
Selective saturation of step-edges as a tool to control the growth of molecular fibres†‡
Maximilian
Dreher
 * and
Gregor
Witte
* and
Gregor
Witte
Molekulare Festkörperphysik, Philipps-University Marburg, D-35032 Marburg, Germany. E-mail: maximilian.dreher@physik.uni-marburg.de
First published on 18th January 2021
Abstract
The concept of bottom-up self-organisation has become a promising alternative for structuring molecular materials, which are hardly accessible by conventional top-down approaches such as lithography due to their limited chemical robustness. While these materials often tend to form three-dimensional, crystalline islands or fibres upon film growth, the size and orientation of such fibres are mainly governed by appropriate preparation conditions as well as microscopic interactions at the interface with the supporting surface. Substrate surface defects such as vacancies or step-edges, which cannot be completely ruled out on real surfaces on the mesoscopic scale, can act as preferred nucleation sites for molecules that leads to parasitic film growth competing with their intrinsic alignment prevailing on an ideal surface. In the present study, we demonstrate for the case of para-quaterphenyl (p-4P) that the presence of azimuthally disordered, fibres on Ag(111) surfaces can be understood as a superposition of step-mediated nucleation and the intrinsic epitaxial fibre growth on ideal surfaces. We validate the concept by purposely exposing the silver substrates briefly to oxygen or even ambient air to passivate the more reactive step-sites, which hampers subsequently grown molecular films to nucleate at these step-edges. This yields a truly epitaxial alignment as well as an enlargement of the fibres present on the whole sample.
Introduction
In modern nanotechnology, self-organisation has become an important tool to fabricate complex, but well-defined mesoscopic architectures that consist of smaller building units such as atoms, molecules or nanoparticles.1–5 Especially, for the emerging fields of organic electronics and molecular nanostructures, top-down structuring strategies such as lithography cannot be used due to a lack of chemical robustness of molecular materials. Even in the case of inorganic materials, the advancing miniaturisation soon will reach structure sizes, which will hardly be accessible using suitable lithography techniques,6,7 therefore requiring other structuring methods. Bottom-up approaches offer an interesting alternative route for micro-structuring future device geometries by precisely controlling the preparation conditions. However, not only external growth parameters have an influence on the resulting morphology, but also the intrinsic interface properties between the supporting substrate and the adlayer play a decisive role.8 A frequently used approach is to use surfaces with high symmetry and to take advantage of an epitaxial relationship between the substrate and the deposited adlayer, which in many cases also leads to a high symmetrical film morphology. Although modern surface science techniques are able to produce highly ordered (substrate) surfaces, defects such as vacancies or steps can never be fully excluded on a large scale due to their entropic contributions. The latter defects cause significant limitations of the diffusion length of adsorbates.9 Moreover, they can act as active adsorption sites creating nucleation centres, which compete with the intrinsic epitaxial driven growth on the idealised, defect-free surface. Across different material classes, defects are sometimes also actively used to induce a specific growth behaviour,10–19 while in general they cause a parasitic film growth, reducing long-range epitaxial ordering and yielding rather undefined interfaces.20–23However, even on idealised substrate surfaces, organic adlayers in particular exhibit pronounced dewetting, which leads to distinct island or fibre formation.24 Especially the latter attracted large interest as model systems for wave guiding applications.25 A prototypical system is the molecular class of para-phenylenes, in which an aggregation in such molecular nanofibres on alkali halides was reported firstly.26 While on insulating substrates such as KCl or (muscovite) mica, these fibres are well aligned along high symmetry azimuth directions, on metal substrates even on single crystalline densely packed surfaces, these fibres do not show such a defined azimuthal alignment as the substrate geometry would suggest, but instead are largely affected by surface steps.27,28 For the growth of para-quaterphenyl films on Au(111), Müllegger et al. have shown that an epitaxial seed layer is formed and the subsequent multilayers form crystalline fibers with preferred azimuthal orientations. However, the fibre alignment is not very perfect because other fiber orientations also occur.28 Very similar film morphologies and the formation of distinct fibres are also found for other rod-like molecules.29–34 The issue of forming fibres with non-discrete azimuthal orientations on metal supports was previously also observed for the case of dinaphthothienothiophene (DNTT) thin films on Ag(111) surfaces. In addition to epitaxial fibres, another set of fibre orientations was identified that could be explained by parasitic fibres whose nucleation and growth is mediated by surface steps. By first exposing the metal substrate briefly to oxygen, a selective saturation of the active step sites was achieved, which successfully suppressed a step-mediated nucleation and the formation of parasitic fibres.21
Here, we examine whether this strategy is also applicable to the alignment of p-4P fibres formed upon growth on Ag(111). Since on metal single crystals (monatomic) surface steps often appear curved, but are actually polylines that consist of short straight segments oriented along high symmetry directions,35,36 we used instead Ag(111) films epitaxially grown on mica, because they reveal long and straight step edges which simplifies the analysis. In the present work, we combined scanning tunnelling microscopy (STM) with X-ray diffraction (XRD) and optical polarisation microscopy to analyse the formation and azimuthal alignment of para-quaterphenyl fibres on bare and oxygen-exposed Ag(111) surfaces. We demonstrate that short exposure to oxygen or ambient air efficiently saturates steps on the silver surface and thereby suppresses a step-mediated nucleation leading to well aligned fibres according to the 6-fold symmetry of the Ag(111) surface without any additional parasitic fibre orientations.
