
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnrichment effects of ionic liquid mixtures at polarized electrode interfaces monitored by potential screening†
Sunghwan
Shin‡
 *,
Francesco
Greco‡
,
Florian
Maier
and
Hans-Peter
Steinrück
*,
Francesco
Greco‡
,
Florian
Maier
and
Hans-Peter
Steinrück
Lehrstuhl für Physikalische Chemie 2, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany. E-mail: sunghwan.shin@fau.de
First published on 26th March 2021
Abstract
The behavior of ionic liquids (ILs) at charged interfaces is pivotal for their application in supercapacitors and electrochemical cells. Recently, we demonstrated for neat ILs that potential screening at polarized electrode interfaces shows a characteristic voltage dependence, as determined in situ by X-ray photoelectron spectroscopy. Herein, we use this fingerprint-type behavior to characterize the nature of the IL/electrode interfaces for IL mixtures of [C8C1Im][Tf2N] and [C8C1Im]Cl on Au and Pt electrodes. For Au, the IL/electrode interfaces are dominated by the Cl− anions, even down to a 0.1 mol% [C8C1Im]Cl content. In contrast, [Tf2N]− anions enrich at the IL/Pt electrode interfaces down to 10 mol% [C8C1Im][Tf2N]; only at lower concentrations does a transition to Cl− enrichment occur. These mixture studies demonstrate that even small concentrations of another IL or contamination, e.g. remaining from synthesis, can strongly influence the situation at charged IL interfaces.
I. Introduction
As a consequence of switching from conventional energy provision towards renewable energy resources with their fluctuating input levels, short-, intermediate- and long-time storage devices are needed to store and deliver electric energy to power grids. While storing generated electricity in various types of batteries is particularly suitable on the time scale of minutes up to hours,1 high-power supercapacitors for intermediate energy storage on the second time scale are highly interesting due to their superior power density, fast charge/discharge rates, and long cycle lifetimes.2 Supercapacitors store energy by charge separation and/or by faradaic redox reactions at the electrolyte/electrode interface. The combination of the electrolyte and electrode determines the characteristics of the capacitor, such as its operating voltage, temperature range, and capacitance. Commercially, organic electrolyte-based supercapacitors are widely used due to their large operating potential window (2.5–2.7 V), which provides a significant improvement in both the energy and power densities as compared to aqueous electrolytes (1.0–1.3 V). However, supercapacitors with organic electrolytes usually have a small specific capacitance and face safety issues concerning toxicity, flammability, and volatility.3The interest in ionic liquids (ILs) as solvent-free electrolytes has grown over time due to their large electrochemical window (3.5–4.0 V), thermal stability, low vapor pressure, and the tunability of their physicochemical properties by choosing certain anion–cation combinations. Recently, IL mixtures have received considerable attention as an additional route to tune the properties of ILs for various applications. For example, the employment of mixtures of [BF4]− and [Tf2N]− with 1-ethyl-3-methylimidazolium ([C2C1Im]+) as a common cation led to symmetric charge storage at the two identical electrodes of a capacitor, which resulted in an expanded operating potential window.4 An IL mixture of tetramethylammonium ([TMA]+) and [C2C1Im]+ with [BF4]− as a common anion on mesoporous carbon electrodes exhibited enhanced power and energy densities. These enhancements were attributed to strong cation–cation interactions between [TMA]+ and [C2C1Im]+.5 Another application field is fuel cells, where binary mixtures of protic ILs exhibited higher activity for hydrogen oxidation and oxygen reduction at the electrodes compared to the neat ILs, and this improvement was attributed to an enhanced proton transfer reaction.6
