
Effect of N-salicylidene hydrazide protonation on the solid state structural diversity of its Cu(II), Ni(II) and Zn(II) complexes†
Ana Belén
Lago
 *ab,
Arantxa
Pino-Cuevas
a,
Rosa
Carballo
a and
Ezequiel M.
Vázquez-López
*a
*ab,
Arantxa
Pino-Cuevas
a,
Rosa
Carballo
a and
Ezequiel M.
Vázquez-López
*a
aDepartamento de Química Inorgánica, Instituto de Investigación Sanitaria Galicia Sur (IISGS)-Universidade de Vigo, 36310 Vigo, Galicia, Spain. E-mail: alagobla@ull.edu.es; ezequiel@uvigo.es
bDepartamento de Química, Facultad de Ciencias, Sección Química Inorgánica, Universidad de la Laguna, 38206 La Laguna, Spain
First published on 16th November 2020
Abstract
We present here ten coordination compounds isolated from reactions of divalent metal ions (CuII, NiII and ZnII) with the Schiff base ligand H2L derived from salicylaldehyde and ω-hydroxy carbonic acid hydrazide. Protonation levels of the ligand has played significant roles in affecting the coordination conformations of the metallosupramolecular compounds in solid state. Four kinds of complexes have been obtained: monomeric [M(HL)2] and dimeric [M2(HL)2(X)2]/[M2(HL)2](X)2 (X = anion) containing monodeprotonated ligand, and polymeric ∞1M(L) and dimeric [Zn2L2] compounds with the doubly deprotonated form of the ligand. The structures of the free ligand H2L and its complexes [Zn(HL)(H2L)]·(NO3)·EtOH (Zn-1a), [Cu2(HL)2(NO3)2] (Cu-3a), [Cu2(HL)2(SiF6)] (Cu-3b) and [Ni2(HL)2(EtOH)(H2O)](NO3)2(Ni-3a) were elucidated by single-crystal X-ray diffraction. The reactivity of [Ni(HL)2] complex in the presence of a competitive ligand was investigated.
Introduction
The preparation of coordination compounds in discrete or extended metallosupramolecular assemblies is of increasing interest both in fundamental research and for possible applications in materials science.1In particular, coordination-directed assembly has evolved into an attractive strategy for the fabrication of functional materials. Metal-coordination bonds are stronger than most supramolecular interactions and they could be reversibly tuned by an appropriate external stimulus.2 Therefore, the ability to modify a given chemical structure by control of metal–ligand interactions on a molecular level could directly impact the chemical and physical properties of the resulting materials.
Metal ion–ligand interactions can be used to construct supramolecular materials with different structures, properties and applications.3 In order to achieve this, the interactions between the metal ions and ligands have to be of the appropriate strength.4 In this sense, the binding constant, i.e., the interactions between the metal ions and the ligands, depends on different factors such as the pH, temperature and the ligand design. In this context, another area of interest for which supramolecular chemistry holds great promise is that of stimuli-responsive materials,2 because environmental variables can have a marked effect on the compositions and topologies of the compounds.5
The coordination chemistry of hydrazine ligands is an intensive area of study, and numerous transition metal complexes of these ligands have been investigated.6 However, controllable synthesis of these compounds is still a great challenge because many factors may affect their self-assembly, in which the coordination geometry of the complex depends upon the coordination geometry preferred by metal, the coordination ability of counteranion, pH value, metal-to-ligand ratio, methods of the crystallization, reaction temperature, solvent system, etc.1c,6b Among these factors, the relationship between the ligands with different degrees of protonation and their resulting solid state metallic structures is underexplored in metallosupramolecular compounds.7 With this concept in mind, Schiff base ligand H2L (Scheme 1) was selected for this study as it contains an salicylidene group that incorporates an OH site, which undergoes acidification and facile ionization upon binding of a metal ion. Moreover, H2L ligand has a covalently linked aliphatic backbone that contains a molecular recognition hydroxyl group on its side-chain. These side-chain functionalized ligands have the potential to be used in the fabrication of supramolecular materials. The use of this kind of tridentate Schiff base ligands in the formation of dynamic metallosupramolecular polymers and constitutional libraries was first studied by the Lehn group.8 In this manuscript, we explored the coordinative behaviour of the N-salicylidene hydrazone ligand towards different first-row transition-metal ions, and a detailed description of their structural arrangements have been done. The possibility of different protonation degrees allowed us to study the effect of the variable coordination behaviour on the solid state structures of the resulting metallosupramolecular compounds.
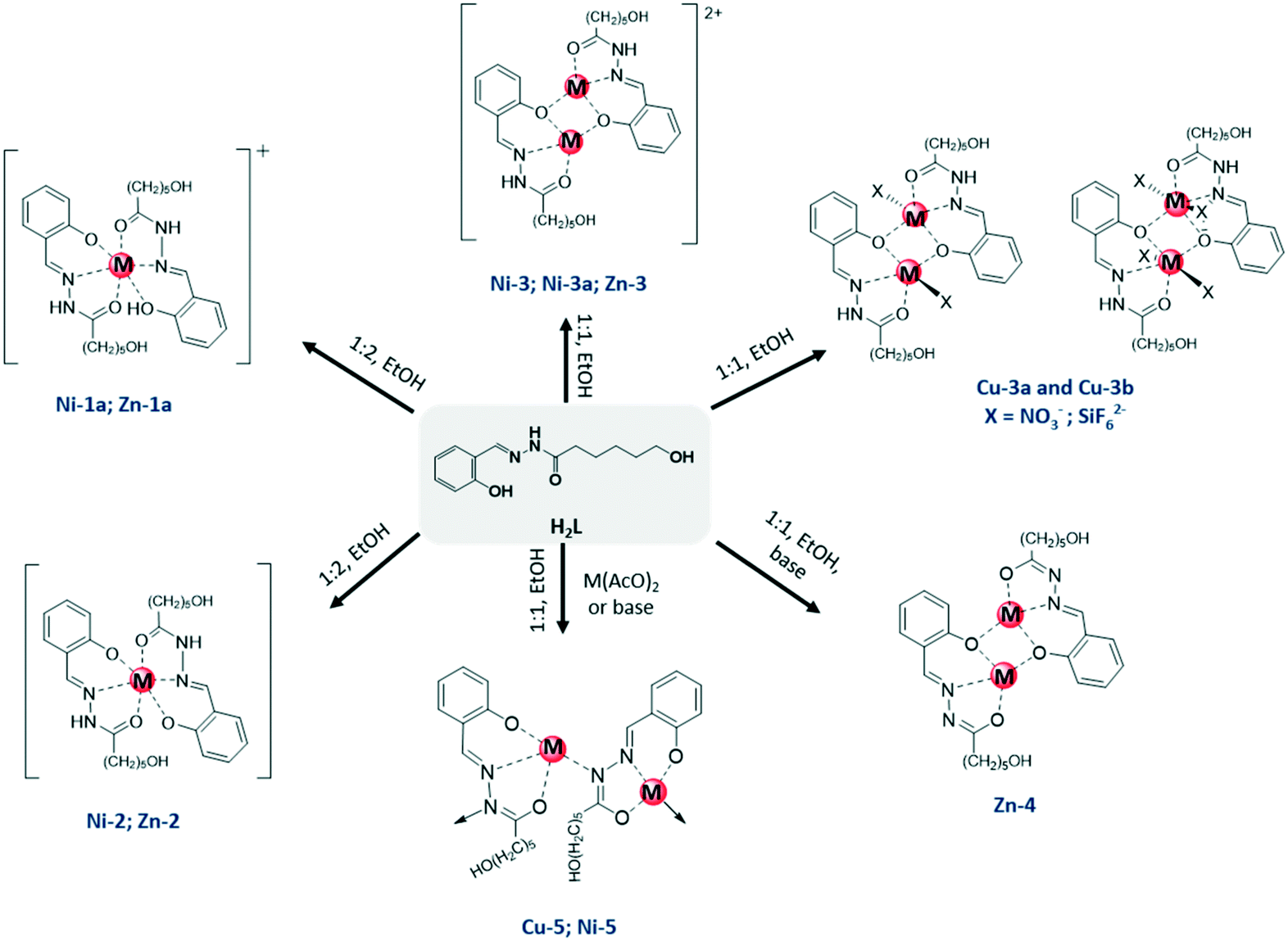 | ||
| Scheme 1 Schematic representation of the compounds isolated showing the mononuclear, dinuclear or polymeric nature of the different complexes. | ||
Results and discussion
The Schiff base ligand H2L with a ω-hydroxy-functionalized side chain was prepared by condensation of salicylaldehyde with the corresponding hydrazide.9 This reaction afforded a tridentate-chelating ligand with a variable protonation degree that allows a behaviour as a mono- or a dianionic ligand. Also, this ligand has two potential binding sites (Scheme 2). The structure of this compound is reported here for the first time.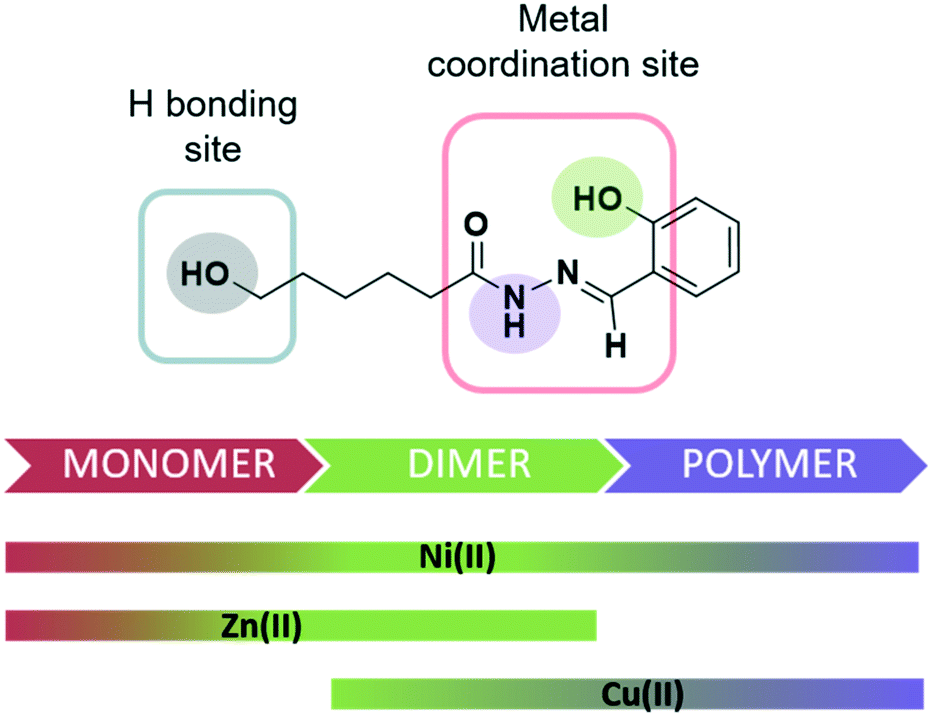 | ||
| Scheme 2 Structural formula of H2L depicting –OH and –NH groups and binding sites. Metal–ligand associations isolated with each metal centre. | ||