Experimental
Sample preparation
The presently used Ag(111) samples consist of 100–200 nm thick silver films that were epitaxially grown by physical vapour deposition under high vacuum conditions onto freshly cleaved and carefully degassed mica substrates.37 After loading into the ultra-high-vacuum (UHV) system, the silver substrates were cleaned in situ by several cycles of Ar+-sputtering (Eion ≈ 800 eV) and annealing (T ≈ 740 K) until a sharp low energy electron diffraction (LEED) pattern (four-grid LEED, Omicron) with low background signal was observed. Corresponding scanning tunnelling microscopy (STM) measurements confirm an excellent long-range ordering and reveal atomically flat terraces extending more than 100 nm. Compared to Ag(111) single crystals, the Ag(111)/mica films have the advantage of revealing very extended atomically flat terraces as well as long straight step edges running primarily along the 〈1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0〉Ag directions (cf. Fig. S1 in the ESI‡). The para-quaterphenyl (p-4P, Sigma-Aldrich, purity: 98%) thin films were prepared under UHV conditions by organic molecular beam deposition (OMBD) from aluminium crucibles of a resistively heated Knudsen cell (T ≈ 450 K). The deposition rate (typical R ≈ 4 Å min−1) was monitored using a quartz crystal microbalance while the Ag substrates were kept at elevated temperatures (see details in the main text) during deposition.
0〉Ag directions (cf. Fig. S1 in the ESI‡). The para-quaterphenyl (p-4P, Sigma-Aldrich, purity: 98%) thin films were prepared under UHV conditions by organic molecular beam deposition (OMBD) from aluminium crucibles of a resistively heated Knudsen cell (T ≈ 450 K). The deposition rate (typical R ≈ 4 Å min−1) was monitored using a quartz crystal microbalance while the Ag substrates were kept at elevated temperatures (see details in the main text) during deposition.
Film characterisation
The crystalline phase and orientation of the p-4P films were analysed by X-ray diffraction measurements (XRD) using a Bruker D8 Discover diffractometer with monochromatised Kα-radiation (λ = 1.542 Å) and a sensitive LynxEye silicon strip detector. The setup was also used for in-plane XRD measurements of the silver substrate to identify their high symmetry azimuth directions. Complementary information on the Ag(111) surface morphology and the initial stage of p-4P film growth (i.e. the seed-layer structure) was obtained in situ by STM (Omicron VT STM XA) operated in constant current mode (typically 300 pA) with etched tungsten tips. Information about the size and azimuthal orientation of crystalline p-4P multilayer fibres was obtained by optical polarisation microscopy (Nikon Eclipse LV-FMA). For this purpose, a series of optical micrographs were acquired in epi-illumination using linearly polarised white light and rotating the angle of the polarisation plane with respect to the 〈11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 〉 high symmetry azimuth directions of the silver substrate, ϕ, in steps of 5°. Due to the optical anisotropy of the p-4P molecules and their anisotropic packing in the crystalline phase, also crystalline domains of the films exhibit an anisotropic absorption. By pixelwise evaluation of the polarisation angle with largest reflectivity, the azimuthal distribution of the molecules inside the fibres can be derived, as described in ref. 38. Since phenylenes exhibit a distinctly anisotropic photo-luminescence characteristics, we have also analysed the azimuthal fluorescence distribution of the fibres by illuminating the films with unpolarised UV-light using a SOLA light source (Lumencor) in combination with a fluorescence filter (Nikon, EX 377/50, DM 409, LP 415 yielding an exciting wavelengths of λexc ≈ 360–380 nm) and acquiring azimuthal dependent polarisation-resolved photo-luminescence (PL) micrographs (λemi > 415 nm) by rotating the polariser in steps of 5°.
〉 high symmetry azimuth directions of the silver substrate, ϕ, in steps of 5°. Due to the optical anisotropy of the p-4P molecules and their anisotropic packing in the crystalline phase, also crystalline domains of the films exhibit an anisotropic absorption. By pixelwise evaluation of the polarisation angle with largest reflectivity, the azimuthal distribution of the molecules inside the fibres can be derived, as described in ref. 38. Since phenylenes exhibit a distinctly anisotropic photo-luminescence characteristics, we have also analysed the azimuthal fluorescence distribution of the fibres by illuminating the films with unpolarised UV-light using a SOLA light source (Lumencor) in combination with a fluorescence filter (Nikon, EX 377/50, DM 409, LP 415 yielding an exciting wavelengths of λexc ≈ 360–380 nm) and acquiring azimuthal dependent polarisation-resolved photo-luminescence (PL) micrographs (λemi > 415 nm) by rotating the polariser in steps of 5°.
Results and discussion
Fig. 1a depicts an optical micrograph of a p-4P film with a nominal thickness of 30 nm grown at a substrate temperature of 340 K on Ag(111) that reveals the presence of narrow fibres with a length of up to 10 μm. While some fibres seem to be oriented along specific azimuthal directions others appear isotropically distributed in the surface plane. The corresponding specular X-ray diffraction data (cf.Fig. 1b) exhibit beside reflections from the silver substrate and the mica support the two p-4P related (201) and (211) reflections. This structural characterisation agrees well with the results obtained before by Müllegger et al. for the growth of p-4P films on Au(111), which can be expected due to the same lattice constant of both noble metals.27,28 Using low current MCP-LEED, they were further able to obtain the structure of the epitaxial p-4P seed layer appearing in several rotational and mirror domains according to the substrate surface symmetry. The molecular arrangements in the identified crystallographic planes are depicted schematically in Fig. 1c and show that in each case the molecular long axes of all molecules are parallel to the silver substrate (and perpendicular to the plane of drawing), while the molecules adopt a herringbone packing in crystalline films. Note that in the gas phase, the phenyl rings of a p-4P molecule adopt a certain dihedral twist around the molecular long axis, while in solid films the molecules are considered flat.39,40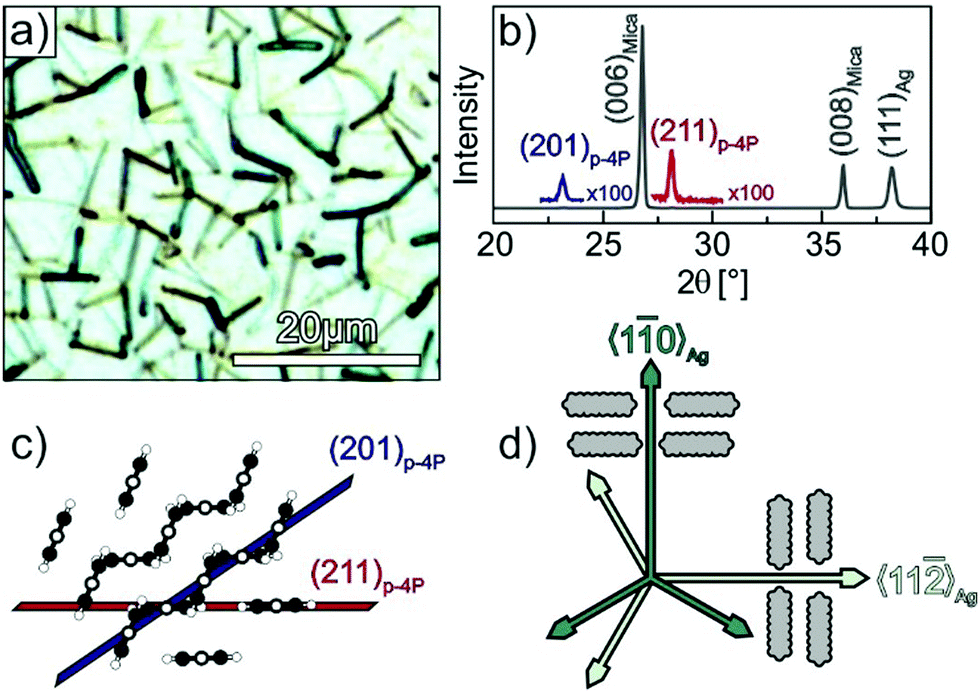 | ||