The behavior of ILs at charged interfaces is relevant to design and develop devices for various electrochemical applications.7 In this context, many experimental and theoretical studies have been developed to achieve a better understanding of the electrochemical double layer (EDL) at IL–electrode interfaces. Typically, the EDL structure of ILs has been investigated by measuring the differential capacitance (DC) in impedance spectroscopy, which shows unique features of ILs such as the bell or camel shape of the DC curves.8–12 These distinctive characteristics are successfully interpreted through mean-field theory,13 which takes into account the lattice saturation effect of IL cations and anions, as well as by computer simulations.14–18 The asymmetric shape of the DC curves, caused by different sizes and shapes of the ions, which is another characteristic feature of ILs, has been studied by impedance measurements, molecular dynamic simulations, and mean-field calculations.8,19,20 Apart from the studies of their DC, several studies have been performed to examine the EDL of ILs at the molecular level. Strong interfacial layering, including oscillation of the ion concentrations and overscreening of the charged layers, has been found by atomic force microscopy21–24 and X-ray reflectivity experiments25 of IL/electrode interfaces under thick IL films, and also by computational studies.26–28 The structural dynamics of EDL formation was studied by XPS using two gold electrodes fabricated on a porous polymer surface.29 The adsorption geometry and the reorientation of ILs in response to the applied potential have also been investigated experimentally using sum frequency generation,30 infrared spectroscopy,31 surface-enhanced Raman spectroscopy,32 scanning tunneling microscopy,33,34 and X-ray photoelectron spectroscopy (XPS) through an ultrathin graphene–carbon nanotube composite window,35 together with computational methods.28,36 The chemical interaction between the electrode and ions in ILs, which is denoted as specific adsorption in electrochemistry, also plays an important role for the EDL structure of ILs. Several experimental studies and simulations show a significant effect of specific adsorption between aromatic imidazolium-rings and a graphite electrode surface, which causes a shorter adsorption distance and a high surface charge density.37,38 In addition, in binary IL mixtures, interfacial enrichment of favored species has been reported in simulations and experimental studies.39,40
The identification of chemical species at the IL/electrode interface is, however, difficult to assess directly. A very powerful method in this context is ultra-high vacuum (UHV)-based angle-resolved XPS, which however can be applied only for ultrathin IL films, due to the limited escape depth of the emitted photoelectrons on the order of a few nm.41 Using this method, the composition of the wetting layer in direct contact with the surface and the growth of successive layers have been studied in great detail.42 Very recently, also ultrathin layers of IL mixtures have been investigated using this approach.43,44 It was found that typically specific cations or anions of the mixture are arranged in a checkerboard structure. Upon successive deposition of individual layers of two different ILs, exchange processes take place which are driven by the interaction strength of the specific ILs with the substrate, yielding interface enrichment effects. The question arises of whether such effects are only occurring in ultrathin films or also in a thicker IL film (larger than 10 nm) on the electrode of an electrochemical cell. Under the latter conditions, angle resolved XPS cannot be directly used to study the chemical composition of the interface, due to the limited electron escape depth.