Different types of complexes based on the M/H2L (M = ZnII, CuII NiII) system were obtained by adjusting the synthetic conditions (Scheme 1). The formula of the metal complexes, the protonation degree of the ligand and the different associations observed are listed in Table 1. In general, compounds were isolated from reactions carried out in ethanol with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2 metal/ligand ratio using the corresponding metal nitrate or acetate as precursors. The same solvent was always used to minimize its influence as structure directing agent in the different compounds. Most of the single crystals were obtained from the mother liquor after adding diethyl ether at 4 °C. Single crystals of Cu-3b resulted from the reaction of the ligand and copper(II) tetrafluoroborate in glass vessel which is a source of the hexafluorosilicato anion, as reported previously.1d,10
:1 or 1:2 metal/ligand ratio using the corresponding metal nitrate or acetate as precursors. The same solvent was always used to minimize its influence as structure directing agent in the different compounds. Most of the single crystals were obtained from the mother liquor after adding diethyl ether at 4 °C. Single crystals of Cu-3b resulted from the reaction of the ligand and copper(II) tetrafluoroborate in glass vessel which is a source of the hexafluorosilicato anion, as reported previously.1d,10
| H2La | NiII | CuII | ZnII | Associationb |
|---|---|---|---|---|
| a Hydrazone ligand. b Association based on hydrazone ligand “a” letter in the label (also “b” in Cu-3b) indicates that the compound was isolated as single crystals. | ||||
| H2L and HL− | [Ni(HL)(H2L)]NO3·EtOH Ni-1a | — | [Zn(HL)(H2L)]NO3·EtOH Zn-1a | Monomer |
| HL− | [Ni(HL)2] Ni-2 | [Zn(HL)2] Zn-2 | ||
| [Ni2(HL)2](NO3)2Ni-3 | [Cu(HL)(NO3)]2Cu-3a | [Zn2(HL)2](NO3)2Zn-3 | Dimer | |
| [Ni2(HL)2(EtOH)(H2O)] (NO3)2Ni-3a | [Cu2(HL)2(SiF6)] Cu-3b | |||
| L2− | [Zn2L2] Zn-4 | |||
| ∞ 1Ni(L) Ni-5 | ∞ 1Cu(L) Cu-5 | Polymer | ||
The reaction of H2L with copper salts (nitrate or tetrafluoroborate) led to the formation of different dimeric species Cu-3a and Cu-3b, in a similar way to that reported previously for perchlorate copper salts.9a On using copper acetate the N–H group was also deprotonated and the previously reported neutral copper(II) polymeric compound ∞1Cu(L) (Cu-5) was obtained.9a
Monomeric and dimeric compounds, i.e., [Ni(HL)2] (Ni-2) and [Ni2(HL)2](NO3)2 (Ni-3), were isolated from reactions of nickel(II) nitrate with a 1:2 and 1:1 metal/ligand ratio, respectively. The bideprotonation of the ligand and formation of the polymeric complex ∞1Ni(L) (Ni-5) was also achieved by addition of a base under the same conditions and also by using nickel acetate. The monomeric zinc compound [Zn(HL)2] (Zn-2) was obtained using the acetate precursor, whereas for the preparation of the dimeric specie [Zn2(HL)2](NO3)2 (Zn-3) was used the nitrate salt in the same synthetic procedure. The presence of an additional base in the reaction led to deprotonation of the amido NH-group, leading to the dianionic ligand L2− in the complex [Zn2(L)2] (Zn-4).
In addition to the above, the isolation of single crystals allowed to identify other species such as [Ni2(HL)2](EtOH)(H2O)](NO3)2 (Ni-3a) or [M(HL)(H2L)](NO3)·EtOH (M = Ni(II) or Zn(II)) (Ni-1a and Zn-1a), where solvent molecules and/or a nitrate anion occupy the first or second coordination sphere around the metal centre. Complexes with mixed neutral H2L and monodeprotonated HL− ligands coordinated to the metal center in an octahedral environment could be isolated in Ni-1a and Zn-1a.
The formation of one or other complex type clearly depends on the stoichiometric ratio used, the coordination preferences of the metal and the deprotonation degree of the ligand. (Scheme 2).
Compounds were characterized by elemental analysis, mass spectrometry (ESI+), infrared spectroscopy and by 1H NMR spectroscopy in the cases of diamagnetic compounds. The ligand-to-metal ratio in each case was confirmed by elemental analysis, which is particularly important to distinguish [M(HL)2] from [M2(HL)2]2+ species. Electrospray ionization (ESI) mass spectrometry was employed to study these compounds in the solution state. For most of the compounds the main peak is assigned to M(HL)2 (M = Zn and Ni), which indicates the stability of the mononuclear species. This peak was not observed for Cu(II) compounds. Similarly, a peak for the dinuclear species [Zn2(HL)2]2+ was also observed in the ESI spectra of zinc complexes, which indicates that the dinuclear species are formed even in dilute solution. Nevertheless, peaks for dinuclear species of nickel compounds were not detected in the ESI mass spectrum, suggesting a lack of robustness of these species in solution.
NMR studies on the free ligand and its zinc complexes were performed in DMSO-d6 solutions due to the low solubility of the compounds in other solvents. The 1H NMR signals due to the –OH and –NH groups in H2L were observed at 11.59, 11.20 and 10.15 ppm and these signals were shifted to slightly higher field in the complexes. The integration was consistent with the monodeprotonated character of the ligand in Zn-2 and Zn-3. The NMR spectra of these complexes also contained signals that are attributed to the free ligand. This behaviour was not observed when MeOH-d4 solvent was used for Zn-3 and this indicates that the compound is pure and probably unstable in DMSO. In the spectra of Zn-4, the absence of the signals between 11.6 and 10.1 ppm is consistent with the dianionic character of the ligand.
The infrared spectrum of the free ligand contains bands centred at 1657, 1623 and 1276 cm−1 attributed to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ν(CN) and ν(C–N), respectively. Upon complex formation the characteristic bands of the N–H and CO stretching vibrations of the ligand disappear and a new strong band is observed at around 1610 cm−1, which is typical of the conjugated –CHN–NC– group in the enolate form of the ligand. A broad band attributed to the O–H stretching vibration of the hydroxyl group of the side chain is observed at around 3300–3700 cm−1 in all compounds. The FT-IR spectra of the compounds display features corresponding to the nitrate anions in the dimeric Cu-3a, Zn-3 and Ni-3 compounds. These bands are not present in the doubly deprotonated compounds.