| Fig. 1 Structural characterisation of p-4P films (d = 30 nm) grown at 340 K on Ag(111)/mica substrates. (a) Optical micrograph and (b) specular X-ray diffraction (Omega-2Theta scan). Panel (c) depicts schematic side views of the p-4P bulk structure where the lattice planes observed in the XRD spectrum are marked. (d) Schematic representation of the azimuthal orientations of the p-4P molecules within monolayer domains, where the long axis of p-4P point either parallel or perpendicular to the 〈10〉Ag direction (proposed by Müllegger et al. for p-4P/Au(111)).27 | ||
Using thick p-4P films, Müllegger et al. performed also in-plane XRD measurements to characterise the azimuthal alignment of the fibres. Interestingly, they found that despite the presence of rotation and mirror domains, the long molecular axes are preferentially oriented along the 〈10〉Ag and 〈11〉Ag high symmetry azimuth directions, which are aligned perpendicular to the fibre directions as schematically depicted in Fig. 1d.27,28
Unfortunately, the low X-ray scattering cross section of the light weighted p-4P molecules hampers detailed in-plane XRD measurements with a laboratory X-ray setup to study the initial stage of fibre formation in thin films. Therefore, we instead have applied polarisation-resolved optical microscopy, taking advantage of the optical anisotropy of p-4P molecules (exhibiting a broad band absorption in the visible region due to excitonic excitations with corresponding transition dipole moment along the molecular long axis41,42) and of the uniaxial packing of their long axis in the crystalline fibres.43 To analyse the azimuthal orientation of molecules and their distribution, a series of optical micrographs were recorded as a function of the azimuthal polarisation direction of the incident linearly polarised light. Here, the azimuthal polarisation angle ϕ is referenced to the 〈11〉Ag azimuth direction, which was identified before on the basis of LEED measurements of the clean Ag(111) surface or in-plane XRD measurements (cf. Fig. S2 in the ESI‡). In a next step, the intensity of each pixel of the micrographs was analysed, and the polarisation angle of maximal reflectivity is displayed in a false colour plot where the colour represents the polarisation angle (as shown in the inset in Fig. 2a). Finally, the frequency of occurrence of maximal pixel intensity as a function of the polarisation angles is determined and represented as a polar diagram.
 | ||
| Fig. 2 (a, c and e) Optical micrographs (in epi-illumination) of p-4P/Ag(111) films with a nominal thickness of 30 nm and a substrate temperature of 340 K during deposition. Before deposition, the Ag/mica substrates were prepared by several sputtering/annealing cycles plus: no further treatment in (a), exposing to 500 L oxygen in (c), bring back to ambient conditions for one minute in (e). On the right half on the images corresponding false colour images are depicted, where colours represent the polarisation angle with maximal pixel intensity. (b, d and f) Corresponding magnified images and polar plots of the angular distribution of the pixels of different polarisation angels. Panel (g) shows the TDM direction of the lowest energetic transition in p-4P together with a top view on the bulk structure of p-4P and their stacking direction inside a “natural” molecular fibre and their alignment with respect to the Ag(111) substrate. | ||
Fig. 2a and b depicts this analysis for a 30 nm p-4P film grown at 340 K on Ag(111). Panel a shows on the left-hand side the optical micrograph while on the right-hand side the corresponding false colours according to the polarisation with maximal fibre reflectivity are superimposed. The magnified micrograph in panel b shows that the individual fibres typically exhibit a homogeneous optical contrast. However, the statistical analysis of the polarisation angle with maximal pixel intensity of the entire micrograph shows a rather broad distribution that reflects significant deviations from preferential azimuthal fibre orientations. To derive the molecular orientation from these data, one has to consider the contrast mechanism. If the E-vector of the incident linearly polarised light is oriented perpendicular to the long molecular axis, no absorption occurs and the pixel has the highest intensity (i.e. maximal reflectivity). The uniform arrangement of the molecular long axes in the crystalline phase of p-4P (cf.Fig. 2g) allows concluding that for E-vectors oriented along the fibres (i.e. perpendicular to the molecular long axis) they appear brightest as they exclude light absorption in the visible range. Although the molecules are uniformly oriented within the individual fibres, the entire sample exhibits a rather isotropic azimuthal distribution of fibre orientations. Thus, despite the excellent long-range order of the Ag(111) surface, this superior ordering seems not to be continued in thicker films as it has been found for other molecular adlayers.44,45 A possible reason for a broad distribution of fibre orientations could be the influence of surface defects such