Herein, we now propose a new approach to indirectly deduce which IL ions are preferentially present at the interface in IL mixtures at applied potentials within the electrochemical stability window, which was confirmed by chronoamperometry (Fig. S1 in the ESI†). This approach is based on recent EDL investigations under well-defined UHV conditions for neat IL electrolytes by using in situ XPS. The measurements were performed with a symmetric two-electrode electrochemical cell setup comprising two wires of an identical metal as electrodes with an identical contact area with the electrolyte.45 At a specific applied cell voltage, Uapplied, the potential screening (PS) at the anode and cathode is determined by the properties of the IL ions at the electrode interfaces. As demonstrated in Fig. 1 and our previous study,45 the change in PS at the anode/cathode can be measured through the binding energy shift (ΔBE) of IL-related core level signals at the IL/vacuum interface by XPS. This allows us to assess the PS response at the anode/cathode in equilibrium under ultraclean conditions in UHV. In the following, we denote the amount of PS relative to Uapplied = 0 V at the anode (plus pole) as Δφanodic and that at the cathode (minus pole) as Δφcathodic; by this definition, the sum Δφanodic + Δφcathodic always equals the applied cell voltage Uapplied.
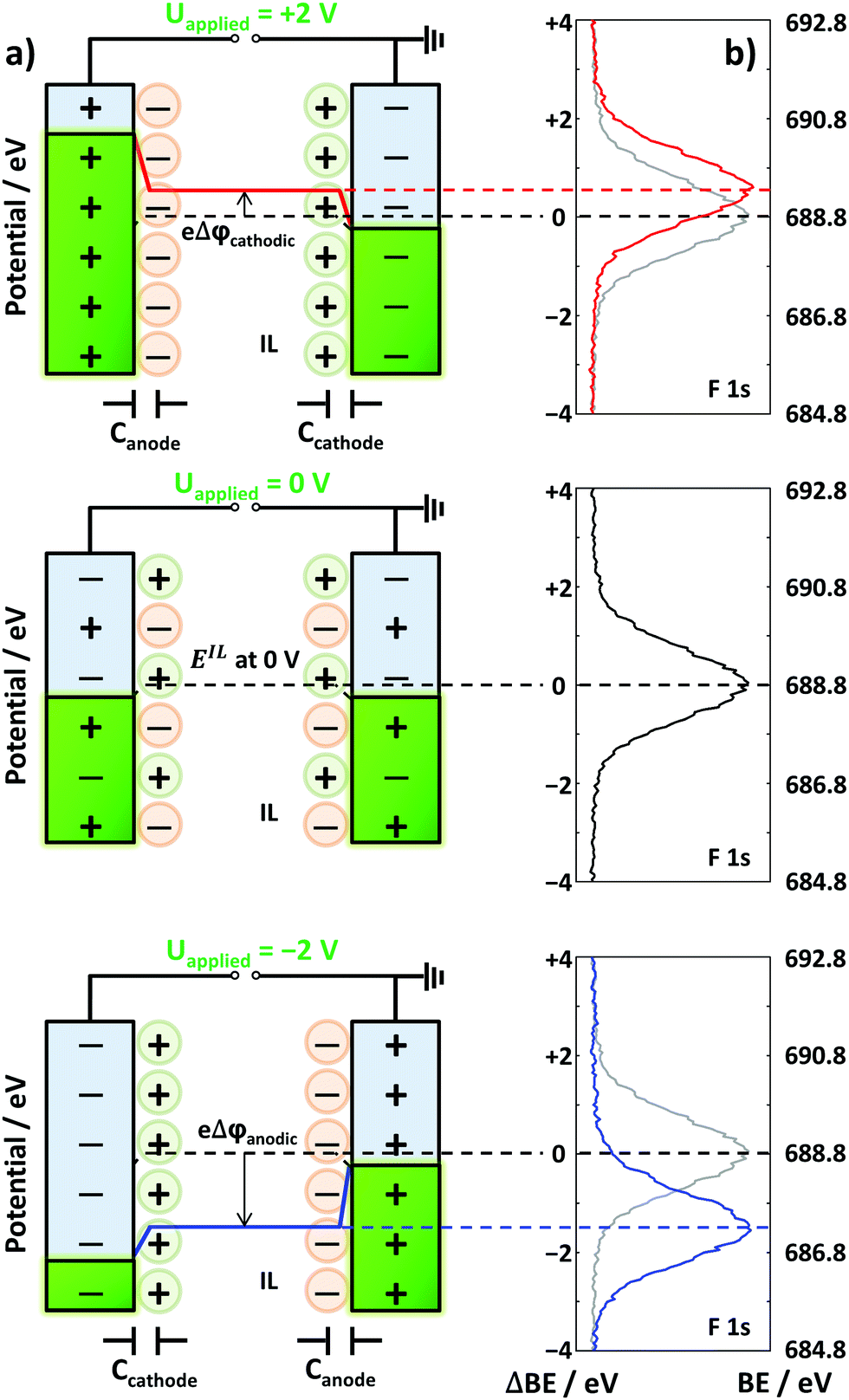 | ||
| Fig. 1 (a) Schematic sketches of the potentials at the IL electrode interfaces and the structure of the EDL, for the electrochemical cell with two Pt electrodes in contact with neat [C8C1Im][Tf2N]. The capacitances at the cathode and anode interfaces are indicated as Ccathode and Canode, respectively (note that for zero applied cell voltage (center), the numbers of anions and cations at the interface may not be exactly equal). (b) XPS spectra of the F 1s region according to Uapplied. The XPS spectrum at 0 V is shown in gray as a reference in the +2 V and −2 V spectra. The potential of the IL (EIL) and the binding energy of F 1s at each applied voltage are indicated by dashed lines. | ||
With this cell setup, we recently observed a very characteristic behavior at the interfaces of certain neat ILs with two identical Au or Pt electrodes.45 The corresponding Δφcathodic curves as a function of Uapplied can be considered as a “fingerprint” signature for certain IL/electrode combinations. The measured behavior ranged from symmetric PS at the cathode and anode to pronounced asymmetric PS at the anode or cathode, along with intermediate behavior. The slope of the Δφcathodicversus applied voltage curve indicates the relative ratio of PS at the electrodes: for symmetric PS the slope is 0.5, that is, an increase of Δφcathodic by 0.5 V per 1.0 V applied cell voltage. For pronounced PS at the anode (or cathode) the slope is close to zero (or one). The observed characteristics reflect the ionic moieties in contact with the respective electrode, that is, the composition and structure of the EDL.
Notably, our method does not allow for determining the specific adsorption geometry of the ions. The idea of the present study rather is to use this fingerprint-type behavior to determine the nature of the IL/electrode interface in mixtures of two ILs A and B, with the same cation. If the mixture shows identical behavior to IL A, then one can conclude that the interface is dominated by the respective ions of IL A, and, if the mixture shows identical behavior to IL B, then one can conclude that the interface is dominated by the respective ions of IL B. A behavior between that of A and B reflects that the interface contains a mixture of the ions of both ILs.
As already emphasized, the structure of the EDLs is determined by the interactions of the IL cations and anions with the metal surface, which depend on their size, orientation, and chemical structure as well as the chemical nature of the electrode. For different IL mixtures, it has been shown by XPS studies of ultrathin layers that typically the ions with the stronger bond to the metal surface preferentially are in direct contact with the substrate, while the less-interacting ions of the same polarity are less present at the interface. While the situation for a specific IL at the IL/electrode interface in an electrochemical cell is not necessarily the same due to ions in the second and further layers, the general conclusion should also hold qualitatively at least.