O), ν(CN) and ν(C–N), respectively. Upon complex formation the characteristic bands of the N–H and CO stretching vibrations of the ligand disappear and a new strong band is observed at around 1610 cm−1, which is typical of the conjugated –CHN–NC– group in the enolate form of the ligand. A broad band attributed to the O–H stretching vibration of the hydroxyl group of the side chain is observed at around 3300–3700 cm−1 in all compounds. The FT-IR spectra of the compounds display features corresponding to the nitrate anions in the dimeric Cu-3a, Zn-3 and Ni-3 compounds. These bands are not present in the doubly deprotonated compounds.
The ligand H2L and its complexes were also characterized by UV-vis spectroscopy and the main results for solid state and solution are included in Table S4.† In EtOH solution four absorption bands were observed in the spectrum of H2L: two more intense bands at 278 and 288 nm and two weaker absorption bands at 217 and 322 nm. The solid diffuse reflectance spectrum of H2L exhibits intense absorptions at 324 and 256 nm. The highest energy band is probably due to a π → π* transition of aromatic rings while the intense band above 300 nm is assigned to the n → π* transition of the hydrazone (azomethine) chromophore.
The diffuse reflectance spectra of the copper complexes contains an asymmetric broad band in the visible region that is characteristic of Cu(II) metal centres. The band at 697 nm in Cu-3b could be attributed to d-d transitions in an octahedral environment, while the band at 598 nm in Cu-5 could be ascribed to the d-d transitions in a square planar environment (Fig. S1†).11
The nickel complexes Ni-2 and Ni-3 show three bands; a broad band at around 940 nm assigned to 3A2g → 3T1g (ν1), a weak shoulder close to 590 nm assigned to 3A2g → 3T1g (ν2) and a strong band at 303 nm assigned to 3A2g → 3T2g (ν3) attributed to d-d transitions in an octahedral environment (see Table S4 and Fig. S2†). In Ni-5, a single d–d band at 468 nm consistent with the presence of square-planar nickel(II) ions in their low-spin state confirms the metal coordination environment in the solid state. (Fig. S3†).12
Structural studies
Structure of the ligand H2L
The molecular structure of H2L is represented in Fig. 1 and selected bond distances and angles are listed in Table S2.† The free molecule is not planar due to the presence of two segments: the planar acylhydrazone group and the aliphatic chain with the hydroxyl group pointed to the opposite side of the hydrazine group. The angle between the planes defined by the acylhydrazone group and the aliphatic chain is 60.49° (Fig. 1).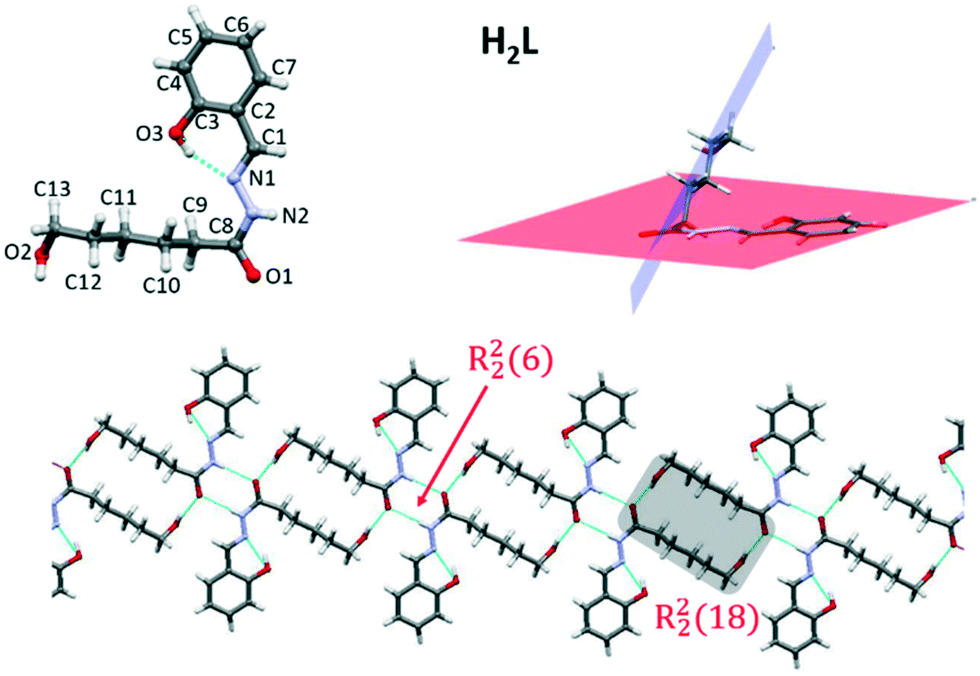 | ||
| Fig. 1 Molecular structure of H2L showing the atomic numbering. Crystal association of the molecules showing details of the intermolecular interactions (bottom) and angle in the molecule (top, right). | ||
The main differences respect to the structures of the complexes are the torsion angles N1–N2–C8–C9 (−1.96°) and C10–C9–C8–N2 (−81.35°), which are far from the usual value of ≈177° found in such complexes. A parameter that highlights differences between the free ligand and its complexes is the intramolecular O3–H⋯N1 hydrogen bond involving the O3–H hydroxyl group as the donor and the N1 of hydrazine group, which leads to a highly stabilized six-membered ring. The structural parameters for this interaction are indicative of a hydrogen bond that is between strong and moderate in nature (ESI†).13 The C8–N2 and C8–O1 bond distances have typical values for the ketonic resonance form. The molecular association in the crystal structure is achieved by means of H-bonds involving the terminal ω-hydroxo oxygen atom O2 and the hydrazone nitrogen N2 as donors and carbonyl O1 oxygen atom as acceptor. The furcated character of these interactions allows the formation of six-membered rings (R22(6)) and also a larger ring (R22(18)) with the aliphatic chains (Fig. 1, bottom) and, consequently, a supramolecular chain is formed.
Structures based on mononuclear [M(HL)(H2L)]+ units
The acidic phenolic hydroxyl group is usually deprotonated upon coordination but an additional base is required to deprotonate the hydrazone group to afford dianionic tridentate-chelating ligands. Nevertheless, H2L was also found to coordinate as a neutral ligand. The mononuclear Zn(II) and Ni(II) (Zn-1a and Ni-1a) compounds were isolated as isotypic single crystals, probably due to the stability of these metal centres in an octahedral environment. Unfortunately, the poor crystallography data obtained for the Ni(II) compound preclude complete structural elucidation.Both compounds crystallize in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and the asymmetric unit contains two independent molecules of the complex together with two nitrate anions and ethanol molecules. The zinc(II) compound is a cationic mononuclear complex with one ligand deprotonated in the phenolic hydroxy group and the second ligand being neutral. The two ligands act as tridentate leading to the formation of six- and five-membered chelate rings. Nitrate anions are in the second coordination sphere and the compound crystallizes as an ethanol solvate. The metal is N2O4 hexa-coordinated by two imino nitrogen atoms, two amido oxygen atoms and two phenoxo oxygen atoms belonging to deprotonated and neutral hydroxyl groups of two different ligands. The metal atom is in a distorted octahedral environment with Zn–O hydrazone distances longer than Zn–O hydroxyl distances.
space group and the asymmetric unit contains two independent molecules of the complex together with two nitrate anions and ethanol molecules. The zinc(II) compound is a cationic mononuclear complex with one ligand deprotonated in the phenolic hydroxy group and the second ligand being neutral. The two ligands act as tridentate leading to the formation of six- and five-membered chelate rings. Nitrate anions are in the second coordination sphere and the compound crystallizes as an ethanol solvate. The metal is N2O4 hexa-coordinated by two imino nitrogen atoms, two amido oxygen atoms and two phenoxo oxygen atoms belonging to deprotonated and neutral hydroxyl groups of two different ligands. The metal atom is in a distorted octahedral environment with Zn–O hydrazone distances longer than Zn–O hydroxyl distances.
The configuration around the Zn(II) can be described by the configuration index [OC-6-22] (Fig. 2a).14 The orientation of the two ligands around the metal cation is almost perpendicular (81°) and the angles between the plane formed by the coordination rings and the aliphatic chain are 84° and 76°.