as step edges or kinks, which can cause parasitic nucleation and lead to deviant fibre growth, as shown before for the case of DNTT-fibres on Ag(111).21 In that case, it was found that a step edge mediated growth could be efficiently suppressed by first exposing the sample to oxygen, which adsorbs preferentially at step edges46,47 and thereby suppresses this specific nucleation. In general, steps on metal surfaces are chemically active sites that have greater binding energies of adsorbates and also show increased catalytic activity, which is used in heterogeneous surface reactions.48 With this knowledge in mind, we applied the previously introduced strategy also to the growth of p-4P films. Therefore, we exposed the freshly sputtered and annealed Ag(111) substrates to 500 L (1 L = 1.33 × 10−6 mbar s) of oxygen (Messer Industriegase GmbH, purity 99.998%) before depositing the p-4P films. A direct confirmation of the uptake of oxygen at the step edges is quite difficult because of their overall low density. Considering the large size of atomically flat terraces (cf. Fig. S1, ESI‡) a density of step sites of less than 0.2% is estimated, which is below the detection limit of XPS. A previous study has further shown that the (111) terraces of the silver surface are barely affected by the exposure to oxygen, since even after a dosage of 3000 L still a sharp (1 × 1) LEED pattern was observed.49 However, we found a distinctly modified growth of p-4P fibres on the oxygen treated Ag(111) surfaces. As shown in Fig. 2c, the molecular films consist of more separated straight fibres. The statistical analysis of the polarisation contrast yields a distinct narrowing of the fibre distribution that are oriented preferentially along the 〈10〉Ag, but also along the 〈11〉Ag directions. Significant adsorption of oxygen on the terraces seems unlikely as we still observed a distinct LEED pattern of the Ag(111) surface after exposure to oxygen (cf. Fig. S5a and b in ESI‡). Moreover, an earlier study showed markedly different growth of p-4P on covered metal surfaces, resulting in an upright molecular orientation and a very different film morphology.50 Interestingly, p-4P fibers on oxygen-treated Ag (111) surfaces not only preferentially grow along the 〈10〉Ag directions, but sometimes also form extended domains with rather uniform fiber orientation. This is e.g. visible in Fig. 2c below the scratch, where a majority domain with a dominant fiber orientation (shown in red) occurs. This leads to an almost absence of specific fiber directions at φ = 90° and 270° in the polarisation analysis (cf.Fig. 2d), while they can be found at other parts of the sample. A pronounced formation of large domains with one of the several surface symmetry-permitted azimuthal alignments was previously also found for epitaxial perfluoropentacene films grown on cleavage planes of KCl(100) and NaF(100),51 where strain of the surfaces can be excluded. Instead, we attribute the occurrence of such majority domains to fluctuations upon growth that cause their spontaneous formation, since they occur in a nonreproducible fashion independently of the presence of additional defects such as scratches.
For comparison, we have also tested a simpler and more rigorous procedure to suppress step-mediated nucleation by exposing the UHV-prepared Ag(111) surface to ambient air for one minute and depositing the p-4P films after the sample had been transferred back into the UHV system. As shown in Fig. 2e and f, the corresponding polarisation analysis yields an exclusive orientation of the long molecular axis perpendicular to the 〈10〉Ag directions, which implies that the fibres themselves are oriented along the 〈10〉Ag directions, hence demonstrating the successful suppression of parasitic fibres. A complementary statistical analysis of the azimuthal orientation of the fibres based on their shape (cf. Fig. S3 in the ESI‡) confirms that most fibres are aligned along the 〈10〉Ag directions. Corresponding specular X-ray diffractograms show a dominance of the (211)p-4P reflection, but still a coexistence of the (201)p-4P reflection. Remarkably, also for the vented silver sample, a distinct (1 × 1) LEED pattern was observed (cf. Fig. S5c and d in ESI‡), thus demonstrating that significant parts of the terraces remain uncovered so that their six-fold symmetry still controls the intrinsic epitaxy of the p-4P fibers. A comparison with the micrograph of p-4P fibres obtained on the oxygen exposed Ag(111) sample (cf.Fig. 2c and d) reveals even larger fibres on the vented surface and therefore suggests that the oxygen exposure might be insufficient to fully saturate all surface defects.
To complement our structural film characterisation and to corroborate the explanation that selective passivation of surface steps suppresses the parasitic step mediated growth of p-4P fibres, additional STM measurements were carried out for the initial stage of film growth. Fig. 3 compares the structure of p-4P seed layers formed on bare and oxygen pre-dosed Ag(111). At densely packed steps of the clean silver substrates, which are oriented along the 〈10〉Ag direction, p-4P molecules are oriented with their long axis along the step and thereby affects the azimuthal orientation of the seed layer that leads upon further growth to parasitic fibres oriented perpendicular to the steps (i.e. along 〈11〉Ag). By contrast, when exposing the silver substrate to oxygen before film deposition, such a step-mediated nucleation is suppressed. Only rather disordered regions are formed next to the step edges, while the molecules adopt the “natural” epitaxial orientation on the ideal Ag(111) surface where their long axis is oriented along the 〈11〉Ag azimuth leading to fibres oriented along the 〈10〉Ag direction.