II. Results and discussion
Mixtures of [C8C1Im][Tf2N] and [C8C1Im]Cl on Au electrodes
To study the competition of two different anions in the PS at the IL/electrode interface, we prepared various IL mixtures with [C8C1Im]+ as a common cation, and [Tf2N]− and Cl− as anions, namely [C8C1Im][Tf2N]xCl1−x. Fig. 2a shows the PS results of neat [C8C1Im][Tf2N], neat [C8C1Im]Cl, and their mixtures on Au electrodes (note that the data of the neat ILs are reproduced from our previous study45). Neat [C8C1Im][Tf2N] (x = 1, red circles) shows within the applied cell voltage range a symmetric behavior, that is, the data for Δφcathodic closely follow the dashed line, which is a slope of 0.5. This indicates similar anodic and cathodic PS for applied voltages from 0 to 2 V. In contrast, neat [C8C1Im]Cl (x = 0, blue squares) shows a much larger Δφcathodic than Δφanodic, as is deduced from a slope of 0.92. This behavior indicates a highly asymmetric situation, with most of the PS occurring at the cathode, while at the anode nearly no changes in PS occur upon applying the voltage. In our previous study, we have assigned this asymmetry for [C8C1Im]Cl mainly to the different sizes of the anion and cation.45 Upon charging the anode more positively by increasing the applied voltage, the additional smaller Cl− counterions in the EDL reside within a smaller distance d at the anode compared to the situation at the cathode, where the larger [C8C1Im]+ counterions compensate the increasing negative charge. Hence, considering the identical contact area A of both electrodes and assuming an identical DC permittivity ε, the corresponding capacitance (C = ε0εA/d) of the EDL at the anode (Canodic) is larger than that of the EDL at the cathode (Ccathodic). Note that in this simple Helmholtz approximation, effects related to differences in ε, multilayer formation (“crowding”), excess charge overcompensation in the next layers (“overscreening”), diffusive layer profiles, charge transfer reactions between the metal and adsorbed ions, ion polarization effects at the interface, etc.46 are neglected.30 The resulting small Δφanodic (∼1/Canodic) consequently leads to a larger value for Δφcathodic (=Uapplied − Δφanodic). Note again that the simplified relation Δφ ∼ d only holds because the contact areas of the anode and cathode are identical in our cell setup. For neat [C8C1Im][Tf2N], the size of [Tf2N]− is similar to that of [C8C1Im]+, and therefore the PS at the cathode and that at the anode side are similar for this IL.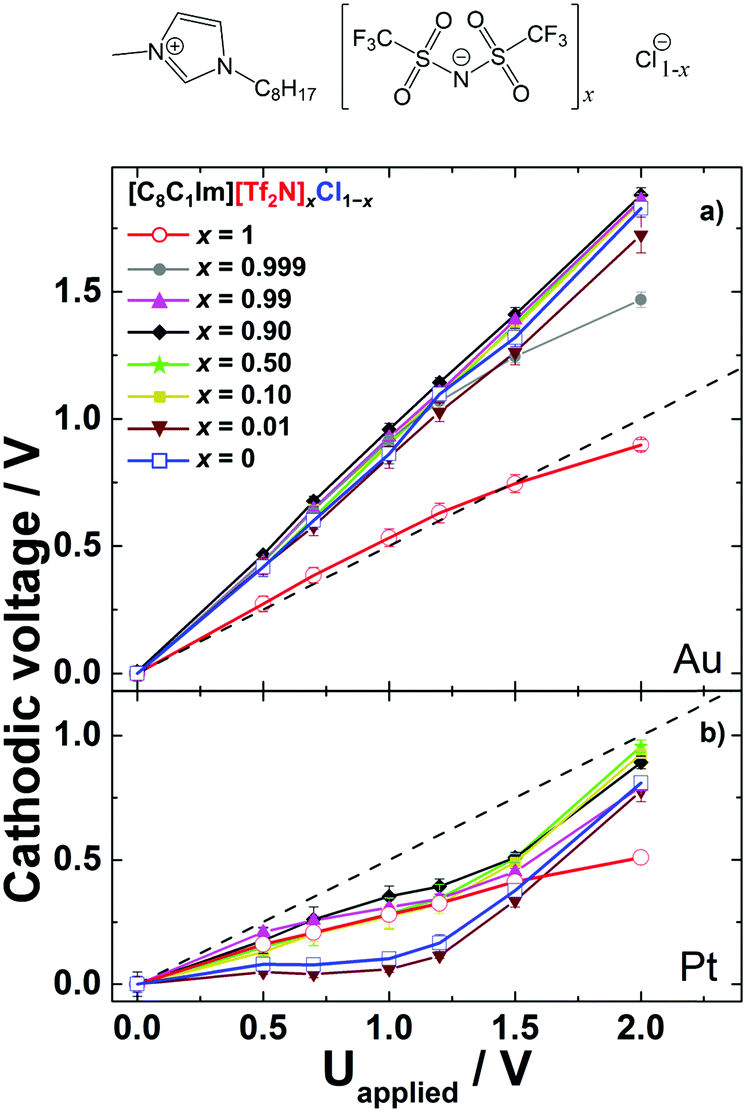 | ||
| Fig. 2 Cathodic voltage of various [C8C1Im][Tf2N]xCl1−x mixtures versus applied voltage with x = 1, 0.999, 0.99, 0.9, 0.5, 0.1, 0.01 and 0, for (a) Pt/Pt electrodes and (b) Au/Au electrodes. The ideal lines for equal potential drops at the anode and cathode interfaces are indicated as dashed straight lines with a slope of +0.5 V/V. The error bars show standard deviations of the measurements. The data for the neat ILs (x = 1 and 0; open symbols) are taken from ref. 45. At the top of the figure, the structure of the ions of the IL mixture is depicted. | ||
To investigate which ions are adsorbed on the electrode in IL mixtures, we investigated a number of different [C8C1Im][Tf2N]xCl1−x mixtures, with x = 0.01, 0.1, 0.5, 0.9, 0.99 and 0.999. Interestingly, up to a +1.5 V applied voltage, the Δφcathodic curves (and consequently, the Δφanodic curves) of all mixtures are virtually identical to that of neat [C8C1Im]Cl (Fig. 2a), even down to only a 0.1 mol% content of [C8C1Im]Cl. This implies that the compositions of the IL/cathode and IL/anode interfaces in the mixtures are the same as for neat [C8C1Im]Cl. In other words, the Cl− anions are extremely interface-active and thus, in all [C8C1Im][Tf2N]xCl1−x mixtures studied here, [Tf2N]− obviously is completely replaced from the electrode interfaces by Cl− at zero applied voltage and up to an applied voltage of 1.5 V. Since the electrode–electrolyte interface region is only a very small fraction (about 10−6) of the total electrolyte volume, even a 0.1 mol% bulk content of [C8C1Im]Cl is enough to replace all [Tf2N]− anions with Cl− at the electrodes. This anion replacement initially occurs without a voltage and is maintained up to a 1.5 V applied cell voltage. We attribute the replacement of [Tf2N]− by Cl− at the Au electrode to a strong attraction (specific adsorption) of Cl− to the Au surface. Preferential enrichment of Cl− anions on Au has previously been reported in electrochemical experiments.47,48 Our arguments imply that up to an applied voltage of 1.5 V the PS at the Au anode occurs mainly within the first monolayer in contact with the electrode, where the [C8C1Im]+ ions are replaced by Cl− at the anode, and the Cl− ions are replaced by [C8C1Im]+ at the cathode.