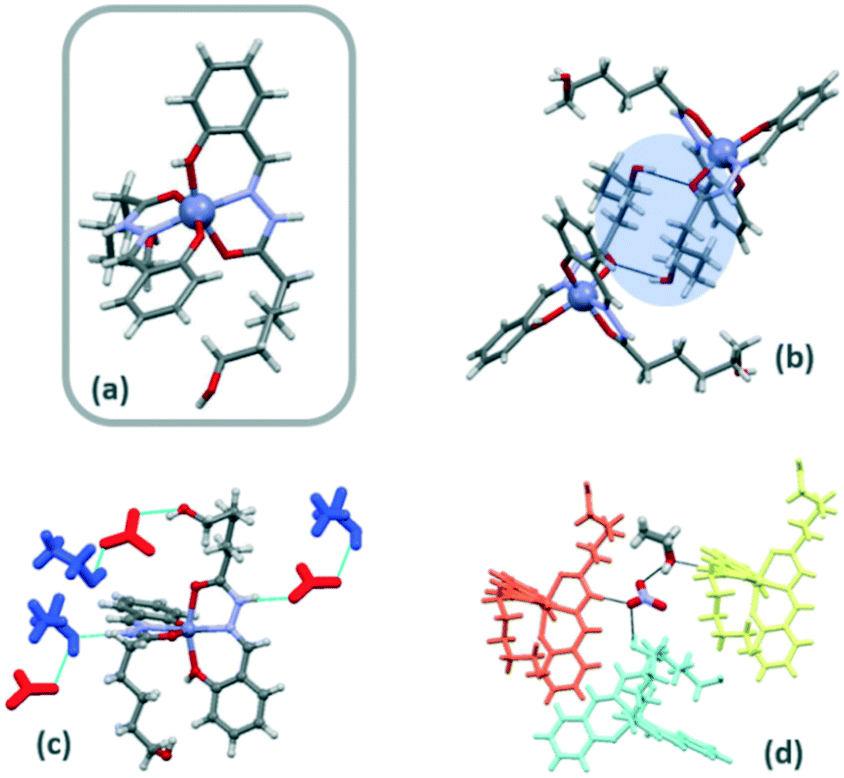 | ||
| Fig. 2 Structure of Zn-1a (a) showing details of the OH⋯O interactions (b) and hydrogen-bonding interactions with nitrate and ethanol molecules (c). Packing of monomers by interactions with the solvate molecules (d). | ||
Nitrate anions and ethanol molecules allow the interactions between complex molecules through different H-bonds. The N–H imine group of H2L is involved in H-bonding interactions as donors with oxygen atoms of nitrate or ethanol molecules. In this context, monomer-nitrate–ethanol-monomer H-bonding interactions are responsible for the supramolecular arrangement (Fig. 2c and d). Moreover, the hydroxyl group of neutral ligands is involved in strong H-bonding interactions with deprotonated hydroxyl groups of ligands of neighbouring monomers. Terminal ω-hydroxyl groups are also involved in hydrogen bonds with amido oxygen atoms of neighbouring ligands to form a ring (R22(18)) with the aliphatic chains (Fig. 2b).
Structures based on dinuclear [M2(HL)2]2+ units
Some complexes of Cu(II) and Ni(II) may be described as being based on {[M2(HL)2]2+} units resulting from the phenoxo-bridging coordination mode of the ligand. The structures observed range from the cationic discrete compound Ni-3a to neutral discrete Cu-3a or polymeric Cu-3b. The main differences in the structures are due to the anions and solvent molecules that complete the coordination sphere and also contribute to the different supramolecular associations.Compound Ni-3a crystallizes in the triclinic P space group and compounds Cu-3a and Cu-3b crystallize in the monoclinic P21/c and C2/m space groups, respectively. The ligand is monodeprotonated to form a dimeric structure in which two metal–ligand units are linked by a μ-phenolate-bridge to generate a M2O2 core. This unit lies on an inversion centre in the three structures. This structural feature has previously been observed in aroylhydrazone complexes15 almost always based on zinc atoms. The M⋯M distances of 2.989, 2.926 and 3.067 Å in Cu-3a, Cu-3b and Ni-3a, respectively, are slightly shorter than those observed previously.9,15 This arrangement is very stable and presents a structural rigidity where the differences in bond distances and angles between each compound are almost negligible.
A dimeric structure based on two di-μ-phenoxo-bridged metal–ligand moieties is also observed with others N-salicylidenehydrazide ligands, especially with copper(II) metal centres (see for example ref. 1d).
All MII cations are {NO3}-tetracoordinated through one amide oxygen, one imino nitrogen and two μ-phenoxo oxygen atoms of HL− ligands to generate five- and six-membered chelate rings. The fifth coordination position in Cu-3a is occupied by one oxygen atom belonging to a nitrate anion and this leads to a square pyramidal geometry (the Addison parameter16 is 0.04) around the metal centre in Cu-3a (Fig. 3a). The metal atom is NO3F2 hexa-coordinated in a distorted octahedral environment with fluoride atoms belonging to SiF62− anions located in trans positions and at longer distances (2.420 Å). In Ni-3a the metal cation is NO5 coordinated with two oxygen atoms belonging to water (2.4119 Å) and ethanol (2.4140 Å) molecules in the same disposition. As expected for copper complexes, the axial positions are strongly affected by the Jahn–Teller effect in Cu-3b. The nitrate anion in Ni-3a is located far from the metal centre and it acts as a ‘glue’ between three different dimeric units where the oxygen atoms of the nitrate anion act as acceptors in different H-bonds (Fig. 3c and d). The SiF62− anions are attached to the dimers by coordination and they act as bridging anions to form a polymeric chain (Fig. 4). The Cu⋯Cu distance across the SiF62− anions is 7.158 Å and the dimeric units are ordered in a parallel manner.
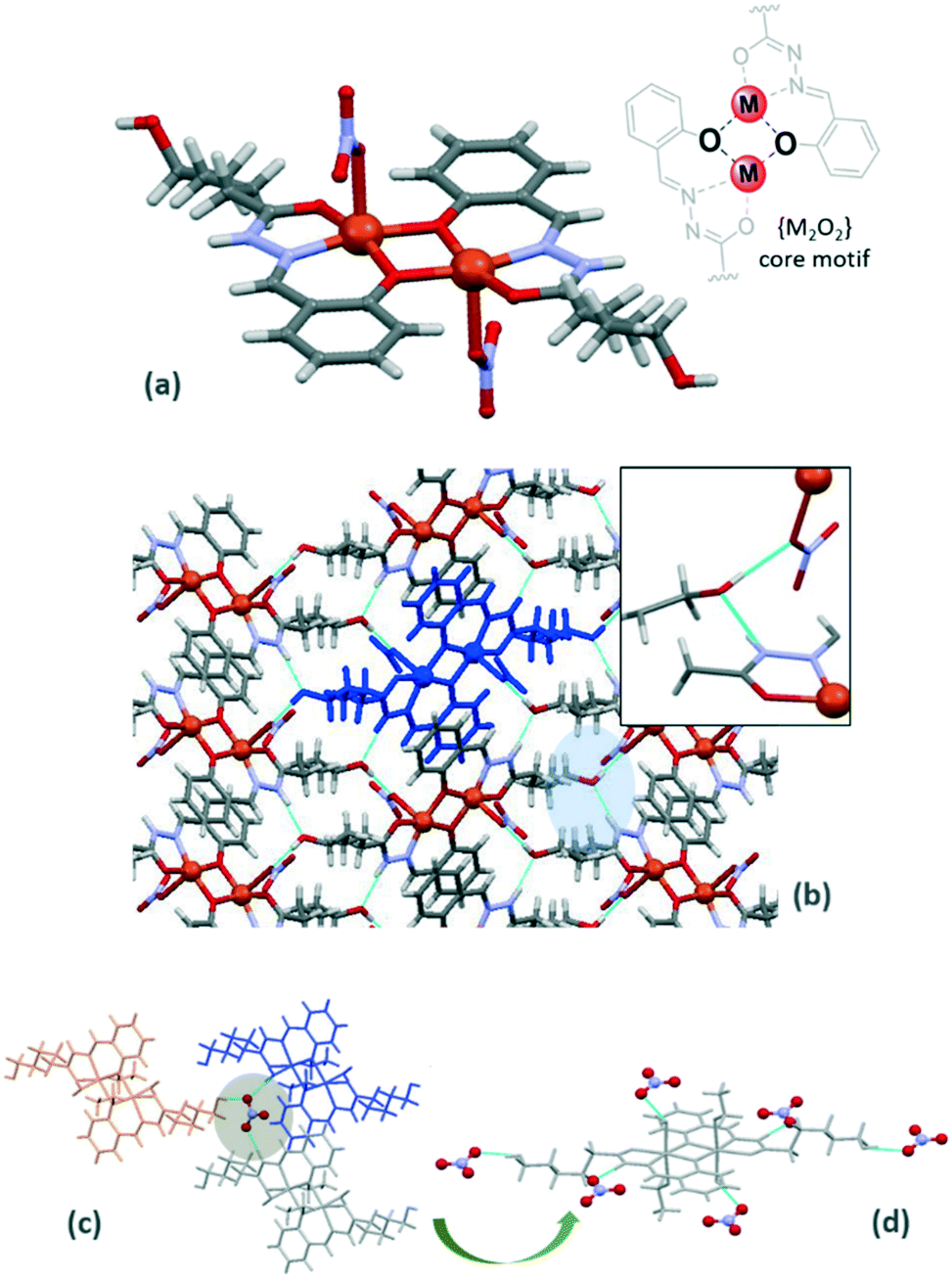 | ||
| Fig. 3 (a) Molecular structure of Cu-3a showing the di-μ-phenolate bridge core motif {M2O2}. (b) Supramolecular arrangement of Cu-3a showing details of the OH⋯O and NH⋯O interactions. Bottom: details of the interactions involving nitrate anions in Ni-3a. (c and d) The different nitrate anions that interact with the dimeric unit. | ||