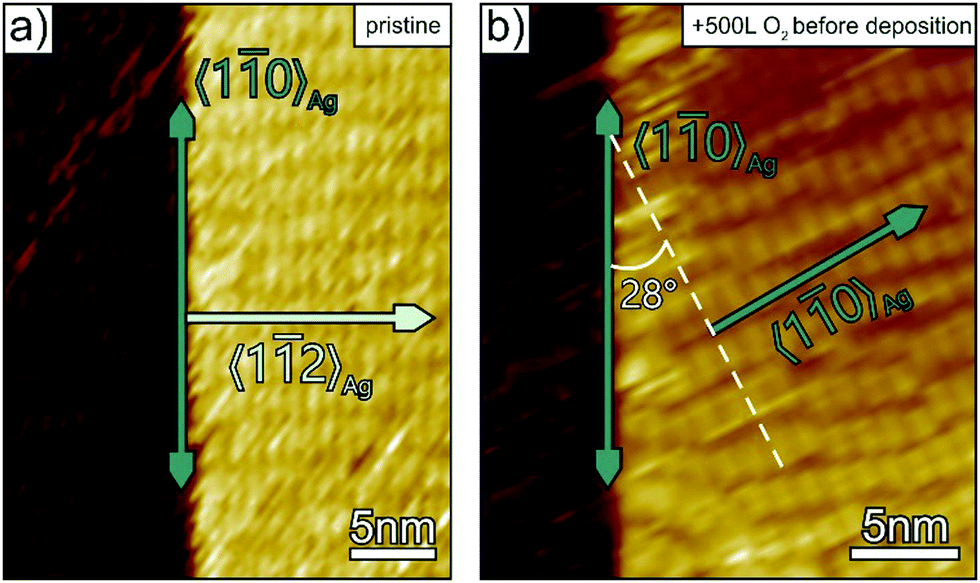 | ||
| Fig. 3 STM micrographs of a p-4P monolayer deposited on (a) pristine and (b) oxygen pre-dosed (500 L) Ag(111)/mica substrates. To reduce admolecule diffusion, the STM measurements were performed at 110 K. Both images exhibit a densely packed silver step oriented along the 〈10〉Ag direction. (Ubias = 1.24 V and It = 300 pA). | ||
Apparently, the presence of surface steps is a critical factor for the domain size of molecular adlayers. In fact, large domains of unidirectional molecular orientation with extensions of several 100 μm are found on cleavage planes of alkali halides or transition metal dichalcogenides, which have a particular lower density of step edges.23,51,52 Another important aspect for the formation of large fibres is the diffusivity of admolecules upon growth. In the case of the somewhat larger oligophenylene p-6P, characteristic fibres with a length of more than 200 μm were observed when growing on mica at 350 K.25 Since such fibre lengths significantly exceed the length of the present p-4P fibres obtained when growing at 340 K (about 10–20 μm, cf.Fig. 2), we have also grown fibres at elevated substrate temperature to examine whether the suppression of step edge nucleation can also be combined with enhanced diffusivity. Fig. 4a and b depicts optical micrographs of such a p-4P film that was deposited at an elevated temperature of 355 K onto an Ag(111) surface that had been pre-dosed by 500 L oxygen. Despite the same nominal thickness of the film compared to the ones depicted in Fig. 2 (grown at 340 K), the fibres are substantially larger with an extension up to 250 μm and reveal shorter side branches at specific angles. As demonstrated before, the corresponding false colour plot in Fig. 4b shows again that each individual fibre consists of molecules with the same azimuthal orientation. Such a pronounced dendritic (so called skeleton) morphology with discrete orientations of side branches is characteristic for the growth of highly crystalline organic films that has been observed before for tetracene53 and perylene54 and is attributed to diffusion limited growth kinetics. The statistical analysis reveals the same trend of a preferential alignment of the fibres along the 〈10〉Ag direction. Complementarily, we prepared also a p-4P film with the same nominal thickness at the same substrate temperature of 355 K, but on a silver substrate, which was stored for three hours in the load lock chamber filled with atmospheric air up to a pressure of 10−6 mbar (yielding a total dosage of 8120 L) before the film growth. The optical micrograph in Fig. 4c also features significant longer fibres. While at first glance some of the fibres seem to be bent, the corresponding false colour plot, which visualises the molecular orientation inside the fibres, reveals them to consist of several straight shorter fibres that are merged together at their ends (cf. white circle in Fig. 4c). The statistical polarisation analysis in Fig. 4d again exhibits a clear preference of the “natural” fibre orientation along the 〈10〉Ag direction. We note that, in contrast to the controlled exposure of the silver sample to pure oxygen, an uptake of other molecules such as water or small hydrocarbons cannot be excluded when exposed to ambient air. In fact, there are several examples of small hydrocarbons that preferentially adsorb on step edges and cause selective passivation.55 Quite inert terraces are an important prerequisite for the demonstrated suppression of parasitic fiber directions, in order to prevent their coating upon small exposures, as this would lead to an adsorption of p-4P molecules in an upright orientation and also result in a significantly changed film morphology.50
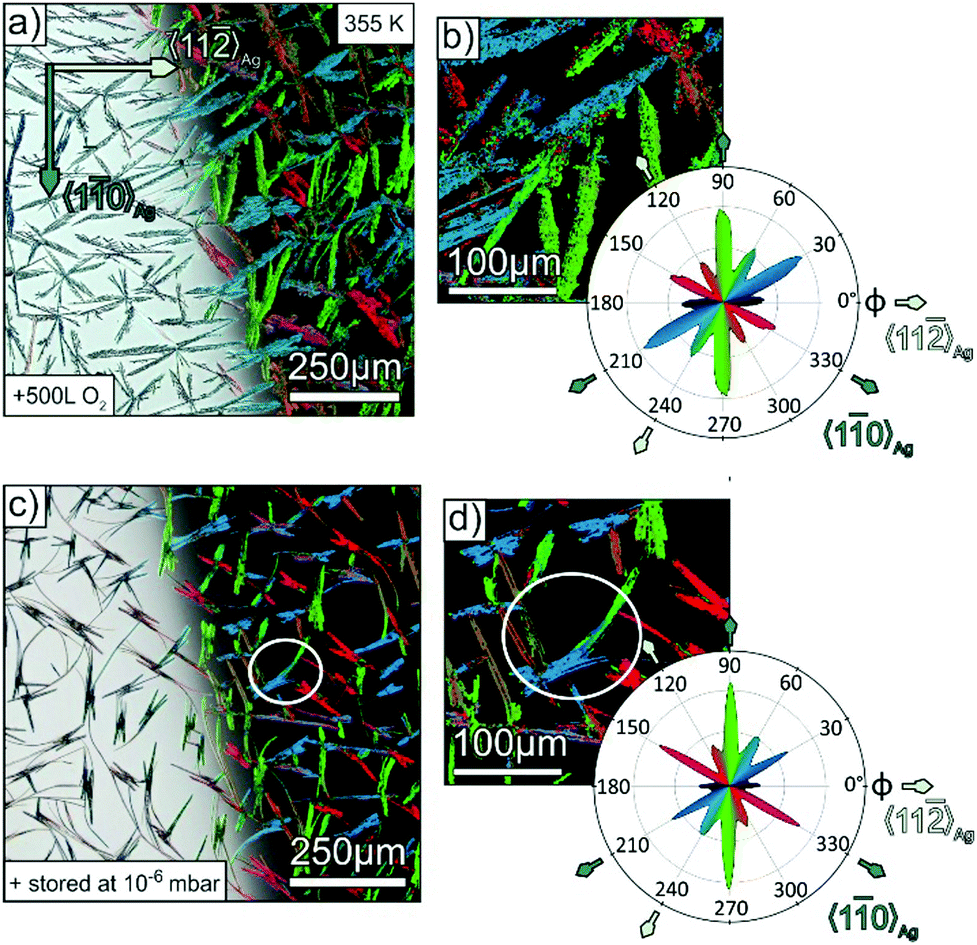 | ||