The pronounced dominance of the [C8C1Im]Cl-like behavior even for x = 0.99 and 0.999 further indicates a very strong energetic driving force for the enrichment of Cl− at the electrodes. The fact that for x = 0.999 the behavior starts to deviate for applied voltages above 1.5 V is tentatively attributed to the very low concentration of Cl− (0.1 mol% Cl−) in the IL mixture, as compared to [Tf2N]−, which starts to play a role at higher voltages, where more anions are required to screen the positive electrode charge.
Mixtures of [C8C1Im][Tf2N] and [C8C1Im]Cl on Pt electrodes
To study the role of the chemical nature of the electrodes, we investigated the same neat ILs as above, [C8C1Im][Tf2N] and [C8C1Im]Cl, and their mixtures [C8C1Im][Tf2N]xCl1−x on Pt electrodes; see Fig. 2b. For applied cell voltages up to 2 V, the Δφcathodic curves for both neat ILs, [C8C1Im][Tf2N] (x = 1, red circles) and [C8C1Im]Cl (x = 0, blue squares), are located below the ideal line, with initial slopes of 0.27 and 0.12 for [C8C1Im][Tf2N] and [C8C1Im]Cl, respectively. While for [C8C1Im][Tf2N] the slope remains unchanged up to 2 V, for [C8C1Im]Cl it strongly increases above ∼1.2 V to a value of 0.81. The small initial slopes below 0.5 imply that the EDL capacitances at the platinum cathode Ccathodic are larger than Canodic for both neat ILs. In our previous work, we tentatively attributed this behavior to the strong chemical interaction of the imidazolium cation with the platinum surface, most likely between the imidazolium ring π orbital and the Pt 5d orbital, which is absent on gold.45 In the case of neat [C8C1Im][Tf2N], the slope of 0.27 thus reflects the smaller distance dcathodic (and thus larger Ccathodic) of the flat-lying imidazolium rings of the additional [C8C1Im]+ counterions at the charged cathode compared to the more bulky [Tf2N]− counterions at the anode. In the case of neat [C8C1Im]Cl with its very small anion, the observed Δφcathodic slope of 0.12 (that is, Ccathodic ≫ Canodic) cannot be simply explained by differences in d, since the size of the Cl− counterions and the vertical extension of a flat-lying imidazolium ring are quite comparable. The situation is complicated by the fact that at applied cell voltages above 1.2 V, the slope of the Δφcathodic curve of [C8C1Im]Cl considerably changes from 0.12 to close to 0.81, similar to the case for [C8C1Im]Cl on gold over the whole cell voltage window. Thus, our simplified description is not sufficient anymore to explain the observed effects. A detailed understanding requires density functional theory calculations describing the chemical interactions of the IL with the metal surface accurately. These are, however, out of the scope of this work. Nevertheless and most importantly, the very characteristic and different behavior for the two ILs again can serve as a fingerprint for characterizing the IL/electrode composition of the corresponding IL mixtures.For the Pt electrode, Δφcathodic curves were measured for [C8C1Im][Tf2N]xCl1−x mixtures with molar fractions of x = 0.99, 0.9, 0.5, 0.1 and 0.01. Interestingly, from x = 1 down to x = 0.1 (90 mol% Cl−), the behavior at low voltages (up to 1.2 V) shows similar Δφcathodic values to neat [C8C1Im][Tf2N], which reflects that, from zero applied potential up to 1.2 V, the [Tf2N]− ion is selectively enriched at the Pt electrode, as compared to Cl−. The reason for the behavior could be a chemical interaction of the SO2 groups of [Tf2N]− with the platinum electrode, which results in specific adsorption of this anion. Only at x = 0.01 is a behavior similar to that of [C8C1Im]Cl observed, indicating that, at very high Cl− concentrations, Cl− starts to dominate the interface behavior.