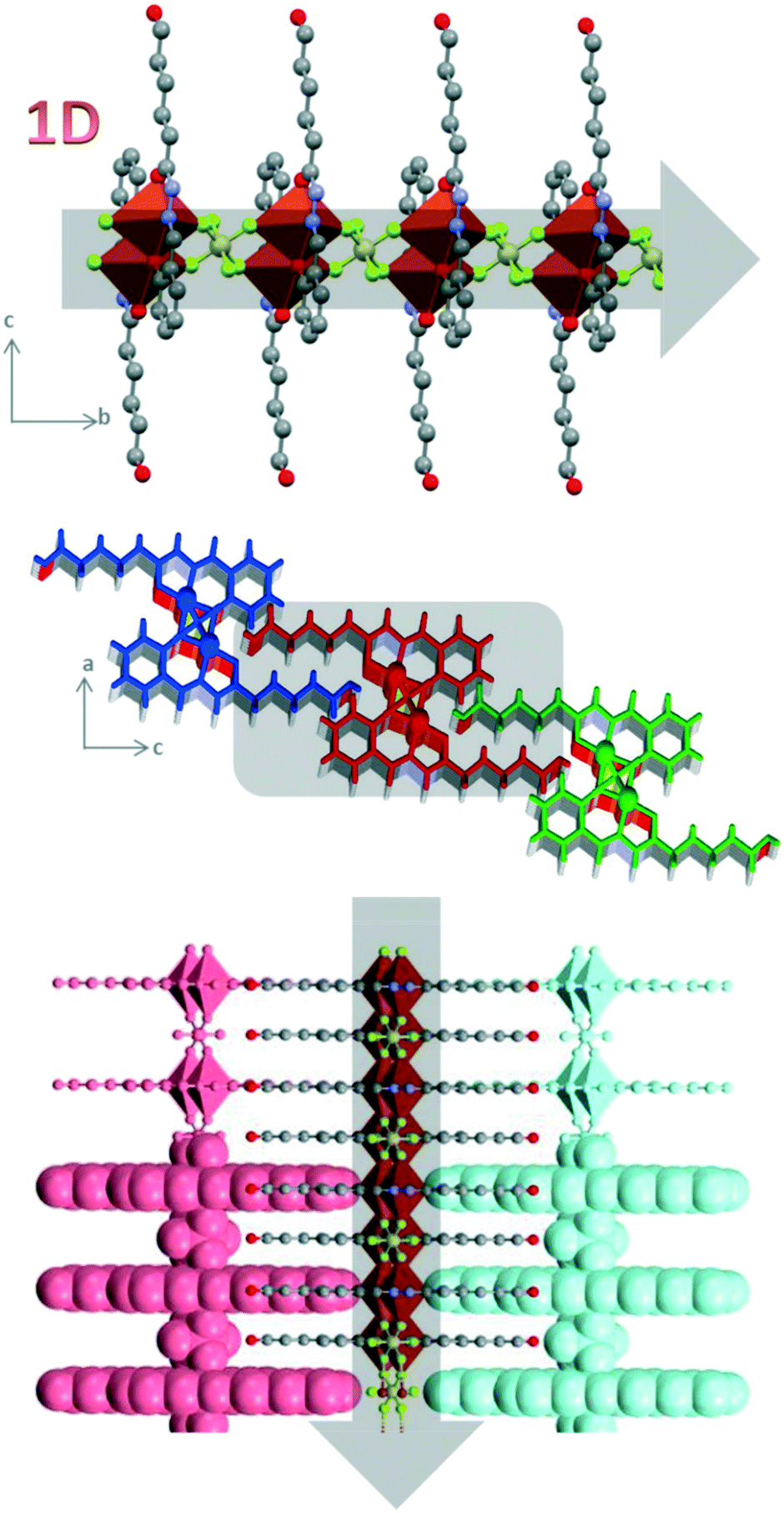 | ||
| Fig. 4 Structure of 1D polymer Cu-3b and interactions between the different chains in the cb plane (top) and ac plane (bottom). | ||
In all structures, the terminal ω-hydroxo group plays a key role in the supramolecular arrangements by allowing interactions with other neighbouring dimeric molecules by means of different H-bonds. For example, this group is involved in hydrogen bonds with the hydrazinic –NH group as the acceptor and with the oxygen atom belonging to a nitrate anion as a donor (Table S3†) to form a layer of dimers in Cu-3a. Furthermore, the terminal ω-hydroxo group is involved in several hydrogen bonds with fluoride atoms of neighbouring chains in Cu-3b, whereas it is also involved in a hydrogen-bond interaction as an acceptor with a coordinated water molecule and as a donor with the free nitrate anion in Ni-3a. The dimers in this structure are connected through different H-bonds to give rise to the final supramolecular arrangement. The system of hydrogen bonds involves the uncoordinated nitrate anion, the terminal ω-hydroxo group and coordinated water molecules.
Structures based on the doubly deprotonated ligand
Roth et al.9a reported the structure of ∞1Cu(L) where the metal centre is coordinated by the phenoxo and oxazine oxygen atoms (trans positions), as well as by the imino nitrogen atom and the deprotonated amido nitrogen atom belonging to a different ligand. This situation results in polymeric structures. Thus, the metal centre is in a square-planar environment and the axial coordination positions remain free. Deprotonation of the NH group in these compounds implies the presence of the enol form in the carboximide fragment and this is reflected in a decrease in the N–C bond length while the C–O carbonyl bond is elongated. The PXRD analysis of the synthesized Ni-5 compound closely matched the simulated pattern generated from the single-crystal data of ∞1Cu(L) (Cu-5) and, consequently, we propose an identical structure for this nickel compound (Fig. S4†).Nevertheless, a powder sample of doubly deprotonated zinc compound Zn-4 gave a different X-ray powder pattern. On the other hand, the bideprotonated nature of Zn-4 could be confirmed by the 1H NMR spectrum and the IR spectrum, which indicates the absence of nitrate anions in the composition, and also an equimolar metal-to-ligand ratio was established by elemental analysis. Bearing in mind the frequent occurrence of the di-μ-hydroxo-bridge core motif {Zn2O2} in zinc complexes of salicylaldehyde ligands,15 we believe that this structural motif could be present in Zn-4 and it probably consists of a dinuclear structure derived from Zn-3 after total deprotonation of both ligands.
Reactivity in solution
In order to gain a deeper understanding of coordination behaviour of this type of compound, we evaluated the reactivity of one of the nickel complexes, [Ni(HL)2] (Ni-2), in the presence of the bidentate ligand (E)-N′-(4-hydroxybenzylidene)-4-hydroxybenzohydrazide (HL*).17 Nickel(II) was selected for this study since it is a metal centre capable of stabilizing different coordination numbers (between 6 and 4) and therefore against bi- or tridentate ligands to form complexes with a 1:2 stoichiometry (Ni:ligand). On the other hand, HL* contains only the O,N-acylhydrazone group, therefore can only act as a bidentate ligand. Consequently, it must form a thermodynamically less estable complex than H2L. In addition, the deprotonation in HL* takes place in the hydrazone group, which is less acidic than the –OH salicylaldehyde group.
In order to study this behaviour, a basic solution of HL* in a mixture of solvents was added to a green suspension of [Ni(HL)2] (Ni-2) in ethanol. The supernatant solution turned orange and orange single crystals of [Ni(L*)2]·(THF)(H2O) (NiL*) were isolated in the middle of the green precipitate of [Ni(HL)2] (Scheme 3). It should be noted that we have not observed a similar process in the case of the Zn and Cu complexes.