| Fig. 4 (a and c) Optical micrographs (in epi-illumination) of p-4P/Ag(111) films with a nominal thickness of 30 nm and a substrate temperature of 355 K during deposition. Before deposition, the Ag/mica substrates were prepared by several sputtering/annealing cycles plus: exposing to 500 L oxygen (a) and storing in the load chamber, which was filled with residual gas up to a pressure of 10−6 mbar of for three hours (about 8120 L) in (e). On the right half side on the images, corresponding false color images are depicted, where colours represent the polarisation angle with maximal pixel intensity. (b and d) Corresponding magnified images and polar plots of the angular distribution of the pixels of different polarisation angels. | ||
Similarly, information on the azimuthal molecule and fibre orientation can also be obtained by polarisation resolved fluorescence microscopy, which is demonstrated in Fig. 5. Here, the distinct blue luminescence of p-4P fibres (cf. Fig. S4 in the ESI‡) is utilised, which is described in detail elsewhere.56 As shown in Fig. 5a, the unpolarised luminescence micrograph clearly shows the individual fibres. Compared to the before discussed absorption/reflection measurements, the PL microscopy offers the advantage of higher sensitivity due to the very low background signal, thereby allowing also an analysis of very thin films. Further information on the molecular direction is again obtained by analysing the polarisation of the PL light, which is governed by the same TDM as the before discussed absorption (cf.Fig. 2g). In fact, the polarisation resolved measurements again reveal distinct azimuthal orientations as exemplarily shown in Fig. 5b for different fixed polariser orientations. Note that different to the contrast mechanism of the reflection measurements, PL measurements directly probe the orientation of the TDM for the emission lines (which is oriented perpendicular to the fibre axis). The statistical analysis nicely confirms the uniform orientation of the p-4P long axes exclusively along the 〈11〉Ag azimuth (i.e. fibre directions along the 〈10〉Ag direction) on Ag(111) samples that had been briefly exposed to air.
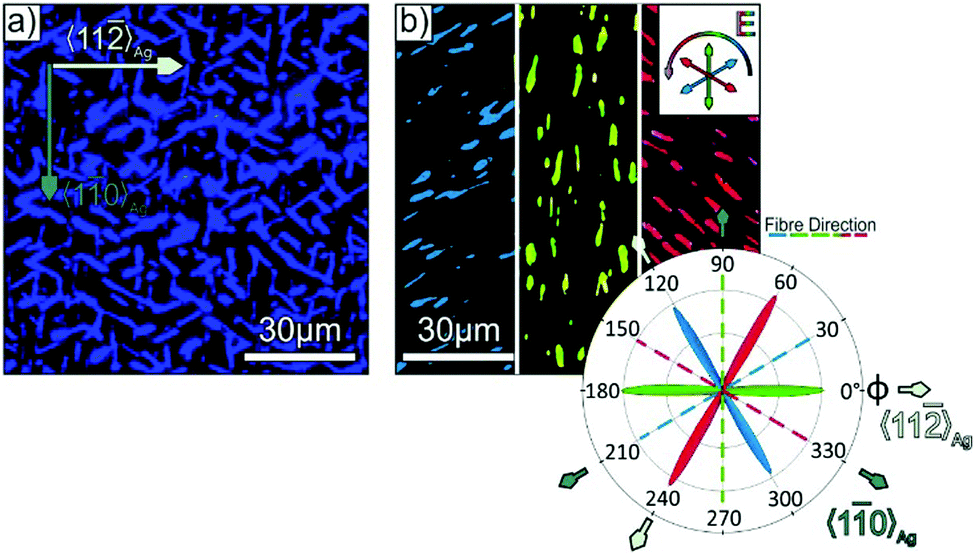 | ||
| Fig. 5 (a) Unpolarised photoluminescence micrograph of a p-4P/Ag(111) film with a nominal thickness of 30 nm, which was exposed briefly to ambient conditions before deposition (same sample as in Fig. 2e and f). (b) False colour image of the micrograph in (a), where the colour represents the orientation of the E-vector of the PL light (note that the image is split into three parts, which are acquired with specific polariser orientations shown in the inset of (b)), together with a polar plot of the relative occurrence of each E-vector with respect to the fibre and substrate alignment. | ||
Conclusions and outlook
Creating controlled nanostructures using bottom-up strategies requires a deep understanding of the initial nucleation and film growth that govern the assembly processes. This situation is even more challenging for molecular nanostructures as they are only weakly (essentially van der Waals) interacting and reveal other peculiarities such as the formation of fibres instead of smooth and contiguous adlayers. Here, we show for the case of p-4P that microscopically influencing the initial nucleation enables control of the direction and size of the formed mesoscopic crystalline fibres. We demonstrate that surface defects such as steps, which occur even on the best metal single crystal surfaces due to their entropic contributions, induce a nucleation of unwanted parasitic fibres whose alignment is different from fibres that are epitaxially grown on ideal Ag(111) surfaces. Our strategy is based on a specific suppression of such step-edge mediated nucleation and subsequent growth of such parasitic fibres through exposure of well-prepared Ag(111) surfaces to oxygen or even brief contact to ambient air. Due to their enhanced chemical reactivity and adsorption energy, the step edges are saturated first, which suppresses the ability to preferentially nucleate organic