B. Verbelen, E. Siemes, A. Ehnbom, C. Räuber, K. Rissanen, D. Wöll and C. Bolm, Org. Lett., 2019, 21, 4293–4297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc04716g |
| This journal is © The Royal Society of Chemistry 2021 |