Again, for applied voltages above 1.2 V, the PS behaviors of the mixtures with x = 0.99, 0.9, 0.5 and 0.1 divert from that of the neat [C8C1Im][Tf2N] and increase with a similar slope (0.78) to the Δφcathodic curve of the neat [C8C1Im]Cl in this applied voltage range. One possible explanation here is that more negative counterions are needed to screen the applied voltage and the packing of the smaller Cl− anions at the IL–anode interface is apparently easier in this applied voltage range; again, electrostatics seems to win over specific chemical interactions, and thus a similar PS behavior to that of the neat [C8C1Im]Cl is observed. An alternative explanation for the behavior of the [C8C1Im][Tf2N]xCl1−x mixtures would be a multilayer EDL model. Up to an applied potential of 1.2 V, also in the mixtures (x = 0.99, 0.9, 0.5 and 0.1) only [Tf2N]− accumulates in the proximate layer at the IL/Pt interface, due to the strong chemical interaction between [Tf2N]− and the Pt electrode.30,49–52 Above 1.2 V, the first layer of the EDL at the anode may be saturated and no more [Tf2N]− ions can approach it. At this point, a multilayer EDL might be required to screen higher voltages. In this multilayer, no specific chemical interaction of the electrode and counterion is possible; therefore Cl− can contribute to the PS on the anode, and thus Δφcathodic increases with a similar slope to that of the neat [C8C1Im]Cl. In contrast to the Pt electrode setup, the [C8C1Im][Tf2N]xCl1−x mixtures on Au do not show the transition behavior in PS at 1.2 V as reported in Fig. 2a. On Au, Cl− is a dominant counterion and the surface number density of Cl− required to saturate the first layer is larger than that of [Tf2N]− on Pt due to the smaller ion size of Cl−. Therefore, the PS occurs within the first layer at the anode EDL solely due to Cl− till 2 V and a contribution from multilayers to PS apparently does not play a role on Au.
III. Conclusion
In this study, we have demonstrated the interfacial enrichment of specific anions in ionic liquid mixtures through a fingerprint-type behavior of potential screening at polarized ionic liquid/electrode interfaces by using in situ XPS. We investigated mixtures of [C8C1Im][Tf2N] and [C8C1Im]Cl and compared their behavior on gold and platinum electrodes. For the Au electrodes, the IL/electrode interface is dominated by the Cl− anions even at a very low concentration of 0.1 mol% [C8C1Im]Cl. This selective enrichment occurs at zero applied voltage and is maintained up to 2 V. In contrast, for the Pt electrode, the IL/electrode interface is enriched with the [Tf2N]− anions up to 90 mol% [C8C1Im]Cl, for applied voltages from zero to ∼1.5 V. These examples demonstrate that the interfacial concentration of counterions at the IL mixture/electrode interfaces is determined by the interaction between the IL and electrode, not by the bulk ratio of the IL mixture.Our results of the enrichment of minority species at the electrode interfaces imply that small concentrations of an added IL or contamination, e.g., remains from the synthesis process, can dominate the entire situation at charged interfaces. This effect applies to various interfacial or electrochemical systems. For example, when tuning the properties of supercapacitors by adapting the stoichiometry of the IL mixture, small concentrations of an added IL can be sufficient to modulate the entire interface, if the added one has a stronger interaction with the electrode. Also, the very small amount of impurities can be remarkably enriched at the electrode interface and change the structures and properties of the EDL. We expect the consequences to be of similar importance to the known surface-enrichment effects at gas/IL or vacuum IL interfaces.43,44 Therefore, the purity of the IL should be of higher concern in electrochemical applications than in bulk IL applications.