molecules. As a consequence, the fibre formation is only controlled by the intrinsic epitaxial growth of the oligophenylene molecules on the ideal (i.e. defect-free) Ag(111) surface.The present results further validate a strategy that had been introduced before to control the oriented growth of DNTT fibres on silver substrates.21 While the present approach of using oxygen to decorate and thereby passivate surface steps is not directly applicable to the more inert gold surfaces, it requires more appropriate molecules. However, this might be achieved by using molecules such as fullerenes, 1,4-diisocyanobenzene or 4,4′-biphenyl dicarboxylic acid, which in the case of low doses initially nucleate at steps55,57,58 and thereby saturate them. In a more abstract context, this study shows the importance of the understanding of initial nucleation phenomena for controlling the structure and morphology of subsequently formed functional layers, a concept that could also be transferred to inorganic semiconductor hetero-structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support by the German Research Foundation (DFG) in the framework of the Collaborative Research Center ‘Structure and Dynamics of Internal Interfaces’ (223848855-SFB 1083, TP A2).Notes and references
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4(7), 3591 CrossRef CAS.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6(8), 557 CrossRef.
- A. Ulman, Chem. Rev., 1996, 96(4), 1533 CrossRef CAS.
- T.-W. Tsai, Q.-R. Huang, S.-M. Peng and B.-Y. Jin, J. Phys. Chem. C, 2010, 114(8), 3641 CrossRef CAS.
- S.-A. Hua, I. P.-C. Liu, H. Hasanov, G.-C. Huang, R. H. Ismayilov, C.-L. Chiu, C.-Y. Yeh, G.-H. Lee and S.-M. Peng, Dalton Trans., 2010, 39(16), 3890 RSC.
- C. Teichert, C. Hofer and G. Hlawacek, Adv. Eng. Mater., 2006, 8(11), 1057 CrossRef CAS.
- L. Liu, Y. Wang, F. Sun, Y. Dai, S. Wang, Y. Bai, L. Li, T. Li, T. Zhang and S. Qin, Microsyst. Nanoeng., 2020, 6, 31 CrossRef CAS.
- V. A. Shchukin and D. Bimberg, Rev. Mod. Phys., 1999, 71(4), 1125 CrossRef CAS.
- K. Kyuno and G. Ehrlich, Surf. Sci., 1997, 383(2-3), L766–L774 CrossRef CAS.
- L. Chen, B. Liu, M. Ge, Y. Ma, A. N. Abbas and C. Zhou, ACS Nano, 2015, 9(8), 8368 CrossRef CAS.
- J. de La Figuera, M. A. Huerta-Garnica, J. E. Prieto, C. Ocal and R. Miranda, Appl. Phys. Lett., 1995, 66(8), 1006 CrossRef CAS.
- E. C. Walter, B. J. Murray, F. Favier, G. Kaltenpoth, M. Grunze and R. M. Penner, J. Phys. Chem. B, 2002, 106(44), 11407 CrossRef CAS.
- D. Kandel and E. Kaxiras, Phys. Rev. Lett., 1995, 75(14), 2742 CrossRef CAS.
- W. Wang, C. Du, D. Zhong, M. Hirtz, Y. Wang, N. Lu, L. Wu, D. Ebeling, L. Li, H. Fuchs and L. Chi, Adv. Mater., 2009, 21(46), 4721–4725 CAS.
- J. Vrijmoeth, H. A. van der Vegt, J. A. Meyer, E. Vlieg and R. J. Behm, Phys. Rev. Lett., 1994, 72(24), 3843 CrossRef CAS.
- H. Matsunami and T. Kimoto, Mater. Sci. Eng., R, 1997, 20(3), 125 CrossRef.
- J. Krug, P. Politi and T. Michely, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61(20), 14037 CrossRef CAS.
- Y. Hwang and N. Shin, Nanoscale, 2019, 11(16), 7701 RSC.
- C. Ratsch and A. Zangwill, Appl. Phys. Lett., 1993, 63(17), 2348 CrossRef CAS.
- U. Martinez, J. Ø. Hansen, E. Lira, H. H. Kristoffersen, P. Huo, R. Bechstein, E. Lægsgaard, F. Besenbacher, B. Hammer and S. Wendt, Phys. Rev. Lett., 2012, 109(15), 155501 CrossRef CAS.
- M. Dreher, D. Kang, T. Breuer and G. Witte, Nanoscale Horiz., 2019, 4(6), 1353 RSC.
- W. L. Ling, T. Giessel, K. Thürmer, R. Q. Hwang, N. C. Bartelt and K. F. McCarty, Surf. Sci., 2004, 570(3), L297–L303 CrossRef CAS.
- D. Günder, K. Watanabe, T. Taniguchi and G. Witte, ACS Appl. Mater. Interfaces, 2020, 12(34), 38757 CrossRef.
- R. Resel, Thin Solid Films, 2003, 433(1-2), 1 CrossRef CAS.
- F. Balzer, V. G. Bordo, A. C. Simonsen and H.-G. Rubahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67(11), 115408 CrossRef.
- H. Yanagi and T. Morikawa, Appl. Phys. Lett., 1999, 75(2), 187 CrossRef CAS.