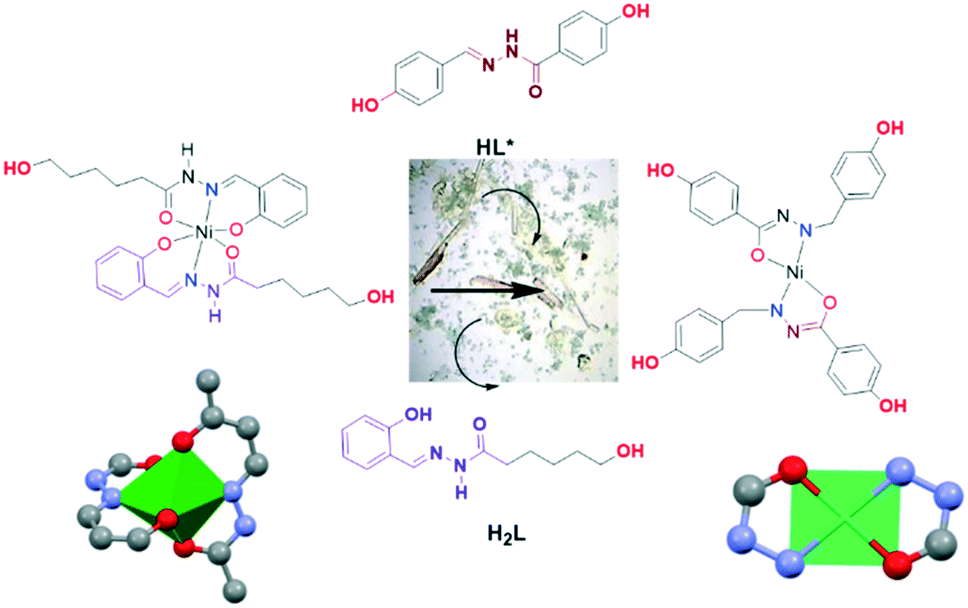 | ||
| Scheme 3 Reactivity of [Ni(HL)2] (Ni-2) in the presence of the bidentate ligand (HL*). | ||
X-ray diffraction on these orange crystals of NiL* ([Ni(L*)2]·(THF)(H2O)) showed (Fig. 5) it is a mononuclear four-coordinated nickel complex with the two monodeprotonated ligands. The compound crystallized as a solvate with THF and water molecules. The bidentate ligand coordinate to nickel(II) through the imine-N and carbonyl-O atoms in a [SP-4-1] geometry with two five-membered chelate rings. The Ni–O and Ni–N bond lengths are shorter than those in the previously discussed dimeric Ni-3a compound. A noteworthy structural feature of this compound is that two phenol rings from two ligands of neighbouring complexes point towards the centre of the nickel chelate ring, which suggests intermolecular metal–π interactions.18 The molecule is almost square and planar with a size around 12 × 11 Å (distances between oxygen phenyl rings). The complex was assembled and stacked through the overlapping of the flat molecules. The intermolecular distance between two metal ions is 5.803 Å. The intermolecular distance between the centroid of the six-membered ring in the ligand moiety and nickel metal ion M+2⋯π is 3.885 Å. The stability of the structure clearly depended on this metal–π supramolecular interactions. The π-interaction of aromatic compounds through electrostatic interactions is considered a promising approach to control the directions and positions of the intermolecular interactions.18a
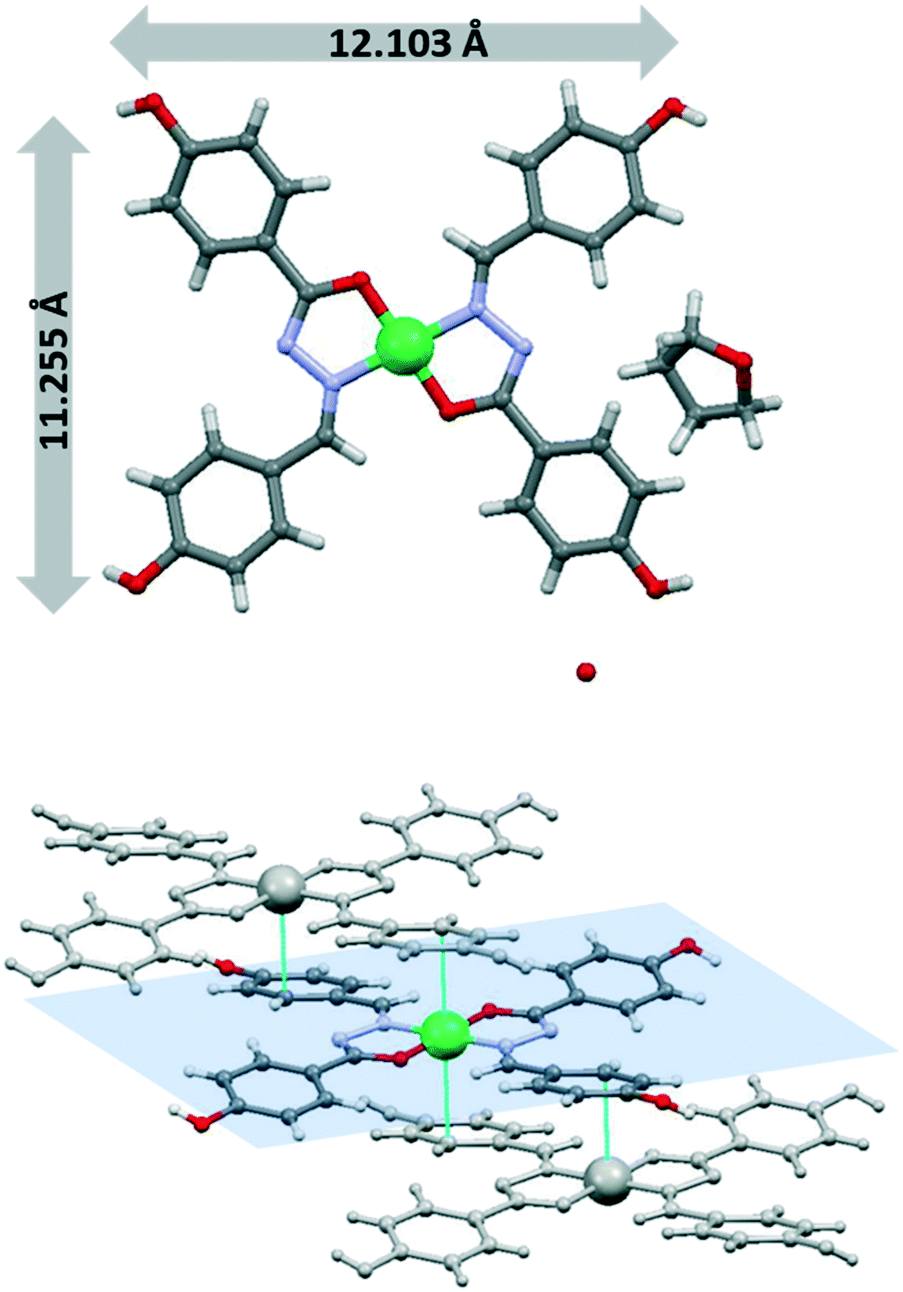 | ||
| Fig. 5 Structure of [Ni(L*)2]·(THF)(H2O) (NiL*) showing details of the Ni+2⋯π interactions. | ||
The interactions of crystallization solvent molecules with the oxygen atoms of phenol groups are co-responsible for the final supramolecular arrangement of the compound through different H-bonding interactions. Work is in progress to deep down into the mechanism of formation of these hydrazone/hydrazonate nickel complexes.
Conclusions
A careful study of the conditions for the reaction of the Schiff base ligand H2L, 6-hydroxy-N-[(2-hydroxyphenyl)methylidene] hexane hydrazide with a variety of divalent atoms allowed the isolation of different kinds of complexes.The nature of these compounds depends on the degree of protonation achieved for the H2L ligand and the stoichiometric relations metal:ligand. In equimolar relationships, without the addition of a base, dinuclear complexes [M2(HL)2]2+ are obtained in which phenyl oxygen acts as a bridge after its deprotonation. Only in the case of using copper is the anion included in the coordination sphere of the metal to reach NC = 5 in [Cu2(HL)2(NO3)2]. In the other cases, the metal is tetracoordinated (M = Zn, Ni).
A different behaviour is observed when a base is added (or the use of metallic acetate as a reagent) in the same molar ratio, since the type of structure formed depends on the nature of the metal. While in the case of Zn, a neutral dinuclear complex is obtained as a consequence of the deprotonation of the hydrazone group, [Zn2L2], with Cu and Ni, coordination polymers are formed in which the [ML] units are associated by M–N2 bridges involving hydrazinic nitrogen. When the metal: ligand stoichiometric ratio is 1:2, neutral complexes [M(HL)2] are obtained (although intermediate species such as [M(HL)(H2L)]+ could also be obtained from mother liquors) in which the metal coordination environment is octahedral.
In all cases, the terminal ω-hydroxo group of H2L plays a crucial role in the supramolecular arrangements of all the structures.