IV. Experimental methods
The PS measurements were performed in a UHV-compatible electrochemical cell developed by our group.45 The cell consists of two identical metallic electrode wires (electrically isolated by polytetrafluoroethene spacers) and a molybdenum sample holder, in which the IL is filled. The distance between the wires was 5 mm. The electrodes are connected to the potentiostat (Keithley 2450) through two contact springs located at the head of the manipulator. Pure Pt (99.99%) and Au (99.995%) wires with a diameter of 0.30 and 0.25 mm, respectively, were purchased from MaTeck. They were cleaned by flame-annealing. The synthesis of ultraclean [C8C1Im][Tf2N] and [C8C1Im]Cl was carried out as described in a previous publication.53 All the IL mixtures were prepared by mixing two ILs with the required mass ratio to achieve the reported molar concentration without the use of co-solvents. The mixtures were sonicated for 1 hour at around 60 °C for homogenous mixing. The molar ratio of the two components of the mixtures was confirmed by XPS quantitative analysis (Tables S1–S15 in the ESI†). All the electrodes were carefully placed in the electrochemical cells in order to have identical contact areas (±5%) with the IL mixtures.To measure the potential screening at the electrodes, XPS spectra were measured in our DASSA (dual analyzer system for surface analysis) setup using a monochromatic Al Kα source and a hemispherical analyzer (detection of photoelectrons in normal emission) with an overall energy resolution of 0.4 eV.54 Data were collected in the F 1s or N 1s regions, due to the strong signal intensities of the corresponding peaks. During XPS, one of the electrodes was grounded through a corresponding connector at the front panel of the source meter. All XPS measurements were performed under non-faradaic conditions, which was confirmed by chronoamperometry (Fig. S1 in the ESI†) using a Keithley 2450 source meter. In addition, we can rule out the production of volatile species such as Cl2 formed by Cl− oxidation at the anode side in our two-electrode electrochemical cell within the applied voltage window from 0–2 V since the resulting formation of Cl2 should lead to a measurable pressure increase in the UHV chamber (base pressure of 5 × 10−10 mbar), which was not observed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. G. and H.-P. S. thank the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme for financial support, in the context of the Advanced Investigator Grant “ILID” to H.-P. S. (Grant Agreement No. 693398-ILID). S. S. thanks the Alexander von Humboldt Foundation for a research fellowship.References
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- F. Wang, X. Wu, X. Yuan, Z. Liu, Y. Zhang, L. Fu, Y. Zhu, Q. Zhou, Y. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816–6854 RSC.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- K. L. Van Aken, M. Beidaghi and Y. Gogotsi, Angew. Chem., Int. Ed., 2015, 54, 4806–4809 CrossRef CAS PubMed.
- X. Wang, A. Y. Mehandzhiyski, B. Arstad, K. L. Van Aken, T. S. Mathis, A. Gallegos, Z. Tian, D. Ren, E. Sheridan, B. A. Grimes, D. E. Jiang, J. Wu, Y. Gogotsi and D. Chen, J. Am. Chem. Soc., 2017, 139, 18681–18687 CrossRef CAS PubMed.
- M. S. Miran, T. Yasuda, M. A. Susan, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2014, 118, 27631–27639 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- V. Lockett, M. Horne, R. Sedev, T. Rodopoulos and J. Ralston, Phys. Chem. Chem. Phys., 2010, 12, 12499–12512 RSC.
- R. Costa, C. M. Pereira and F. Silva, Phys. Chem. Chem. Phys., 2010, 12, 11125–11132 RSC.
- V. Lockett, R. Sedev, J. Ralston, M. Horne and T. Rodopoulos, J. Phys. Chem. C, 2008, 112, 7486–7495 CrossRef CAS.
- M. M. Islam, M. T. Alam and T. Ohsaka, J. Phys. Chem. C, 2008, 112, 16568–16574 CrossRef CAS.
- B. Roling, M. Drüschler and B. Huber, Faraday Discuss., 2012, 154, 303–311 RSC.
- A. A. Kornyshev, J. Phys. Chem. B, 2007, 111, 5545–5557 CrossRef CAS PubMed.
- S. Lamperski, C. W. Outhwaite and L. B. Bhuiyan, J. Phys. Chem. B, 2009, 113, 8925–8929 CrossRef CAS PubMed.
- M. V. Fedorov, N. Georgi and A. A. Kornyshev, Electrochem. Commun., 2010, 12, 296–299 CrossRef CAS.
- G. Feng and P. T. Cummings, J. Phys. Chem. Lett., 2011, 2, 2859–2864 CrossRef CAS.
- Y. Shim and H. J. Kim, ACS Nano, 2010, 4, 2345–2355 CrossRef CAS PubMed.
- P. Wu, J. Huang, V. Meunier, B. G. Sumpter and R. Qiao, ACS Nano, 2011, 5, 9044–9051 CrossRef CAS PubMed.
- M. V. Fedorov and A. A. Kornyshev, J. Phys. Chem. B, 2008, 112, 11868–11872 CrossRef CAS PubMed.
- Y. Lauw, M. D. Horne, T. Rodopoulos, A. Nelson and F. A. Leermakers, J. Phys. Chem. B, 2010, 114, 11149–11154 CrossRef CAS PubMed.
- X. Zhang, Y. X. Zhong, J. W. Yan, Y. Z. Su, M. Zhang and B. W. Mao, Chem. Commun., 2012, 48, 582–584 RSC.
- T. Cui, A. Lahiri, T. Carstens, N. Borisenko, G. Pulletikurthi, C. Kuhl and F. Endres, J. Phys. Chem. C, 2016, 120, 9341–9349 CrossRef CAS.
- Y. X. Zhong, J. W. Yan, M. G. Li, X. Zhang, D. W. He and B. W. Mao, J. Am. Chem. Soc., 2014, 136, 14682–14685 CrossRef CAS PubMed.
- T. A. Petach, A. Mehta, R. Marks, B. Johnson, M. F. Toney and D. Goldhaber-Gordon, ACS Nano, 2016, 10, 4565–4569 CrossRef CAS PubMed.
- M. Mezger, H. Schroder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schoder, V. Honkimaki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424–428 CrossRef CAS PubMed.