- S. Müllegger, I. Salzmann, R. Resel and A. Winkler, Appl. Phys. Lett., 2003, 83(22), 4536 CrossRef.
- S. Müllegger, I. Salzmann, R. Resel, G. Hlawacek, C. Teichert and A. Winkler, J. Chem. Phys., 2004, 121(5), 2272 CrossRef.
- M. Schiek, A. Lützen, K. Al-Shamery, F. Balzer and H.-G. Rubahn, Surf. Sci., 2006, 600(18), 4030 CrossRef CAS.
- M. Schiek, A. Lützen, R. Koch, K. Al-Shamery, F. Balzer, R. Frese and H.-G. Rubahn, Appl. Phys. Lett., 2005, 86(15), 153107 CrossRef.
- C. Simbrunner, G. Hernandez-Sosa, M. Oehzelt, T. Djuric, I. Salzmann, M. Brinkmann, G. Schwabegger, I. Watzinger, H. Sitter and R. Resel, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(11), 115443 CrossRef.
- C. Simbrunner, D. Nabok, G. Hernandez-Sosa, M. Oehzelt, T. Djuric, R. Resel, L. Romaner, P. Puschnig, C. Ambrosch-Draxl, I. Salzmann, G. Schwabegger, I. Watzinger and H. Sitter, J. Am. Chem. Soc., 2011, 133(9), 3056 CrossRef CAS.
- C. Simbrunner, F. Quochi, G. Hernandez-Sosa, M. Oehzelt, R. Resel, G. Hesser, M. Arndt, M. Saba, A. Mura, G. Bongiovanni and H. Sitter, ACS Nano, 2010, 4(10), 6244 CrossRef CAS.
- C. Simbrunner, G. Schwabegger, R. Resel, T. Dingemans and H. Sitter, Cryst. Growth Des., 2014, 14(2), 442 CrossRef CAS.
- E. Bauer, Rep. Prog. Phys., 1994, 57(9), 895 CrossRef CAS.
- M. Ondrejcek, W. Swiech, G. Yang and C. P. Flynn, J. Vac. Sci. Technol., B, 2002, 20(6), 2473 CrossRef CAS.
- L. Fernandez, S. Thussing, A. Mänz, J. Sundermeyer, G. Witte and P. Jakob, Phys. Chem. Chem. Phys., 2017, 19(3), 2495 RSC.
- F. Balzer and M. Schiek, Springer Ser. Mater. Sci., 2015, 217, 151 CAS.
- B. Toudic, P. Limelette, G. Froyer, F. Le Gac, A. Moréac and P. Rabiller, Phys. Rev. Lett., 2005, 95(21), 215502 CrossRef CAS.
- P. Puschnig and C. Ambrosch-Draxl, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60(11), 7891 CrossRef CAS.
- V. A. Postnikov, N. I. Sorokina, O. A. Alekseeva, V. V. Grebenev, M. S. Lyasnikova, O. V. Borshchev, N. M. Surin, E. A. Svidchenko, S. A. Ponomarenko and A. E. Voloshin, Crystallogr. Rep., 2018, 63(1), 139 CrossRef CAS.
- T. Mikami and H. Yanagi, Appl. Phys. Lett., 1998, 73(5), 563 CrossRef CAS.
- Y. Delugeard, J. Desuche and J. L. Baudour, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32(3), 702 CrossRef.
- R. Félix, T. Breuer, P. Rotter, F. Widdascheck, B. Eckhardt, G. Witte, K. Volz and K. I. Gries, Cryst. Growth Des., 2016, 16(12), 6941 CrossRef.
- M. Kothe, F. Widdascheck and G. Witte, J. Phys. Chem. C, 2019, 123(10), 6097 CrossRef CAS.
- H. Brune, J. Wintterlin, J. Trost, G. Ertl and J. Wiechers, J. Chem. Phys., 1993, 99(3), 2128 CrossRef CAS.
- H.-Y. Su, Z. Zeng, X.-H. Bao and W.-X. Li, J. Phys. Chem. C, 2009, 113(19), 8266 CrossRef CAS.
- Introduction to Surface Chemistry and Catalysis, ed. G. A. Somorjai and Y. Li, Wiley, 2010 Search PubMed.
- H. A. Engelhardt and D. Menzel, Surf. Sci., 1976, 57(2), 591 CrossRef CAS.
- R. Resel, M. Oehzelt, T. Haber, G. Hlawacek, C. Teichert, S. Müllegger and A. Winkler, J. Cryst. Growth, 2005, 283(3–4), 397 CrossRef CAS.
- T. Breuer and G. Witte, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(15), 155428 CrossRef.
- M. Dreher, D. Günder, S. Zörb and G. Witte, Chem. Mater., 2020, 32(20), 9034 CrossRef CAS.
- M. Voigt, S. Dorsfeld, A. Volz and M. Sokolowski, Phys. Rev. Lett., 2003, 91(2), 26103 CrossRef CAS.
- G. Witte, K. Hänel, S. Söhnchen and C. Wöll, Appl. Phys. A: Mater. Sci. Process., 2006, 82(3), 447 CrossRef CAS.
- L. Kormoš, P. Procházka, T. Šikola and J. Čechal, J. Phys. Chem. C, 2018, 122(5), 2815 CrossRef.
- F. Balzer and H.-G. Rubahn, Surf. Sci., 2004, 548(1-3), 170 CrossRef CAS.
- L. Tang, X. Zhang and Q. Guo, Surf. Sci., 2010, 604(15–16), 1310 CrossRef CAS.
- J. A. Boscoboinik, F. C. Calaza, Z. Habeeb, D. W. Bennett, D. J. Stacchiola, M. A. Purino and W. T. Tysoe, Phys. Chem. Chem. Phys., 2010, 12(37), 11624 RSC.
Footnotes |
| † Dedicated to J. P. Toennies on his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: Additional XRD, LEED, STM data, and PL-spectra as well as optical micrographs. See DOI: 10.1039/d0cp06725c |
| This journal is © the Owner Societies 2021 |