Moreover, compound [Ni(HL)2] (Ni-2) exhibits a structural transformation from an octahedral metal coordination environment to a four-coordination environment through the addition of a competitive bidentate hydrazone ligand (HL*). The single crystals of this new complex were isolated from the initial non-reactive complex, and the X-ray diffraction study showed a highly stacked arrangement of the complex molecules.
The structural diversity observed in the complexes together with the available molecular recognition hydroxyl group in the ligand open the possibility to use these compounds in the fabrication of new supramolecular materials.
Experimental section
Materials and physical measurements
Metal salts and solvents were obtained commercially and were used as supplied. Elemental analyses (C, H, N) were carried out on a Fisons EA-1108 microanalyser. IR spectra were recorded from KBr discs (4000–400 cm−1) with a Jasco FT/IR-6100 spectrophotometer. 1H NMR spectra were obtained on a Bruker AMX 400 spectrometer. Mass spectra were recorded on an FTMS Bruker APEXIII system under ESI conditions. UV-vis spectra were recorded on a Jasco V-670 spectrophotometer.The Schiff base ligand H2L, 6-hydroxy-N-[(2-hydroxyphenyl)methylidene]hexane hydrazide, was prepared as described previously.9 The structure of H2L was elucidated by X-ray diffraction studies on single crystals obtained by slow evaporation (one month) of an ethanolic solution of the compound. The bidentate ligand (E)-N′-(4-hydroxybenzylidene)-4-hydroxybenzohydrazide (HL*) was prepared as described previously.17
Synthesis of complexes with the monodeprotonated ligand
Data for Ni-2: yield: 17 mg (38%). Anal. found: C, 56.25; H, 6.18; N, 9.90; C26H34N4O6Ni requires: C, 56.04; H, 6.15; N, 10.05. IR (KBr, cm−1) = 3414 br, 2934 m, 2860 m (OH–NH), 1630 m + 1607 vs ((CO) amide I), 1557 s, ((CN) amide II), 1457 m, (ν(CN) amide III). MS-ESI: m/z (%) = 251 (01) |H2L|+, 385 (03) |Ni(HL)(DMSO)|+, 557 (100) |Ni(H2L)2|+, 807 (20) |Ni(H2L)2 + H2L|+, 1114 (01) |Ni2(H2L)4|+.
Data for Ni-3: yield: 46 mg (57%). Anal. found: C, 41.99; H, 5.59; N, 10.07 C28H42N6O14Ni2 requires: C, 41.83; H, 5.27; N, 10.45. IR (KBr, cm−1) = 2935 m, 2862 m (OH–NH), 1607 m ((CO) amide I), 1557 s ((CN) amide II), 1457 m (ν(CN) amide III), 1338 s, 1319 m, 1286 m (ν(NO3−)). MS-ESI: m/z (%) = 251 (100) |H2L|+, 385 (03) |Ni(HL)(DMSO)|+, 523 (86) |Na(H2L)2|+, 557 (16) |Ni(H2L)2|+.
Data for Zn-2: yield: 23 mg (41%). Anal. found: C, 55.65; H, 6.19; N, 9.80; C26H34N4O6Zn requires: C, 55.37; H, 6.08; N, 9.93. IR (KBr, cm−1) = 3015 m, 2933 m, 2857 m (OH–NH), 1633 m + 1609 vs ((CO) amide I), 1569 s, 1542 m ((CN) amide II), 1466 m, 1439 s, 1386 w, 1332 w, 1305 s, (ν(CN) amide III), 1386 m (C–O). MS-ESI: m/z (%) = 251 (22) |H3L|+, 313 (27) |Zn2(HL)2 +H|+, 391 (24) |Zn(HL)(DMSO)|+, 471 (02) |Zn(HL)(DMSO)2|+, 563 (100) |Zn(H2L)2|. 1H-NMR ([D6]-DMSO, 400 MHz): δ (ppm) = 1.2–1.7 (m, 6H, H10, H11 and H12); 2.2 (t, 2H, H9); 3.4 (m, 2 H, H13); 4.4 (br. s, 1 H, O(2)–H); 6.3–7.7 (m, 4H, CHarom); 8.24 and 8.32 (s, total 1 H, H1); 11.2, 11.6,12.3 (all s, total 1 H, NH).
Data for Zn-3: yield: 0.055 g (73%). Anal. found: C, 41.05; H, 4.72; N, 11.01; C26H34N6O12Zn2 requires: C, 41.45; H, 4.55; N, 11.16. IR (KBr, cm−1) = 3565 w, 3190 w, 3015w, 2922 w, 2883 w (OH–NH), 1610 w + 1599 vs ((CO) amide I), 1573 m, 1551 m ((CN) amide II), 1384 vs ((C–O)), 1297 vs, 1277 vs (NO3). MS-ESI: m/z (%) = 251 (100) |H2L|+, 313 (3) |Zn2(HL)2|+, 391 (3) |Zn(HL)(DMSO)|+, 563 (100) |Zn(H2L)2|+. 1H-NMR ([D6]-DMSO, 400 MHz): δ (ppm) = 1.3–1.7 (m, 6H, H10, H11 and H12); 2.2 (t, 2H, H9); 3.4 (m, 2 H, H13); 4.4 (br. s, 1H, O(2)–H); 6.4–7.2 (m, 4H, CHarom); 8.18 and 8.22 (s, 1H, H1); 10.1, 11.2, 11.6,12.2, 12.8 (all s, total 1 H, NH).
Data for Cu-3a: yield: 0.110 g (43%). Anal. found: C, 41.02; H, 4.83; N, 11.22; C26H34N6O12Cu2 requires: C, 41.66; H, 4.57; N, 11.21. IR (KBr, cm−1) = 3175 m, 3088 m, 2942 m, 2865 m (OH–NH), 1635 w + 1604 vs ((CO) amide I), 1581 s, 1552 m ((CN) amide II), 1384 vs ((C–O)), 1280 vs (NO3), 1276 m (ν(C–N) amide III). MS-ESI: m/z (%) = 251 (14) |H2L|+, 312 (84) |Cu(HL)|+, 390 (08) |Cu(HL)(DMSO)|+, 562 (19) |Cu(HL)2|+, 623 (11) |Cu2(L)2|+.
Synthesis of complexes with the bideprotonated ligand
Data for Zn-4: yield: 0.020 g (32%). Anal. found: C, 50.09; H, 5.23; N, 9.30; C13H16N2O3Zn requires: C, 49.78; H, 5.14; N, 8.93. IR (KBr, cm−1) = 3370 br, 2859 w, 2859 m (OH–NH), 1617 s ((CO) amide I), 1598 s, 1518 vs ((CN) amide II), 1474 m, 1442 s, (ν(CN) amide III), 1400 m (C–O). MS-ESI: m/z (%)= 251 (100) |H2L|+, 313 (38) |Zn2(HL)2|+, 391 (32) |Zn(HL)(DMSO)|+, 563 (54) |Zn(H2L)2|+. 1H-NMR ([D6]-DMSO, 400 MHz): δ (ppm) = 1.4–1.6 (m, 6H, H10, H11 and H12); 2.1–2.2 (t, 2H, H9); 3.4 (m, 2 H, H13); 4.4 (br. s, 1 H, O(2)–H); 6.5–7.3 (m, 4H, CHarom); 8.34 (s, 1H, H1).
Data for Ni-5: yield: 17 mg (55%). Anal. found: C, 50.62; H, 5.61; N, 9.31; C13H16N2O3Ni requires: C, 50.87; H, 5.25; N, 9.13. IR (KBr, cm−1) = 3412 br, 2936 m, 2855 m (OH–NH), 1608 s ((CO) amide I), 1535 vs, ((CN) amide II), 1470 m, 1459 m, 1407 m, (ν(CN) amide III), 1374 s (C–O). MS-ESI: m/z (%) = 251 (10) |H2L|+, 385 (43) |Ni(HL)(DMSO)|+, 557 (100) |Ni(H2L)2|+, 807 (62) |Ni(H2L)2 + H2L|+, 1114 (02) |Ni2(H2L)4|+. 1H-NMR ([D6]-DMSO, 400 MHz): δ (ppm) = 1.1–1.7 (m, 6H, H10, H11 and H12); 2.0 (t, 2H, H9); 3.4 (m, 2H, H13); 4.3, 4.2 (s, 1H, O(2)–H); 6.4–6.7 (m, 4H, CHarom); 7.8 (s, 1H, H1).