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Phys. Rev. Lett., 2011, 106, 046102 CrossRef PubMed.
- C. Noh and Y. Jung, Phys. Chem. Chem. Phys., 2019, 21, 6790–6800 RSC.
- J. Vatamanu, O. Borodin and G. D. Smith, J. Am. Chem. Soc., 2010, 132, 14825–14833 CrossRef CAS PubMed.
- M. T. Camci, P. Aydogan, B. Ulgut, C. Kocabas and S. Suzer, Phys. Chem. Chem. Phys., 2016, 18, 28434–28440 RSC.
- S. Baldelli, Acc. Chem. Res., 2008, 41, 421–431 CrossRef CAS PubMed.
- O. Brummel, F. Faisal, T. Bauer, K. Pohako-Esko, P. Wasserscheid and J. Libuda, Electrochim. Acta, 2016, 188, 825–836 CrossRef CAS.
- V. O. Santos, Jr., M. B. Alves, M. S. Carvalho, P. A. Suarez and J. C. Rubim, J. Phys. Chem. B, 2006, 110, 20379–20385 CrossRef PubMed.
- R. Wen, B. Rahn and O. M. Magnussen, Angew. Chem., Int. Ed., 2015, 54, 6062–6066 CrossRef CAS PubMed.
- Y. Z. Su, Y. C. Fu, J. W. Yan, Z. B. Chen and B. W. Mao, Angew. Chem., Int. Ed., 2009, 48, 5148–5151 CrossRef CAS PubMed.
- P. Wang, Y. Li, L. Wang, J. Kłos, Z. Peng, N. Kim, H. Bluhm, K. Gaskell, P. Liu and S. B. Lee, EcoMat, 2020, 2(2), e12023 CrossRef CAS.
- C. Pinilla, M. G. Del Popolo, J. Kohanoff and R. M. Lynden-Bell, J. Phys. Chem. B, 2007, 111, 4877–4884 CrossRef CAS PubMed.
- X. Si, S. Li, Y. Wang, S. Ye and T. Yan, ChemPhysChem, 2012, 13, 1671–1676 CrossRef CAS PubMed.
- Q. Zhang, Y. N. Han, Y. L. Wang, S. H. Ye and T. Y. Yan, Electrochem. Commun., 2014, 38, 44–46 CrossRef CAS.
- A. Fang and A. Smolyanitsky, J. Phys. Chem. C, 2019, 123, 1610–1618 CrossRef CAS.
- M. T. Alam, M. M. Islam, T. Okajima and T. Ohsaka, J. Phys. Chem. C, 2009, 113, 6596–6601 CrossRef CAS.
- M. Lexow, F. Maier and H.-P. Steinrück, Adv. Phys.: X, 2020, 5, 1761266 CAS.
- M. Lexow, T. Talwar, B. S. J. Heller, B. May, R. G. Bhuin, F. Maier and H.-P. Steinrück, Phys. Chem. Chem. Phys., 2018, 20, 12929–12938 RSC.
- M. Lexow, B. S. J. Heller, G. Partl, R. G. Bhuin, F. Maier and H.-P. Steinrück, Langmuir, 2019, 35, 398–405 CrossRef CAS PubMed.
- M. Lexow, B. S. J. Heller, F. Maier and H.-P. Steinrück, ChemPhysChem, 2018, 19, 2978–2984 CrossRef CAS PubMed.
- F. Greco, S. Shin, F. J. Williams, B. S. J. Heller, F. Maier and H.-P. Steinrück, ChemistryOpen, 2019, 8, 1365–1368 CrossRef CAS PubMed.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- R. Hayes, N. Borisenko, B. Corr, G. B. Webber, F. Endres and R. Atkin, Chem. Commun., 2012, 48, 10246–10248 RSC.
- Z. C. Shi and J. Lipkowski, J. Electroanal. Chem., 1996, 403, 225–239 CrossRef.
- A. Ejigu and D. A. Walsh, J. Phys. Chem. C, 2014, 118, 7414–7422 CrossRef CAS.
- M. Chu, M. Miller and P. Dutta, ACS Cent. Sci., 2016, 2, 175–180 CrossRef CAS PubMed.
- S. Baldelli, J. Phys. Chem. Lett., 2013, 4, 244–252 CrossRef CAS PubMed.
- N. Zhang, X.-R. Wang, Y.-X. Yuan, H.-F. Wang, M.-M. Xu, Z.-G. Ren, J.-L. Yao and R.-A. Gu, J. Electroanal. Chem., 2015, 751, 137–143 CrossRef CAS.
- C. Kolbeck, T. Cremer, K. R. Lovelock, N. Paape, P. S. Schulz, P. Wasserscheid, F. Maier and H.-P. Steinrück, J. Phys. Chem. B, 2009, 113, 8682–8688 CrossRef CAS PubMed.
- I. Niedermaier, C. Kolbeck, H.-P. Steinrück and F. Maier, Rev. Sci. Instrum., 2016, 87, 045105 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp04811a |
| ‡ Contributed equally to this work. |
| This journal is © the Owner Societies 2021 |