Cu(L) (Cu-5)
A solution of H2L (0.1 mmol) in ethanol (5 mL) was added to a solution of Cu(AcO)2·2H2O (0.1 mmol) in ethanol (5 mL). The green suspension was stirred for 1 d and heated under reflux for 10 h. The resulting green precipitate was filtered off, washed with ethanol and dried under vacuum.Data for Cu-5: yield: 14 mg (45%). Anal. found: C, 50.19; H, 5.37; N, 8.68; C13H16N2O3Cu requires: C, 50.07; H, 5.17; N, 8.98. IR (KBr, cm−1) = 3395 br, 2938 m, 2859 m (OH–NH), 1619 s + 1604 m ((CO) amide I), 1537 s, 1497 vs ((CN) amide II), 1470 m, 1440 m, (ν(CN) amide III), 1381 vs (C–O). MS-ESI: m/z (%) = 251 (12) |H2L|+, 312 (100) |Cu(HL)|+, 390 (63) |Cu(HL)(DMSO)|+, 562 (28) |Cu(HL)2|+, 623 (05) |Cu2(L)2|+.
Crystallography
Crystallographic data for H2L, Cu-3a, Cu-3b, Ni-3a and NiL* were collected on a microfocal Bruker Smart 6000 CCD diffractometer at 293 K using graphite monochromated Cu-Kα radiation (λ = 1.54178 Å) and data for Zn-1a were collected on a Bruker Smart 1000 CCD diffractometer at 100 K and 293 K, using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Data were corrected for Lorentz and polarization effects. The frames were integrated with the Bruker SAINT19 software package and the data were corrected for absorption using the program SADABS.20 The structures were solved by direct methods using the program SHELXS.21 All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2 using the program SHELXL.21 Hydrogen atoms were inserted at calculated positions and were constrained with isotropic thermal parameters. Drawings were produced with MERCURY22 and special computations for the crystal structure discussions were carried out with PLATON.23 Powder X-ray diffraction data were obtained using Cu-Kα radiation and were collected on a Siemens D-5000 diffractometer over the range 5.0–45.0° in steps of 0.20° (2θ) with a count time per step of 5.0 s. The structural data have been deposited with the Cambridge Crystallographic Data Centre (CCDC) with the reference numbers included in Table S1.† Selected bond lengths and angles and hydrogen bond distances are listed in Tables S2 and S3.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from ERDF (EU) and MEC (Spain) (Spain) (research projects CTQ2017-90802-REDT and PID2019-110218RBI00) are gratefully acknowledged. We thank the Structural Determination Service of the Universidade de Vigo – CACTI for X-ray diffraction measurements.Notes and references
- (a) P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC; (b) G. Mahmoudi, A. A. Khandart, F. A. Afkhami, B. Miroslaw, A. V. Gurbanov, F. I. Zubkov, A. Kennedy, A. Franconetti and A. Frontera, CrystEngComm, 2019, 21, 108–117 RSC; (c) A. Finelli, N. Hérault, A. Crochet and K. Fromm, Cryst. Growth Des., 2018, 18, 1215–1226 CrossRef CAS; (d) S. Rodríguez-Hermida, A. B. Lago, R. Carballo, O. Fabelo and E. M. Vázquez-López, Chem. – Eur. J., 2015, 21, 6605–6616 CrossRef; (e) Y. Song, Y. Liu, T. Qi and G. L. Li, Angew. Chem., Int. Ed., 2018, 57, 13838 CrossRef CAS.
- (a) J. H. Callahan, Inorg. Chim. Acta, 2000, 297, 79–87 CrossRef; (b) C. D. Stewart, H. Arman, H. Bawazir and G. T. Musie, Inorg. Chem., 2014, 53, 10974–10988 CrossRef CAS; (c) X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963 RSC.
- (a) S. Saha, N. Biswas, A. Sasmal, C. J. Gómez-García, E. Garribba, A. Bauza, A. Frontera, G. Pilet, G. M. Rosair, S. Mitra and C. R. Choudhury, Dalton Trans., 2018, 47, 16102–16118 RSC; (b) C. Zhang, R. S. Patil and J. L. Atwood, Adv. Inorg. Chem., 2018, 71, 247 CrossRef CAS; (c) E. Constable, Coord. Chem. Rev., 2008, 252, 842 CrossRef CAS.
- V. A. Friese and D. G. Kurth, Coord. Chem. Rev., 2008, 252, 199–211 CrossRef CAS.
- (a) D. G. Kurth, Sci. Technol. Adv. Mater., 2008, 9(1), 014103 CrossRef; (b) O. Gómez-Paz, R. Carballo, A. B. Lago and E. M. Vázquez-López, Chemistry, 2020, 2, 33–49 CrossRef.
- (a) S. Rodríguez-Hermida, A. B. Lago, L. Cañadillas-Delgado, R. Carballo and E. M. Vázquez-López, Cryst. Growth Des., 2013, 13, 1193–1205 CrossRef; (b) X. Zhou, H. Fang, Y. Ge, Z. Zhou, Z. Gu, X. Gong, G. Zhao, Q. Zhan, R. Zeng and Y. Cai, Cryst. Growth Des., 2010, 10, 4014–4022 CrossRef CAS.
- (a) H. Lv, H. Li, L. Xin and F. Guo, CrystEngComm, 2020, 22, 2406–2413 RSC; (b) A. B. Lago, R. Carballo, O. Fabelo, N. Fernández-Hermida, F. Lloret and E. M. Vázquez-López, CrystEngComm, 2013, 15, 10550–10562 RSC.
- (a) C.-F. Chow, S. Fuji and J.-M. Lehn, Angew. Chem., Int. Ed., 2007, 46, 5007–5010 CrossRef CAS; (b) J. G. Hardy, X. Cao, J. Harrowfield and J.-M. Lehn, New J. Chem., 2012, 36, 668 RSC; (c) C.-F. Chow, S. Fujii and J.-M. Lehn, Chem. – Asian J., 2008, 3, 1324–1335 CrossRef CAS.
- (a) A. Roth, A. Buchholz and W. Plass, Z. Anorg. Allg. Chem., 2007, 633, 383–392 CrossRef CAS; (b) S. Nica, A. Buchholz, M. Rudolph, A. Schweitzer, M. Wächtler, H. Breitzke, G. Buntkowsky and W. Plass, Eur. J. Inorg. Chem., 2008, 2350–2359 CrossRef CAS.
- (a) Q.-H. Jin, L.-L. Zhou, L.-J. Xu, Y.-Y. Zhang, C.-L. Zhang and X.-M. Lu, Polyhedron, 2010, 29, 317–327 CrossRef CAS; (b) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2010, 133, 858–870 CrossRef; (c) W. Chen, J. Chu, I. Mutikainen, J. Reedijk, U. Turpeinen and Y.-F. Song, CrystEngComm, 2011, 13, 7299–7304 RSC.
- A. B. Lago, R. Carballo, S. Rodríguez-Hermida and E. M. Vázquez-López, CrystEngComm, 2013, 15, 1563–1570 RSC.
- (a) L. Sacconi, F. Mani and A. Bencini, in Comprehensive Coordination Chemistry I, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, UK, 1987, vol. 5 Search PubMed; (b) F. Meyer and H. Kozlowski, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, UK, 2004, vol. 6, p. 247 Search PubMed.
- P. Barbazán, R. Carballo, B. Covelo, C. Lodeiro, J. C. Lima and E. M. Vázquez-López, Eur. J. Inorg. Chem., 2008, 1713–2720 Search PubMed.
- A. von Zelewsky, Stereochemistry of Coordination Compounds, Wiley, Chichester, 1996 Search PubMed.
- (a) Z. Xu, X. Zhang, W. Zhang, Y. Gao and Z. Zeng, Inorg. Chem. Commun., 2011, 14, 1569–1573 CrossRef CAS; (b) L. Parrilha, R. P. Vieira, A. P. Rebolledo, I. C. Mendes, L. M. Lima, E. J. Barreiro, O. E. Piro, E. E. Castellano and H. Beraldo, Polyhedron, 2011, 30, 1891–1898 CrossRef; (c) J. J. Vittal and X. Yang, Cryst. Growth Des., 2002, 2, 259–262 CrossRef CAS; (d) A. K. Singh, M. Yadav, S. K. Singh, S. Sunkari and S. Pandey, Inorg. Chim. Acta, 2010, 363, 995–1000 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- C.-M. Li and H.-Y. Ban, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o1465 CrossRef CAS.
- (a) T. Kusakawa, T. Goto and A. Hori, CrystEngComm, 2020, 22, 3090–3094 RSC; (b) R. Carballo, B. Covelo, N. Fernández-Hermida, A. B. Lago and E. M. Vázquez López, J. Mol. Struct., 2009, 936, 87–91 CrossRef CAS.
- Siemens SAINT, Version 4, Software Reference Manual, Siemens Analytical X-Ray Systems, Inc., Madison, WI, USA, 1996 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, MERCURY, New Software for Visualising Crystal Structures, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1858827–1858832. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01204a |
| This journal is © The Royal Society of Chemistry 2021 |